

Abstract
Elevated testosterone levels during prenatal life lead to hyperandrogenism and insulin resistance in adult females. This study evaluated whether prenatal testosterone exposure leads to the development of insulin resistance in adult male rats in order to assess the influence of gonadal hormones on glucose homeostasis in these animals. Male offspring of pregnant rats treated with testosterone propionate or its vehicle (control) were examined. A subset of male offspring was orchiectomized at 7 wk of age and reared to adulthood. At 24 wk of age, fat weights, plasma testosterone, glucose homeostasis, pancreas morphology, and gastrocnemius insulin receptor (IR) beta levels were examined. The pups born to testosterone-treated mothers were smaller at birth and remained smaller through adult life, with levels of fat deposition relatively similar to those in controls. Testosterone exposure during prenatal life induced hyperinsulinemia paralleled by an increased HOMA-IR index in a fasting state and glucose intolerance and exaggerated insulin responses following a glucose tolerance test. Prenatal androgen-exposed males had more circulating testosterone during adult life. Gonadectomy prevented hyperandrogenism, reversed hyperinsulinemia, and attenuated glucose-induced insulin responses but did not alter glucose intolerance in these rats. Prenatal androgen-exposed males had decreased pancreatic islet numbers, size, and beta-cell area along with decreased expression of IR in gastrocnemius muscles. Gonadectomy restored pancreatic islet numbers, size, and beta-cell area but did not normalize IRbeta expression. This study shows that prenatal testosterone exposure leads to a defective pancreas and skeletal muscle function in male offspring. Hyperinsulinemia during adult life is gonad-dependent, but glucose intolerance appears to be independent of postnatal testosterone levels.
Keywords: developmental programing, glucose intolerance, gonadectomy, hyperinsulinemia, insulin resistance, pancreas
INTRODUCTION
Cardiovascular, renal, and metabolic diseases are inextricably linked and are the leading causes of mortality and morbidity in the United States. Among metabolic dysfunctions, type II diabetes/insulin resistance ranks first. With a 23% surge in prevalence over the last decade, it now affects more than 29.1 million people [1]. Despite increased efforts to prevent, treat, and control type II diabetes and its sequelae, the prevalence has not decreased. The pathogenesis of type II diabetes remains unclear, and consequently, treatment is currently based on using drugs, with an emphasis on improving insulin synthesis or sensitivity rather than on treating its causative factors.
Genetic animal models, such as Lepob/ob mice or Zucker diabetic fatty rats, have contributed greatly to the understanding of type II diabetes pathophysiology. However, these genetically altered strains have an abnormal leptin function prior to the development of insulin resistance, thus making it difficult to determine precisely the contributing mechanisms. Also, genetic studies do not truly reflect the actual type II diabetes setting in the general population, partly because insulin resistance is impacted by nongenetic factors, such as exercise, diet, and endocrine hormones. Recent studies show that type II diabetes has developmental origins; that is, an adverse intrauterine environment during the critical period of fetal development causes long-term structural and functional effects in the developing fetus, predisposing it to an increased risk for development of metabolic dysfunction during adult life [2–6]. Several animal studies have confirmed the observation that an unfavorable fetal environment causing fetal malnutrition—induced by restriction of food, protein, or uteroplacental blood flow—leads not only to low birth weight but also to metabolic dysfunction in adult offspring [7–10].
Recently, attention has focused on maternal androgen exposure because the number of pregnant women with elevated circulating testosterone levels and their problems with low birth weight and adverse adult health consequences are rapidly increasing. Higher androgen levels are reported in several obstetric pathological conditions that lead to fetal growth restriction, such as preeclampsia [11–13], maternal polycystic ovary syndrome (PCOS) [14, 15], obesity [16, 17], stress [18, 19], and smoking [20–22]. In addition, pregnant African-American mothers have higher serum testosterone levels and a greater frequency of low-birth-weight babies [23–25]. Moreover, the highest rates of adult metabolic dysfunction are also concentrated in these populations [26]. In addition, dysregulated factors, such as hypoxia [27], retinoids [28], tumor-necrosis factor alpha [29], lipid radicals [30], insulin [11], and leptin [31], during preeclampsia, which is known to down-regulate aromatase [32–35], could contribute for maternal androgen increase.
Testosterone is an important regulator of growth and differentiation during fetal development [36, 37]. Prenatal androgen excess in pregnant women, as well as in animals, is consistently associated with low birth weight [38–40] and with postnatal catch-up growth that is known to amplify adult life outcomes [41]. In addition to its effect on fetal growth, elevated testosterone levels during pregnancy have lasting effects in the offspring, causing reproductive and endocrine disturbances in adult life [42, 43]. In fact, prenatal exposure to testosterone was consistently shown to lead to hyperandrogenemia in both male and female offspring [44, 45], consistent with reports of boys and girls born to mothers with PCOS and higher testosterone levels [46, 47]. In female offspring, it has been reported that prenatal testosterone treatment leads to the development of hypertension [48, 49], insulin resistance [3, 4, 50, 51], visceral adiposity [2, 52], and increased triglycerides and cholesterol [50, 53] during adult life. Although prenatal testosterone exposure leads to the development of these cardinal features of PCOS in adult female rats, whether a similar phenotype occurs in adult males is unclear. Because fetal programming studies have shown sex-related differences in the occurrence of postnatal outcomes, with a more pronounced effect in males than in females [9, 54, 55], and because gonadal steroids are key regulators of metabolic function [45, 56], we tested whether prenatal testosterone exposure leads to glucose intolerance in adult males and assessed the influence of gonadal hormones on glucose homeostasis in these animals
MATERIALS AND METHODS
Animals
Timed-pregnant Sprague-Dawley rats were purchased from Harlan Laboratories on Day 12 of pregnancy. Rats were maintained on a 12L:12D photoperiod in a temperature-controlled room (23°C) and provided with ad libitum food and water. The experimental procedures are in accordance with the National Institutes of Health guidelines (NIH Publication 85–23, revised 1996) for care and use of animals and approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch. On Day 15 of pregnancy, rats were divided into control and treatment groups (n = 8 in each group). The control group received vehicle (sesame oil) subcutaneously, and the treatment group received testosterone propionate (TP; 0.5 mg/kg) subcutaneously from Day 15 to Day 19 of gestation. Dose and duration of TP were selected to mimic increases in testosterone levels in pregnant women with preeclampsia [56]. This treatment leads to an increase in plasma testosterone levels to 2.2 ± 0.23 (mean ± SEM) ng/ml in TP-treated dams compared to 1.0 ± 0.25 (mean ± SEM) ng/ml in vehicle-treated control dams (n = 8 in each group; P < 0.05). Rats were allowed to deliver normally, and birth weight was recorded within 12 h of birth. The number of pups per control and TP-treated mother were kept at 10 to ensure equal nutrient access for each pup. When possible, the male:female ratio was also kept equal. On weaning, pups were separated from their mothers; only male pups were used in the present study. Pups were allowed to eat a regular diet ad libitum. At 7 wk of age, control and TP-exposed offspring were sham-operated or orchiectomized as previously described [44] and reared up to 24 wk of age. Body weight was monitored weekly. Changes in plasma testosterone levels and glucose homeostasis were measured at 24 wk of age. Then, the rats were killed, and skeletal muscle (gastrocnemius) and pancreases were collected for protein analysis and histopathology. At the same time, retroperitoneal and subcutaneous fat pads were excised and weighed.
Plasma Testosterone Levels
Plasma testosterone levels in the samples were measured using an ELISA kit (ADI-900-065; Enzo Life Sciences) as described previously [57]. The minimum detectable concentration of testosterone is 6 pg/ml. The intra- and interassay coefficients of variation for testosterone were each lower than 5%.
