

Abstract
Objective:
In this study, we evaluated whether variations within genes specifically associated with dyslexia, namely KIAA0319, DCDC2, and CNTNAP2, were associated with greater damage of language-related regions in patients with frontotemporal dementia (FTD) and primary progressive aphasia (PPA) in particular.
Methods:
A total of 118 patients with FTD, 84 with the behavioral variant of FTD (bvFTD) and 34 with PPA, underwent neuropsychological examination, genetic analyses, and brain MRI. KIAA0319 rs17243157 G/A, DCDC2 rs793842 A/G, and CNTNAP2 rs17236239 A/G genetic variations were assessed. Patients were grouped according to clinical phenotype and genotype status (GA/AA or GG). Gray matter (GM) and white matter (WM) differences were assessed by voxel-based morphometry and structural intercorrelation pattern analyses.
Results:
Patients carrying KIAA0319 A* (GA or AA) showed greater GM and WM atrophy in the left middle and inferior temporal gyri, as compared with KIAA0319 GG (p < 0.001). The effect of KIAA0319 polymorphism was mainly reported in patients with PPA. In patients with PPA carrying at-risk polymorphism, temporal damage led to loss of interhemispheric and intrahemispheric GM and WM structural association. No effect of DCDC2 and CNTNAP2 was found.
Conclusions:
Genes involved in dyslexia susceptibility, such as KIAA0319, result in language network vulnerability in FTD, and in PPA in particular.
Frontotemporal lobar degeneration is a heterogeneous neurodegenerative entity encompassing different clinical syndromes in which language impairment is a key symptom.1–4 The evidence from genotype–phenotype correlation studies showing the identical molecular deficit determining different phenotypes5 promoted the hypothesis that specific susceptibility factors interacting with the underlying neurodegenerative process may drive the disease toward different clinical variants. Thus, the phenotypic variability in frontotemporal dementia (FTD) and the selective involvement of specific networks, i.e., the language network, seem to reflect the interaction between individual susceptibility factors and the underlying neurodegenerative process.
Similarly, previous studies have reported an increased frequency of learning and language disabilities in patients with primary progressive aphasias (PPAs) and in their first-degree relatives, claiming that PPA may represent a late-onset presentation of an antecedent selective vulnerability of language network.6,7 This hypothesis was further corroborated by the identification of language-related genes (i.e., FOXP2 gene).8–10
While the FOXP2 gene has been linked to the acquisition of spoken language, other candidate genes were identified as specifically associated with dyslexia, namely KIAA0319, DCDC2, and CNTNAP2.11–13 Their genetic polymorphisms have been demonstrated to affect brain volume and activation of language-related brain areas and to drive hemispheric asymmetry.14–16
It might be hypothesized that these genes modulated regional brain atrophy in FTD, magnifying brain damage in the language-related regions. The present study aimed to test whether variations within dyslexia-related genes were associated with greater vulnerability of language networks in patients with FTD, with particular focus on PPAs.
METHODS
Setting and participants.
Patients fulfilling criteria for FTD1,17 were consecutively recruited from the Centre for Aging Brain and Neurodegenerative Disorders, University of Brescia, Italy, from December 2001 to July 2014. All patients underwent somatic and neurologic evaluation, routine laboratory examination, and a complete mental status evaluation. Each patient was screened for monogenic inherited disease, such as GRN, MAPT, or C9orf72 mutations.
Stringent exclusion criteria were applied as follows: (1) cerebrovascular disorders, previous stroke, hydrocephalus, and intracranial mass documented by MRI; (2) history of traumatic brain injury or another neurologic disease; (3) relevant medical problems (e.g., poorly controlled diabetes or hypertension; cancer within the past 5 years; clinically important hepatic, renal, cardiac, or pulmonary disorders); (4) history of major depressive disorder, bipolar disorder, schizophrenia, substance abuse disorder, or mental retardation according to DSM-IV criteria; (5) CSF β-amyloid/tau amyloid profile of Alzheimer disease type (available in almost 60% of patients) to avoid confounding effects of different neuropathologic substrates; and (6) logopenic variant of PPA (lvPPA).2,17
A comprehensive neuropsychological and behavioral assessment, including Basic Activities of Daily Living and Instrumental Activities of Daily Living, was carried out.18,19 The neuropsychological testing was performed by a standardized neuropsychological battery including Mini-Mental State Examination, Frontotemporal Dementia-modified Clinical Dementia Rating scale (FTD-modified CDR), Raven Colored Progressive Matrices, Controlled Oral Word Association Test and Category Fluency, Clock Drawing Test, Rey Complex Figure Copy and Recall, Story Recall Test, Digit Span, Trail Making Test A and B, Token Test, and De Renzi Imitation Test.20–22 In addition, the Aachener Aphasie Test was further administered to patients with PPA (data not shown).
