

Abstract
Objective:
To identify the genetic cause of isolated inclusion body myopathy (IBM) with autosomal dominant inheritance in 2 families.
Methods:
Genetic investigations were performed using whole-exome and Sanger sequencing of the heterogeneous nuclear ribonucleoprotein A1 gene (hnRNPA1). The clinical and pathologic features of patients in the 2 families were evaluated with neurologic examinations, muscle imaging, and muscle biopsy.
Results:
We identified a missense p.D314N mutation in hnRNPA1, which is also known to cause familial amyotrophic lateral sclerosis, in 2 families with IBM. The affected individuals developed muscle weakness in their 40s, which slowly progressed toward a limb-girdle pattern. Further evaluation of the affected individuals revealed no apparent motor neuron dysfunction, cognitive impairment, or bone abnormality. The muscle pathology was compatible with IBM, lacking apparent neurogenic change and inflammation. Multiple immunohistochemical analyses revealed the cytoplasmic aggregation of hnRNPA1 in close association with autophagosomes and myonuclei. Furthermore, the aberrant accumulation was characterized by coaggregation with ubiquitin, sequestome-1/p62, valosin-containing protein/p97, and a variety of RNA-binding proteins (RBPs).
Conclusions:
The present study expands the clinical phenotype of hnRNPA1-linked multisystem proteinopathy. Mutations in hnRNPA1, and possibly hnRNPA2B1, will be responsible for isolated IBM with a pure muscular phenotype. Although the mechanisms underlying the selective skeletal muscle involvement remain to be elucidated, the immunohistochemical results suggest a broad sequestration of RBPs by the mutated hnRNPA1.
Multisystem proteinopathy (MSP) is an inherited pleiotropic degenerative disorder that can affect muscle, bone, and/or the nervous system. MSP is genetically heterogeneous and has been associated with mutations in VCP,1 hnRNPA1,2 hnRNPA2B1,2 SQSTM1/p62,3 and MATR3 genes.4,5
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are ubiquitously expressed RNA-binding proteins (RBPs) that are involved in messenger RNA metabolism and transport. In 2013, novel missense mutations in the prion-like domain (PrLD) of hnRNPA1 and hnRNPA2B1 genes were identified in patients with MSP/amyotrophic lateral sclerosis (ALS),2 which were later designated as MSP3 and MSP2, respectively.6 Subsequent to the discovery of hnRNPA1 and hnRNPA2B1 mutations, screening of these genes resulted in no additional mutation in Dutch,7 French,8 and Italian9 patients with ALS, frontotemporal dementia, inclusion body myopathy (IBM), or MSP, which suggests that MSP3 and MSP2 are rarer than expected.
We identified an MSP3-linked hnRNPA1 mutation by exome sequencing and segregating study in 2 unrelated Japanese families that presented with dominantly inherited IBM without bone or CNS involvement. The pure muscular phenotype is distinct from that of previously described MSP3 cases and is believed to be a novel MSP3 phenotype. In this study, we report clinical, genetic, and histopathologic features of the 2 pedigrees.
METHODS
Patients.
Family 1 included 6 patients (4 male and 2 female) in 3 successive generations; family 2 included 11 patients (7 male and 4 female) in 4 successive generations (figure 1A). Both families were of Japanese ancestry; no consanguineous or international mating was found. Of all patients, 6 individuals (II-13, III-1, III-2, and III-6 in family 1; IV-1 and IV-2 in family 2) were physically and neurologically examined; 4 of them (III-1 and III-2 in family 1; IV-1 and IV-2 in family 2) were also evaluated by electrophysiology, muscle imaging, and biochemical testing. In addition, 4 patients (III-1 and III-2 in family 1; IV-1 and IV-4 in family 2) underwent muscle biopsy.
Figure 1. Family pedigrees, muscle imaging, and genetic investigation.

(A) Family pedigrees are shown. Filled-in symbols indicate individuals with muscle weakness. Empty symbols indicate unaffected individuals without any medical history or related complaint of muscle weakness, dementia, or bone disease. Asterisks indicate individuals whose DNA was used for this study. In family 1, DNA was used for whole-exome sequencing and segregating study. In family 2, it was used for Sanger sequencing. Arrows and arrowheads indicate individuals who underwent clinical examination and muscle biopsy, respectively. (B) Muscle CT was evaluated 8 years after onset in patient III-1 and 10 years after onset in the other patients. The images show slices of the trunk (first row), proximal arms (second row), and proximal (third row) and distal legs (fourth row). At the level of the trunk, all of the patients exhibited moderate-to-severe atrophy in the paraspinal (P) muscles. At the level of the proximal arms, the biceps brachii (BB) was commonly affected, although the triceps brachii (TB) and brachioradialis (BR) were spared. At the level of the proximal legs, the biceps femoris (BF), semimembranosus (SM), adductor magnus (AM), and vastus intermedius (VI) were predominantly affected. Less predominant atrophy was observed in the sartorius (SA), semitendinosus (ST), vastus lateralis (VL), and vastus medialis (VM) muscles. The atrophy of the semitendinosus muscle of patient IV-1 appeared asymmetrical. The rectus femoris (RF) and gracilis (G) muscles were relatively spared in patients III-1 and III-2, whereas they appeared to be affected later in patients IV-1 and IV-2. At the level of the distal legs, the soleus (S) was severely affected in all patients. In contrast, the peroneus longus (PL) gastrocnemius (GC), and tibialis posterior (TP) muscles were relatively spared. The tibialis anterior (TA) and extensor digitorum longus (EDL) muscles were affected to varying degrees in each individual. (C) The sequence of the identified hnRNPA1 mutation and its conservation among species are shown. Sanger sequencing confirmed the heterozygous G to A substitution (indicated by arrows) at the position chr12: 54,677,628, which corresponds to c. 940G>A in exon 9 (NM_031157). The substitution leads to p.D314N (NP_112420), and this amino acid is conserved among species.