Glucose Tolerance Test
Rats were fasted overnight for 14 h and then given glucose (2 g/kg) using oral gavage. Blood was collected before (time 0) and 30, 60, 90, 120, 180, and 240 min after oral gavage. Blood glucose levels were measured using an automatic glucometer (StatStrip; REF42214; Nova Biosciences).
Insulin Measurements
Insulin was measured using a rat insulin ELISA kit (10-1250-01; Mercodia) following the manufacturer's instruction. Briefly, heparinized blood was centrifuged for 15 min at 3000 rpm and 4°C for plasma isolation, and 10 μl of plasma or calibrators and 100 μl of enzyme conjugate were mixed and incubated for 2 h at room temperature with constant shaking. After washing, 200 μl of 3,3′,5,5′-tetramethylbenzidine substrate were added and incubated for 15 min for color development. The reaction was then stopped by adding 50 μl of stop solution. Absorbance was read at 450 nm using a FLUOstar Omega plate reader (BMG Labtech Gmbh), and the results were calculated with cubic spline regression fit using FLUOstar Omega data analysis software. The sensitivity of the insulin assay is 0.07 g/L, with an inter-and intra-assay precision of 3.3 and 1.8, respectively.
Plasma C-Peptide Levels
C-peptide in the plasma was measured using a solid-phase two-site enzyme immunoassay (10-1172-01; Rat C-Peptide ELISA; Mercodia). Ten microliters of plasma or calibrators and 50 μl of assay buffer were mixed and incubated for 1 h at room temperature with constant shaking. After washing, 100 μl of enzyme conjugate were added, followed by a 1-h incubation at room temperature with constant shaking. Two hundred microliters of substrate TMB were added after a final wash and incubated for 15 min on the bench. The reaction was then stopped by adding 50 μl of stop solution. Absorbance was read at 450 nm using a FLUOstar Omega plate reader, and the results were calculated with cubic spline regression fit using FLUOstar Omega data analysis software. The sensitivity of the C-peptide assay is 27.5 pmol/L−1, with an inter- and intra-assay precision of 2.9 and 4.4, respectively.
Homeostatic Model Assessment
Insulin resistance (homeostatic model assessment [HOMA]-IR) and insulin sensitivity (HOMA-IS) were calculated as described by Hsing et al. [59] and Park et al. [60] as follows:
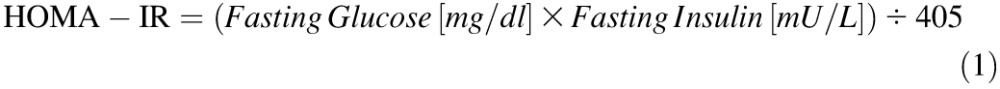 |
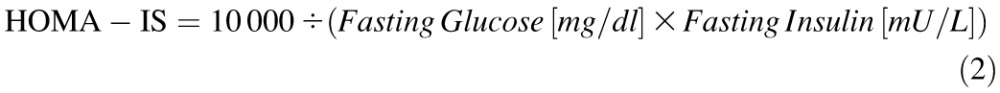 |
Pancreas Morphology and Immunohistochemistry
Pancreatic tissue was processed as previously described [61]. In brief, the pancreas was dissected and fixed in 4% formaldehyde before being embedded in paraffin. Then, 5-μm sections (Histopathology Core Facility, University of Texas Medical Branch) were deparaffinized and rehydrated, followed by heat-induced antigen retrieval. Three pancreatic sections per male rat, separated by at least 200 μm, were stained with hematoxylin-and-eosin. Sections were also immunostained for insulin (1:300; ab181547; Abcam), and secondary incubation was performed at room temperature for 1 h using anti-rabbit fluorescein isothiocyanate (FITC; ab97050; Abcam). Sections were imaged with an Olympus IX71 microscope at 10× magnification. Four random fields were selected per section as described by Huang et al. [62]. In each pancreatic section, the number of islets, mean islet area, fluorescent intensity, and area of insulin immune-reactivity were measured using ImageJ software (http:/rsh.info.nih.gov/ij/). Islet number was defined as the number of islets per microscopic field. The number of islets was expressed per square micrometer of the total area of the pancreas. For islet size, images of islets were traced and analyzed with the use of ImageJ software. Islet size was examined as the total islet area divided by the total number of islets. The islet size was examined by analyzing four clusters per pancreatic section.
Apoptosis and Proliferation
Slides were blocked with 10% normal serum with 1% bovine serum albumin in Tris-buffered saline and treated with rabbit anti-Ki67 antibody (ab15580; Abcam), followed by FITC-secondary antibody (ab97050; Abcam). TUNEL assay was performed using an in situ apoptosis detection kit (S7110; Millipore) according to manufacturer's instructions. Sections were double labeled with insulin to help identify events that occurred within endocrine versus exocrine tissue. Negative controls were developed with the same protocol, less the addition of the primary antibodies. The number of apoptotic and Ki67-positive events was manually counted and divided by total surface area scanned using ImageJ.
Western Blot Analysis
A protocol similar to those used in our previous studies was followed [8]. In brief, gastrocnemius muscles were weighed and homogenized in a 1× radio-immunoprecipitation assay buffer (4°C; Cell Signaling Technology) containing a protease inhibitor tablet (11836170001; Roche) and Phosphatase Inhibitor Cocktails 2 and 3 (P5726 and P0044; Sigma). Lysates were then sonicated using three bursts of 5 sec each at 30% power and were spun at 14 000 × g for 10 min. Total proteins were quantified using a BCA Protein Assay Kit (23227; Thermo Scientific). Proteins (30 μg) in supernatant were suspended in NuPAGE LDA Sample Buffer and Reducing Agent (Invitrogen) and resolved on 4%–12% precasted gradient polyacrylamide gels (NP0301BOX; NuPAGE Life Technologies) alongside Precision Plus Standard (161-0375; Kaleidoscope; Bio-Rad Laboratories). Furthermore, proteins in gel were transferred to polyvinylidene difluoride membranes (Millipore) by electroblotting. Membranes were blocked with 5% nonfat dried milk for 1 h, then incubated with primary antibodies at 4°C overnight. Antibodies for insulin receptor (IR) β (1:500, catalog no. 610109) were obtained from BD Bioscience. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies (1:2500; catalog no. ab9485) were obtained from Abcam. After incubation and washing, membranes were incubated with secondary antibodies (anti-mouse and anti-rabbit conjugated with horseradish peroxidase) at 1:12 000 dilutions for 1 h and detected with an ECL detection kit (WBKLS0100; Millipore). After development, a densitometric analysis was performed using ImageJ software.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software. Data are presented as the mean ± SEM. Comparisons between the two groups were performed using unpaired Student t-tests. Comparisons between multiple groups were performed using ANOVA, followed by Newman-Keuls tests. When two factors were involved, statistical analyses were performed with a two-way ANOVA, followed by Bonferroni tests. Differences were considered to be statistically significant at P < 0.05. The term “n” refers the number of litters studied.
RESULTS
Birth Weight and Postnatal Growth
Testosterone administration during pregnancy in rats caused fetal growth restriction. Male pups were 11% smaller at birth compared to corresponding controls (n = 8 litters in each group) (Fig. 1A). The length of gestation was not significantly affected by prenatal TP treatment (control, 22 ± 0.3 days; T, 22 ± 0.7 days; n = 8 in each group), and no significant differences were noted in the mean litter size between the control (13.4 ± 1.8) and TP-treated (12.8 ± 1.6) dams. These results are consistent with those in our previous publications [45, 63, 64].
FIG. 1.

Effect of prenatal testosterone exposure on birth weight and growth rate changes. Birth weight (A) was measured within 12 h of birth, and growth rates (B) were monitored each week up to 24 wk of age in intact and orchiectomized males of control and prenatally TP-exposed groups. Bars with different superscript letters differ significantly (P < 0.05). All data are expressed as the mean ± SEM (n = 6–8 animals in each group). OCX, orchiectomy.