The lvPPA diagnosis was supported by the pattern of atrophy in posterior/presylvian or parietal regions at the MRI examination; β-amyloid 42 and total tau CSF determinations were also considered. Behavioral disturbances were evaluated by using the Frontal Behavioral Inventory and Neuropsychiatry Inventory.23,24
Standard protocol approvals, registrations, and patient consents.
Informed consent was obtained for blood collection and genetic analyses from each patient. The work conformed to the Helsinki Declaration and was approved by the local Ethics Committee of Brescia Hospital, Italy.
Genetic analyses.
Total genomic DNA was isolated from peripheral blood according to standard procedures. Three single nucleotide polymorphisms located within KIAA0319/TTRAP/THEM2 locus (rs17243157 G/A), DCDC2 (rs793842 A/G), and CNTNAP2 (rs17236239 A/G) genes were evaluated. Primers for each polymorphism are reported in table e-1 at Neurology.org/ng.
The amplification protocols were designed as follows: 5 minutes at 95°C for the first cycle, denaturation at 95°C for 30 seconds, annealing ranging from 59°C to 66°C for 30 seconds (depending on the analyzed polymorphism), extension at 72°C for 30 seconds for the subsequent 35 cycles, and a final extension at 72°C for 5 minutes. The PCR products were analyzed on 2% agarose with 0.005% of ethidium bromide to reveal the reaction and to verify their size.
To evaluate rs17243157 and rs793842 polymorphisms, denaturing high performance liquid chromatography (DHPLC) analysis on the WAVE nucleic acid fragment analysis system was performed (Transgenomic, Santa Clara, CA). Samples with an altered DHPLC profile were purified with Microcon Centrifugal filter devices (Amicon Bioseparations; Millipore Corp, Billerica, MA) and sequenced. Nucleotide direct sequencing was performed on genomic DNA for both strands by the ABI 3500xl DNA analyzer (Applied Biosystems, Foster City, CA) and analyzed using Chromas (Technelysium Pty Ltd, South Brisbane, Australia).
To assess rs17236239, direct sequencing was performed for both strands from purified PCR on the ABI 3500 DNA genetic analyzer (Applied Biosystems), according to the manufacturer's instructions, and analyzed using Chromas (Technelysium Pty Ltd). Genotype analysis was performed blinded to clinical diagnoses.
MRI data acquisition and preprocessing.
In the present study, brain images were collected using 2 different MR scanners: (1) 1.5T MR scanner (Siemens Symphony, Erlangen, Germany), equipped with a circularly polarized transmit–receive coil to acquire 3D magnetization-prepared rapid gradient echo (MPRAGE) T1-weighted scan (repetition time [TR] = 2,010 milliseconds, echo time [TE] = 3.93 milliseconds, matrix = 1× 1× 1×, in-plane field of view [FOV] = 250 × 250 mm2, slice thickness = 1 mm, flip angle = 15°); and (2) 1.5T MR scanner (Siemens Avanto) to acquire 3D MPRAGE T1-weighted scan (TR = 2,050 milliseconds, TE = 2.56 milliseconds, matrix = 1× 1× 1×, in-plane FOV = 256 × 256 mm2, slice thickness = 1 mm, flip angle = 15°).
Preprocessing and statistical analyses were performed using the Statistical Parametric Mapping (SPM8) software package (Wellcome Department of Imaging Neuroscience, London, England, http://www.fil.ion.ucl.ac.uk/spm) running on MATLAB 7.1 (MathWorks, Natick, MA). For voxel-based morphometry (VBM) analysis, the “Segment” module, as previously described, was used.25 Images were spatially normalized to a reference stereotactic template (Montreal Neurological Institute [MNI]) and smoothed by a gaussian kernel of 10 × 10 × 10 mm full width at half maximum.
MRI data analyses.
