

Abstract
Synaptic GABAA receptors are primary mediators of rapid inhibition in the brain and play a key role in the pathophysiology of epilepsy and other neurologic disorders. The δ-subunit GABAA receptors are expressed extrasynaptically in the dentate gyrus and contribute to tonic inhibition, promoting network shunting as well as reducing seizure susceptibility. However, the neurosteroid structure-function relationship at δGABAA receptors within the native hippocampus neurons remains unclear. Here we report a structure-activity relationship for neurosteroid modulation of extrasynaptic GABAA receptor–mediated tonic inhibition in the murine dentate gyrus granule cells. We recorded neurosteroid allosteric potentiation of GABA as well as direct activation of tonic currents using a wide array of natural and synthetic neurosteroids. Our results shows that, for all neurosteroids, the C3α-OH group remains obligatory for extrasynaptic receptor functional activity, as C3β-OH epimers were inactive in activating tonic currents. Allopregnanolone and related pregnane analogs exhibited the highest potency and maximal efficacy in promoting tonic currents. Alterations at the C17 or C20 region of the neurosteroid molecule drastically altered the transduction kinetics of tonic current activation. The androstane analogs had the weakest modulatory response among the analogs tested. Neurosteroid potentiation of tonic currents was completely (approximately 95%) diminished in granule cells from δ-knockout mice, suggesting that δ-subunit receptors are essential for neurosteroid activity. The neurosteroid sensitivity of δGABAA receptors was confirmed at the systems level using a 6-Hz seizure test. A consensus neurosteroid pharmacophore model at extrasynaptic δGABAA receptors is proposed based on a structure-activity relationship for activation of tonic current and seizure protection.
Introduction
GABAA receptors are responsible for the majority of inhibitory currents in the brain. Structurally, they are pentameric channels made from various subunits (α1–α6, β1–β4, γ1–γ3, δ, ε, θ, ρ1–ρ3). In adult neurons, GABAA channels conduct chloride ions into the cell and hyperpolarize the neuronal membrane. GABAA receptors are categorized into synaptic (primarily γ-containing) and extrasynaptic (primarily δ-containing) receptors. Synaptic receptors produce phasic currents in response to vesicular release of GABA. Extrasynaptic receptors, which are expressed in many brain regions including the hippocampus, thalamus, and cerebellum, generate nondesensitizing tonic currents that are continuously gated by extracellular GABA. Tonic current sets the baseline and shunting inhibition via continuous chloride conductance in specific neurons. The dentate gyrus is a critical region for controlling inputs from afferent neuronal pathways (Coulter and Carlson, 2007). Dentate gyrus granule cells (DGGCs) highly express δ-subunit GABAA receptors that modulate network inhibition within the hippocampus (Glykys et al., 2008; Carver and Reddy, 2013). Neurosteroids have been identified as endogenous modulators of GABAA receptors and brain function (Reddy and Kulkarni, 2000; Reddy and Rogawski, 2000, 2009; Reddy et al., 2004; Reddy, 2006, 2010).
Neurosteroids are positive allosteric modulators of GABAA receptors (Akk et al., 2007; Hosie et al., 2007; Mitchell et al., 2008; Chisari et al., 2009). There are two discrete binding sites for neurosteroids, including an allosteric site within the α-subunit transmembrane domain and a site of direct activation at the α-β subunit interface (Hosie et al., 2006, 2007, 2009). Previous structure-activity relationship (SAR) studies at synaptic γGABAA receptors show that the C3α-OH steroid structure is essential for the binding affinity and the receptor-enhancing function by neurosteroids (Harrison et al., 1987; Mitchell et al., 2008). Apart from 5α-H stereoselectivity, the C17 or C20 ketone group is important for high-potency positive allosteric modulation (Kokate et al., 1994; Upasani et al., 1997; Covey et al., 2000; Reddy and Jian, 2010; Qian et al., 2014). In addition, the lipophilic properties of neurosteroids contribute to the potency and efficacy of receptor binding (Chisari et al., 2009). However, there is little information on SARs for neurosteroids at extrasynaptic δGABAA receptors.
Neurosteroids bind to all GABAA receptors, but δ-containing receptors exhibit preferential sensitivity at extracellular GABA concentrations (Brown et al., 2002; Meera et al., 2011). The allosteric binding of neurosteroid to low-efficacy δGABAA receptors induces a pronounced conformational change, greater channel opening, and nondesensitizing tonic inhibition (Bianchi and Macdonald, 2003). Increased δGABAA receptor expression enhances neurosteroid sensitivity through greater potentiation of tonic current (Sanna et al., 2009; Wu et al., 2013; Carver et al., 2014). Conversely, deficient δGABAA receptor expression reduces the sensitivity to neurosteroids (Mihalek et al., 1999; Spigelman et al., 2003; Stell et al., 2003; Pandit et al., 2013). Mutations to the Gabrd gene result in dysfunctional GABAA receptor currents, and these mutations have been linked with clinical cases of generalized epilepsies (Dibbens et al., 2004; Feng et al., 2006). Diminished δGABAA receptor–mediated tonic inhibition exerts a significant effect on network excitability and seizure susceptibility. Therefore, characterization of neurosteroid SARs for extrasynaptic δGABAA receptors is essential for novel therapeutic agents for epilepsy and related brain disorders.
In this study, we investigated the SAR of neurosteroids at extrasynaptic δGABAA receptors in the dentate gyrus, a key brain region for the pathophysiology of epilepsy and brain disorders. We examined a library of structurally distinct natural and synthetic neurosteroids to assess fractional tonic currents in native, murine DGGCs with hippocampus slice electrophysiology (Table 1). We also correlated the SAR of neurosteroid protection against 6-Hz stimulation seizures. Our results demonstrate the key structural features and a consensus pharmacophore pocket for neurosteroid efficacy at extrasynaptic δGABAA receptors in the brain.
TABLE 1.
Chemical structures of neurosteroids tested for extrasynaptic activity
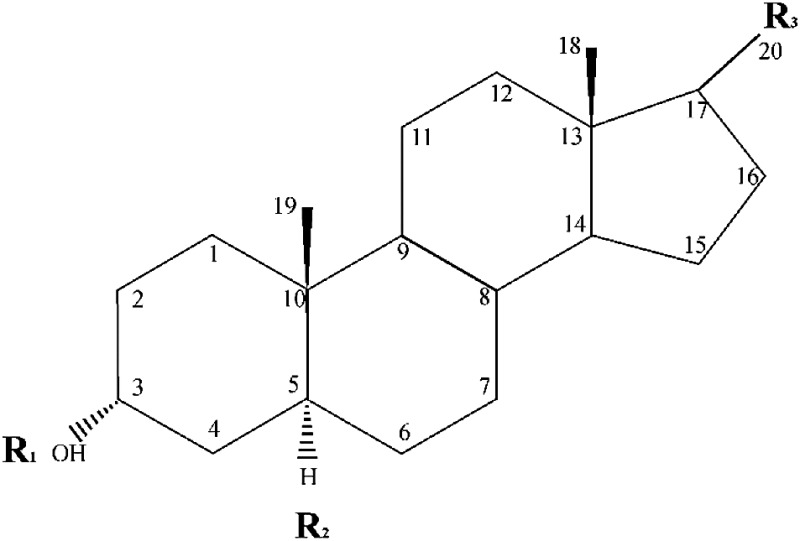
| Steroid | C3 Position (R1) | C5 Position (R2) | C17 Position (R3) | Key Structural Significance |
|---|---|---|---|---|
| 3α 5α-AP | α-OH | α-H | -COCH3 | 3α-hydroxyl, 5α-reduction,17-acetyl |
| GX | α-OH β-CH3 | α-H | -COCH3 | 3β-methyl |
| 3α 5β-AP | α-OH | β-H | -COCH3 | 5β-reduction |
| 3β5α -AP | β-OH | α-H | -COCH3 | 3β-hydroxyl |
| THDOC | α-OH | α-H | -COCH2OH | 21-hydroxyl |
| ALFX | α-OH | α-H | -COCH3 | 11-carbonyl |
| AD | α-OH | α-H | β-OH | 17-hydroxyl |
| AN | α-OH | α-H | =O | 17-carbonyl |
| ETIO | α-OH | β-H | =O | 17-carbonyl, 5β-reduction |
| ORG-20599 | α-H | α-H | -COCH2Cl | 2β-(4-morpholinyl), 17-acyl chloride |
Materials and Methods
Animals.
Adult female C57BL/6 mice aged 2 to 3 months were used in this study. Wild-type (WT) and GABAA receptor δ-subunit knockout mice (Gabrd−/−, δKO) were used (Mihalek et al., 1999; Carver et al., 2014) for experiments. All strains were maintained on a hybrid C57BL/6-129SV background. All mice were housed four to a cage in an environmentally controlled animal facility with a 12-hour light/dark cycle with access to food and water ad libitum. The animals were cared for in strict compliance with the guidelines outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animal procedures were performed in a protocol approved by the Texas A&M University Health Science Center Institutional Animal Care and Use Committee.
Hippocampal Slice Preparation.
Transverse slices (300-μm thickness) of hippocampus were prepared using standard techniques from adult female mice in the diestrus I stage (Carver et al., 2014). Only animals fitting these criteria were investigated to control for the cycle-related changes to level of δ-specific expression in the hippocampus (Wu et al., 2013). Mice were anesthetized with isoflurane, and brains were excised rapidly and placed in cold (3.5°C) artificial cerebrospinal fluid (aCSF) buffer containing 0.3 mM kynurenic acid (Tocris Bioscience, Minneapolis, MN). The aCSF buffer was composed of 126 mM NaCl, 3 mM KCl, 0.5 mM CaCl2, 5 mM MgCl2, 26 mM NaHCO3, 1.25 mM NaH2PO4, and 11 mM glucose (pH adjusted to 7.35–7.40 with 95% O2/5% CO2; osmolarity, 305–315 mOsm/kg). Hippocampal slices were cut with a Vibratome in 3.5°C aCSF (model 1500 with a 900 Refrigeration System; Leica Microsystems, Inc., Bannockburn, IL). For each electrophysiology experiment, two to four animals were used for each group and drug concentration tested.
Dissociation of Neurons.
Hippocampus DGGC dissociation was prepared by the standard enzymatic technique described previously (Reddy and Jian, 2010; Wu et al., 2013). Briefly, dentate gyrus region slices were incubated in aCSF for 1 hour, transferred to protease XXIII solution (3 mg/ml; Sigma-Aldrich, St. Louis, MO), and then acutely dissociated by fire-polished pipette trituration. The suspension of freshly dispersed cells was plated onto the recording chamber for electrophysiology experiments.
Recording of GABA-Gated Currents.
Electrophysiological recordings in dissociated cells were performed in the whole-cell patch-clamp configuration (Reddy and Jian, 2010; Wu et al., 2013) with an inverted microscope with phase contrast and differential interference contrast optics (model IX71; Olympus, Tokyo, Japan). Physiologic bath solution for dissociated neurons contained 140 mM NaCl, 3 mM KCl, 10 mM HEPES, 2 mM MgCl2, 2 mM CaCl2, and 16 mM glucose (pH adjusted to 7.4 with NaOH; osmolarity, 315–325 mOsm/kg). Recording pipette tip resistances were 3–5 MΩ for single-cell recording and were filled with a cesium pipette solution containing 124 mM CsCl, 20 mM tetraethylammonium, 2 mM MgCl2, 10 mM EGTA, 10 mM HEPES, 0.1 mM GTP, 4 mM ATP (pH adjusted to 7.2 with CsOH; osmolarity, 295–305 mOsm/kg). Currents were recorded using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). The membrane capacitance, series resistance, and input resistance of the recordings were monitored by applying a 5-mV (100-millisecond) depolarizing voltage step from a holding potential of −70 mV for dissociated cells. Signals were low-pass filtered at 2 kHz and digitized at 10 kHz with a Digidata 1440A system (Molecular Devices, Sunnyvale, CA). The current values were normalized to cell capacitance (an index of cell size) and expressed as current density (pA/pF). For whole-cell current from isolated single cells, fractional potentiation produced by allosteric modulator was calculated as IA/IGABA, where IGABA was the response of peak amplitude at the application of GABA (3 μM) and IA is the response of peak amplitude at the coapplication of GABA and the allosteric drug (0.01–1 μM). For fast application of test drugs, the perfusion pipette was positioned < 200 µm away from the cell in the chamber. GABA and allopregnanolone (AP) were applied using a multichannel perfusion system (Automate Scientific, Berkeley, CA). A 2-minute wash with bath solution was instituted after each drug trial to prevent receptor desensitization.