Standard protocol approvals, registrations, and patient consents.
This study was approved by the Ethics Committee of the Tohoku University School of Medicine; all individuals provided informed consent prior to their inclusion in the study.
Muscle histopathology and immunohistochemistry.
Biopsied skeletal muscles were rapidly frozen with isopentane cooled with liquid nitrogen; a section of tissues was fixed in 2.5% glutaraldehyde, postfixed with 1% OsO4, embedded in epoxy resin, and subjected to light and electron microscopy according to standard procedures. We performed single and multiple immunohistochemistry as previously described (e-Methods at Neurology.org/ng).10,11 Primary and secondary antibodies used in this study are listed in table e-1. Biopsied muscle specimens from patients without signs of neuromuscular diseases were used as controls.
Genetic analysis.
Exome sequencing.
Exome sequencing was performed on 6 family members in family 1 (figure 1A), 4 of whom were affected. Exon capture was performed using the SureSelect Human All Exon kit v5 (Agilent Technologies, Santa Clara, CA). Exon libraries were sequenced using the Illumina HiSeq 2500 platform according to the manufacturer's instructions (Illumina, San Diego, CA). Paired 101-base pair reads were aligned to the reference human genome (UCSChg19) using the Burrows-Wheeler Alignment Tool.12 Likely PCR duplicates were removed using the Picard program (http://broadinstitute.github.io/picard/). Single nucleotide variants (SNVs) and indels were identified using Genome Analysis Toolkit v1.6 software.13 SNVs and indels were annotated against the RefSeq database and dbSNP135 with the ANNOVAR program.14
Sanger sequencing.
To confirm that mutations identified by exome sequencing segregated with the disease, we performed Sanger sequencing. PCR was performed with the primers shown in table e-1. PCR products were purified using a MultiScreen PCR plate (Millipore, Billerica, MA) followed by sequencing using 3500xL genetic analyzer (Thermo Fisher Scientific, Waltham, MA).
RESULTS
Clinical features.
Clinical, laboratory, and electrophysiologic data of patients III-1 and III-2 in family 1 and patients IV-1 and IV-2 in family 2 are summarized in table 1. Clinical and pathologic findings of patient III-1 in family 1, who presented with slowly progressive weakness and atrophy in the scapular, proximal, and distal lower limb muscles since his 40s, were described in our previous report,15 Although his symptoms initially mimicked distal myopathy with rimmed vacuoles,15 the affected muscle distribution later advanced to a typical limb-girdle pattern.
Table 1.
Clinical, laboratory, and electrophysiologic data of the affected individuals
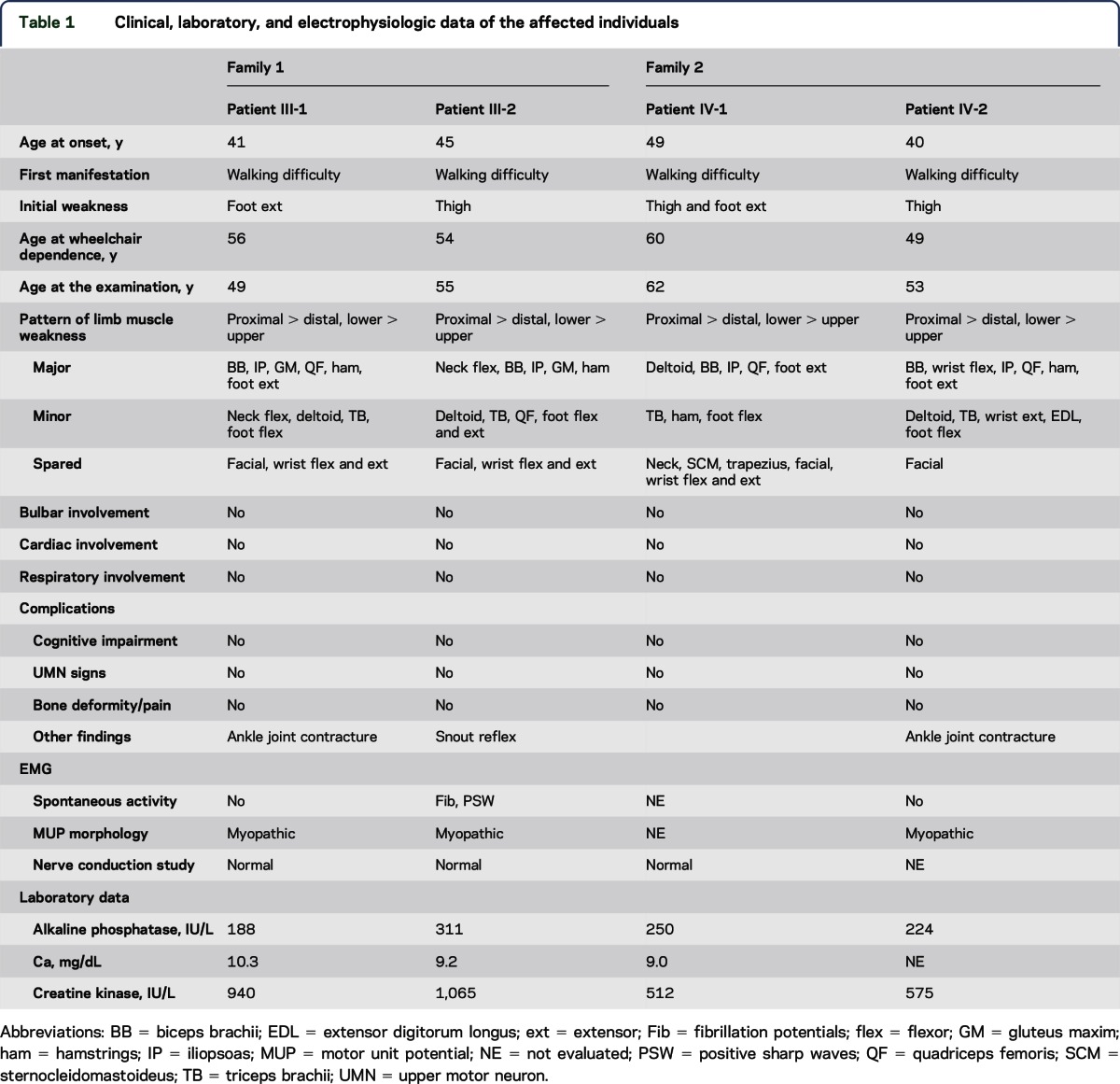
Patient III-2 in family 1 noticed muscle weakness in his thighs while walking at age 45. The muscle weakness gradually spread to proximal upper limbs, trunk, and distal lower limbs. He required a walking aid at age 52 and became wheelchair dependent at age 54. No developmental disability, skeletal deformity, or contracture was found. Neurologic examination revealed no abnormality in his cranial nerves or his sensory, autonomic, and coordination systems. Snout reflex was positive only in this patient (table 1); however, other frontal release signs and cognitive/behavioral impairment were not detected. His weakness and atrophy were predominant in neck and limb-girdle muscles (table 1). Fasciculation was not observed in these