Gray matter (GM) and white matter (WM) differences according to genotype status for each polymorphism were evaluated by VBM analyses in all FTD groups (behavioral variant of FTD [bvFTD] and PPAs together) and in PPAs, respectively. An analysis of covariance model design was applied by entering 4 groups, i.e., (1) bvFTD carrying at-risk genotype, (2) bvFTD not carrying at-risk genotype, (3) PPA carrying at-risk genotype, and (4) PPA not carrying at-risk genotype, and the covariates of no interest, i.e., age, sex, disease severity (as measured by FTD-modified CDR), presence of monogenic disease, and scanner type. Total intracranial volume, i.e., GM + WM + CSF, was introduced in all the statistical analyses as a global nuisance variable to avoid the confounding effect on cortical atrophy.
Correlation analysis between cognitive performances and regional GM and WM atrophy clusters was carried out. For this, the clusters of GM and WM atrophy obtained in the previous analysis (comparison between the at-risk group and not-at-risk group) were selected as regions of interest (ROIs), and the mean density was calculated for each patient. Linear correlation analysis between ROIs and neuropsychological test scores was computed.
Finally, structural covariance analysis was further performed by means of VBM. This approach relies on the assumption that functionally correlated brain regions show a greater concordance in GM and WM volumes as a result of mutually trophic influences or common experience-related plasticity.26,27 For this, the clusters of GM and WM atrophy obtained in the previous analysis (comparison between the at-risk group and not-at-risk group) were used as “seed” regions, exploring the pattern of covariance between the GM volume of each “seed” and the GM volume throughout the whole brain. For all the aforementioned analyses, the threshold was established at p < 0.001 (uncorrected for multiple comparisons) and a cluster size threshold was set at 200 voxels.
Statistical analysis.
The data were analyzed using SPSS 16.0 software (http://www.spss.com). Patients were analyzed according to clinical phenotype (bvFTD or PPA) or genotype status for each evaluated polymorphism. Genotype distribution and allele frequencies were computed by the χ2 test. Sociodemographic and clinical data were assessed with the Mann–Whitney test and χ2 test, as appropriate. Spearman correlation analyses were carried out and Pearson correlation coefficients were computed.
RESULTS
Patients.
A total of 118 patients fulfilled inclusion/exclusion criteria and entered the present study, namely 84 patients with bvFTD and 34 with PPA (24 agrammatic variant PPA and 10 semantic variant PPA). Demographic and clinical characteristics are reported in table 1. No differences between bvFTD and PPA were found, with the exception of sex and Frontal Behavioral Inventory scores.
Table 1.
Demographic and clinical characteristics of patients with FTD
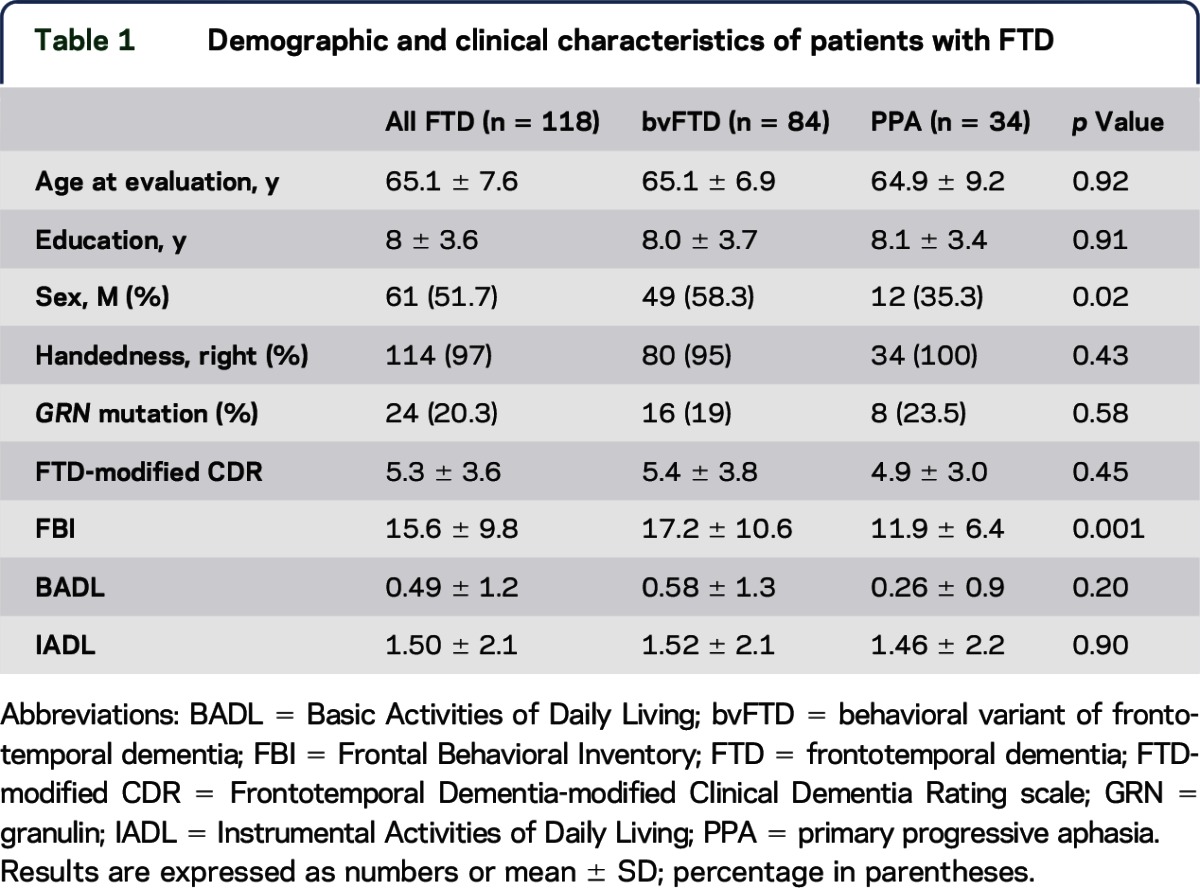