Patch-Clamp Slice Electrophysiology.
Electrophysiological recordings were performed in the whole-cell patch-clamp configuration (Reddy and Jian, 2010; Wu et al., 2013). Hippocampal slices (300 µm) were maintained in continuously oxygenated aCSF at 32°C in a holding chamber for 60 minutes, and then recordings were made at room temperature. Hippocampal DGGCs were visually identified with an Olympus BX51 microscope equipped with a ×40 water-immersion objective, infrared-differential interference contrast optics, and video camera. Physiologic bath solution for the slices comprised 124 mM NaCl, 3 mM KCl, 1.5 mM MgSO4, 2.4 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose (pH adjusted to 7.4 with NaOH; osmolarity, 295–305 mOsm/kg). The recording pipette tip resistances were 4–6 MΩ for slice recordings and were filled with a cesium pipette solution containing 124 mM CsCl, 20 mM tetraethylammonium-chloride, 2 mM MgCl2, 10 mM EGTA, 10 mM HEPES, 0.1 mM GTP, 4 mM ATP, and 5 mM lidocaine QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide] (pH adjusted to 7.2 with CsOH; osmolarity, 295–305mOsm/kg). The current values were normalized to cell capacitance (pA/pF, as an index of current density).
Tonic Current Recording and Analysis.
The GABAA receptor–mediated tonic current recording and analysis were made as described previously (Carver et al., 2014). Tonic current of GABAA receptors was recorded in the presence of tetrodotoxin (TTX; 0.5 μM, Na+ channel blocker and inhibition of action potential-evoked neurotransmitter release), d,l-2-amino-5-phosphonovaleric acid (40 μM, N-methyl-d-aspartate channel blocker; Sigma-Aldrich), and 6,7-dinitroquinoxaline-2,3-dione (10 μM, non–N-methyl-d-aspartate glutamate receptor blocker). After establishing a baseline for the holding current in the presence of GABA, neurosteroid was applied to the bath perfusion. Excessive tissue accumulation of steroid was prevented by sustaining a high rate of perfusion (2 ml/min). The competitive antagonist, SR-95531 (gabazine or 4-[6-imino-3-(4-methoxyphenyl)pyridazin-1-yl] butanoic acid hydrobromide; 50 µM), was added to perfusion at the conclusion of slice recordings to confirm block of GABAergic currents. Sigmoidal curves were fitted to the concentration-response data for neurosteroid to determine relative efficacy and potency of receptor modulation. Off-line current analysis was performed with pClamp10.2 software (Molecular Devices) and in-house software. Averaged amplitude of tonic current shift in conductance was measured. The GABAA receptor tonic current was expressed as the outward shift in holding current after the application of gabazine (50 µM). Itonic was measured and averaged in 100 milliseconds for each epoch with 1-second intervals between epochs for 30 epochs. The measurements were taken 30 seconds before and 2 to 3 minutes after application of a drug. Itonic change for a single cell was normalized to membrane capacitance (pA/pF).
6-Hz Seizure Test.
The 6-Hz model of partial seizures was used according to a previously described protocol (Barton et al., 2001; Kaminski et al., 2004; Reddy et al., 2015). WT mice were stimulated with a constant-current device delivered via corneal stimulation for 0.2-millisecond–duration monopolar rectangular pulses at 6 Hz for 3 seconds (World Precision Instruments, Sarasota, FL). To determine the CC50 value (current causing seizures in 50% of animals), different current intensities in the range of 6–44 mA were administered to control groups of animals. Current intensity at 32 mA was determined to elicit seizures in 100% population of male and female C57BL/6-129SV control mice. A fixed current of 32 mA was delivered as the standardized intensity for comparing all mice across structure-activity responses of drugs. Ocular anesthetic (0.5% tetracaine) was administered as an antipruritic to the corneas 10 minutes before stimulation. Immediately prior to stimulation, the corneal electrodes were wetted with 0.9% saline solution. Mice were manually restrained during the 6-Hz stimulation, and they were then immediately released into an observation chamber. Seizures were frequently preceded by a brief period of locomotor convulsion and hunched posture. Animals then exhibited various combinations of stunned posture, rearing (bipedal standing), forelimb automatic movements, clonus, twitching of vibrissae, and Straub tail. The seizure activity duration ranged from 20 to 60 seconds in untreated, control animals. After completion of the seizure, mice resumed normal exploratory behavior. Protection from seizure activity was designated as the experimental end point of anticonvulsant treatment. Animals were considered to be protected if they resumed exploratory behavior within 10 seconds of the stimulation. Eight to 10 animals were used for each sex and dose of anticonvulsant drug treatment.
Plasma AP Level Determination.
For studies to assay plasma AP, mice were injected with vehicle or 1–10 mg/kg AP (s.c.). Fifteen minutes after injection, mice were anesthetized with isoflurane and approximately 0.5 ml of carotid blood was collected in heparinized tubes. The plasma was separated by centrifugation at 12,000 rpm for 10 minutes at 4°C. Plasma concentrations of AP were analyzed by liquid chromatography/mass spectrometry using 3β-methyl-allopregnanolone as internal standard (Reddy et al., 2004, 2012).
Drugs and Reagents.
All chemicals used in the electrophysiology studies were purchased from Sigma-Aldrich unless otherwise specified. Stock concentrations of GABA, SR-95531, d,l-2-amino-5-phosphonovaleric acid, 6,7-dinitroquinoxaline-2,3-dione, TTX, THIP (4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol hydrochloride, also known as gaboxadol), and DS2 (4-chloro-N-[2-(2-thienyl)imidazo[1,2-a]pyridin-3-yl]benzamide) were dissolved in water. AP (3α-hydroxy-5α-pregnan-20-one), THDOC (3α,21-dihydroxy-5α-pregnan-20-one), ganaxolone (GX; 3α-hydroxy-3β-methyl-5α-pregnan-20-one), 5β-AP (3α-hydroxy-5β-pregnan-20-one), 3β-AP (3β-hydroxy-5α-pregnan-20-one), androstanediol (AD; 3α-hydroxy-5α-androstane-17β-diol), androsterone (AN; 3α-hydroxy-5α-androstane-17-one), etiocholanolone (ETIO; 3α-hydroxy-5β-androstane-17-one), alfaxolone (ALFX; 3α-hydroxy-5α-pregnan-11,20-dione), and ORG-20599 [2β,3α,5α-21-chloro-3-hydroxy-2-(4-morpholinyl)pregnan-20-one] were prepared in dimethylsulfoxide for electrophysiology experiments. The concentration of dimethylsulfoxide in final solution was less than 1%. For behavioral experiments, neurosteroid was dissolved in 0.9% NaCl and suspended in 20% β-cyclodextrin. Neurosteroid dosage selection was based on previous studies in seizure models (Reddy and Rogawski, 2001, 2002; Reddy, 2004; Reddy et al., 2004, 2010). Drugs were injected subcutaneously in adult animals in a volume equaling 1% of total body weight. Neurosteroids were acquired from Steraloids (Newport, RI). Kynurenic acid, DS2, GX, and THIP were acquired from Tocris Bioscience. TTX was acquired from Calbiochem. Midazolam [8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine] was diluted from 5 mg/ml solution (Akorn, Inc., Lake Forest, IL).
Statistical Analysis.
Group data were expressed as means ± S.E.M. Statistical comparisons of electrophysiology group data were performed using an independent two-tailed t test followed by Tukey’s honestly significant difference test post hoc. In all statistical tests, the criterion for statistical significance was P < 0.05, unless otherwise specified. The concentration of allosteric modulator producing half of the maximal increase in the amplitude of the GABAergic response (EC50) was determined by fitting the concentration-response relationships to the following nonlinear sigmoid Hill function: I/Imax = [1 + (EC50 / [A])n]−1, where A is the allosteric modulator concentration, Imax is the current evoked in the presence of a maximal potentiating concentration of allosteric modulator, I is the current produced in the presence of a concentration A, EC50 is the concentration of A required to produce half of its own maximal potentiating effect, and n is the Hill coefficient. Concentration-response curve data were subjected to nonlinear, logistic fitting. For 6-Hz drug treatments, ED50 values (dose protecting 50% of mice from seizures) and 95% confidence intervals were determined by nonlinear curve fitting of the percentage protection data at different doses between 0% and 100% protection.
Results
Characteristics of GABA-Gated Whole-Cell Currents in Acutely Dissociated WT and δKO DGGCs.
To determine the contribution of the δ subunit on GABA-gated current in native neurons, we characterized whole-cell GABA concentration responses from acutely dissociated DGGCs in WT and δKO mice. Native DGGCs of δKO mice exhibited marginally greater GABA potency than WT mice (Fig. 1A). WT DGGCs had a mean GABA EC50 of 27.2 ± 6.5 µM and a Hill coefficient of 1.24 ± 0.24 (n = 11), whereas δKO DGGCs had an EC50 of 15.3 ± 1.8 µM and a Hill coefficient of 1.21 ± 0.12 (n = 7) (EC50: P = 0.1738; Hill coefficient: P = 0.9261). The mean GABA-evoked current values (as percentage of Imax GABA) were significantly greater in δKO DGGCs at 3 μM (P = 0.0257), 10 μM (P = 0.0014), and 50 μM (P = 0.0064) than in WT cells. We previously determined that in the condition of robustly increased δ-subunit expression, potency was unaffected in dissociated DGGCs, and IGABA was greater in CA1 cells with lower expression of the δ subunit (Wu et al., 2013). All cells displayed GABAA receptor current saturation at 1 mM GABA. The mean peak currents achieved by GABA at DGGCs were −844 ± 62 pA/pF in WT and −878 ± 158 pA/pF in δKO.
Fig. 1.

Reduced GABAergic inhibition from dissociated DGGCs in δKO mice by patch-clamp recordings. (A) GABA or THIP concentration-response curve of dissociated WT and δKO DGGCs. Each data point was fit according to the maximum GABA-gated current at the saturating concentration (1000 μM). EC50 and Hill coefficient values are noted in the text. At 10 μM GABA, δKO responses were significantly greater than WT. THIP displayed partial agonist activity at DGGCs. (B) Percent desensitization of GABAergic macrocurrent kinetics due to rapid application of 10 μM GABA (−5 log M), 100 μM GABA (−4 log M), or 1000 μM GABA (−3 log M). δKO cells had significantly greater percent desensitization than WT (n = 14–17 cells per group). (C) Representative traces of dissociated DGGC response to potentiation of 3 μM GABA (EC10) by AP in WT and δKO DGGC. (D) Concentration-response quantification of fold potentiation by AP. δKO cells had a significantly reduced response to AP compared with WT. n = 8–10 cells per group. *P < 0.05; **P < 0.01 by the independent t test. P values are cited in the text. All cells dissociated from diestrus, WT female mice dentate gyrus. Recordings in whole-cell mode, voltage clamped at −70 mV.