muscles. Tendon reflexes and muscle tonus were diffusely decreased. The serum creatine kinase (CK) level was moderately elevated (approximately 1,000 IU/L); both alkaline phosphatase and calcium levels were within normal ranges. Cardiac and respiratory functions were normal. Needle EMG revealed mild spontaneous activity, such as positive sharp waves and fibrillation potentials, in the affected biceps brachii and tibialis anterior muscles. However, motor unit potentials generally indicated a myogenic pattern, characterized by normal duration, low amplitude, early recruitment, and reduced interference patterns. Brain CT did not indicate any pathologic atrophy. Furthermore, x-rays of the skull, spine, and pelvis did not reveal bone disorganization resembling Paget disease of bone (PDB).
Clinical profiles of patients IV-1 and IV-2 in family 2 were similar to each other. They experienced walking difficulty, particularly in climbing stairs, around their 40s. They became wheelchair dependent 10 years after the clinical onset. Their weakness was detected mainly in the iliopsoas, quadriceps femoris, hamstrings, tibialis anterior, and biceps brachii muscles. Tendon reflexes were generally decreased. No other abnormality was evident on physical and neurologic examination. Their serum CK levels were mildly elevated (approximately 500 IU/L); alkaline phosphatase levels were within the normal range. X-rays did not reveal bone disorganization. Needle EMG in patient IV-2 indicated a generally myogenic pattern without spontaneous activity in the affected muscles. Motor and sensory nerve conduction in patient IV-1 was normal.
Muscle imaging.
Muscle CT revealed a similar distribution of affected muscles among patients in families 1 and 2, presenting a remarkable amyotrophy in the paraspinal, biceps brachii, biceps femoris, semimembranosus, adductor magnus, vastus intermedius, and soleus muscles (figure 1B). Less predominant atrophy was distributed in the sartorius, semitendinosus, vastus lateralis and medialis, tibialis anterior, and extensor digitorum longus muscles. This muscle atrophy was essentially symmetrical. The triceps brachii, brachioradialis, rectus femoris, gracilis, peroneus longus, gastrocnemius, and tibialis posterior muscles were relatively spared.
Muscle histopathology.
In family 1, the obvious finding in patients III-1 and III-2 was rimmed vacuoles, which were found in 7.0% and 3.7% of myofibers, respectively. The rimmed vacuoles were mainly identified in atrophic fibers (figure 2C). In addition, chronic myopathic changes, including increased endomysial fibrosis, highly increased variability in the size of myofibers, and increased central nuclei, were found (figure 2A). There was a small number of regenerated fibers. In contrast, inflammatory cell infiltration and apparent neurogenic change were absent. Immunohistochemistry revealed that α-sarcoglycan, α-dystroglycan, caveolin-3, emerin, and dystrophin were normally expressed (data not shown). Dysferlin expression was decreased in patient III-115 and normal in patient III-2 (data not shown). Electron microscopic examination of the patient III-2 specimen revealed the presence of membrane-bound vacuoles containing myeloid bodies (figure 3, A and B). The vacuoles were located among myofibrils and were often adjacent to irregularly shaped or indented myonuclei (figure 3, C–F). We could not find nuclear or cytoplasmic tubulofilamentous inclusions in this study.
Figure 2. Muscle histopathology.