No difference of genotype distribution and allele frequency of KIAA0319 (rs17243157 G/A), DCDC2 (rs793842 A/G), and CNTNAP2 (rs17236239 A/G) polymorphisms between bvFTD and PPA was observed (table e-2).
MRI analyses in patients with FTD.
We evaluated the effect of studied functional polymorphisms on GM damage, i.e., KIAA0319 (GG vs GA/AA), DCDC2 (GG vs GA/AA), and CNTNAP2 (GG vs GA/AA), by VBM. As reported in figure 1A and table e-3, patients carrying KIAA0319 A* (GA or AA) showed greater GM atrophy in the left inferior and middle temporal gyri as compared with KIAA0319 GG.
Figure 1. Gray matter atrophy in patients with FTD according to KIAA0319 genotype.

(A) Gray matter (GM) atrophy in patients with FTD carrying KIAA0319 A* vs KIAA0319 GG (A*<GG). (B) Mean scores obtained by plotting middle temporal gyrus values in the 4 considered groups. (C) Correlation analysis between the GM volume of the middle temporal gyrus and semantic fluency. The results are superimposed on a 2D-standardized T1 brain template. p < 0.001 uncorrected, clusters >200 voxels. Color bar: T values. bvFTD = behavioral variant of frontotemporal dementia; FTD = frontotemporal dementia; PPA = primary progressive aphasia.
As shown in figure 1B, the main effect in the left temporal region was driven more by PPA than by bvFTD. Patients with PPA carrying KIAA0319 A* had the greatest damage in the left middle temporal gyrus compared with patients who had bvFTD carrying KIAA0319 A* and those carrying the GG genotype. The same results were obtained when the left inferior temporal gyrus was considered (data not shown).
GM damage of the aforementioned atrophic regions was related to the decreasing semantic fluency scores (r = 0.37, p < 0.001, figure 1C). No other association between GM damage and neuropsychological tests was found.
The analysis of WM damage in patients with FTD grouped according to KIAA0319 genotype led to comparable results. KIAA0319 A* carriers showed greater WM involvement of the left middle temporal gyrus, as compared with KIAA0319 GG (figure 2A and table e-3). As for GM damage, this effect was mainly due to PPA KIAA0319 A* (figure 2B). WM atrophy of the left middle temporal gyrus was related to the decreasing semantic fluency (r = 0.34, p < 0.001) and short story (r = 0.38, p < 0.001) scores (figure 2C).
Figure 2. White matter atrophy in patients with FTD according to KIAA0319 genotype.

(A) White matter atrophy in patients with FTD carrying KIAA0319 A* vs KIAA0319 GG (A*<GG). (B) Mean scores obtained by plotting middle temporal gyrus values in the 4 considered groups. (C) Correlation analysis between the gray matter volume of the middle temporal gyrus and short story/semantic fluency. The results are superimposed on a 2D-standardized T1 brain template, p < 0.001 uncorrected, clusters >200 voxels. Color bar: T values. bvFTD = behavioral variant of frontotemporal dementia; FTD = frontotemporal dementia; PPA = primary progressive aphasia.