THIP concentration responses were also measured in dissociated WT and δKO DGGCs, and 1 mM GABA was perfused at the end of recording in comparison of THIP responses to maximal IGABA. THIP displayed partial agonist activity on the whole-cell GABAA receptor current of DGGCs. Maximum efficacy of THIP-gated response in WT (ITHIP/IGABA: 0.78 ± 0.08) was marginally higher than δKO DGGCs (ITHIP/IGABA: 0.60 ± 0.03), but these values were not significantly different (n = 6 to 7 cells per group, P = 0.0823). In addition, the THIP EC50 response was 400.8 ± 21.5 μM for WT and 204.9 ± 11.9 μM for δKO DGGCs. Change to THIP potency therefore mirrored the increased GABA agonist potency in cells lacking the δ subunit. Previous THIP concentration-response curves in Xenopus oocytes indicate that δ-containing receptors only display THIP selectivity in the absence of γ, and this selectivity is also present in receptors of α4β3 composition (Ebert et al., 2001; Stórustovu and Ebert, 2006). Multiple reports in the literature indicate that γGABAA receptors possess THIP potencies in the 100- to 1000-μM range (Stórustovu and Ebert, 2006; Meera et al., 2011). In native DGGCs, the observed THIP concentration response we report is consistent with a pharmacological profile of partial agonism of γ2-containing receptors, in which potency (EC50) is much larger than with δGABAA receptors alone (Stórustovu and Ebert, 2006; Meera et al., 2011). Furthermore, Meera et al. (2011) report a greater THIP-dependent maximal current achieved by the γGABAA receptors compared with δGABAA receptors. In support of this, our findings indicate that the lack of δGABAA receptors in DGGCs decreases the maximum efficacy of THIP. In dissociated DGGC recordings, the relative lack of difference in THIP sensitivity between WT and δKO neurons may signify that the THIP whole-cell current response is largely driven by γGABAA receptors rather than δGABAA receptors.
We also measured the percent desensitization of the GABAA chloride channel macrocurrents after barrel-pipette application of GABA. In the DGGCs, 0.1–10 μM GABA displayed little or no desensitization. However, higher-concentration 100–1000 μM GABA displayed peak current responses that rapidly desensitized and were curve fitted by the Chebyshev method with a single-exponent decay function. We observed that δKO DGGC macrocurrents displayed significantly greater channel desensitization than WT DGGCs in the range of 100–1000 μM GABA (Fig. 1B). In addition, the mean 10%–90% decay time for the 1 mM GABA-evoked macrocurrent was significantly greater for WT DGGCs (7.53 ± 0.40 seconds) than δKO DGGCs (4.60 ± 0.50 seconds; P = 0.0002; n = 7–10 cells per group).
Allosteric Potentiation by 3α5α-AP Is Diminished in Dissociated δKO DGGCs.
Pharmacological responses to the 3α5α-AP potentiation of GABAA receptors were obtained in whole-cell current recordings from dissociated DGGCs. A GABA EC10 (3 μM) bath concentration was perfused to determine control current responses. Subsequently, increasing concentrations of 3α5α-AP were coapplied with GABA to determine allosteric potentiation (Fig. 1C). GABAergic current potentiation by 3α5α-AP was represented by the mean fold enhancement of the GABA EC10 response. 3α5α-AP (0.1–1 μM) did not significantly potentiate currents in the absence of GABA, signifying a lack of direct activation of receptors at submicromolar concentrations in DGGCs. 3α5α-AP enhanced GABAergic whole-cell current in a concentration-dependent manner in WT DGGCs. δKO DGGCs exhibited significantly reduced 3α5α-AP potentiation of GABA current compared with WT responses (Fig. 1D). At 0.3, 0.5, and 1 μM 3α5α-AP, WT cells displayed significantly enhanced GABAergic currents compared with δKO counterparts (P < 0.05). Furthermore, only 1 μM AP elicited potentiation significantly greater than the GABA control response in δKO cells (P = 0.008).
Extrasynaptic δ-Subunit GABAA Receptor GABA-Gated Tonic Currents.
To derive the basal tonic current attributable to extrasynaptic receptors, we examined exogenous GABA responses in patch-clamp recordings from DGGCs in the slice. After GABA application, the competitive antagonist gabazine (50 μM) was applied to block all GABAergic current. The difference between holding current level before and after gabazine-induced blockade was determined as the Itonic shift (see the Materials and Methods). In WT DGGCs, GABA increased Itonic in a concentration-dependent manner (Fig. 2). Mean Itonic was not significantly different between 0.3 μM and 1 µM GABA, which is within the physiologic range of extracellular GABA (Lerma et al., 1986). Current desensitization was observed at 10 μM, but not at lower concentrations within slice recordings. In contrast with δ-containing neurons, δKO exhibited significantly attenuated Itonic responses to GABA (0.1–3 μM). Itonic responses by δKO cells in this GABA range were not significantly different than responses without exogenous GABA. Only 10 μM GABA significantly increased current in δKO above the endogenous condition (P = 0.0092). In the presence of 10 μM GABA, Itonic was not significantly different between WT and δKO (P = 0.1260) but was marginally greater in WT DGGCs.
Fig. 2.

Concentration-related responses of GABA on extrasynaptic δGABAA receptor–mediated tonic inhibition. (A) Representative whole-cell, voltage-clamp recordings of tonic current in DGGCs in response to GABA. After recording, gabazine was applied to measure total tonic shift. (B) GABA concentration-response curves for normalized tonic current shift (pA/pF) in WT and δKO DGGCs. (C) Percent tonic current contribution of δ-subunit–containing and non–δ-containing receptors relative to the 3 μM GABA response. Percentage values were determined from the mean values of WT and δKO cells in (B). GABA concentrations less than 10 μM did not elicit significant responses in δKO DGGCs, thereby denoting majority tonic current contribution by δ-containing GABAA receptors. Recordings in whole-cell mode, voltage clamped at −65 mV. n = 6–10 cells per group and concentration. * p < 0.05.
The highest GABA concentration at which δKO Itonic was not significantly different than the response without exogenous GABA represented the δ-containing, extrasynaptic, EC100. Therefore, EC100 denotes the GABA ceiling at which synaptic, non–δ-containing receptors do not significantly contribute to the hyperpolarization of tonic current of DGGCs in the slice. Based on this criterion, 3 μM GABA was designated as the extrasynaptic EC100 (Fig. 2C). However, we previously reported that 3 μM GABA is capable of modulating miniature inhibitory postsynaptic current (mIPSC) amplitude and kinetics (Carver et al., 2014), and it likely supersedes the physiologic range of extracellular GABA (Lerma et al., 1986). Therefore, to delineate the synaptic/extrasynaptic receptor classes, we selected 1 μM GABA as a baseline response for further tonic current studies to explore modulation by neurosteroids. Furthermore, 10 μM GABA demonstrated higher-efficacy modulation of non–δ-containing receptors, which contribute to the Itonic response above the extrasynaptic GABA EC100 level in both WT and δKO DGGCs.
Obligatory Role of the δ Subunit for Neurosteroid Allosteric Modulation of Tonic Currents.
To confirm the relative contribution of δ-containing receptors to total tonic current, we performed patch-clamp experiments measuring neurosteroid-mediated Itonic in WT and δKO DGGCs in the slice. Reduced tonic current and decreased neurosteroid sensitivity owing to δKO is well documented (Mihalek et al., 1999; Stell et al., 2003; Glykys et al., 2008; Carver et al., 2014). From the tonic response to GABA alone, we observed that 10 μM GABA elicited strong hyperpolarizing responses representative of continuous, nondesensitizing tonic current, even in δKO DGGCs (Fig. 2B). However, the modulatory effects on high-efficacy receptors have not been thoroughly explored. Therefore, we first assessed 3α5α-AP responses in WT and δKO DGGCs to determine the extent of δ-subunit–specific allosteric modulation of extrasynaptic function (Fig. 3). Nondesensitizing, exogenous GABA (1 μM) was applied to continuously gate extrasynaptic receptors prior to allosteric modulation by neurosteroid. Rate of change was monitored in real time and current recordings were allowed to reach an asymptotic, steady-state level of neurosteroid perfusion before complete block by gabazine to determine total Itonic shift (pA/pF). The mean Itonic response from 0.01 μM 3α5α-AP plus 1 μM GABA application (0.86 ± 0.17 pA/pF, n = 8 cells) was greater than, but not significantly different than, 1 μM GABA in WT DGGCs (0.66 ± 0.22 pA/pF, n = 9 cells; P = 0.4912). Increasing concentrations of 3α5α-AP (0.1–3.0 μM) elicited enhanced, potentiated Itonic responses. In δKO granule cells, 3α5α-AP modulation was significantly diminished (Fig. 3). Only 3μM AP plus GABA significantly potentiated current within δKO DGGCs, but the Itonic response of WT neurons remained significantly greater than δKO (P = 0.0008, n = 5 to 6 cells per group). The criterion for determining the δ-containing, extrasynaptic EC100 for allosteric modulation by 3α5α-AP was the highest concentration when Itonic in δKO DGGCs was not significantly different than condition of baseline 1 μM GABA. We investigated the relative percent contribution of δ-containing and non–δ-containing receptors to 3α5α-AP–mediated Itonic using 1 μM AP as the EC100 for allosteric activation (Fig. 3C). The δ-containing receptors contributed the majority of the tonic current to modulation by 3α5α-AP. These results suggest that neurosteroid ≤ 1 μM is selective for δ-containing, extrasynaptic receptors in modulation of tonic current. Since a plateau response was not reached in the range of neurosteroid tested, the data could not be fit to a Hill curve and potency could not be determined in this way. However, the aforementioned EC100 represents maximum efficacy for this allosteric mechanism of action.
Fig. 3.

Contribution of δ-containing extrasynaptic GABAA receptors to total tonic inhibition elicited by AP allosteric potentiation. (A) Representative tonic current recordings from patch-clamped DGGCs in the slice. AP was applied to the bath in addition to 1 μM GABA to measure allosteric enhancement of tonic current. Then 1 μM GABA, AP, and gabazine were sequentially applied in perfusion to measure tonic current shift. (B) AP concentration-response curves from WT (squares) and δKO (circles) DGGCs. Tonic current (pA/pF) is normalized to cell capacitance as a measure of current density. δKO DGGCs displayed highly attenuated tonic currents compared with WT. (C) Percent contribution of allosteric AP tonic current from δ-containing and non–δ-containing receptors, relative to 1 μM allosteric AP, which was determined to be the constrained maximum allosterically-active concentration. DGGCs from brain slice of diestrus female mice, recorded in whole-cell mode, voltage clamped at −65 mV. n = 6–8 cells per concentration and group. * p < 0.05.
Lack of Tonic Current Response and Neurosteroid Sensitivity in Dentate Gyrus Interneurons.
To investigate the tonic inhibition in distinct cell types within the dentate gyrus, we recorded Itonic in a specific population of interneurons. Hippocampal interneurons have been reported to possess GABAA receptor–mediated tonic conductance, which can affect network excitability and the inhibitory drive to principal cells (Semyanov et al., 2003). Molecular layer (ML) interneurons express the α1βδ isoform extrasynaptically, but the contribution to network tonic conductance is not very well understood (Sun et al., 2004; Glykys et al., 2007). The α1βδ receptor isoform exhibits very little GABA-mediated current, but it can be significantly potentiated by neurosteroids (Zheleznova et al., 2008, 2009). To better understand neurosteroid sensitivity of δ-containing extrasynaptic receptors, we compared tonic currents between WT DGGCs and ML interneurons in the slice (Fig. 4). The cell types were visually identified by location and morphology using differential interference contrast microscopy within the slice. We measured Itonic in the presence of 1 μM GABA plus 3 μM 3α5α-AP to maximize the channel potentiation (Fig. 4A). Mean baseline Itonic with GABA alone was not significantly different between DGGCs and ML interneurons. DGGCs exhibited significantly greater Itonic sensitivity to 3α5α-AP than ML interneurons (Fig. 4B). The mean enhancement of tonic current elicited by 3α5α-AP was a 12.1- ± 3.1-fold increase in DGGC, whereas ML interneuron enhancement was only 4.3- ± 1.2-fold (P = 0.0409). These results are consistent with previous reports demonstrating that ML interneurons exhibit significantly less GABAergic tonic conductance than DGGCs (Glykys et al., 2008). Therefore, α1βδ and α4βδ receptors display differential properties in tonic current modulation of neurosteroids.