Hematoxylin & eosin (A, B), modified Gomori trichrome (C, D), Nicotinamide adenine dinucleotide–tetrazolium reductase (E, F), cytochrome c oxidase (COX) (G, H), serial, and ATPase pH4.5 (I, J) staining of the biopsied muscle samples from the biceps brachii of patient III-2 in family 1 (left panels, A, C, E, G, I) and the triceps brachii of patient IV-1 in family 2 (right panels, B, D, F, H, J) are shown. Hypertrophic fibers larger than 100 μm and angulated or rounded atrophic fibers are shown (A, B). Highly increased central nuclei and a fraction of fibers with pyknotic nuclear clumps are present (B). Rimmed vacuoles are located in atrophic fibers, which tend to make small groups (C, D). Disorganization of the myofibrillar network is observed (F). COX staining does not display complete COX-deficient myofibers (G, H). On ATPase pH4.5, the type distribution is almost equal, and atrophic fibers are observed in both type 1 and type 2 fibers (I, J). Type grouping is essentially negative except for the subtle finding of several type 1 fibers making a small group (I, J). Scale bars = 100 μm.
Figure 3. Ultrastructural analysis of muscle.

Ultrastructural findings of patient III-2 in family 1 are shown. The enclosed section of E is enlarged in F. The autophagic vacuoles containing myeloid bodies and glycogen granules are located among myofibrils (A, B), and neighboring myonuclei (C–F). Myonuclei (indicated by N) located in subsarcolemma (C) or sarcoplasm (D) are irregularly shaped and have indentation (an arrow). Scale bars are embedded in each panel.
Muscle pathology of patients IV-1 and IV-4 in family 2 was essentially identical to that observed in patients in family 1. Rimmed vacuoles were detected in atrophic fibers along with a chronic myopathic change (figure 2D). Nicotinamide adenine dinucleotide–tetrazolium reductase staining disclosed disorganization of the intermyofibrillar network (figure 2F). Inflammation and apparent neurogenic changes were not detected.
Genetic investigation.
We identified heterozygous nonsynonymous variants, which were common among patients and absent in unaffected individuals, using whole-exome sequencing. After filtering against the 1000 Genomes (http://www.1000genomes.org/) and Human Genetic Variation Databases (http://www.genome.med.kyoto-u.ac.jp/SnpDB/), 3 novel (i.e., absent according to the 2 aforementioned databases) variants were identified: c.76G>A, p.D26N in HIST1H2BJ encoding histone H2B type J; c.202C>T, p.R68W in HIST1H4I encoding histone H4I; and c.940G>A, p.D314N in hnRNPA1. Because the p.D314N and p.D314V mutations in hnRNPA1 are known to cause familial ALS and MSP3, respectively,2 we then performed segregation analysis on this candidate through Sanger sequencing in 6 family members analyzed by exome sequencing. This candidate (c.940G>A in hnRNPA1 [NM_031157]) was detected in all patients (n = 4) and was not detected in any of the unaffected family members (n = 2) or in 190 ethnically matched control individuals (380 chromosomes) (figure 1C). These results suggested that the rare mutation identified in hnRNPA1 segregated with the disease in family 1.
All previously reported mutations in hnRNPA1 and hnRNPA2B1 were consistently located on the core PrLD of the hnRNPs.2 We then analyzed the sequence-encoding core PrLDs of hnRNPA1 (nucleotides 697–972 in NM_031157, which encode codons 233–324 in NP_112420) and hnRNPA2B1 (nucleotides 796–909 in NM_002137, which encode codons 266–303 in NP_002128) in patient III-1 in family 1 and patients IV-1 and IV-2 in family 2. Sanger sequencing confirmed that p.D314N in hnRNPA1 is the only mutation across the above-mentioned sequence in patient III-1 in family 1 and revealed that patients IV-1 and IV-2 in family 2 also harbor the same mutation.
Multiple immunohistochemistry.
To define hnRNPA1 protein localization with its related proteins, we performed multiple immunofluorescence in biopsied muscle specimens from patient III-2 in family 1; samples from other patients in the 2 families were unavailable. The major finding was sarcoplasmic, subsarcolemmal, and perinuclear aggregation of hnRNPA1 in atrophic fibers and in fibers with rimmed vacuoles (figure 4, A–H). Atrophic fibers frequently retained transactive response DNA-binding protein 43 kDa (TDP-43)/ubiquitin (Ub) double-positive cytoplasmic aggregates with the nuclear depletion of TDP-43 (figure 4, I–L). In association with multiple rimmed vacuoles, the mislocalized TDP-43 was occasionally phosphorylated at Ser409/Ser410 residues; it also colocalized with sequestome-1 (SQSTM1/p62) and hnRNPA1 (figure 4, M–P). The rimmed vacuoles were Ub- and SQSTM1/p62-positive; some were stained with anti-SQSTM1/p62 phosphorylated at the Ser403 residue (figure 4, Q–T). The MSP2-linked gene product hnRNPA2B1 was also detected in Ub-positive aggregates in atrophic or rimmed vacuole–carrying fibers (figure 4, U–X). hnRNPA1 and A2B1 were generally colocalized (figure e-1, I–L); a discrete aggregation of the 2 hnRNPs occurred infrequently (figure e-1, M–P). Mislocalization and deprivation of hnRNPA1 or hnRNPA2B1 from the myonuclei were rarely found in extremely atrophied fibers (figure e-1, M–P). Neonatal myosin heavy chain–positive regenerative fibers also exhibited the hnRNPA1 aggregation pathology (data not shown).
Figure 4. Multiple immunofluorescence for hnRNPA1 and related proteins.