The prenatally TP-exposed males maintained a smaller body weight throughout adult life (Fig. 1B). Orchiectomy attenuated body weight gain in both groups, but orchiectomized prenatally TP-exposed males maintained smaller body weights compared to orchiectomized controls (n = 6–7, P < 0.05) (Fig. 1B).
Fat Data
At 24 wk of age, the retroperitoneal and subcutaneous fat deposition in TP-exposed males was similar to that in controls (n = 6 in each group) (Fig. 2). Orchiectomy did not affect fat deposition in TP-exposed males but significantly increased fat accumulation in both retroperitoneal and subcutaneous fat in controls (n = 7 in each group, P < 0.05) (Fig. 2). Thus, retroperitoneal and subcutaneous fat deposition was significantly lower in orchiectomized TP-exposed males compared to orchiectomized controls (P < 0.05) (Fig. 2).
FIG. 2.

Effect of prenatal testosterone exposure on fat deposition. Retroperitoneal (A) and subcutaneous (B) fat accumulation was measured at 24 wk of age in intact and orchiectomized males of control and prenatally TP-exposed groups. Bars with different superscript letters differ significantly (P < 0.05). All data are expressed as the mean ± SEM (n = 6–7 animals in each group). OCX, orchiectomy.
Plasma Testosterone Levels
Plasma testosterone levels were significantly higher (1.5-fold) in testes-intact, TP-exposed males (n = 7, P < 0.05) (Fig. 3) compared with intact controls (n = 7), and testosterone levels were markedly reduced following orchiectomy in both TP-exposed males (n = 7) and controls (n = 8; P < 0.05) compared with their intact counterparts (Fig. 3). However, testosterone levels were significantly higher (2-fold) in orchiectomized TP-exposed males (n = 7, P < 0.05) compared with orchiectomized controls (n = 7).
FIG. 3.
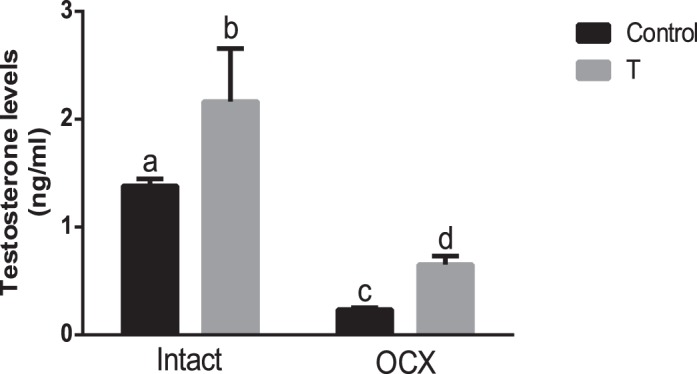
Effect of prenatal testosterone exposure on plasma testosterone levels of adult males. Plasma testosterone levels were measured in intact and orchiectomized control and prenatally TP-exposed males. Blood was collected through cardiac puncture following CO2 inhalation at 24 wk of age. Bars with different superscript letters differ significantly (P < 0.05). All data are expressed as the mean ± SEM (n = 7–8 animals in each group). OCX, orchiectomy.
Fasting Plasma Glucose, Insulin, and C-Peptide Levels
Fasting glucose levels in TP-exposed males (7.27 ± 0.34 mmol/L, n = 6) were similar to those in controls (7.36 ± 0.39 mmol/L, n = 6). Orchiectomy did not alter fasting glucose levels in both TP-exposed males (7.24 ± 0.22 mmol/L, n = 7) and in controls (7.04 ± 0.13 mmol/L, n = 8) compared with their intact counterparts (Fig. 4A). Thus, fasting glucose levels were not significantly different between orchiectomized TP-exposed males (n = 7) and orchiectomized controls (n = 8).
FIG. 4.

Effect of prenatal testosterone exposure on fasting glucose (A), insulin (B), and C-peptide (C) levels measured in intact and orchiectomized control and prenatally TP-exposed males. Animals were fasted overnight, and blood was collected through the retro-orbital plexus following CO2 inhalation at 24 wk of age. Bars with different superscript letters differ significantly (P < 0.05). All data are expressed as the mean ± SEM (n = 6–8 animals in each group). OCX, orchiectomy.
Fasting insulin levels in TP-exposed males (149.38 ± 28.99 pmol/L, n = 6) were significantly higher than in controls (70.21 ± 18.92 pmol/L, n = 6, P < 0.05). Orchiectomy negated the marked increase in fasting insulin levels observed in TP-exposed males (80.18 ± 24.51 pmol/L, n = 7, P < 0.05) but did not have a significant effect in controls (65.91 ± 9.52 pmol/L, n = 8) (Fig. 4B). Thus, no significant difference in fasting insulin levels was found between orchiectomized TP-exposed males and orchiectomized controls (Fig. 4B).
To determine whether the increased plasma insulin levels in TP-exposed rats were a consequence of increased secretion from β cells, we also determined levels of C-peptide, which is cosecreted with insulin from β cells at a 1:1 molar ratio, as a reliable measurement of secretion [65]. Fasting C-peptide levels in plasma were higher in TP-exposed males (453.58 ± 57.68 pmol/L, n = 6, P < 0.05) than in controls (242.32 ± 19.23 pmol/L, n = 6). Orchiectomy normalized C-peptide levels in TP-exposed males (232.97 ± 36.19 pmol/L, n = 7, P < 0.05) but did not have any effect in controls (273.57 ± 24.53 pmol/L, n = 8) (Fig. 4C). No significant difference in C-peptide levels was found between orchiectomized TP-exposed males and orchiectomized controls (Fig. 4C).
HOMA-IR and HOMA-IS
An index of insulin resistance, HOMA-IR was significantly higher in the TP-exposed males (7.03 ± 1.28, n = 7, P < 0.05) compared to their controls (3.35 ± 0.82, n = 6) (Fig. 5A). Orchiectomy significantly decreased HOMA-IR in the TP-exposed group (3.84 ± 1.26, n = 7, P < 0.05) but did not have a significant effect in controls compared with their intact counterparts (2.98 ± 0.46, n = 8) (Fig. 5A). Thus, no significant difference in HOMA-IR was found between orchiectomized TP-exposed males and orchiectomized controls (Fig. 5A).
FIG. 5.

Effect of prenatal testosterone exposure on HOMA-IR (A) and HOMA-IS (B) measured in intact and orchiectomized control and prenatally TP-exposed males. Bars with different superscript letters differ significantly (P < 0.05). All data are expressed as the mean ± SEM (n = 6–8 animals in each group). OCX, orchiectomy.
The HOMA-IS was the opposite: insulin sensitivity in the TP-exposed males (13.36 ± 2.70, n = 7, P < 0.05) was lower than that in the controls (34.72 ± 10.16, n = 7) (Fig. 5B). Orchiectomy significantly increased HOMA-IS in the TP-exposed group (32.41 ± 7.65, n = 7, P < 0.05) but did not have a significant effect in controls (30.17 ± 4.90, n = 8) (Fig. 5B) compared with their intact counterparts. Thus, no significant difference in HOMA-IS was found between orchiectomized TP-exposed males and orchiectomized controls (Fig. 5B).
Glucose Tolerance Test
To further characterize the effects of prenatal TP exposure on glucose handling, glucose tolerance was assessed using an oral glucose tolerance test. After administration of glucose, plasma glucose levels in TP-exposed males were significantly higher than in controls at 30 min (12.71 ± 1.14 vs. 9.17 ± 0.32 mmol/L, P < 0.05) and 60 min (10.83 ± 0.55 vs. 8.42 ± 0.30 mmol/L, P < 0.05) (Fig. 6A), followed by no difference at other time points. Orchiectomy did not affect glucose responses in both TP-exposed males and controls compared to their respective testes-intact groups (n = 6 in each group) (Fig. 6B). Thus, the orchiectomized TP-exposed males maintained significantly higher plasma glucose levels than in controls at 30 min (12.94 ± 1.14 vs. 9.00 ± 0.54 mmol/L, P < 0.05) and 60 min (10.96 ± 0.69 vs. 8.28 ± 0.49 mmol/L, P < 0.05), followed by no difference at other time points (Fig. 6B).