The inverse comparisons (greater GM/WM volume in GG<A*) did not show any clusters above the pre-established threshold. No differences of GM and WM atrophy in patients with FTD grouped according to DCDC2 (GG vs GA/AA) and CNTNAP2 (GG vs GA/AA) genotypes were found at the pre-established threshold.
MRI analyses in PPAs.
As the effect of KIAA0319 polymorphism was mainly found in PPAs, further analyses were carried out in this group.
Patients with PPA carrying KIAA0319 A* had greater involvement of both GM and WM in the left middle temporal gyrus as compared with KIAA0319 GG (figure 3 and table e-3). The inverse comparisons did not yield results at the pre-established threshold.
Figure 3. Gray and white matter atrophy in patients with PPA according to KIAA0319 genotype.

(A) Gray matter (GM) atrophy in patients with primary progressive aphasia (PPA) carrying KIAA0319 A* vs KIAA0319 GG (A*<GG). (B) Correlation analysis between the GM volume of the middle temporal gyrus and semantic fluency/short story. (C) White matter (WM) atrophy in patients with PPA carrying KIAA0319 A* vs KIAA0319 GG (A*<GG). (D) Correlation analysis between the WM volume of the middle temporal gyrus and semantic fluency.
Structural correlation analysis to assess interhemispheric and intrahemispheric connectivity according to KIAA0319 genotype was carried out. In patients with PPA carrying at-risk KIAA0319 A* genotype, decreased structural GM correlation between the left middle temporal gyrus (peak MNI coordinates x, y, z: −56, 2, −20) and other both interhemispheric and intrahemispheric brain regions was highlighted, as compared with patients with PPA carrying KIAA0319 GG. The same pattern was seen for WM density (figure 4 and table e-4).
Figure 4. Gray and white matter structural connectivity analyses in patients with PPA according to KIAA0319 genotype.

Pattern of gray matter (A) and white matter (C) structural correlation of the middle temporal gyrus with other regions of the brain in primary progressive aphasia (PPA) KIAA0319 A* vs PPA KIAA0319 GG. Linear correlation of decreased gray matter (B) and white matter (D) structural association between brain regions in PPA KIAA0319 A* (dashed lines) vs PPA KIAA0319 GG (continuous lines). The results are superimposed on a 3D-standardized T1 brain template. p < 0.001 uncorrected, voxel threshold = 100. Color bar: T values.
DISCUSSION
In the present study, we tested whether genetic variations in specific dyslexia-related genes were involved in modulating the clinical phenotypes in an extensively characterized cohort of patients with FTD.
Dyslexia is known to run in families and has been associated with a number of susceptibility genes.7 It might be hypothesized that in some cases, such genetic predisposition may interfere with initial development of language; in others, the effect may remain dormant for decades but resurface as PPA or as FTD with language disturbances in patients with an ongoing neurodegenerative disease.7 This is even supported by the evidence that different neuropathologic mechanisms can cause PPA, and on the other hand, that an identical neuropathologic entity may lead to PPA, bvFTD, or, in other cases, amnestic dementia. Thus, susceptibility factors interact with the neurodegenerative process to determine the anatomical location.
In our sample, the majority of patients with PPA were women, which suggests the presence of sex-related vulnerability, as already reported in previous studies in which a male predominance in the semantic subtype and female prevalence in the nonfluent subtype were observed.28,29 Moreover, GRN mutations were overrepresented in patients with language variants, which reflects a peculiar role of the GRN gene in language networks.30
To further explore genetic susceptibility to language impairment in FTD, functional genetic variations within 3 genes involved in language disabilities, in particular KIAA0319 rs17243157 G/A, DCDC2 rs793842 A/G, and CNTNAP2 rs17236239 A/G, were analyzed with respect to their effect on brain structures and the correlation with cognitive performances. It is noteworthy that, of all 3 genes, only the KIAA0319 A* (GA or AA) genotype was demonstrated to influence brain damage in language areas, therefore supporting the specificity of the results. No specific patterns of greater atrophy were identified as associated with DCDC2 and CNTNAP2 polymorphisms, although we cannot exclude the possibility that with a larger patient sample or different approaches, an effect of these genes could also be found in patients with FTD.