Representative microphotographs of transverse cryosections from the biopsied skeletal muscle of patient III-2 in family 1. (A–H) Aberrant subsarcolemmal/perinuclear aggregation (arrowheads) and increased sarcoplasmic retention of heterogeneous nuclear ribonucleoprotein (hnRNPA1) were mainly evident in atrophic fibers. Inset in H is a higher magnification of the boxed area (scale bar = 10 μm). The subsarcolemmal/perinuclear hnRNPA1 aggregates were often colocalized with ubiquitin (Ub, A–D, arrowheads) and SQSTM1/p62 (E–H, arrows). Note the close association of hnRNPA1/Ub double-positive aggregation with the rimmed vacuole (A–D, arrows). (I–L) In the atrophic fibers, transactive response DNA binding protein 43 kDa (TDP-43)/Ub double-positive aggregation (arrowheads) was also observed with the cytoplasmic mislocalization and nuclear depletion of TDP-43 (arrows). (M–P) The aberrant aggregation of hnRNPA1 was occasionally triple-labeled with phosphorylated TDP-43 and sequestome-1/p62 (SQSTM1/p62), closely adjacent to rimmed vacuoles (arrows). (Q–X) The rimmed vacuoles were often related to Ub, phosphorylated SQSTM1/p62 (Q–T, arrows), and hnRNPA2B1 (U–X, asterisks indicate a rimmed vacuole–carrying fiber). TO-PRO-3: nuclear staining (C, G, K, W). Scale bars = 50 μm.
On the basis of the aforementioned findings with hnRNPA1, hnRNPA2B1, TDP-43, SQSTM1/p62, and Ub pathology, we further examined the possible coaggregation/mislocalization of MSP/ALS-linked proteins. Fused in sarcoma/translated in liposarcoma (FUS/TLS), TATA-binding protein–associated factor 2N, and Ewing sarcoma breakpoint region 1, known as FET family proteins harboring the PrLD, were colocalized with Ub- or SQSTM1/p62-positive sarcoplasmic aggregates in atrophic fibers (figure 5, A–H) and in rimmed vacuoles (figure 5, I–L). However, FUS/TLS aggregation was substantially minimal; nuclear clearance of the FET proteins was lacking. Valosin-containing protein (VCP)/p97 was also colocalized with SQSTM1/p62-positive hnRNPA1 aggregates closely related to rimmed vacuoles (figure 5, M–P). Furthermore, matrin-3 formed subsarcolemmal SQSTM1/p62-labeled clumps (figure 5, Q–T). However, loss of matrin-3 immunoreactivity in the myonuclei was essentially absent. All aberrant aggregations observed in patient III-2 in family 1 were negative in control specimens (data not shown).
Figure 5. Multiple immunohistochemistry for various RNA-binding proteins and VCP/p97.

Representative microphotographs of transverse cryosections from the specimens of patient III-2 in family 1. (A–D) Note the perinuclear and subsarcolemmal aggregation of fused in sarcoma/translated in liposarcoma (FUS/TLS) with sparse staining in cytoplasm (arrows), whereas FUS/TLS mislocalization was scarcely observed. (E–H) In atrophic fibers (asterisks), diffuse cytoplasmic expression and extranuclear/subsarcolemmal aggregation of TATA-binding protein–associated factor 2N (TAF15) were frequently found. The aggregation is partially colabeled with ubiquitin (arrowheads). (I–L) In addition to heterogeneous nuclear ribonucleoprotein (hnRNP) A1 and A2B1, Ewing sarcoma breakpoint region 1 (EWSR1) and sequestome-1/p62 (SQSTM1/p62) double-positive aggregation was observed in rimmed vacuoles (arrows). (M–P) Note the multisystem proteinopathy 1–linked valosin-containing protein (VCP)/p97, SQSTM1/p62, and hnRNPA1 triple-labeled aggregates in the rimmed vacuoles (arrows). (Q–T) In atrophic fibers, amyotrophic lateral sclerosis/distal myopathy–linked matrin-3 was aberrantly involved in the subsarcolemmal SQSTM1/p62-positive aggregates (arrowheads). Scale bars = 50 μm.
DISCUSSION
We found that the missense mutation c.940G>A, p.D314N in hnRNPA1 segregated with IBM, an additional pure muscular phenotype of MSP3, in 2 unrelated Japanese families. The identified mutation in hnRNPA1 was considered to be pathogenic for the following reasons: (1) exome sequencing revealed that this was one of the best candidate mutations after filtering single nucleotide polymorphisms and indels; (2) no pathogenic variants were detected among known causative genes of dominantly inherited myopathy with rimmed vacuoles, such as VCP,1 MYH2,16 MYOT,17 DES,18 CRYAB,19 TTN,20 PABP2,21 and MATR3,4 in exome sequencing; (3) it was common between 2 independent families with dominantly inherited IBM; (4) it was not detected in 190 ethnically matched control individuals; and (5) this mutation and the substitute on the identical residue are known to be causative of familial ALS and MSP3, respectively.2
All patients included in this study had nearly identical clinical features, such as autosomal dominant inheritance with probable high penetrance, late adult-onset slowly progressive myopathy with predominant limb-girdle weakness, absence of cognitive/motor neuron/bone tissue involvement, mild-to-moderate elevation of serum CK, and histologic findings compatible with IBM (table 1, figure 2). The p.D314N mutation in hnRNPA1 was reported to cause familial ALS in a previous report.2 In contrast to that report, motor neuron disease was fundamentally negative in our patients. The different substitute on the identical residue p.D314V in hnRNPA1 was linked to late-onset autosomal dominant limb-girdle muscular dystrophy accompanied by PDB.2,22 The muscular phenotype caused by the hnRNPA1 p.D314V mutation is similar to that of our patients with regard to age at onset, progression course, and affected muscle distribution. However, the bone involvement in patients with p.D314V mutations accentuates the phenotypic difference between the previously reported cases and the cases presented in this study. In this context, the isolated muscle involvement is extremely distinct from that of the previously reported cases and is believed to be a novel pure phenotype of MSP3.