Fig. 4.

ML interneurons exhibit less tonic current modulation elicited by AP compared with DGGCs. (A) Representative traces for 1 μM GABA plus 3 μM AP application in slice recording of extrasynaptic, tonic current. Traces from dentate gyrus granule cell (black trace) and ML interneuron (gray trace) are superimposed to show contrast, with equal scale. The dashed line describes the holding current level after gabazine block of GABAergic current. The DGGC exhibited a robust response to 3 μM AP (Δ279 pA), whereas the ML interneuron cell had reduced tonic current response (Δ41 pA). (B) Quantification of tonic current due to 1 µM GABA or 3 μM AP allosteric modulation of 1 µM GABA. Interneurons had significantly less normalized tonic current than granule cells (*P < 0.01 versus GABA control; #P < 0.01 versus DGGC plus AP). Mean tonic shift (pA) was also significantly different for AP potentiation (DGGC: 223.7 ± 26.6 pA; ML interneuron: 28.3 ± 1.8 pA; P < 0.01). Cells were recorded from adult WT female (diestrous stage) hippocampus slices, voltage clamped (−65 mV) in whole-cell mode. n = 5 to 6 cells per group.
Obligatory Role of the δ Subunit for Neurosteroid Activation of Tonic Currents at Extremely Low GABA Levels.
We examined GABAA receptor tonic currents activated by neurosteroid in the absence of exogenous GABA. The concentration of neurosteroid able to directly gate GABAA receptors (≥1 μM) is greater than the observed physiologic range of endogenous neurosteroids (0.1–0.3 μM) (Majewska et al., 1986; Carver and Reddy, 2013). However, exogenous or therapeutic administration of neurosteroids can reach sufficiently high concentrations within the brain to achieve direct activation of GABAA receptors, anticonvulsant activity, sedation, and motor impairment (Reddy, 2003). True “direct” activation by neurosteroid cannot be adequately measured in the slice owing to the low concentration of GABA persisting in the slice; extracellular concentrations of GABA have been proposed to be approximately 0.2 μm in cerebellar slice preparations (Santhakumar et al., 2006). Nevertheless, we explored a range of applied 3α5α-AP without exogenous GABA to assess the modulation of Itonic in WT DGGC in a minimal-GABA slice condition (Fig. 5). 3α5α-AP (0.3–3.0 μM) application significantly enhanced Itonic above the baseline control level. The concentration response of neurosteroid without exogenous GABA was reduced in efficacy compared with the allosteric curve (with 1 μM GABA). Itonic owing to application of 3 μM 3α5α-AP with 1 μM GABA (7.25 ± 0.40 pA/pF) was significantly greater than perfusion of 3 μM 3α5α-AP alone in WT neurons (4.54 ± 0.56 pA/pF; P = 0.0046, n = 5–7 cells per group). Next, we investigated the relative percent contribution of δ-containing and non–δ-containing receptors to 3α5α-AP–mediated tonic current. We compared current conductance values as a percentage of 3 μM 3α5α-AP in activation of extrasynaptic receptors without exogenous GABA. We demonstrate that at submicromolar concentrations, 3α5α-AP can enhance and potentiate Itonic in a low GABA environment. However, accumulation of neurosteroid within the neuronal membrane likely contributes to this binding activity, as enhancement of tonic current proceeds gradually (Akk et al., 2005).
Fig. 5.

Contribution of δ-containing extrasynaptic GABAA receptors to total tonic inhibition elicited by AP without exogenous GABA. (A) Representative tonic current recordings from patch-clamped WT and δKO DGGCs in a slice with AP (0.3 or 1.0 μM). AP alone (no GABA) followed by gabazine was applied to measure tonic current shift of extrasynaptic receptors by AP in low GABA conditions. The bottom superimposed trace displays same-scale, 3 μM AP-dependent tonic currents from a WT DGGC (black trace) and a δKO DGGC (gray trace). The dashed line indicates the holding current level upon gabazine block of GABAergic current. In addition, the increased root mean square noise of channel conductance is apparent in the WT recordings, owing to δ-containing receptor activation. (B) AP concentration-response curves from WT (squares) and δKO (circles) DGGCs. Neurosteroid was applied to bath solution alone, without any exogenous GABA to measure directly activating extrasynaptic currents. (C) Relative percent contribution of δ-subunit and non–δ-subunit receptors to activating tonic current by AP, relative to WT 3 μM AP alone.
In δKO DGGCs, a small fraction of residual, extrasynaptic current remains, due to extrasynaptic α5-containing receptors (Glykys et al., 2008). However, the neurosteroid affinities for extrasynaptic α5- and γ-containing receptors are much lower than δ-containing receptors (Brown et al., 2002; Stell et al., 2003). The residual tonic current response was measured in δKO DGGCs during application of 3α5α-AP without exogenous GABA. Itonic recordings of 3α5α-AP alone displayed striking attenuation of response in δKO compared with WT DGGCs (Fig. 5). In prolonged application of neurosteroid to allow membrane accumulation, we did not observe any gradual enhancement of extrasynaptic current by 1 μM 3α5α-AP in δKO cells. At 3 μM 3α5α-AP, δKO tonic current was significantly increased; however, non–δ-containing receptors only contributed 29.0% of the tonic current achieved by δ-containing receptors from WT. Taken together, these results suggest an obligatory role of δ-subunit transduction for the neurosteroid activation of tonic currents in slice conditions without exogenous GABA.
SAR of Neurosteroid Allosteric Potentiation of Tonic Currents.
To identify the key neurosteroid structures contributing to GABAA receptor tonic inhibition, we compiled a library of neurosteroids with distinct structural features (Table 1). Each neurosteroid was evaluated for its ability to potentiate tonic current. Concentration-dependent, normalized tonic current responses are shown in Fig. 6.
Fig. 6.

Neurosteroid SARs on extrasynaptic δGABAA receptor–mediated tonic inhibition. Concentration-response curves of neurosteroids as measured by tonic current from DGGCs (pA/pF) from patch-clamp recordings. (A) Endogenous pregnane neurosteroid responses. Lines represent nonlinear curve fit by the neurosteroids 3α5α-AP (solid), 3α5β-AP (dash), THDOC (dot), and 3β5α-AP (dash-dot). (B) Synthetic neurosteroid response curves in comparison between 3α5α-AP (solid), GX (dash), ALFX (dash-dot), and ORG-20599 (dot). (C) Androstane neurosteroid responses to AD (squares), AN (circles), and ETIO (triangles) exhibited relative decrease to potency and efficacy for extrasynaptic receptors. (D) Tonic current responses for the δ-selective compounds THIP (stars) via direct activation and DS2 (triangles) allosteric modulation compared with AP (squares). Both WT (closed symbols) and δKO (open symbols) DGGC responses are depicted. The AP concentration response is from WT DGGCs. To note, + 1 μM GABA denotes allosteric modulation by coapplication of the drug compound with GABA in the bath perfusion, whereas THIP was applied alone in bath solution. (A–C) On the x-axis, + 1 μM GABA denotes allosteric modulation by coapplication of the drug compound with GABA in the bath perfusion. All data were derived from female (diestrus I) WT DGGCs. All cells were voltage clamped in whole-cell mode at −65 mV. n = 4–10 cells per concentration and drug.
Aqueous concentrations of neurosteroid in perfusion reflect levels that are circulating within the extracellular fluid; however, the membrane concentration of steroid drives the binding interactions with GABAA receptors (Chisari et al., 2009). In examination of structure-activity within the slice, our findings reflect a gradual access of neurosteroid to receptors on the order of minutes rather than instantaneous or rapid access. Furthermore, this preparation allows for possible accumulation of neurosteroid in surrounding tissue at the time of receptor modulation, more analogous to that of a physiologic state. The concentration-dependent responses we report here convey the pharmacodynamic activity and specificity of neurosteroids.
The concentration response of 3α5α-AP displayed significantly greater functional efficacy than other endogenous neurosteroids tested (Fig. 6A). THDOC, derived from deoxycorticosterone, modulated significantly less tonic current in the 0.1- to 0.3-μM range compared with 3α5α-AP. However, at 1.0–3.0 μM, THDOC and 3α5α-AP potentiated tonic current to similar levels.
The α or β orientation of the C5 position affects the shape of the A-ring of the steroid molecule (Hosie et al., 2007) and may influence receptor affinity. Several previous studies report no change in the SAR of 5β- compared with 5α-pregnane neurosteroids (Prince and Simmonds, 1992; Xue et al., 1997). By contrast, we observed significantly less tonic current modulation by the 3α5β-AP stereoisomer, because 3α5α-AP maintained nearly 2-fold greater tonic current response. 3α5β-AP displayed a reduced tonic current response at all concentrations tested. These findings are consistent with prior reports of reduced 5β efficacy (Kokate et al., 1994; Hosie et al., 2006). The 3β5α-AP steroid, which was previously observed to have reduced efficacy on whole-cell or synaptic currents (Gee et al., 1988; Kokate et al., 1994), was not functionally active on tonic currents within the slice, even at 10 μM when applied with GABA (0.51 ± 0.14 pA/pF, mean tonic shift 15.7 ± 4.3 pA).
Next, synthetic neurosteroids were compared in their ability to enhance extrasynaptic tonic current (Fig. 6B). GX differs from 3α5α-AP by the addition of a C3-position β-methyl group. The ORG-20599 compound includes a C2-position β-4-morpholinyl group as well as modification to the 17β group by acyl chloride. Compared with synthetic neurosteroid structures, 3α5α-AP had similar response profiles to GX and ORG-20599. However, ALFX, which includes a C11-position ketone, displayed reduced tonic currents and an overall decrease in δ-mediated extrasynaptic receptor efficacy.
The androstane neurosteroids display weakly positive allosteric modulation of GABAA receptors (Reddy and Jian, 2010) but have not been explored with regard to extrasynaptic structure-activity. The 17β-ketosteroids AN and ETIO are the main metabolites of testosterone. AD is derived from dihydrotestosterone, the 5α-reduced metabolite of testosterone. We evaluated tonic currents from DGGCs in response to AD, AN, and ETIO (Fig. 6C). Compared with 3α5α-AP (1 μM), the mean tonic currents of AD, AN, and ETIO were smaller by a factor of 3.1, 4.5, and 4.9, respectively. Overall, these steroids have lesser potency on δ-containing extrasynaptic receptors than the pregnane neurosteroids, suggesting weaker binding affinity and/or transduction.
To compare the extrasynaptic, tonic current contribution of each structurally distinct neurosteroid, we analyzed the differences in fold potentiation of the GABA (1 μM) response (Fig. 6, A–C; Table 2). At the 0.1-μM concentration, 3α5α-AP (2.5-fold) displayed significantly greater potentiation; GX (1.6-fold) and ORG-20599 (1.9-fold) showed moderate potentiation of extrasynaptic GABA. At 0.3- to 3-μM concentrations, GX and ORG-20599 exhibited comparable potentiation of tonic current to the responses by 3α5α-AP. Interestingly, 0.1 μM THDOC did not elicit potentiation of tonic current, indicating that the C21-hydroxylation modification confers lower extrasynaptic receptor efficacy or potency. This was confirmed by the diminished response of THDOC at higher concentrations, compared with the pregnane neurosteroids with C17 structural features similar to 3α5α-AP (see Table 1). At 0.3 μM and above, all pregnane neurosteroids tested except for 3β5α-AP exhibited significant potentiation of the extrasynaptic GABA response. Androstane neurosteroids did not significantly enhance GABAergic tonic current at concentrations under 1 μM, and only modestly potentiated current at 3 μM.