FIG. 6.

Effect of prenatal testosterone exposure on blood glucose clearance following oral glucose tolerance test in intact (A) and orchiectomized (B) control and prenatally TP-exposed males and on glycemia AUC (C) generated from the glucose tolerance test. Blood samples were collected at 0, 30, 60, 90, 120, 180, and 240 min following oral glucose (2 g/kg) administration. The overall plasma glucose levels are expressed as the AUC calculated by the trapezoidal method. Bars with different superscript letters differ significantly (P < 0.05). All data are expressed as the mean ± SEM (n = 6–7 animals in each group). OCX, orchiectomy.
Area-under-the-curve (AUC) analysis (mmol/L × 240 min) of glucose responses revealed glucose intolerance in TP-exposed males (811.82 ± 86.54, n = 6, P < 0.05) compared with controls (496.82 ± 47.96, n = 6), which was maintained after gonadectomy (Fig. 6C). Glycemia AUC was significantly higher in orchiectomized TP-exposed males (864.45 ± 74.04, n = 7, P < 0.05) compared with orchiectomized controls (509.43 ± 115.05, n = 7) (Fig. 6C).
Insulin Responses
In response to an oral glucose challenge, TP-exposed males exhibited an increase in plasma insulin levels compared to controls at 30 min (272.34 ± 8.25 vs. 162.63 ± 16.87 pmol/L, P < 0.05), followed by no difference at other time points (Fig. 7A). Orchiectomy significantly decreased plasma insulin levels at 30 min in TP-exposed males (156.71 ± 30.48, n = 7, P < 0.05) and was without any effect in controls (137.78 ± 24.05, n = 7) compared to their respective testis-intact counterparts (Fig. 7B). Thus, following an oral glucose challenge, no significant difference in insulin responses was found between orchiectomized TP-exposed males and orchiectomized controls (Fig. 7B).
FIG. 7.

Effect of prenatal testosterone exposure on insulin responses following oral glucose tolerance test in intact (A) and orchiectomized (B) control and prenatally TP-exposed males and on insulin AUC (C) generated from the glucose tolerance test. Blood samples were collected at 0, 30, 60, 90, 120, 180, and 240 min following oral glucose (2 g/kg) administration. The overall plasma glucose levels are expressed as the AUC calculated by the trapezoidal method. Bars with different superscript letters differ significantly (P < 0.05). All data are expressed as the mean ± SEM (n = 6–8 animals in each group). OCX, orchiectomy.
The overall plasma insulin responses, expressed as insulin AUC after oral glucose administration, were greater in TP-exposed males (18 398 ± 1762.34 pmol/L, n = 6, P < 0.05) compared with controls (11 335.75 ± 1498.35 pmol/L, n = 6). Orchiectomy significantly reduced insulin AUC in the TP-exposed group (11 761.4 ± 2317.94 pmol/L, n = 8, P ≤ 0.05) but had no effect in controls (12 712.6 ± 1977.62 pmol/L, n = 6) (Fig. 7C). Thus, insulin AUC was not significantly different between orchiectomized TP-exposed males and orchiectomized controls (Fig. 7C).
Pancreatic Islets
To elucidate how prenatal testosterone exposure could affect the development of the pancreas, sections of pancreas were analyzed for islet numbers, size, and β-cell area. The number of pancreatic islets were significantly lower in TP-exposed males (1.5 ± 0.64, n = 5, P < 0.05) (Fig. 8) compared to controls (4.0 ± 0.57, n = 5). Also, islet size, (i.e., mean islet area) was significantly smaller in TP-exposed males (76.68 ± 35.84 per mm2 section, n = 5, P < 0.05) compared to controls (160.90 ± 24.38 per mm2 section, n = 5) (Fig. 8). Orchiectomy significantly increased the number and size of pancreatic islets in TP-exposed males but did not alter them in controls (n = 5 in each group) (Fig. 8). The number and size of pancreatic islets were not significantly different between orchiectomized TP-exposed males and orchiectomized controls (Fig. 8).
FIG. 8.

Effect of prenatal testosterone exposure on pancreatic histology. Pancreatic sections from intact and orchiectomized control and prenatally TP-exposed males were stained with hematoxylin and eosin. A) Representative islets from each group. Original magnification ×40. B) The number of islets and mean islet area were determined as described in Materials and Methods. Values are presented as the mean ± SEM (n = 5 animals in each group). Different superscript letters indicate significant differences (P < 0.05).
The intensity of insulin staining in TP-exposed males was greater compared to that in controls (44.61 ± 2.05 vs. 17.49 ± 0.86, n = 5), whereas orchiectomy decreased the staining intensity in TP-exposed males (19.97 ± 2.42, n = 5) but did not affect controls (20.29 ± 1.00, n = 5) (Fig. 9, A and B). The intensity of insulin staining was not significantly different between orchiectomized TP-exposed males and orchiectomized controls (Fig. 9, A and B).
FIG. 9.

Effect of prenatal testosterone exposure on β-cell area. Pancreatic sections from intact and orchiectomized control and prenatally TP-exposed males were stained with anti-insulin antibody. A) Representative immunofluorescence images of insulin. Original magnification ×40. B and C) Insulin staining intensity (green fluorescence) per nuclei (blue fluorescence) (B) and β-cell area (C) relative to total pancreas area were quantified from pancreatic sections. Four clusters per pancreatic section and three pancreatic sections per animal were examined. Values are presented as the mean ± SEM (n = 5 animals in each group). Different superscript letters indicate significant differences (P < 0.05).
The relative β-cell area was significantly decreased in TP-exposed males compared to controls (1.55% ± 0.14% vs. 2.90% ± 0.14%, n = 5 in each group, P < 0.05) (Fig. 9C); however, orchiectomy significantly increased β-cell area in TP-exposed males (3.27% ± 0.23%, n = 5, P < 0.05) but did not affect controls (2.92% ± 0.18%, n = 5, P < 0.05). Thus, no significant difference in β-cell area was found between orchiectomized TP-exposed males and orchiectomized controls (Fig. 9C).
Apoptosis and Proliferation
None of the groups exhibited positive TUNEL staining, and no significant difference in the number of Ki67-positive cells was found between groups (data not shown).
Expression of IRβ protein
To determine whether the glucose dysregulation in TP-exposed rats is due to altered ability of the cells to interact with insulin, expression of the β subunit of IR in gastrocnemius muscle was examined. IRβ protein expression in TP-exposed males was significantly lower compared to controls (n = 5, P < 0.05) (Fig. 10). Orchiectomy did not alter IRβ levels in both TP-exposed males (n = 7) and controls (n = 8) compared to their intact counterparts, and thus the IRβ levels were significantly lower in the orchiectomized TP-exposed males (P < 0.05) compared to orchiectomized controls (Fig. 10).
FIG. 10.