KIAA0319 functional polymorphism was shown to determine GM atrophy in the left inferior and middle temporal gyri and WM involvement in the middle temporal gyrus and to be associated with the semantic fluency test and the short story scores. Of note, these results were stronger in patients with PPA than in those with bvFTD. Furthermore, in patients with PPA carrying the at-risk KIAA0319 polymorphism, damage of the left temporal lobe showed an interhemispheric and intrahemispheric breakdown with the rest of the brain.
The areas found to be affected by KIAA0319 genetic variation are known to be one of the hubs of phonological, articulatory planning, and syntactic processes and are most notably affected in patients with dyslexia.31 Accordingly, several independent studies reported that KIAA0319 allelic variants were associated with different reading and spelling abilities,13,32 being able to reduce KIAA0319 gene expression, and affecting neuronal migration in the developing neocortex.14 In healthy controls, KIAA0319 polymorphism was shown to influence asymmetric functional activation of superior temporal sulcus, suggesting its role in modulation and lateralization of phonemic process.15
Our results further contribute to understanding of the effect of KIAA0319 on language networks in patients with FTD and have major relevance for those who present prominent language deficits at disease onset. A major limitation of the study is the lack of neuropathologic confirmation. Indeed, the results we reported are not necessarily about PPA caused by a specific neuropathologic substrate, and further studies could explore whether the dyslexia genes may also influence brain atrophy in other neurodegenerative conditions, including lvPPA and Alzheimer dementia. Unfortunately, we have no reliable information on learning disabilities in these patients,33 in part because the diagnosis of learning disabilities is relatively recently recognized.
These findings shed light on genetic predisposition in defining how a disease process becomes distributed asymmetrically and how it targets language networks. Indeed, the identification of a possible genetic risk factor for language deficits in FTD represents a possible molecular target for the development of disease biomarkers or future therapeutic strategies.
Supplementary Material
GLOSSARY
- bvFTD
behavioral variant of frontotemporal dementia
- DHPLC
denaturing high performance liquid chromatography
- DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, 4th edition
- FOV
field of view
- FTD
frontotemporal dementia
- FTD-modified CDR
Frontotemporal Dementia-modified Clinical Dementia Rating scale
- GM
gray matter
- lvPPA
logopenic variant of PPA
- MNI
Montreal Neurological Institute
- MPRAGE
3D magnetization-prepared rapid gradient echo
- PPA
primary progressive aphasia
- ROI
region of interest
- TE
echo time
- TR
repetition time
- VBM
voxel-based morphometry
- WM
white matter
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, and/or acquisition of data, and/or analysis and interpretation of data. Donata Paternicó: first draft of the manuscript, imaging analysis, and interpretation of data. Enrico Premi: imaging analysis and interpretation of data. Antonella Alberici: first draft of the manuscript, patient cohort, and analysis and interpretation of data. Silvana Archetti: genetic analysis and critical revision of the manuscript for content. Elisa Bonomi: genetic analysis and critical revision of the manuscript for content. Vera Gualeni: imaging analysis and critical revision of the manuscript for content. Roberto Gasparotti: critical revision of the manuscript for content. Alessandro Padovani: patient cohort and critical revision of the manuscript for content. Barbara Borroni: first draft of the manuscript, patient cohort, analysis and interpretation of data, and study conceptualization and design.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harciarek M, Cosentino S. Language, executive function and social cognition in the diagnosis of frontotemporal dementia syndromes. Int Rev Psychiatry 2013;25:178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies RR, Kipps CM. Lobar atrophy in frontotemporal dementia: diagnostic and prognostic implications. Curr Alzheimer Res 2011;8:261–265. [DOI] [PubMed] [Google Scholar]
- 5.Josephs KA, Hodges JR, Snowden JS, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122:137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogalski E, Johnson N, Weintraub S, Mesulam M. Increased frequency of learning disability in patients with primary progressive aphasia and their first-degree relatives. Arch Neurol 2008;65:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogalski E, Weintraub S, Mesulam MM. Are there susceptibility factors for primary progressive aphasia? Brain Lang 2013;127:135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcus GF, Fisher SE. FOXP2 in focus: what can genes tell us about speech and language? Trends Cogn Sci 2003;7:257–262. [DOI] [PubMed] [Google Scholar]