TABLE 2.
Neurosteroid-mediated potentiation of tonic currents by 1 uM GABA
Values derived from fold potentiation of mean, normalized tonic current response to 1 uM GABA, (INS/IGABA). INS is the mean, normalized tonic current response to neurosteroid at the given concentration plus 1 uM GABA from Figure 6 Tonic current responses were recorded from voltage-clamped (−65 mV) DGGCs from WT, female (diestrous stage) mice. All mean values are representative of four to eight cells per neurosteroid and concentration.
| Compound | Neurosteroid Potentiation |
|||
|---|---|---|---|---|
| 0.1 μM | 0.3 μM | 1.0 μM | 3.0 μM | |
| AP | 2.50 | 2.94 | 4.36 | 10.98 |
| 5β-AP | 0.76 | 1.88 | 2.24 | 6.47 |
| 3β-AP | 0.76 | 0.76 | 0.76 | 0.77 |
| THDOC | 1.01 | 1.94 | 3.52 | 9.24 |
| GX | 1.61 | 2.76 | 3.47 | 8.89 |
| ALFX | 0.88 | 1.32 | 1.98 | 7.30 |
| ORG-20599 | 1.88 | 3.32 | 4.70 | 9.41 |
| AD | 0.94 | 1.09 | 1.30 | 2.95 |
| AN | 0.92 | n.d. | 1.47 | 2.15 |
| ETIO | 0.91 | n.d. | 1.20 | 1.97 |
Interestingly, 3 μM pregnane neurosteroids displayed exponentially greater potentiation of tonic currents. These results suggest that the high-concentration neurosteroid is able to directly open GABAA receptors that are low occupancy and thus produce a biphasic effect of channel activation. Therefore, 1 μM neurosteroid was sufficient to elicit the maximum channel response of the allosteric activation of extrasynaptic receptors. This was consistent in our extensive concentration-response study of both allosteric potentiation and direct activation by AP in determination of δ-containing receptor contribution to tonic current (Figs. 5 and 6). However, relative efficacy of neurosteroid to potentiate functional inhibition was determined for each structure based on normalized data for the entire range of concentrations tested (0.1–3 μM). Higher concentrations exceed the physiologic range of relevance of this study beyond that of feasible limits (Gee et al., 1988; Reddy and Rogawski, 2012), but pharmacological doses of neurosteroids remain salient to understanding their therapeutic application in the brain.
Tonic Current Responses of Nonsteroidal, δ-Selective Compounds.
We investigated DS2 as a comparative δ-selective agent and nonsteroidal, allosteric modulator (Wafford et al., 2009; Jensen et al., 2013). DS2 selectivity was previously determined in recombinant cells expressing α4βδ receptors and thalamic relay neurons; however, it has not been analyzed in tonic currents from native DGGCs in the slice. In extrasynaptic function as an allosteric modulator, DS2 was less potent in modulation and affinity than the pregnane class of neurosteroids, represented by the prototypical 3α5α-AP (Fig. 6D). In δKO DGGCs, DS2 did not significantly potentiate tonic current greater than the 1-μM GABA baseline responses (10 μM DS2 plus 1 μM GABA: 7.3 ± 4.3 pA; 1 μM GABA: 6.9 ± 2.1 pA; n = 6 to 7 cells per group, P = 0.9317). These responses confirm the δ selectivity of DS2 in native DGGCs in the slice. Previous studies established conservation of residues on the α4 subunit necessary for neurosteroid binding (Hosie et al., 2006; Jensen et al., 2013). In mutation of these residues, Jensen et al. (2013) showed that the GABA-enhancing action of DS2 did not rely on these residues, whereas the mutation of the α4 subunit resulted in 3α5α-AP insensitivity. Thus, the decreased potency of DS2 compared with neurosteroids suggests a lower affinity for extrasynaptic GABAA receptors and a different binding site from neurosteroids.
THIP is a selective low-efficacy modulator of δ-containing receptors at submicromolar concentrations (Mortensen et al., 2010). THIP (no GABA in bath perfusion) was investigated for tonic current modulation and compared in its potency and efficacy with 3α5α-AP and DS2 (Fig. 6D). The concentration response of THIP in the 0.1- to 1-μM range was less efficacious than 3α5α-AP but more potent than DS2 allosteric modulation. However, 1 μM THIP tonic current in δKO DGGCs (0.32 ± 0.08 pA/pF) was significantly lower compared with WT DGGCs (2.57 ± 0.48 pA/pF, n = 6 to 7 cells per group, P = 0.0013). An increased concentration of THIP (10 μM) in δKO (2.68 ± 0.41 pA/pF) produced tonic currents comparable to 1 μM THIP in WT cells (2.87 ± 0.44 pA/pF). These results indicate a single order of magnitude, rightward shift of THIP affinity for δ-deficient DGGCs. Decreased THIP sensitivity is consistent with previous reports of loss of sensitivity in which functional extrasynaptic receptors are absent either through α4 knockout (Chandra et al., 2006) or δ knockout (Meera et al., 2011). The THIP tonic current responses here (0.3 μM THIP: 1.5 ± 0.2 pA/pF; 1.0 μM: 2.6 ± 0.5 pA/pF) are within range of a recent report of THIP tonic current modulation in rodent DGGCs (0.5 μM THIP, 1.8 ± 0.2 pA/pF) (Hoestgaard-Jensen et al., 2014).
The Benzodiazepine Midazolam Is Not an Effective Modulator of Extrasynaptic Tonic Currents in the Dentate Gyrus.
Classic 1,4-benzodiazepines allosterically potentiate γ-containing receptors at the α-γ interface (Derry et al., 2004) and are relatively insensitive at δ-containing receptors (Möhler et al., 2002; Herd et al., 2008). Previous studies have demonstrated the lack of tonic current modulation by midazolam at δ-subunit–containing extrasynaptic receptors (Jia et al., 2005). To further elucidate the receptor subunit contribution to tonic currents in DGGCs, we explored midazolam-dependent currents in the slice (Fig. 7). In tonic current recordings, we observed no significant differences between baseline (1 μM GABA) and holding current in the presence of midazolam at any of the concentrations tested (0.03–0.30 μM). These results suggest that midazolam is insensitive at extrasynaptic, δ-containing receptors in the dentate gyrus.
Fig. 7.

The benzodiazepine midazolam does not modulate δGABAA receptor–mediated tonic currents in DGGCs. (A) Representative traces of tonic current recordings from WT DGGCs perfused with 1 μM GABA and the absence or presence of midazolam (MDZ) and subsequent block of tonic current with 50 μM gabazine. (B) Concentration-response curve of midazolam as measured by normalized tonic current (pA/pF) from patch-clamp recordings. All data were derived from female (diestrus I) WT DGGCs. All cells were voltage clamped in whole-cell mode at −65 mV. n = 4–6 cells per concentration.
SAR of Neurosteroid Antiseizure Actions in the 6-Hz Seizure Test.
To study the in vivo anticonvulsant effects of neurosteroids, we used the 6-Hz model of partial seizures. The 6-Hz model is highly sensitive to antiepileptic drugs, which act to primarily increase GABA-mediated inhibition. Previous studies have used 6-Hz stimulation to characterize the anticonvulsant profiles of neurosteroids within mice (Barton et al., 2001; Kaminski et al., 2004; Reddy et al., 2015). We first determined incidence of seizures as a function of current intensity in the mice (Fig. 8A). The CC50 value (current intensity in which 50% of animals experienced seizure) was 16.5 mA for WT male mice and 10.4 mA for WT female mice. Current intensity of 32 mA elicited seizures in 100% of the population of WT male and female control mice injected with vehicle, and further drug studies were conducted at this level of current.
Fig. 8.

Neurosteroid SAR for seizure protection in the 6-Hz seizure test. (A) Percentage of animals responding to 6-Hz stimulation as a function of current intensity delivered through shock. All animals had 100% seizure activity at 32 mA. The dashed line perpendicular to the fitted data represents the current intensity at which 50% of animals experienced seizures. (C) Dose-response curves for neurosteroid modulation of 32 mA, 6-Hz–induced seizures in WT female mice. Neurosteroids were applied (s.c.) 15 minutes prior to stimulation. Seven structurally distinct pregnane-class neurosteroids were compared in seizure protection, using potency (ED50, see Table 3) as the main index of anticonvulsant activity in vivo (3β-AP not shown because it did not provide full protection, even at the 100-mg/kg dose). n = 10 animals per group. AFX, alfaxolone (3α-hydroxy-5α-pregnan-11,20-dione).
Adult female WT mice were treated with neurosteroids 15 minutes before 6-Hz electrical stimulation at 32 mA (Fig. 8B). Motor toxicity was determined prior to stimulation. After stimulation, incidence of seizure protection was recorded as an all-or-nothing event (see the Materials and Methods for scoring criteria). Dose-response curves were derived for each neurosteroid drug, and potency was determined as ED50 (effective dose that is protective for 50% of animals). The potency of 5α- and 5β-AP was similar in protection within the 6-Hz model, supporting previous evidence (Kaminski et al., 2004). Higher bioavailability of 5β-AP could contribute to the anticonvulsant effect in vivo despite less overall potentiation of GABAergic current in the slice (Fig. 6A) (Porcu et al., 2009). At elevated concentrations that activate both synaptic and extrasynaptic receptor types, it is feasible that the direct binding of 5β-AP to receptors overcomes the reduced effects on tonic current (Hosie et al., 2006). In the 6-Hz model, 3β-AP failed to protect against seizures in all animals tested up to a dose of 100 mg/kg, consistent with a previous report (Kokate et al., 1994). GX treatment provided improved seizure protection potency compared with 5α-AP (Fig. 8). Our data support that GX has a similar SAR to AP, although GX may have improved pharmacokinetic distribution and bioavailability in the brain.
Previous study of motor toxicity with 3α5α-pregnane steroids demonstrated that motor impairment is low at doses that confer seizure protection (Kokate et al., 1994). Reports have demonstrated that treatment with 5α-reduced compounds produces a lower incidence of motor toxicity compared with the 5β epimers (Kokate et al., 1994). Subcutaneous treatment with the maximum efficacy dose did not exhibit a plateau or full motor toxicity. The experimental rate of absorption for subcutaneous injection of steroid is slower than other routes of administration, but we did not observe significant toxicity in animals at maximally effective doses. A previous study of loss of righting reflex as a measure of AP’s anesthetic effect found minimal impairment at 10 mg/kg (s.c.) in WT male and female mice (Reddy and Zeng, 2007). Therefore, the ED100 may be representative of binding and modulating a more specific, rather than global, GABAA receptor population.
To correlate activity of therapeutically relevant doses of neurosteroid, we measured plasma levels of AP after exogenous administration in mice. Blood plasma was collected 15 minutes after AP (1–10 mg/kg, s.c.) or vehicle (0 mg/kg) was administered. Plasma AP levels were measured with liquid chromatography/mass spectrometry as described previously (Reddy and Mohan, 2011; Reddy et al., 2012). The equivalent molar concentrations used in Itonic experiments and the 6-Hz dose-response study were plotted and compared with the measured plasma concentrations of AP (Fig. 9). The window of AP acting allosterically exerted little seizure protection in the 6-Hz model. Full protection from 6-Hz seizures required a therapeutic concentration of AP that elicits the directly activating activity at GABAA receptors. Therefore, these findings with pharmacological doses of neurosteroids are physiologically or clinically relevant to extrasynaptic function.