The affected muscle distribution revealed by muscle imaging is similar to that of previously reported patients with MSP322 and MSP4,3 particularly in the significant involvement of biceps femoris and soleus with relatively spared rectus femoris and tibialis posterior muscles. However, the distribution pattern is highly diverse even in VCP-linked MSP123,24 and also in various rimmed vacuolar myopathies including IBM with GNE mutations and sporadic inclusion body myositis. Nonetheless, the aforementioned resemblance, at least among the cases with MSP3, suggests a possible association between myotoxicity by the mutant hnRNPA1 and vulnerability in each part of muscles. A possible common feature of the vulnerable muscles, such as expression level of mutated/associated proteins, muscle fiber–type composition, and energy metabolism, remains to be clarified.
Muscle histopathology of the 2 families was compatible with IBM. Common features were increased rimmed vacuoles, absence of inflammation and apparent neurogenic change, and slight muscle fiber necrosis and regeneration. The involvement of neurogenic atrophy was not completely excluded because of angulated fibers (figure 2A), pyknotic nuclear clumps (figure 2B), and several type 1 fibers making a small group (figure 2, I and J), whereas our specimen lacked the findings of large group atrophy or apparent fiber type grouping. These pathologic findings share a close resemblance to those of IBM with the p.D314V hnRNPA1 mutation.22 We have clearly identified autophagic vacuoles containing myeloid bodies, which often neighbored irregularly shaped or indented myonuclei. Similar pathologic findings were also reported in patients with a mutation in MATR3.4,5 In addition, enlarged myonuclear pores and amorphous materials along the nuclear membrane were reported in a patient with MSP3 with the p.D314V hnRNPA1 mutation.22 Therefore, the question of whether this perinuclear pathology reflects possible dysfunction of RBPs and myonuclei is of interest.
Cytoplasmic retention and subsarcolemmal/perinuclear aggregation of the hnRNPA1 protein in degenerating myofibers were prominent features revealed by immunohistochemistry (figure 4). The distribution of hnRNPA1-postitive aggregates corresponds closely to clusters of autophagic vacuoles ultrastructurally. Moreover, the aggregated hnRNPA1 frequently coexisted with Ub and an autophagic substrate, (Ser403-phosphorylated) SQSTM1/p62. These congruent events suggest an aberrantly increased burden triggered by the hnRNPA1 mutation on the intrinsic protein degradation system, including selective autophagy.25
TDP-43 pathology comprising cytoplasmic aggregation and nuclear exclusion was also abundant in our study (figure 4), consistent with previous observations in patients carrying the p.D314V hnRNPA1 mutation.2 Despite the resemblance, mislocalization or clearance of myonuclear hnRNPA1/A2B1 was barely found in our specimens (figure e-1), suggesting sarcoplasmic pathology rather than defective physiologic function in myonuclei as a possible primary event by mutant hnRNPA1. Nonetheless, the cytoplasmic aggregation and nuclear depletion of hnRNPA1, hnRNPA2B1, and TDP-43 are, to a varying degree, pathologic hallmarks in common with muscle degeneration in MSP1–3 and in sporadic inclusion body myositis.26,27
Unexpectedly, as unreported observations, other PrLD-harboring proteins, such as FET family proteins, also constituted cytoplasmic aggregates in the degenerating muscle fibers to a lesser extent on FUS/TLS (figure 5). Furthermore, even proteins without a PrLD, such as ALS/distal myopathy-linked4,5 matrin-3 and MSP1-linked VCP/p97, formed cytoplasmic coaggregation with hnRNPA1 in this study (figure 5), suggesting broad involvement of RBPs and their regulators. Recent reports uncovered a direct interaction among hnRNPA1, hnRNPA2B1, and TDP-43.28 They have also suggested the essential role of VCP/p97 in the autophagic clearance of excess assembly of nontranslating messenger ribonucleoprotein complexes to prevent pathogenic ribonucleoprotein aggregates.29,30 Together with the results of previous reports, whichever is mutated, ribonucleoprotein granules' hyperassembly and their defective clearance may be a major pathomechanism leading to widespread sequestration of the MSP/ALS-linked proteins and eventual muscle fiber degeneration.
The sarcolemmal dysferlin expression was varied in the presented cases, ranging from decreased to normal immunoreactivity. Although comorbid loss of dysferlin in biopsied muscles is occasionally observed in dystrophinopathy, sarcoglycanopathy, caveolinopathy, calpainopathy, and sporadic inclusion body myositis,31 loss of physiologic function of dysferlin has not been reported in MSPs to date. Thus, the possible association between dysferlin and hnRNPA1 remains to be elucidated.
We have reported dominantly inherited isolated IBM as a novel phenotype of MSP3. Muscle weakness as an isolated symptom is estimated to account for approximately 30% of all symptoms in MSP1.24,32 In addition to the rare involvement of bone reported in Asian patients with MSP1,33 PDB itself is infrequent in the Japanese population.34 Therefore, such ethnic differences in genetic background may be associated with the Japanese MSP3 phenotype. The selective involvement of skeletal muscles reported in this study should be confirmed, particularly by pathologic analyses as a cumulative case study. Nevertheless, the present results suggest that mutations in hnRNPA1, and possibly hnRNPA2B1, will be identified in patients with a pure muscular phenotype specifically presenting etiology-unknown IBM, as was the case with hnRNP-associated genes reported in Welander distal myopathy and limb-girdle muscular dystrophy 1G.35–37 The precise evaluation of these cases will help to elucidate the disease frequency, penetrance, genotype–phenotype correlation, and natural history of patients with MSPs. The unresolved issue will be challenged by the development of cellular and animal models strictly reflecting MSPs.