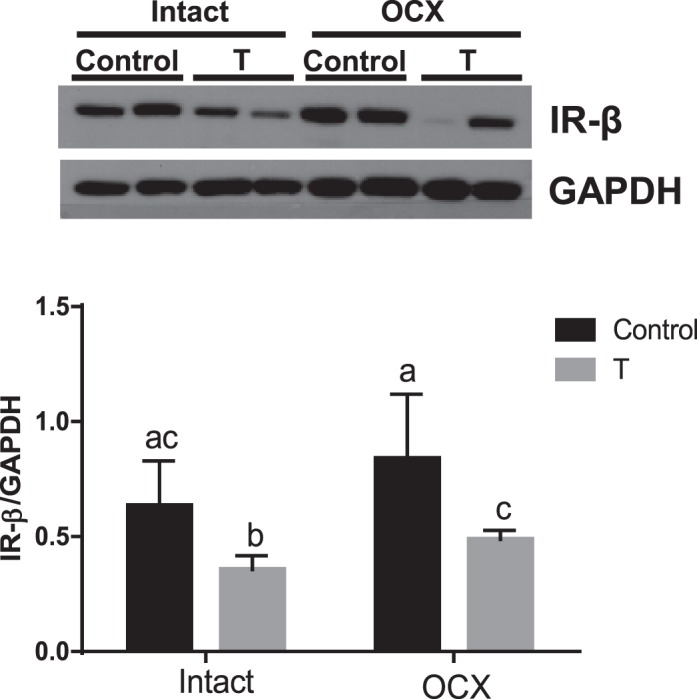
Effect of prenatal testosterone exposure on IRβ protein expression. IR-β levels were determined in gastrocnemius muscle from intact and orchiectomized control and prenatally TP-exposed males. Representative Western blots for IR-β (top) and blot density obtained from densitometric scanning of IR-β normalized to GAPDH (bottom) are shown. Values are presented as the mean ± SEM (n = 5–8 animals in each group). Different superscript letters indicate significant differences (P < 0.05).
DISCUSSION
The major findings of the present study are that elevated maternal testosterone levels during pregnancy, at clinically relevant concentrations, lead to hyperinsulinemia paralleled by a decrease in pancreatic islet number, size, β-cell area, and insulin resistance in adult male offspring. In addition, following oral glucose administration, the prenatally TP-exposed rats exhibit glucose intolerance in adult life, with higher plasma glucose levels in spite of exaggerated insulin responses. Hyperandrogenism appears to be the underlying factor inducing hyperinsulinemia and to be partially responsible for insulin resistance because peripubertal gonadectomy reverses hyperinsulinemia; restores pancreatic islet number, size, and β-cell area; and improves insulin sensitivity in adult offspring. However, gonadectomy does not restore the impaired glucose homeostasis induced by an oral glucose tolerance test. Decreased IRβ expression in the skeletal muscles may contribute, in part, to the observed glucose intolerance in this model of developmental programming.
Mounting evidence indicates that elevated testosterone levels during pregnancy may have adverse effects on fetal growth and thereby increase the potential for an increased risk of metabolic dysfunction in adult life [45, 48, 64, 66]. Prenatal testosterone exposure is shown to cause fetal growth restriction leading to low-birth-weight offspring [40, 57, 63, 67]. Consistently, the present study showed that elevated maternal testosterone leads to a significant reduction in pup birth weight. Whether the increase in fetal testosterone levels directly contributes to growth restriction is not known. However, fetal growth restriction may be secondary to the impact of prenatal testosterone treatment on placental physiology, as elevated testosterone has been shown to alter placental development and differentiation and restrict nutrient transfer to fetuses [63, 68]. These low-birth-weight testosterone males do not exhibit catch-up growth (present study), contrary to earlier reports in which the prenatal testosterone-exposed females showed early postnatal catch-up growth between 8 and 16 wk of age [40, 64]. Furthermore, our study shows that prenatal testosterone exposure does not alter adiposity, as the subcutaneous and peritoneal fat deposition was comparable to that in controls, which is in contrast to prenatal testosterone-exposed adult females that show increased visceral adiposity [2]. This difference suggests that postnatal growth responses and fat deposition following prenatal testosterone exposure may vary depending on the sex of the offspring, with only females exhibiting catch-up growth and adiposity.
Consistent with evidence showing that boys and girls of mothers with PCOS are often hyperandrogenic [46, 47], we and others using this model of prenatal androgen exposure have shown that adult males and females tend to produce higher testosterone levels than controls [44, 45, 69, 70]. Normally, testes are the primary source of testosterone, and consistently, we show that orchiectomy of control and TP-exposed males significantly decreases testosterone production. However, the testosterone levels in orchiectomized TP-exposed males were significantly higher than in orchiectomized controls, suggesting increased testosterone production from nontestes sources [71].
Although glucose levels were maintained, the TP-exposed males had an increased fasting circulating insulin level paralleled by an increase in HOMA-IR and reduction in HOMA-IS, suggesting insulin resistance. Upon glucose administration, TP-exposed males, unlike controls, exhibited glucose intolerance with elevated plasma glucose (higher glycemia AUC) in spite of exaggerated insulin responses (higher insulin AUC). Thus, although the basal glucose levels seem unaltered with prenatal testosterone exposure, the induced responses are severely altered, leading to metabolic disturbances, which contrasts with the prenatal testosterone-exposed adult females, in which no differences in glucose tolerance are reported but changes in fasting hyperinsulinemia are seen [2]. Presence of estradiol may contribute to less severe phenotype in females [2, 72]. Hyperinsulinemia is often observed with obesity and insulin resistance [73, 74]; however, the lack of alteration in fat deposition in TP-exposed males compared to controls suggests that adiposity may not be the contributory factor in mediating hyperinsulinemia. The observation of higher HOMA-IR and increased glycemia AUC in TP-exposed males suggests that hyperinsulinemia may be secondary or compensatory to altered insulin sensitivity at peripheral organs. Surprisingly, the smaller number and size of islets and β-cell area in TP-exposed males do not substantiate the observed hyperinsulinemia in these animals. Analysis of C-peptide levels confirms that TP-exposed males are indeed hyperinsulinemic and clarifies that the increase in insulin levels may not be due to deficits in clearance but may be related to increased synthesis. Consistently, the increased intensity of insulin staining in the smaller islets of TP-exposed males suggests that these males produce higher insulin content per β-cell area. This leads to the question of whether such pathophysiology is a potential nidus for β cell dysfunction and eventual burnout and, maybe, analogous to a prediabetic state in humans [75]. The finding that orchiectomy of TP-exposed males reduces both testosterone and hyperinsulinemia suggests that elevated testosterone may play a critical role in provoking increased insulin secretion. The finding that orchiectomy does not affect glucose intolerance following oral glucose administration but reverses exaggerated glucose-induced insulin responses further confirms a role for elevated testosterone in facilitating higher insulin responses. However, orchiectomy restoring the number and size of islets and β-cell area suggests that elevated testosterone in TP-exposed males has a role in maintaining smaller and hypersecretory β cells. Evidence indicates that testosterone has a paradoxical ability to induce apoptosis, inhibit proliferation [76], and expand islets [77], as well as to improve islet function by increasing insulin gene expression, transcription [78], and secretion [78, 79] in vivo in rats and in vitro in pancreatic islets [78]. Thus, we suggest that hyperandrogenism may be an underlying factor for hyperinsulinemia and insulin resistance in the prenatally TP-exposed adult males. Murine studies have demonstrated severely limited β-cell proliferation with advancing age [80, 81]. The lack of increased proliferation of β cells in our hyperinsulinemic TP-exposed males illustrates this as well; the adaptation to produce more insulin appears to come from hypersensitive islets rather than an increased number or area. The almost complete absence of apoptosis in these same pancreata, however, attests to the resilience and long lifespan of β cells that have also been previously reported [82]. Future studies should examine testosterone-exposed males at ages earlier than 6 mo to examine how prenatal androgens alter the establishment and expansion of islets. Also, studies are warranted to examine if restoration of testosterone restores hyperinsulinemia and insulin resistance in orchiectomized male rats exposed to testosterone during prenatal life.
Although orchiectomy decreased HOMA-IR and increased HOMA-IS, glucose intolerance was persistent in orchiectomized TP-exposed males, suggesting that glucose dysregulation is independent of postnatal testosterone levels and may be due to the permanent programming effect induced by the prenatal insult. The question arises as to how androgens provoke glucose intolerance and insulin resistance. The presence of reduced IRβ protein levels in the skeletal muscle, which is the major glucose utilizer and contributor of insulin resistance [83] in both intact and orchiectomized TP-exposed males, suggests that prenatal testosterone exposure induces reduced insulin signaling, which may contribute to glucose intolerance.