Fig. 9.

Correlation between neurosteroid levels and tonic inhibition and seizure protection. Blood plasma was collected 15 minutes after vehicle or AP injection (1–10 mg/kg, s.c.). Liquid chromatography/mass spectrometry was used to measure AP levels (circles, left y-axis) and was compared with the percentage of animals protected from 6-Hz–induced seizures (triangles, right y-axis). The dotted lines indicate the interpolated, equivalent 0.3 μM (96 ng/ml), 1 μM (318 ng/ml), and 3 μM (955 ng/ml) concentrations of AP with average AP-dependent potentiation of Itonic in parentheses (current shift in pA, see Table 3). n = 5–8 animals per dose.
Discussion
The principal finding of this study is the identification of obligatory structural features of neurosteroids for functional activation of extrasynaptic δGABAA receptors that mediate tonic inhibition in the dentate gyrus. This was clearly evident from observations using a library of neurosteroid structures at a wide concentration range to assess both physiologic and clinically relevant modulation of extrasynaptic receptors. The tonic current of DGGCs is highly (approximately 95%) dependent on δ-containing receptors (Stell et al., 2003; Glykys et al., 2008; Carver et al., 2014). Synaptic and extrasynaptic receptors differ in their GABA affinity (Mortensen et al., 2012), desensitization rate, agonist efficacy (Bianchi and Macdonald, 2002, 2003), and neurosteroid sensitivity (Brown et al., 2002; Wohlfarth et al., 2002). We focused on the physiologically relevant tonic conductance properties of native DGGCs for characterization of SARs. Our findings support an essential role of the δ subunit in neurosteroid modulation of tonic inhibition.
Neurosteroids are uniquely sensitive to the channel function conferred by the δ subunit that enables continuous, inhibitory conductance (Bianchi and Macdonald, 2003). Within in vitro expression systems, GABAA receptors have distinct gating properties in low-efficacy and high-efficacy states. It is unclear whether the overall changes in efficacy during recombinant expression of δGABAA receptors are truly representative of the neurosteroid response within native neurons. The tonic current recorded from brain slice reflects physiologic activation of localized extrasynaptic receptors, rather than measurement of aggregate responses that are inclusive of all membrane GABAA receptors. We ascertained the extrasynaptic contribution of δ- and non–δ-containing receptors in modulation of tonic current by neurosteroids. 3α5α-AP was more potent in tonic current modulation than the δ-selective agents THIP and DS2, and δKO attenuated such δ-dependent tonic current (Fig. 6D). Deletion of δ critically diminished 3α5α-AP allosteric potentiation and activation effects without exogenous GABA across the entire range of its activity (Figs. 4 and 6). Therefore, the low- to high-efficacy transduction produced by neurosteroid is δ-subunit mediated under physiologic conditions. These findings are consistent with the significance of δGABAA receptors to tonic inhibition and neurosteroid sensitivity (Wu et al., 2013; Carver et al., 2014).
In this SAR study, 3α5α-AP was used as a prototypical allosteric modulator of GABAA receptor function. 3α5β-AP displayed an overall reduced efficacy on tonic current function, indicating a strong influence of the C5 structure on efficacy. 3β5α-AP was ineffective in modulating extrasynaptic receptors, exhibiting the motif of inactivity for 3β-reduced steroids (Puia et al., 1990). This further suggests an essential role for the 3α-hydroxyl in extrasynaptic activity. Interestingly, THDOC displayed significantly lower tonic current modulation compared with 3α5α-AP at submicromolar concentrations, suggesting a disparity in extrasynaptic efficacy between C20 structural moieties. This could be related to the decreased lipophilicity of THDOC compared with 3α5α-AP affecting either membrane access or receptor affinity (Akk et al., 2009). ALFX exhibited reduced tonic current compared with 3α5α-AP, suggesting that the C11 ketone might interfere with the coordinated binding at the hydrophobic pocket in the transmembrane domain of the receptor. It is likely that differences in neurosteroid sensitivity could be due to experimental conditions (Stell et al., 2003). Thus, the conserved C3α and C17 groups may be influenced by other structural factors in extrasynaptic receptor interaction.
Weaker tonic modulation was demonstrated by androstane neurosteroids. The androstane 17-β functional groups (see Table 1) suggest a more polar C17 position, resulting in weaker affinity for the hydrophobic binding pocket (Reddy and Jian, 2010). Therefore, structural differences in electronegativity from the C17 position may be crucial to the binding activity for extrasynaptic receptors. The ORG-20599 compound exhibited high extrasynaptic efficacy and affinity greater than 3α5α-AP. The morpholinyl conjugation in the A-ring served to facilitate the hydrogen bonding of the 3α group rather than steric hindrance of binding. However, ORG-20599 possessed very weak potency in 6-Hz antiseizure function in vivo that could be related to pharmacokinetic dissimilarity compared with previous reports of comparable potency to 3α5α-AP at anesthetic doses (Hill-Venning et al., 1996).
Higher concentrations of neurosteroids directly activate GABAA receptors (Puia et al., 1990). We described 3α5α-AP’s capacity to activate extrasynaptic receptors in native WT and δKO DGGCs in conditions lacking exogenous GABA. Compared with allosteric activity with 1 μM GABA, 3α5α-AP alone was less efficacious in potentiation of tonic current. In conditions without GABA, δ-containing receptors accounted for 71% of the total AP-dependent tonic current, indicating substantial contribution of δ to the direct activation by neurosteroid (Fig. 5C). Other data suggest that higher concentrations of neurosteroid may block the GABAA receptor channel pore, inhibiting chloride conductance (Brown et al., 2002). By contrast, we observed extensive potentiation at higher neurosteroid concentrations as well as directly activating effects. However, we were unable to demonstrate differences in the kinetic action of neurosteroids between direct gating (neurosteroid alone) and allosteric potentiation (Shu et al., 2004). This could be attributable to our gradual perfusion methodology in the slice. A different perfusion set-up may allow kinetic determinations. Nevertheless, the slice preparation enables membrane accumulation of neurosteroid and likely represents measurement of nonaqueous access to affect receptor activity in both allosteric and direct gating function. Therefore, the differences in efficacy at the two neurosteroid binding sites reflect the significance of δ-subunit transduction.
Previous reports indicate that high-efficacy synaptic receptors are sensitive to direct activation by neurosteroids (Kokate et al., 1994; Reddy and Rogawski, 2002). We discovered a role for other GABAA receptors that may contribute to tonic conductance in facilitation of high-efficacy states. Within δKO DGGCs, 10 µM GABA or 3 µM neurosteroid elicited modest hyperpolarizing, tonic responses. Despite δ deficiency, the high-efficacy modulation signifies a biphasic response of the native receptor population in which non-δ receptors are being activated to influence tonic current. Neurosteroid doses required to achieve full seizure protection likely modulate these high-efficacy channels in addition to δ-containing receptors (Fig. 9). The subunit configuration and location of these high-efficacy receptors remains unclear but may relate to the compensational shift of other subunit expression within the germline δKO mouse strain model (Peng et al., 2002). Changes to neurosteroid sensitivity are also relevant to the hyperexcitability of epileptic granule cells (Kobayashi and Buckmaster, 2003) and the decreased expression of the δ subunit in epileptic hippocampus (Whissell et al., 2015). Large neurosteroid accumulation appears to overcome the desensitization rate of synaptic or perisynaptic receptors to provide for increased hyperpolarizing tone. A large reduction in tonic conductance may lead to lack of shunting inhibition for δ-deficient neurons, and this likely presents a model for hyperexcitability within the hippocampus in which neurosteroids may still modulate current at higher concentrations. A recent concatamer-receptor study reported that α4βδ and α1βγ2 GABAA receptors have largely similar structure-activity profiles for neurosteroids (Shu et al., 2012). However, we demonstrate the localized influence of neurosteroids that is specific to native, extrasynaptic, δ-containing receptors and further reveals a divergent role of tonic inhibition in antiseizure control.
The effects of δ-subunit–specific mutations on the transduction of the neurosteroid interaction are yet to be determined. The Q241 and N407/Y410 site-specific residues of the α1 subunit were determined to be crucial for neurosteroid binding (Hosie et al., 2007). More recently, T235W and Q240W mutations in the α4 subunit rendered δ-containing receptors (α4βδ) insensitive to positive allosteric modulation by 3α5α-AP (Jensen et al., 2013). Therefore, the higher affinity for the α4 subunit binding interface and δ-dependent channel transduction greatly contribute to neurosteroid binding. In support of this, we demonstrate that α1βδ-containing ML interneurons have lower sensitivity to neurosteroid modulation than DGGCs (Fig. 4).
We attempted to correlate the SARs of neurosteroids for tonic inhibition with seizure-protective effects in the mouse 6-Hz seizure model. We noted a moderate correlation between action on δGABA receptors and in vivo antiseizure activity. As evident from Table 3 and Fig. 9, for many neurosteroids, such as AP and GX, the correlation between tonic inhibition and seizure protection is reasonably strong. The modest correlation with some molecules is attributable to many factors. The net effect of a specific neurosteroid to total GABAergic inhibition depends on its structural feature for efficient binding to synaptic and extrasynaptic receptors and also its bioavailability to reach the target sites in the brain (Visser et al., 2002). For example, GX performed much better than AP because of a plausible better bioavailability. We unexpectedly found that global δKO mice are highly insensitive to 6-Hz seizure induction, which precluded further attempts to clarify the role of δGABARs and in vivo antiseizure activity. This is not surprising because many constitutive transgenic mouse strains exhibit such phenotype as a result of development defects. Nevertheless, it is thought that extrasynaptic receptors contribute greatly to the overall net inhibition and seizure resistance, which is affected by many factors that ultimately govern the extent of seizure protection.
TABLE 3.
Correlation between neurosteroid modulation of tonic current and anticonvulsant profiles in the 6-Hz seizure model in mice
E1 μM values represent the mean normalized tonic current responses of drug at 1 μM concentration coapplied with 1 μM GABA. The GABA 1 μM tonic current was 0.66 ± 0.22 pA/pF, 19.6 pA. E3μMvalues represent the mean normalized tonic current responses of drug at 3 μM concentration coapplied with 1 μM GABA. The GABA 3 μM tonic current was 1.35 ± 0.23 pA/pF, 33.5 pA. EF values represent the effective functional concentration of drug (nanomolar) required to double or triple the 1 μM GABA response. The 6-Hz test ED50 values represent the dose in milligrams per kilogram that protected 50% of animals in the 6-Hz seizure stimulation test. 95% confidence intervals are listed in parentheses, according to a normal distribution.
| Compound | E1 μM | E1 μM | E3 μM | E3 μM | EF(2-Fold GABA) | EF(3-Fold GABA) | 6-Hz Test ED50 |
|---|---|---|---|---|---|---|---|
| pA/pF | pA | pA/pF | pA | nM | nM | mg/kg | |
| 3α5αAP | 2.88 ± 0.50 | 100.6 | 7.25 ± 0.40 | 223.7 | 80 | 290 | 4.2 (2.7–5.8) |
| GX | 2.29 ± 0.41 | 64.0 | 5.87 ± 0.50 | 176.1 | 290 | 650 | 1.5 (1.3–1.7) |
| 3α5β-AP | 1.48 ± 0.13 | 44.4 | 4.27 ± 0.25 | 181.7 | 780 | 1380 | 7.7 (6.6–8.8) |
| 3β5α-AP | 0.50 ± 0.12 | 15.3 | 0.51 ± 0.14 | 15.7 | >10,000 | >10,000 | >100 |
| THDOC | 2.32 ± 0.43 | 66.6 | 6.10 ± 0.68 | 183.0 | 410 | 760 | 5.0 (2.6–7.4) |
| ALFX | 1.31 ± 0.19 | 40.9 | 4.82 ± 0.53 | 144.6 | 990 | 1500 | 8.8 (6.1–11.4) |
| ORG-20599 | 3.10 ± 0.54 | 86.4 | 6.21 ± 0.42 | 186.3 | 120 | 310 | 18.6 (16.6–20.6) |
| AD | 0.86 ± 0.19 | 33.2 | 1.95 ± 0.29 | 49.9 | 1710 | 3120 | 44.0 (30.2–58.8) |
We analyzed the correlation between plasma levels of neurosteroids and seizure protection due likely to tonic inhibition. Based on plasma AP levels subsequent to AP treatment (Fig. 9), the estimated threshold plasma concentration for seizure protection (25%) is in the range of 500–700 ng/ml (1.6–2.2 μM). This concentration exceeds neurosteroid potentiation of receptors occurring only via an allosteric binding mechanism of action and includes direct activation. The estimated plasma concentration producing 50% seizure protection is in the range of 1050–1150 ng/ml (3.3–3.6 μM), and this involves an exponential increase in tonic current modulation by AP. Higher concentrations represent direct activation of GABAA receptors in the ability to achieve full anticonvulsant protection by AP. This pharmacokinetics/pharmacodynamics relationship is consistent with the anticonvulsant profile of neurosteroids (Reddy and Rogawski, 2002; Reddy, 2004, 2011, 2014; Reddy et al., 2004, 2010; Reddy and Woodward, 2004).