Supplementary Material
ACKNOWLEDGMENT
The authors appreciate the cooperation of the patients and their families and are grateful to Drs. Kazuo Kobayashi, Hideki Mizuno, Takafumi Hasegawa, Ohito Tano, and Hiroshi Kuroda for their fruitful discussions and Yoko Tateda, Kumi Kato, Riyo Takahashi, Naoko Shimakura, Risa Ando, Maya Narisawa, Miyuki Tsuda, Makiko Nakagawa, Mami Kikuchi, and Kiyotaka Kuroda for technical assistance. Moreover, the support of the Biomedical Research Core of Tohoku University Graduate School of Medicine is also gratefully acknowledged.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- CK
creatine kinase
- FUS/TLS
fused in sarcoma/translated in liposarcoma
- hnRNP
heterogeneous nuclear ribonucleoprotein
- IBM
inclusion body myopathy
- MSP
multisystem proteinopathy
- PDB
Paget disease of bone
- PrLD
prion-like domain
- RBP
RNA-binding protein
- SNV
single nucleotide variant
- SQSTM1/p62
sequestome-1/p62
- TDP-43
transactive response DNA binding protein 43 kDa
- Ub
ubiquitin
- VCP
valosin-containing protein
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Rumiko Izumi: principal author, designed the study, analyzed and interpreted the data, drafted the manuscript. Hitoshi Warita: coauthor, designed the study, performed the immunohistochemical study of biopsied muscle specimens, and revised the manuscript. Tetsuya Niihori and Yoko Aoki: coauthors, designed the study, analyzed and interpreted the data, and revised the manuscript. Toshiaki Takahashi, Satomi Mitsuhashi, and Ichizo Nishino: coauthors, acquired and provided data. Maki Tateyama: coauthor, performed the light and electron microscopic study of the biopsied muscles, and revised the manuscript. Naoki Suzuki and Masashi Aoki: coauthors, revised the manuscript. Ayumi Nishiyama: coauthor, performed the Sanger sequencing. Matsuyuki Shirota, Ryo Funayama, and Keiko Nakayama: coauthors, analyzed and interpreted the data.
STUDY FUNDING
This study was supported by an Intramural Research Grant (26-8) for Neurological and Psychiatric Disorders of NCNP; the grant on Research on Rare and Intractable Diseases (H26-intractable disease 037 and 082) from the Ministry of Health, Labour and Welfare of Japan; the Ministry of Health, Labour and Welfare of Japan (H26-Nanchitou(Nan)-Ippan-079); Grants-in-Aid for research on rare and intractable diseases; the Research Committee on Establishment of Novel Treatments for Amyotrophic Lateral Sclerosis; Grants-in-Aid from the Research Committee of CNS Degenerative Diseases from the Japanese Ministry of Health, Labor, and Welfare; Grants-in-Aid for Scientific Research (25293199 and 26461288); Grant-in-Aid for Challenging Exploratory Research (26670436) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
DISCLOSURE
Dr. Izumi reports no disclosures. Dr. Warita has received research support from Grant-in-Aid for Scientific Research (26461288, 25293199, and 23591229) and Grant-in-Aid for Challenging Exploratory Research (26670436) from Japan Society for the Promotion of Science (JSPS), Japan. Dr. Niihori and Dr. Takahashi report no disclosures. Dr. Tateyama has received honoraria for lecturing from Daiichi Sankyo company and has received research support from the Japan Society of the Promotion of Science (KAKENHI 25461265). Dr. Suzuki and Dr. Nishiyama report no disclosures. Dr. Shirota has received research support from MEXT, JSPS KAKENHI (26730148), and JST (14533504). Dr. Funayama has a patent pending on the quantitative ChIP-seq technology and has received research support from the grant on Research on Rare and Intractable Diseases from the Ministry of Health, Labour and Welfare (Japan), an Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP, and Grant-in-Aid for Young Scientists (B) from Japan Society for the Promotion of Science (JSPS). Dr. Nakayama and Dr. Mitsuhashi report no disclosures. Dr. Nishino serves on the scientific advisory board for Nobelpharma, Ultragenyx, and Genzyme; serves as an editorial board member of Neuromuscular Disorders, Neurology and Clinical Neuroscience, eNeurologicalSci, Skeletal Muscle, Journal of Neuromuscular Diseases, Journal of the Neurological Sciences, Rinsho Shinkeigaku, Chinese Journal of Contemporary Neurology and Neurosurgery, and Therapeutic Advances in Neurological Disorders; has a patent pending on the therapeutic pharmaceutical agent for diseases associated with decrease in function of GNE protein, food composition, and food additive; has received honoraria from Genzyme Japan; and has received research support from Genzyme Japan, Novartis Japan, Astellas, JSPS (KAKENHI 26293214), AMED (Itaku(Nan)-Ippan-081), NCNP Intramural Research Grant (26-8), NCNP Intramural Research Grant (23-5), and the Ministry of Health, Labour, and Welfare. Dr. Y. Aoki has received research support from the Japanese Foundation for Pediatric Research, the Japanese Ministry of Health Labor and Welfare, and Japan Society for the Promotion of Science (JSPS), Japan; and has served on the editorial board of the Journal of Human Genetics. Prof. M. Aoki has received research grants from the Japanese Ministry of Health Labor and Welfare, Grants-in-Aids for Scientific Research, an Intramural Research Grant from NCNP, and Grants-in-Aids for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–381. [DOI] [PubMed] [Google Scholar]