In conclusion, the present study shows that prenatal testosterone excess leads to development of defective pancreatic morphology, generating alterations in plasma insulin levels during adult life, which may contribute to the development of hyperinsulinemia and insulin resistance. Although mechanisms linking prenatal testosterone to insulin resistance in later life are likely to be complex, the present study underscores the importance of gonadal hyperandrogenism. In addition, we show that prenatal testosterone excess induces the direct programming effect by decreasing skeletal muscle IRβ levels that are independent of postnatal testosterone levels. Future studies are warranted to dissect out the testosterone-mediated programming mechanisms in utero as well as signaling events that lead to the development of insulin resistance in adult life. Understanding these mechanisms is pivotal for the development of novel strategies to prevent and treat type II diabetes and related metabolic dysfunctions.
Footnotes
Financial support from the National Institutes of Health (NIH) through grant HL102866 is greatly appreciated. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
REFERENCES
- Centers for Disease Control and Prevention (CDC) National diabetes statistics report [Internet], 2014. Atlanta, GA: Centers for Disease Control and Prevention http://www.cdc.gov/diabetes/pubs/statsreport14.htm. Accessed 17 February 2014. [Google Scholar]
- Demissie M, Lazic M, Foecking EM, Aird F, Dunaif A, Levine JE. Transient prenatal androgen exposure produces metabolic syndrome in adult female rats. Am J Physiol Endocrinol Metab. 2008;295:E262–E268. doi: 10.1152/ajpendo.90208.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner JR, Dumesic DA, Kemnitz JW, Abbott DH. Timing of prenatal androgen excess determines differential impairment in insulin secretion and action in adult female rhesus monkeys. J Clin Endocrinol Metab. 2000;85:1206–1210. doi: 10.1210/jcem.85.3.6453. [DOI] [PubMed] [Google Scholar]
- Padmanabhan V, Veiga-Lopez A, Abbott DH, Recabarren SE, Herkimer C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology. 2010;151:595–605. doi: 10.1210/en.2009-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland AV, Nunemaker CS, Keller SR, Moenter SM. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol. 2010;207:213–223. doi: 10.1677/JOE-10-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Moeller J, Patel D, Ye W, Pease A, Kinns J, Padmanabhan V. Developmental programming: impact of prenatal testosterone excess on insulin sensitivity, adiposity, and free fatty acid profile in postpubertal female sheep. Endocrinology. 2013;154:1731–1742. doi: 10.1210/en.2012-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander BT. Fetal programming of hypertension. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1–R10. doi: 10.1152/ajpregu.00417.2005. [DOI] [PubMed] [Google Scholar]
- Blesson CS, Sathishkumar K, Chinnathambi V, Yallampalli C. Gestational protein restriction impairs insulin-regulated glucose transport mechanisms in gastrocnemius muscles of adult male offspring. Endocrinology. 2014;155:3036–3046. doi: 10.1210/en.2014-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T, Nishina H, Hanson MA, Poston L. Dietary restriction in pregnant rats causes gender-related hypertension and vascular dysfunction in offspring. J Physiol. 2001;530:141–152. doi: 10.1111/j.1469-7793.2001.0141m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker CJ, Arch JR, Cawthorne MA. Fetal origins of insulin resistance and obesity. Proc Nutr Soc. 2005;64:143–151. doi: 10.1079/pns2005417. [DOI] [PubMed] [Google Scholar]
- Acromite MT, Mantzoros CS, Leach RE, Hurwitz J, Dorey LG. Androgens in preeclampsia. Am J Obstet Gynecol. 1999;180:60–63. doi: 10.1016/s0002-9378(99)70150-x. [DOI] [PubMed] [Google Scholar]
- Ghorashi V, Sheikhvatan M. The relationship between serum concentration of free testosterone and pre-eclampsia. Endokrynol Pol. 2008;59:390–392. [PubMed] [Google Scholar]
- Salamalekis E, Bakas P, Vitoratos N, Eleptheriadis M, Creatsas G. Androgen levels in the third trimester of pregnancy in patients with preeclampsia. Eur J Obstet Gynecol Reprod Biol. 2006;126:16–19. doi: 10.1016/j.ejogrb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Codner E, Escobar-Morreale HF. Clinical review: hyperandrogenism and polycystic ovary syndrome in women with type 1 diabetes mellitus. J Clin Endocrinol Metab. 2007;92:1209–1216. doi: 10.1210/jc.2006-2641. [DOI] [PubMed] [Google Scholar]
- Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Perez-Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002;17:2573–2579. doi: 10.1093/humrep/17.10.2573. [DOI] [PubMed] [Google Scholar]
- Evans DJ, Hoffmann RG, Kalkhoff RK, Kissebah AH. Relationship of androgenic activity to body fat topography, fat cell morphology, and metabolic aberrations in premenopausal women. J Clin Endocrinol Metab. 1983;57:304–310. doi: 10.1210/jcem-57-2-304. [DOI] [PubMed] [Google Scholar]
- Pasquali R, Casimirri F, Cantobelli S, Labate AM, Venturoli S, Paradisi R, Zannarini L. Insulin and androgen relationships with abdominal body fat distribution in women with and without hyperandrogenism. Horm Res. 1993;39:179–187. doi: 10.1159/000182732. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Bergman K, Fisk NM, O'Connor TG, Glover V. Amniotic fluid testosterone: relationship with cortisol and gestational age. Clin Endocrinol (Oxf) 2007;67:743–747. doi: 10.1111/j.1365-2265.2007.02955.x. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Bergman K, O'Connor TG, Glover V. Maternal antenatal anxiety and amniotic fluid cortisol and testosterone: possible implications for foetal programming. J Neuroendocrinol. 2008;20:489–496. doi: 10.1111/j.1365-2826.2008.01659.x. [DOI] [PubMed] [Google Scholar]
- Kandel DB, Udry JR. Prenatal effects of maternal smoking on daughters' smoking: nicotine or testosterone exposure? Am J Public Health. 1999;89:1377–1383. doi: 10.2105/ajph.89.9.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlau-Hansen CH, Thulstrup AM, Olsen J, Ernst E, Andersen CY, Bonde JP. Maternal smoking in pregnancy and reproductive hormones in adult sons. Int J Androl. 2008;31:565–572. doi: 10.1111/j.1365-2605.2007.00807.x. [DOI] [PubMed] [Google Scholar]
- Rizwan S, Manning JT, Brabin BJ. Maternal smoking during pregnancy and possible effects of in utero testosterone: evidence from the 2D:4D finger length ratio. Early Hum Dev. 2007;83:87–90. doi: 10.1016/j.earlhumdev.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Hall WD, Ferrario CM, Moore MA, Hall JE, Flack JM, Cooper W, Simmons JD, Egan BM, Lackland DT, Perry M Jr, Roccella EJ. Hypertension-related morbidity and mortality in the southeastern United States. Am J Med Sci. 1997;313:195–209. doi: 10.1097/00000441-199704000-00002. [DOI] [PubMed] [Google Scholar]
- Hoyert DL, Freedman MA, Strobino DM, Guyer B. Annual summary of vital statistics: 2000. Pediatrics. 2001;108:1241–1255. doi: 10.1542/peds.108.6.1241. [DOI] [PubMed] [Google Scholar]
- Huisman HW, Schutte AE, Van Rooyen JM, Malan NT, Malan L, Schutte R, Kruger A. The influence of testosterone on blood pressure and risk factors for cardiovascular disease in a black South African population. Ethn Dis. 2006;16:693–698. [PubMed] [Google Scholar]