Based on the neurosteroid SARs at extrasynaptic receptors, we designed a consensus pharmacophore map (Fig. 10). It reveals a critical requirement for distinct chemical functional groups for the channel transduction interface facilitated by the δ subunit. This pharmacophore map is derived based on the correlation between neurosteroid modulation of tonic current and anticonvulsant profiles in the 6-Hz seizure model. The salient features of the proposed pharmacophore model are as follows. First, the C3 and C17 regions remain critical to the δ efficacy of neurosteroids. Second, the C5 region indicates a stereoselectivity for neurosteroid interaction at extrasynaptic sites, similar to that of synaptic receptor binding. Third, the C20 branch of the pregnane neurosteroids provides coordination of the hydrophobic pocket affinity that yields effects on both potency and efficacy. Fourth, reduced potency is evident in the case of C17 androstane ring structured neurosteroids. Finally, a ketone group attached to the C11 position (e.g., ALFX) is detrimental to δ-subunit–containing receptor efficacy.
Fig. 10.

A schematic pharmacophore model for the neurosteroid structure-functional activity at extrasynaptic δGABAA receptors. The key neurosteroid structure features are illustrated with relative percent efficacy and receptor affinity. These key features are derived from electrophysiological and behavioral studies of neurosteroid activity on the putative extrasynaptic function of δ-containing GABAA receptors. Values listed are approximate range of neurosteroid potentiation of Itonic as a percentage of the AP prototypical Itonic responses in electrophysiology slice recordings.R1, C3-position stereoselectivity of the hydroxyl group; R2, C3-position β-conjugation in conjunction with the 3α-OH group; R3, C5-position stereoselectivity of the hydrogen bond upon 5α- or 5β-reductase activity; R4, C11-position conjugation of double-bonded oxygen in the case of ALFX and alfadolone synthetic neurosteroids; R5, C17-position β-conjugation necessary for efficacy, C20-position ketone for pregnane neurosteroids (AP, THDOC), or C17-position for androstane neurosteroids (AD, AN, ETIO).
In conclusion, our results provide strong evidence that neurosteroids exhibit greater sensitivity at extrasynaptic δGABAA receptors, as evident from extensive tonic current recordings in native DGGCs in in vitro electrophysiology and in an in vivo 6-Hz seizure model. Our SAR study uncovers the key structural features and a consensus pharmacophore pocket for neurosteroid efficacy at extrasynaptic δGABAA receptors in the hippocampus.
Acknowledgments
The authors thank Bill Griffith, Mendell Rimer, and Warren Zimmer for input on the manuscript.
Abbreviations
- 3β-AP
3β-hydroxy-5α-pregnan-20-one
- 3β-Me-AP
3α-hydroxy-3β-methyl-5β-pregnan-20-one
- 5β-AP
3α-hydroxy-5β-pregnan-20-one
- aCSF
artificial cerebrospinal fluid
- AD
androstanediol (5α-androstan-3α,17β-diol)
- ALFX
alfaxolone (3α-hydroxy-5α-pregnan-11,20-dione)
- AN
androsterone
- AP
allopregnanolone (3α-hydroxy-5α-pregnan-20-one)
- DGGC
dentate gyrus granule cell
- δKO
GABAA receptor δ-subunit knockout
- DS2
4-chloro-N-[2-(2-thienyl)imidazo[1,2-a]pyridin-3-yl]benzamide
- ETIO
etiocholanolone
- GABA-AR
γ-aminobutyric acid type A receptor
- GX
ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one)
- ML
molecular layer
- ORG-20599
2β,3α,5α-21-chloro-3-hydroxy-2-(4-morpholinyl)pregnan-20-one
- QX-314
N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide
- SAR
structure-activity relationship
- SR-95531
4-[6-imino-3-(4-methoxyphenyl)pyridazin-1-yl] butanoic acid hydrobromide
- THDOC
allotetrahydrodeoxycorticosterone (3α,21-dihydroxy-5α-pregnan-20-one)
- THIP
gaboxadol (4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-3-ol)
- TTX
tetrodotoxin
- WT
wild type
Authorship Contributions
Participated in research design: Carver, Reddy.
Conducted experiments: Carver, Reddy.
Performed data analysis: Carver, Reddy.
Wrote or contributed to the writing of the manuscript: Carver, Reddy.
Footnotes
This research was partly supported by the National Institutes of Health National Institute of Neurologic Disorders and Stroke [Grant R01NS051398 (to D.S.R.)]. This research was also partly supported by the CounterACT Program, National Institutes of Health Office of the Director, and the National Institutes of Health National Institute of Neurologic Disorders and Stroke [Grant U01NS083460 (to D.S.R.)]. The views expressed in this article are those of the authors and do not reflect the official policy of the National Institutes of Health or the U.S. Government or the Texas A&M University.
References
- Akk G, Covey DF, Evers AS, Steinbach JH, Zorumski CF, Mennerick S. (2007) Mechanisms of neurosteroid interactions with GABA(A) receptors. Pharmacol Ther 116:35–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Covey DF, Evers AS, Steinbach JH, Zorumski CF, Mennerick S. (2009) The influence of the membrane on neurosteroid actions at GABA(A) receptors. Psychoneuroendocrinology 34 (Suppl 1):S59–S66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S. (2005) Neurosteroid access to the GABAA receptor. J Neurosci 25:11605–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ME, Klein BD, Wolf HH, White HS. (2001) Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res 47:217–227. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. (2002) Slow phases of GABA(A) receptor desensitization: structural determinants and possible relevance for synaptic function. J Physiol 544:3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. (2003) Neurosteroids shift partial agonist activation of GABA(A) receptor channels from low- to high-efficacy gating patterns. J Neurosci 23:10934–10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. (2002) Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol 136:965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM, Reddy DS. (2013) Neurosteroid interactions with synaptic and extrasynaptic GABA(A) receptors: regulation of subunit plasticity, phasic and tonic inhibition, and neuronal network excitability. Psychopharmacology (Berl) 230:151–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM, Wu X, Gangisetty O, Reddy DS. (2014) Perimenstrual-like hormonal regulation of extrasynaptic δ-containing GABAA receptors mediating tonic inhibition and neurosteroid sensitivity. J Neurosci 34:14181–14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra D, Jia F, Liang J, Peng Z, Suryanarayanan A, Werner DF, Spigelman I, Houser CR, Olsen RW, Harrison NL, et al. (2006) GABAA receptor α4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc Natl Acad Sci USA 103:15230–15235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisari M, Eisenman LN, Krishnan K, Bandyopadhyaya AK, Wang C, Taylor A, Benz A, Covey DF, Zorumski CF, Mennerick S. (2009) The influence of neuroactive steroid lipophilicity on GABAA receptor modulation: evidence for a low-affinity interaction. J Neurophysiol 102:1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Carlson GC. (2007) Functional regulation of the dentate gyrus by GABA-mediated inhibition. Prog Brain Res 163:235–243. [DOI] [PubMed] [Google Scholar]
- Covey DF, Nathan D, Kalkbrenner M, Nilsson KR, Hu Y, Zorumski CF, Evers AS. (2000) Enantioselectivity of pregnanolone-induced gamma-aminobutyric acid(A) receptor modulation and anesthesia. J Pharmacol Exp Ther 293:1009–1016. [PubMed] [Google Scholar]
- Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, et al. (2004) GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet 13:1315–1319. [DOI] [PubMed] [Google Scholar]
- Derry JM, Dunn SM, Davies M. (2004) Identification of a residue in the gamma-aminobutyric acid type A receptor alpha subunit that differentially affects diazepam-sensitive and -insensitive benzodiazepine site binding. J Neurochem 88:1431–1438. [DOI] [PubMed] [Google Scholar]
- Ebert B, Mortensen M, Thompson SA, Kehler J, Wafford KA, Krogsgaard-Larsen P. (2001) Bioisosteric determinants for subtype selectivity of ligands for heteromeric GABA(A) receptors. Bioorg Med Chem Lett 11:1573–1577. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Kang JQ, Song L, Dibbens L, Mulley J, Macdonald RL. (2006) δ subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of α4β2δ GABAA receptors. J Neurosci 26:1499–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee KW, Bolger MB, Brinton RE, Coirini H, McEwen BS. (1988) Steroid modulation of the chloride ionophore in rat brain: structure-activity requirements, regional dependence and mechanism of action. J Pharmacol Exp Ther 246:803–812. [PubMed] [Google Scholar]
- Glykys J, Mann EO, Mody I. (2008) Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J Neurosci 28:1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Peng Z, Chandra D, Homanics GE, Houser CR, Mody I. (2007) A new naturally occurring GABAa receptor subunit with high sensitivity to ethanol. Nat Neurosci 10:40–48. [DOI] [PubMed] [Google Scholar]
- Harrison NL, Majewska MD, Harrington JW, Barker JL. (1987) Structure-activity relationships for steroid interaction with the gamma-aminobutyric acidA receptor complex. J Pharmacol Exp Ther 241:346–353. [PubMed] [Google Scholar]
- Herd MB, Haythornthwaite AR, Rosahl TW, Wafford KA, Homanics GE, Lambert JJ, Belelli D. (2008) The expression of GABAA β subunit isoforms in synaptic and extrasynaptic receptor populations of mouse dentate gyrus granule cells. J Physiol 586:989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill-Venning C, Peters JA, Callachan H, Lambert JJ, Gemmell DK, Anderson A, Byford A, Hamilton N, Hill DR, Marshall RJ, et al. (1996) The anaesthetic action and modulation of GABAA receptor activity by the novel water-soluble aminosteroid ORG 20599. Neuropharmacology 35:1209–1222. [DOI] [PubMed] [Google Scholar]
- Hoestgaard-Jensen K, Dalby NO, Krall J, Hammer H, Krogsgaard-Larsen P, Frolund B, Jensen AA. (2014) Probing α4βδ GABAA receptor heterogeneity: differential regional effects of a functionally selective α4β1δ/α4β3δ receptor agonist on tonic and phasic inhibition in rat brain. J Neurosci 34:16256–16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Clarke L, da Silva H, Smart TG. (2009) Conserved site for neurosteroid modulation of GABA A receptors. Neuropharmacology 56:149–154. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. (2006) Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444:486–489. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, Smart TG. (2007) Neurosteroid binding sites on GABA(A) receptors. Pharmacol Ther 116:7–19. [DOI] [PubMed] [Google Scholar]
- Jensen ML, Wafford KA, Brown AR, Belelli D, Lambert JJ, Mirza NR. (2013) A study of subunit selectivity, mechanism and site of action of the delta selective compound 2 (DS2) at human recombinant and rodent native GABA(A) receptors. Br J Pharmacol 168:1118–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F, Pignataro L, Schofield CM, Yue M, Harrison NL, Goldstein PA. (2005) An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J Neurophysiol 94:4491–4501. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Livingood MR, Rogawski MA. (2004) Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice. Epilepsia 45:864–867. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. (2003) Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci 23:2440–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokate TG, Svensson BE, Rogawski MA. (1994) Anticonvulsant activity of neurosteroids: correlation with γ-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther 270:1223–1229. [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V, Martín del Río R. (1986) In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res 384:145–155. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. (1986) Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232:1004–1007. [DOI] [PubMed] [Google Scholar]