- 2.Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013;495:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 2015;85:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senderek J, Garvey SM, Krieger M, et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet 2009;84:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson JO, Pioro EP, Boehringer A, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 2014;17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benatar M, Wuu J, Fernandez C, et al. Motor neuron involvement in multisystem proteinopathy: implications for ALS. Neurology 2013;80:1874–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seelen M, Visser AE, Overste DJ, et al. No mutations in hnRNPA1 and hnRNPA2B1 in Dutch patients with amyotrophic lateral sclerosis, frontotemporal dementia, and inclusion body myopathy. Neurobiol Aging 2014;35:1956 e1959–e1911. [DOI] [PubMed] [Google Scholar]
- 8.Le Ber I, Van Bortel I, Nicolas G, et al. hnRNPA2B1 and hnRNPA1 mutations are rare in patients with “multisystem proteinopathy” and frontotemporal lobar degeneration phenotypes. Neurobiol Aging 2014;35:934 e935–e936. [DOI] [PubMed] [Google Scholar]
- 9.Calini D, Corrado L, Del Bo R, et al. Analysis of hnRNPA1, A2/B1, and A3 genes in patients with amyotrophic lateral sclerosis. Neurobiol Aging 2013;34:2695 e2611–e2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizuno H, Warita H, Aoki M, Itoyama Y. Accumulation of chondroitin sulfate proteoglycans in the microenvironment of spinal motor neurons in amyotrophic lateral sclerosis transgenic rats. J Neurosci Res 2008;86:2512–2523. [DOI] [PubMed] [Google Scholar]
- 11.Tateyama M, Fujihara K, Itoyama Y. Dendritic cells in muscle lesions of sarcoidosis. Hum Pathol 2011;42:340–346. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki N, Aoki M, Mizuno H, et al. Late-onset distal myopathy with rimmed vacuoles without mutation in the GNE or dysferlin genes. Muscle Nerve 2005;32:812–814. [DOI] [PubMed] [Google Scholar]
- 16.Darin N, Kyllerman M, Wahlstrom J, Martinsson T, Oldfors A. Autosomal dominant myopathy with congenital joint contractures, ophthalmoplegia, and rimmed vacuoles. Ann Neurol 1998;44:242–248. [DOI] [PubMed] [Google Scholar]
- 17.Hauser MA, Horrigan SK, Salmikangas P, et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet 2000;9:2141–2147. [DOI] [PubMed] [Google Scholar]
- 18.Horowitz SH, Schmalbruch H. Autosomal dominant distal myopathy with desmin storage: a clinicopathologic and electrophysiologic study of a large kinship. Muscle Nerve 1994;17:151–160. [DOI] [PubMed] [Google Scholar]
- 19.Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 1998;20:92–95. [DOI] [PubMed] [Google Scholar]
- 20.Udd B, Partanen J, Halonen P, et al. Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol 1993;50:604–608. [DOI] [PubMed] [Google Scholar]
- 21.Brais B, Bouchard JP, Xie YG, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 1998;18:164–167. [DOI] [PubMed] [Google Scholar]
- 22.Kottlors M, Moske-Eick O, Huebner A, et al. Late-onset autosomal dominant limb girdle muscular dystrophy and Paget's disease of bone unlinked to the VCP gene locus. J Neurol Sci 2010;291:79–85. [DOI] [PubMed] [Google Scholar]
- 23.Kimonis VE, Mehta SG, Fulchiero EC, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A 2008;146A:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul Disord 2009;19:308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011;44:279–289. [DOI] [PubMed] [Google Scholar]
- 26.Pinkus JL, Amato AA, Taylor JP, Greenberg SA. Abnormal distribution of heterogeneous nuclear ribonucleoproteins in sporadic inclusion body myositis. Neuromuscul Disord 2014;24:611–616. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita S, Kimura E, Tawara N, et al. Optineurin is potentially associated with TDP-43 and involved in the pathogenesis of inclusion body myositis. Neuropathol Appl Neurobiol 2013;39:406–416. [DOI] [PubMed] [Google Scholar]
- 28.Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 2005;280:37572–37584. [DOI] [PubMed] [Google Scholar]
- 29.Shorter J, Taylor JP. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis 2013;1:e25200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013;153:1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cacciottolo M, Nogalska A, D'Agostino C, Engel WK, Askanas V. Dysferlin is a newly identified binding partner of AβPP and it co-aggregates with amyloid-β42 within sporadic inclusion-body myositis (s-IBM) muscle fibers. Acta Neuropathol 2013;126:781–783. [DOI] [PubMed] [Google Scholar]
- 32.Mehta SG, Khare M, Ramani R, et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet 2013;83:422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi Z, Hayashi YK, Mitsuhashi S, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol 2012;19:501–509. [DOI] [PubMed] [Google Scholar]
- 34.Takata S, Hashimoto J, Nakatsuka K, et al. Guidelines for diagnosis and management of Paget's disease of bone in Japan. J Bone Miner Metab 2006;24:359–367. [DOI] [PubMed] [Google Scholar]
- 35.Hackman P, Sarparanta J, Lehtinen S, et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol 2013;73:500–509. [DOI] [PubMed] [Google Scholar]
- 36.Klar J, Sobol M, Melberg A, et al. Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum Mutat 2013;34:572–577. [DOI] [PubMed] [Google Scholar]
- 37.Vieira NM, Naslavsky MS, Licinio L, et al. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G). Hum Mol Genet 2014;23:4103–4110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.