- Clark LT, El-Atat F. Metabolic syndrome in African Americans: implications for preventing coronary heart disease. Clin Cardiol. 2007;30:161–164. doi: 10.1002/clc.20003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Kamat A, Mendelson CR. Hypoxia prevents induction of aromatase expression in human trophoblast cells in culture: potential inhibitory role of the hypoxia-inducible transcription factor Mash-2 (mammalian achaete-scute homologous protein-2) Mol Endocrinol. 2000;14:1661–1673. doi: 10.1210/mend.14.10.0539. [DOI] [PubMed] [Google Scholar]
- Zhu SJ, Li Y, Li H, Wang YL, Xiao ZJ, Vihko P, Piao YS. Retinoic acids promote the action of aromatase and 17beta-hydroxysteroid dehydrogenase type 1 on the biosynthesis of 17beta-estradiol in placental cells. J Endocrinol. 2002;172:31–43. doi: 10.1677/joe.0.1720031. [DOI] [PubMed] [Google Scholar]
- Diaz L, Noyola-Martinez N, Barrera D, Hernandez G, Avila E, Halhali A, Larrea F. Calcitriol inhibits TNF-α-induced inflammatory cytokines in human trophoblasts. J Reprod Immunol. 2009;81:17–24. doi: 10.1016/j.jri.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Milczarek R, Sokolowska E, Hallmann A, Kaletha K, Klimek J. NADPH- and iron-dependent lipid peroxidation inhibit aromatase activity in human placental microsomes. J Steroid Biochem Mol Biol. 2008;110:230–235. doi: 10.1016/j.jsbmb.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Christensen A. Hormone and enzyme assays in pregnancy. I. Studies on the placental and the tissue cystine-aminopeptidase activity in peripheral plasma from non-pregnant and pregnant women, and in plasma from the umbilical cord. Acta Endocrinol (Copenh) 1974;76:189–200. [PubMed] [Google Scholar]
- Zhang C, Williams MA, Sanchez SE, King IB, Ware-Jauregui S, Larrabure G, Bazul V, Leisenring WM. Plasma concentrations of carotenoids, retinol, and tocopherols in preeclamptic and normotensive pregnant women. Am J Epidemiol. 2001;153:572–580. doi: 10.1093/aje/153.6.572. [DOI] [PubMed] [Google Scholar]
- Williams MA, Woelk GB, King IB, Jenkins L, Mahomed K. Plasma carotenoids, retinol, tocopherols, and lipoproteins in preeclamptic and normotensive pregnant Zimbabwean women. Am J Hypertens. 2003;16:665–672. doi: 10.1016/s0895-7061(03)00897-5. [DOI] [PubMed] [Google Scholar]
- Cackovic M, Buhimschi CS, Zhao G, Funai EF, Norwitz ER, Kuczynski E, Lockwood CJ, Buhimschi IA. Fractional excretion of tumor necrosis factor-α in women with severe preeclampsia. Obstet Gynecol. 2008;112:93–100. doi: 10.1097/AOG.0b013e31817c4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Mishra S, Sehgal A, Prasad R. Alterations in lipid peroxidation and antioxidant status in pregnancy with preeclampsia. Mol Cell Biochem. 2008;313:37–44. doi: 10.1007/s11010-008-9739-z. [DOI] [PubMed] [Google Scholar]
- de Zegher F, Devlieger H, Eeckels R. Fetal growth: boys before girls. Horm Res. 1999;51:258–259. doi: 10.1159/000023382. [DOI] [PubMed] [Google Scholar]
- Viger RS, Silversides DW, Tremblay JJ. New insights into the regulation of mammalian sex determination and male sex differentiation. Vitam Horm. 2005;70:387–413. doi: 10.1016/S0083-6729(05)70013-3. [DOI] [PubMed] [Google Scholar]
- Hotchkiss AK, Lambright CS, Ostby JS, Parks-Saldutti L, Vandenbergh JG, Gray LE., Jr Prenatal testosterone exposure permanently masculinizes anogenital distance, nipple development, and reproductive tract morphology in female Sprague-Dawley rats. Toxicol Sci. 2007;96:335–345. doi: 10.1093/toxsci/kfm002. [DOI] [PubMed] [Google Scholar]
- Carlsen SM, Jacobsen G, Romundstad P. Maternal testosterone levels during pregnancy are associated with offspring size at birth. Eur J Endocrinol. 2006;155:365–370. doi: 10.1530/eje.1.02200. [DOI] [PubMed] [Google Scholar]
- Manikkam M, Crespi EJ, Doop DD, Herkimer C, Lee JS, Yu S, Brown MB, Foster DL, Padmanabhan V. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology. 2004;145:790–798. doi: 10.1210/en.2003-0478. [DOI] [PubMed] [Google Scholar]
- Eriksson J, Forsen T, Tuomilehto J, Osmond C, Barker D. Fetal and childhood growth and hypertension in adult life. Hypertension. 2000;36:790–794. doi: 10.1161/01.hyp.36.5.790. [DOI] [PubMed] [Google Scholar]
- Padmanabhan V, Manikkam M, Recabarren S, Foster D. Prenatal testosterone excess programs reproductive and metabolic dysfunction in the female. Mol Cell Endocrinol. 2006;246:165–174. doi: 10.1016/j.mce.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord. 2007;8:127–141. doi: 10.1007/s11154-007-9046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XY, Li ZL, Wu CY, Liu YM, Lin H, Wang SH, Xiao WF. Endocrine traits of polycystic ovary syndrome in prenatally androgenized female Sprague-Dawley rats. Endocr J. 2010;57:201–209. doi: 10.1507/endocrj.k09e-205. [DOI] [PubMed] [Google Scholar]
- Chinnathambi V, Balakrishnan M, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure leads to hypertension that is gonadal hormone-dependent in adult rat male and female offspring. Biol Reprod. 2012;86:137. doi: 10.1095/biolreprod.111.097550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent SC, Gnatuk CL, Kunselman AR, Demers LM, Lee PA, Legro RS. Hyperandrogenism and hyperinsulinism in children of women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab. 2008;93:1662–1669. doi: 10.1210/jc.2007-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sir-Petermann T, Maliqueo M, Codner E, Echiburu B, Crisosto N, Perez V, Perez-Bravo F, Cassorla F. Early metabolic derangements in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92:4637–4642. doi: 10.1210/jc.2007-1036. [DOI] [PubMed] [Google Scholar]
- King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endocrinol Metab. 2007;292:E1837–E1841. doi: 10.1152/ajpendo.00668.2006. [DOI] [PubMed] [Google Scholar]
- Blesson CS, Chinnathambi V, Hankins GD, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure induces hypertension in adult females via androgen receptor-dependent protein kinase Cδ-mediated mechanism. Hypertension. 2014;65:683–690. doi: 10.1161/HYPERTENSIONAHA.114.04521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexanderson C, Eriksson E, Stener-Victorin E, Lystig T, Gabrielsson B, Lonn M, Holmang A. Postnatal testosterone exposure results in insulin resistance, enlarged mesenteric adipocytes, and an atherogenic lipid profile in adult female rats: comparisons with estradiol and dihydrotestosterone. Endocrinology. 2007;148:5369–5376. doi: 10.1210/en.2007-0305. [DOI] [PubMed] [Google Scholar]
- Recabarren SE, Padmanabhan V, Codner E, Lobos A, Duran C, Vidal M, Foster DL, Sir-Petermann T. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. Am J Physiol Endocrinol Metab. 2005;289:E801–E806. doi: 10.1152/ajpendo.00107.2005. [DOI] [PubMed] [Google Scholar]
- Eisner JR, Dumesic DA, Kemnitz JW, Colman RJ, Abbott DH. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res. 2003;11:279–286. doi: 10.1038/oby.2003.42. [DOI] [PubMed] [Google Scholar]
- Nilsson C, Niklasson M, Eriksson E, Bjorntorp P, Holmang A. Imprinting of female offspring with testosterone results in insulin resistance and changes in body fat distribution at adult age in rats. J Clin Invest. 1998;101:74–78. doi: 10.1172/JCI1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco MC, Dantas AP, Akamine EH, Kawamoto EM, Fortes ZB, Scavone C, Tostes RC, Carvalho MH, Nigro D. Enhanced oxidative stress as a potential mechanism underlying the programming of hypertension in utero. J Cardiovasc Pharmacol. 2002;40:501–509. doi: 10.1097/00005344-200210000-00002. [DOI] [PubMed] [Google Scholar]
- Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1131–R1136. doi: 10.1152/ajpregu.00037.2003. [DOI] [PubMed] [Google Scholar]
- Varlamov O, Bethea CL, Roberts CT., Jr Sex-specific differences in lipid and glucose metabolism. Front Endocrinol (Lausanne) 2014;5:241. doi: 10.3389/fendo.2014.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CJ, Hotchkiss A, Ostby JS, LeBlanc GA, Gray LE., Jr Effects of prenatal testosterone propionate on the sexual development of male and female rats: a dose-response study. Toxicol Sci. 2002;65:71–86. doi: 10.1093/toxsci/65.1.71. [DOI] [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Yallampalli U, Yallampalli C. Protein restriction during pregnancy induces hypertension in adult female rat offspring: influence of estradiol. Br J Nutr. 2012;107:665–673. doi: 10.1017/S0007114511003448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsing AW, Gao YT, Chua S Jr, Deng J, Stanczyk FZ. Insulin resistance and prostate cancer risk. J Natl Cancer Inst. 2003;95:67–71. doi: 10.1093/jnci/95.1.67. [DOI] [PubMed] [Google Scholar]
- Park JM, Bong HY, Jeong HI, Kim YK, Kim JY, Kwon O. Postprandial hypoglycemic effect of mulberry leaf in Goto-Kakizaki rats and counterpart control Wistar rats. Nutr Res Pract. 2009;3:272–278. doi: 10.4162/nrp.2009.3.4.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Georgia S, Tschen SI, Nakayama K, Nakayama K, Bhushan A. Essential role of Skp2-mediated p27 degradation in growth and adaptive expansion of pancreatic β cells. J Clin Invest. 2007;117:2869–2876. doi: 10.1172/JCI32198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HH, Farmer K, Windscheffel J, Yost K, Power M, Wright DE, Stehno-Bittel L. Exercise increases insulin content and basal secretion in pancreatic islets in type 1 diabetic mice. Exp Diabetes Res. 2011;2011:481427. doi: 10.1155/2011/481427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Chinnathambi V, Gao H, Hankins GD, Yallampalli C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod Biol Endocrinol. 2011;9:1–12. doi: 10.1186/1477-7827-9-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Yallampalli U, Balakrishnan M, Yallampalli C. Fetal programming of adult hypertension in female rat offspring exposed to androgens in utero. Early Hum Dev. 2011;87:407–414. doi: 10.1016/j.earlhumdev.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tura A, Ludvik B, Nolan JJ, Pacini G, Thomaseth K. Insulin and C-peptide secretion and kinetics in humans: direct and model-based measurements during OGTT. Am J Physiol Endocrinol Metab. 2001;281:E966–E974. doi: 10.1152/ajpendo.2001.281.5.E966. [DOI] [PubMed] [Google Scholar]
- Chinnathambi V, Yallampalli C, Sathishkumar K. Prenatal testosterone induces sex-specific dysfunction in endothelium-dependent relaxation pathways in adult male and female rats. Biol Reprod. 2013;89:1. doi: 10.1095/biolreprod.113.111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CJ, LeBlanc GA, Gray LE., Jr Interactive effects of vinclozolin and testosterone propionate on pregnancy and sexual differentiation of the male and female SD rat. Toxicol Sci. 2004;78:135–143. doi: 10.1093/toxsci/kfh018. [DOI] [PubMed] [Google Scholar]
- Beckett EM, Astapova O, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programing: impact of testosterone on placental differentiation. Reproduction. 2014;148:199–209. doi: 10.1530/REP-14-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recabarren SE, Lobos A, Figueroa Y, Padmanabhan V, Foster DL, Sir-Petermann T. Prenatal testosterone treatment alters LH and testosterone responsiveness to GnRH agonist in male sheep. Biol Res. 2007;40:329–338. [PubMed] [Google Scholar]
- Birch RA, Padmanabhan V, Foster DL, Unsworth WP, Robinson JE. Prenatal programming of reproductive neuroendocrine function: fetal androgen exposure produces progressive disruption of reproductive cycles in sheep. Endocrinology. 2003;144:1426–1434. doi: 10.1210/en.2002-220965. [DOI] [PubMed] [Google Scholar]
- Parker L, Lai M, Wolk F, Lifrak E, Kim S, Epstein L, Hadley D, Miller J. Orchiectomy does not selectively increase adrenal androgen concentrations. J Clin Endocrinol Metab. 1984;59:547–550. doi: 10.1210/jcem-59-3-547. [DOI] [PubMed] [Google Scholar]
- Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150:2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- German JP, Wisse BE, Thaler JP, Oh I, Sarruf DA, Ogimoto K, Kaiyala KJ, Fischer JD, Matsen ME, Taborsky GJ, Jr, Schwartz MW, Morton GJ. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes. 2010;59:1626–1634. doi: 10.2337/db09-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, Botezelli JD, Asadi A, Hoffman BG, Kieffer TJ, Bamji SX, Clee SM, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16:723–737. doi: 10.1016/j.cmet.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Costes S, Langen R, Gurlo T, Matveyenko AV, Butler PC. β-Cell failure in type 2 diabetes: a case of asking too much of too few? Diabetes. 2013;62:327–335. doi: 10.2337/db12-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RJ, Qiu SD, Wang HX, Tian H, Wang LR, Huo YW. Androgen receptor: a new player associated with apoptosis and proliferation of pancreatic beta-cell in type 1 diabetes mellitus. Apoptosis. 2008;13:959–971. doi: 10.1007/s10495-008-0230-9. [DOI] [PubMed] [Google Scholar]
- Nicol LE, O'Brien TD, Dumesic DA, Grogan T, Tarantal AF, Abbott DH. Abnormal infant islet morphology precedes insulin resistance in PCOS-like monkeys. PLOS ONE. 2014;9:e106527. doi: 10.1371/journal.pone.0106527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto S, Fernandez-Mejia C, Romero-Navarro G, Morales-Peza N, Diaz-Sanchez V. Testosterone effect on insulin content, messenger ribonucleic acid levels, promoter activity, and secretion in the rat. Endocrinology. 2001;142:1442–1447. doi: 10.1210/endo.142.4.8069. [DOI] [PubMed] [Google Scholar]
- Grillo ML, Jacobus AP, Scalco R, Amaral F, Rodrigues DO, Loss ES, Wassermann GF. Testosterone rapidly stimulates insulin release from isolated pancreatic islets through a non-genomic dependent mechanism. Horm Metab Res. 2005;37:662–665. doi: 10.1055/s-2005-870575. [DOI] [PubMed] [Google Scholar]
- Rankin MM, Kushner JA. Adaptive β-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58:1365–1372. doi: 10.2337/db08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes. 2009;58:1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Hughes SJ, Igoillo-Esteve M, Hoppa MB, Sayyed F, van de Laar L, Gunter JH, de Koning EJ, Walls GV, Gray DW, Johnson PR, Hansen BC, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia. 2010;53:321–330. doi: 10.1007/s00125-009-1562-x. [DOI] [PubMed] [Google Scholar]
- Fatani S, Abubakari AR, Itua I, Wong C, Thomas C, Naderali EK. Effects of diet-induced obesity on protein expression in insulin signaling pathways of skeletal muscle in male Wistar rats. Int J Gen Med. 2012;5:573–582. doi: 10.2147/IJGM.S31819. [DOI] [PMC free article] [PubMed] [Google Scholar]