- Meera P, Wallner M, Otis TS. (2011) Molecular basis for the high THIP/gaboxadol sensitivity of extrasynaptic GABA(A) receptors. J Neurophysiol 106:2057–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi Z, Lagenaur C, Tretter V, Sieghart W, Anagnostaras SG, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Zhiwei, DeLorey TM, Olsen RW, Homanics GE. (1999) Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor delta subunit knockout mice. Proc Natl Acad Sci USA 96:12905–12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell EA, Herd MB, Gunn BG, Lambert JJ, Belelli D. (2008) Neurosteroid modulation of GABAA receptors: molecular determinants and significance in health and disease. Neurochem Int 52:588–595. [DOI] [PubMed] [Google Scholar]
- Möhler H, Fritschy JM, Rudolph U. (2002) A new benzodiazepine pharmacology. J Pharmacol Exp Ther 300:2–8. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Ebert B, Wafford K, Smart TG. (2010) Distinct activities of GABA agonists at synaptic- and extrasynaptic-type GABAa receptors. J Physiol 588:1251–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Patel B, Smart TG. (2012) GABA potency at GABAa receptors found in synaptic and extrasynaptic zones. Front Neurosci 6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S, Jeong JA, Jo JY, Cho HS, Kim DW, Kim JM, Ryu PD, Lee SY, Kim HW, Jeon BH, et al. (2013) Dual mechanisms diminishing tonic GABAA inhibition of dentate gyrus granule cells in Noda epileptic rats. J Neurophysiol 110:95–102. [DOI] [PubMed] [Google Scholar]
- Peng Z, Hauer B, Mihalek RM, Homanics GE, Sieghart W, Olsen RW, Houser CR. (2002) GABA(A) receptor changes in δ subunit-deficient mice: altered expression of α4 and γ2 subunits in the forebrain. J Comp Neurol 446:179–197. [DOI] [PubMed] [Google Scholar]
- Porcu P, O’Buckley TK, Alward SE, Marx CE, Shampine LJ, Girdler SS, Morrow AL. (2009) Simultaneous quantification of GABAergic 3α,5α/3α,5β neuroactive steroids in human and rat serum. Steroids 74:463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince RJ, Simmonds MA. (1992) 5 β-pregnan-3 β-ol-20-one, a specific antagonist at the neurosteroid site of the GABAA receptor-complex. Neurosci Lett 135:273–275. [DOI] [PubMed] [Google Scholar]
- Puia G, Santi MR, Vicini S, Pritchett DB, Purdy RH, Paul SM, Seeburg PH, Costa E. (1990) Neurosteroids act on recombinant human GABAA receptors. Neuron 4:759–765. [DOI] [PubMed] [Google Scholar]
- Qian M, Krishnan K, Kudova E, Li P, Manion BD, Taylor A, Elias G, Akk G, Evers AS, Zorumski CF, et al. (2014) Neurosteroid analogues. 18. Structure-activity studies of ent-steroid potentiators of γ-aminobutyric acid type A receptors and comparison of their activities with those of alphaxalone and allopregnanolone. J Med Chem 57:171–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS. (2003) Pharmacology of endogenous neuroactive steroids. Crit Rev Neurobiol 15:197–234. [DOI] [PubMed] [Google Scholar]
- Reddy DS. (2004) Testosterone modulation of seizure susceptibility is mediated by neurosteroids 3alpha-androstanediol and 17beta-estradiol. Neuroscience 129:195–207. [DOI] [PubMed] [Google Scholar]
- Reddy DS. (2006) Physiological role of adrenal deoxycorticosterone-derived neuroactive steroids in stress-sensitive conditions. Neuroscience 138:911–920. [DOI] [PubMed] [Google Scholar]
- Reddy DS. (2010) Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res 186:113–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS. (2011) Role of anticonvulsant and antiepileptogenic neurosteroids in the pathophysiology and treatment of epilepsy. Front Endocrinol (Lausanne) 2:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS. (2014) Neurosteroids and their role in sex-specific epilepsies. Neurobiol Dis 72:198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Castaneda DC, O’Malley BW, Rogawski MA. (2004) Anticonvulsant activity of progesterone and neurosteroids in progesterone receptor knockout mice. J Pharmacol Exp Ther 310:230–239. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Gangisetty O, Briyal S. (2010) Disease-modifying activity of progesterone in the hippocampus kindling model of epileptogenesis. Neuropharmacology 59:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Gould J, Gangisetty O. (2012) A mouse kindling model of perimenstrual catamenial epilepsy. J Pharmacol Exp Ther 341:784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Jian K. (2010) The testosterone-derived neurosteroid androstanediol is a positive allosteric modulator of GABAA receptors. J Pharmacol Exp Ther 334:1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Kulkarni SK. (2000) Development of neurosteroid-based novel psychotropic drugs. Prog Med Chem 37:135–175. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Mohan A. (2011) Development and persistence of limbic epileptogenesis are impaired in mice lacking progesterone receptors. J Neurosci 31:650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Rogawski MA. (2000) Enhanced anticonvulsant activity of ganaxolone after neurosteroid withdrawal in a rat model of catamenial epilepsy. J Pharmacol Exp Ther 294:909–915. [PubMed] [Google Scholar]
- Reddy DS, Rogawski MA. (2001) Enhanced anticonvulsant activity of neuroactive steroids in a rat model of catamenial epilepsy. Epilepsia 42:337–344. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Rogawski MA. (2002) Stress-induced deoxycorticosterone-derived neurosteroids modulate GABA(A) receptor function and seizure susceptibility. J Neurosci 22:3795–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Rogawski MA. (2009) Neurosteroid replacement therapy for catamenial epilepsy. Neurotherapeutics 6:392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Rogawski MA. (2012) Neurosteroids – endogenous regulators of seizure susceptibility and role in the treatment of epilepsy, in Jasper’s Basic Mechanisms of the Epilepsies, 4th ed (Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV. eds) pp 984–1002, Oxford University Press, Bethesda, MD. [Google Scholar]
- Reddy DS, Woodward R. (2004) Ganaxolone: a prospective overview. Drugs Future 29:227–242. [Google Scholar]
- Reddy SD, Younus I, Clossen BL, Reddy DS. (2015) Antiseizure activity of midazolam in mice lacking δ-subunit extrasynaptic GABA-A receptors. J Pharmacol Exp Ther 353:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Zeng YC. (2007) Differential anesthetic activity of ketamine and the GABAergic neurosteroid allopregnanolone in mice lacking progesterone receptor A and B subtypes. Methods Find Exp Clin Pharmacol 29:659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna E, Mostallino MC, Murru L, Carta M, Talani G, Zucca S, Mura ML, Maciocco E, Biggio G. (2009) Changes in expression and function of extrasynaptic GABAA receptors in the rat hippocampus during pregnancy and after delivery. J Neurosci 29:1755–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Hanchar HJ, Wallner M, Olsen RW, Otis TS. (2006) Contributions of the GABAA receptor α6 subunit to phasic and tonic inhibition revealed by a naturally occurring polymorphism in the α6 gene. J Neurosci 26:3357–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu HJ, Bracamontes J, Taylor A, Wu K, Eaton MM, Akk G, Manion B, Evers AS, Krishnan K, Covey DF, et al. (2012) Characteristics of concatemeric GABA(A) receptors containing α4/δ subunits expressed in Xenopus oocytes. Br J Pharmacol 165:2228–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM. (2003) GABA uptake regulates cortical excitability via cell type-specific tonic inhibiton. Nature Neurosci 6:484–490. [DOI] [PubMed] [Google Scholar]
- Shu HJ, Eisenman LN, Jinadasa D, Covey DF, Zorumski CF, Mennerick S. (2004) Slow actions of neuroactive steroids at GABAA receptors. J Neurosci 24:6667–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spigelman I, Li Z, Liang J, Cagetti E, Samzadeh S, Mihalek RM, Homanics GE, Olsen RW. (2003) Reduced inhibition and sensitivity to neurosteroids in hippocampus of mice lacking the GABAA receptor δ subunit. J Neurophysiol 90:903–910. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. (2003) Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc Natl Acad Sci USA 100:14439–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stórustovu SI, Ebert B. (2006) Pharmacological characterization of agonists at δ-containing GABAA receptors: Functional selectivity for extrasynaptic receptors is dependent on the absence of γ2. J Pharmacol Exp Ther 316:1351–1359. [DOI] [PubMed] [Google Scholar]
- Sun C, Sieghart W, Kapur J. (2004) Distribution of α1, γ2, and δ subunits of GABAa receptors in hippocampal granule cells. Brain Res 1029:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upasani RB, Yang KC, Acosta-Burruel M, Konkoy CS, McLellan JA, Woodward RM, Lan NC, Carter RB, Hawkinson JE. (1997) 3 α-Hydroxy-3 β-(phenylethynyl)-5 β-pregnan-20-ones: synthesis and pharmacological activity of neuroactive steroids with high affinity for GABAA receptors. J Med Chem 40:73–84. [DOI] [PubMed] [Google Scholar]
- Visser SA, Gladdines WW, van der Graaf PH, Peletier LA, Danhof M. (2002) Neuroactive steroids differ in potency but not in intrinsic efficacy at the GABA(A) receptor in vivo. J Pharmacol Exp Ther 303:616–626. [DOI] [PubMed] [Google Scholar]
- Wafford KA, van Niel MB, Ma QP, Horridge E, Herd MB, Peden DR, Belelli D, Lambert JJ. (2009) Novel compounds selectively enhance δ subunit containing GABAa receptors and increase tonic currents in thalamus. Neuropharmacology 56:182–189. [DOI] [PubMed] [Google Scholar]
- Whissell PD, Lecker I, Wang DS, Yu J, Orser BA. (2015) Altered expression of δGABAA receptors in health and disease. Neuropharmacology 88:24–35. [DOI] [PubMed] [Google Scholar]
- Wohlfarth KM, Bianchi MT, Macdonald RL. (2002) Enhanced neurosteroid potentiation of ternary GABA(A) receptors containing the δ subunit. J Neurosci 22:1541–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Gangisetty O, Carver CM, Reddy DS. (2013) Estrous cycle regulation of extrasynaptic δ-containing GABA(A) receptor-mediated tonic inhibition and limbic epileptogenesis. J Pharmacol Exp Ther 346:146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue BG, Whittemore ER, Park CH, Woodward RM, Lan NC, Gee KW. (1997) Partial agonism by 3α,21-dihydroxy-5β-pregnan-20-one at the γ-aminobutyric acidA receptor neurosteroid site. J Pharmacol Exp Ther 281:1095–1101. [PubMed] [Google Scholar]
- Zheleznova N, Sedelnikova, Weiss DS. (2008) α1β2δ, a silent GABAa receptor: recruitment by tracazolate and neurosteroids. Br J Pharmacol 153:1062–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheleznova NN, Sedelnikova A, Weiss DS. (2009) Function and modulation of delta-containing GABA(A) receptors. Psychoneuroendocrinology 34 Suppl 1:S67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]