- 9.Padovani A, Cosseddu M, Premi E, et al. The speech and language FOXP2 gene modulates the phenotype of frontotemporal lobar degeneration. J Alzheimers Dis 2010;22:923–931. [DOI] [PubMed] [Google Scholar]
- 10.Premi E, Pilotto A, Alberici A, et al. FOXP2, APOE, and PRNP: new modulators in primary progressive aphasia. J Alzheimers Dis 2012;28:941–950. [DOI] [PubMed] [Google Scholar]
- 11.Meng H, Smith SD, Hager K, et al. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci U S A 2005;102:17053–17058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet 2014;22:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harold D, Paracchini S, Scerri T, et al. Further evidence that the KIAA0319 gene confers susceptibility to developmental dyslexia. Mol Psychiatry 2006;11:1085–1091, 1061. [DOI] [PubMed] [Google Scholar]
- 14.Paracchini S, Thomas A, Castro S, et al. The chromosome 6p22 haplotype associated with dyslexia reduces the expression of KIAA0319, a novel gene involved in neuronal migration. Hum Mol Genet 2006;15:1659–1666. [DOI] [PubMed] [Google Scholar]
- 15.Pinel P, Fauchereau F, Moreno A, et al. Genetic variants of FOXP2 and KIAA0319/TTRAP/THEM2 locus are associated with altered brain activation in distinct language-related regions. J Neurosci 2012;32:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darki F, Peyrard-Janvid M, Matsson H, Kere J, Klingberg T. Three dyslexia susceptibility genes, DYX1C1, DCDC2, and KIAA0319, affect temporo-parietal white matter structure. Biol Psychiatry 2012;72:671–676. [DOI] [PubMed] [Google Scholar]
- 17.McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol 2001;58:1803–1809. [DOI] [PubMed] [Google Scholar]
- 18.Katz S, Ford AB, Moskowitz RW, Jackson BA, Jaffe MW. Studies of illness in the aged. The index of ADL: a standardized measure of biological and psychosocial function. JAMA 1963;185:914–919. [DOI] [PubMed] [Google Scholar]
- 19.Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist 1969;9:179–186. [PubMed] [Google Scholar]
- 20.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 21.Knopman DS, Kramer JH, Boeve BF, et al. Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain 2008;131:2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lezak MD. Neuropsychological Assessment: Oxford: Oxford U Press; 1976. [Google Scholar]
- 23.Kertesz A, Davidson W, Fox H. Frontal Behavioral Inventory: diagnostic criteria for frontal lobe dementia. Can J Neurol Sci 1997;24:29–36. [DOI] [PubMed] [Google Scholar]
- 24.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308–2314. [DOI] [PubMed] [Google Scholar]
- 25.Borroni B, Alberici A, Cercignani M, et al. Granulin mutation drives brain damage and reorganization from preclinical to symptomatic FTLD. Neurobiol Aging 2012;33:2506–2520. [DOI] [PubMed] [Google Scholar]
- 26.Fornito A, Zalesky A, Breakspear M. The connectomics of brain disorders. Nat Rev Neurosci 2015;16:159–172. [DOI] [PubMed] [Google Scholar]
- 27.Montembeault M, Joubert S, Doyon J, et al. The impact of aging on gray matter structural covariance networks. Neuroimage 2012;63:754–759. [DOI] [PubMed] [Google Scholar]
- 28.Johnson JK, Diehl J, Mendez MF, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol 2005;62:925–930. [DOI] [PubMed] [Google Scholar]
- 29.Rogalski E, Rademaker A, Weintraub S. Primary progressive aphasia: relationship between gender and severity of language impairment. Cogn Behav Neurol 2007;20:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohrer JD, Crutch SJ, Warrington EK, Warren JD. Progranulin-associated primary progressive aphasia: a distinct phenotype? Neuropsychologia 2010;48:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Binder JR, Desai RH, Graves WW, Conant LL. Where is the semantic system? A critical review and meta-analysis of 120 functional neuroimaging studies. Cereb Cortex 2009;19:2767–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paracchini S, Steer CD, Buckingham LL, et al. Association of the KIAA0319 dyslexia susceptibility gene with reading skills in the general population. Am J Psychiatry 2008;165:1576–1584. [DOI] [PubMed] [Google Scholar]
- 33.Rogalski EJ, Rademaker A, Wieneke C, Bigio EH, Weintraub S, Mesulam MM. Association between the prevalence of learning disabilities and primary progressive aphasia. JAMA Neurol 2014;71:1576–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.