

Abstract
With the technology of reprogramming somatic cells by introducing defined transcription factors that enables the generation of “induced pluripotent stem cells (iPSCs)” with pluripotency comparable to that of embryonic stem cells (ESCs), it has become possible to use this technology to produce various cells and tissues that have been difficult to obtain from living bodies. This advancement is bringing forth rapid progress in iPSC-based disease modeling, drug screening, and regenerative medicine. More and more studies have demonstrated that phenotypes of adult-onset neurodegenerative disorders could be rather faithfully recapitulated in iPSC-derived neural cell cultures. Moreover, despite the adult-onset nature of the diseases, pathogenic phenotypes and cellular abnormalities often exist in early developmental stages, providing new “windows of opportunity” for understanding mechanisms underlying neurodegenerative disorders and for discovering new medicines. The cell reprogramming technology enables a reverse engineering approach for modeling the cellular degenerative phenotypes of a wide range of human disorders. An excellent example is the study of the human neurodegenerative disease amyotrophic lateral sclerosis (ALS) using iPSCs. ALS is a progressive neurodegenerative disease characterized by the loss of upper and lower motor neurons (MNs), culminating in muscle wasting and death from respiratory failure. The iPSC approach provides innovative cell culture platforms to serve as ALS patient-derived model systems. Researchers have converted iPSCs derived from ALS patients into MNs and various types of glial cells, all of which are involved in ALS, to study the disease. The iPSC technology could be used to determine the role of specific genetic factors to track down what’s wrong in the neurodegenerative disease process in the “disease-in-a-dish” model. Meanwhile, parallel experiments of targeting the same specific genes in human ESCs could also be performed to control and to complement the iPSC-based approach for ALS disease modeling studies. Much knowledge has been generated from the study of both ALS iPSCs and ESCs. As these methods have advantages and disadvantages that should be balanced on experimental design in order for them to complement one another, combining the diverse methods would help build an expanded knowledge of ALS pathophysiology. The goals are to reverse engineer the human disease using ESCs and iPSCs, generate lineage reporter lines and in vitro disease models, target disease related genes, in order to better understand the molecular and cellular mechanisms of differentiation regulation along neural (neuronal versus glial) lineages, to unravel the pathogenesis of the neurodegenerative disease, and to provide appropriate cell sources for replacement therapy.
Keywords: Motor neurons, Glia, Lou Gehrig disease, Induced pluripotent stem cells, CRISPR
1. Introduction
There has been tremendous interest to apply pluripotent stem cell technologies for disease modeling, drug screening and regenerative medicine, as well as to determine genetic factors contributing to disease onset and treatment response as a means of improving health outcomes. The focus of this review article is amyotrophic lateral sclerosis (ALS) (Boillee et al., 2006a; Brown, 1997; Cole and Siddique, 1999), a devastating neurodegenerative disease with a worldwide prevalence of 4–6 per 100,000 people. ALS affects lower motor neurons (MNs) in brainstem and spinal cord, upper MNs in motor cortex, and the corticospinal tract, resulting in progressive weakness and atrophy of skeletal muscles. Death results from respiratory failure within three years on average of initial diagnosis. Despite the selective functional deficiency due to MN loss, recent evidence has implicated glial cells (astrocytes and oligodendrocytes) and microglia as contributors to MN death (Beers et al., 2006; Frakes et al., 2014; Ilieva et al., 2009; Kang et al., 2010; Maragakis and Rothstein, 2006). Although several mechanisms have been proposed to likely contribute to sporadic disease pathogenesis, the etiology of selective MN death in this disease remains elusive. As a result, there exists no effective treatment for ALS.
The groundbreaking development of a cellular reprogramming technology, through which “induced pluripotent stem cells (iPSCs)” could be derived from easily accessible somatic cells such as dermal fibroblasts by forced expression of defined pluripotency-inducing “reprogramming” factors, has provided an unprecedented approach that enables generation of patient-specific cells for cell-specific pathogenesis studies and for cell-based therapeutic developments (Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007). Patient-derived iPSCs have been used to investigate the key pathogenic processes of ALS, using the reprogramming technology to “de-differentiate” patient-specific skin fibroblasts back to stem cells, and then “re-differentiate” them into specific neural lineages to create appropriate in vitro models of “disease in a dish” (Bilican et al., 2012; Donnelly et al., 2013; Kiskinis et al., 2014; Wainger et al., 2014). Such a “de- and re-differentiation” approach is to make a “cellular U-turn” and is ideal to track down what’s wrong in the neurodegenerative disease process. The iPSC technology has proven useful for the generation of individual cell lines from different patients to study the nature of the disease. To complement the iPSC-based approach, researchers have also performed gene targeting to knock-in disease-relevant mutations in human embryonic stem cells (ESCs) for disease modeling and for comparative mechanistic studies. By taking advantage of the ESC/iPSC-based platform to determine the key pathogenic events in disease progression and pathogenic development, researchers are investigating how the patients’ iPSCs take various lineages (neuronal/glial versus non-neural), monitor the development of the neurodegenerative disease phenotypes, and determine their regulatory role in ALS development, in hope to gain new insights into the pathogenesis and treatment of the neurodegenerative disease. This approach thus combines a reverse engineering concept with molecular studies in order to identify key mechanisms in the neuronal versus glial causes of ALS.
2. Disease modeling for ALS using human pluripotent stem cells (PSCs)
Both the molecular mechanisms of neurodegeneration in ALS and the reasons accounting for the selective vulnerability of MNs in ALS remain poorly understood. Several pathogenic mechanisms have been taken into consideration, including impaired RNA metabolism, aberrant protein mis-folding, mitochondrial alterations, defective axonal transport, excitotoxicity and local inflammation (Beers et al., 2006; Beers et al., 2008; Cozzolino et al., 2013; Fischer et al., 2011; Israelson et al., 2015; Ling et al., 2013; Magrane and Manfredi, 2009; Maragakis and Rothstein, 2006; Wong et al., 1995). The discovery of causative genes has given new inputs to the field. After the first report of ALS causative mutations in the gene encoding the Cu/Zn-dependent antioxidant enzyme superoxide dismutase-1 (SOD1), researchers have started to investigate non-cell-autonomous mechanisms linked to the development of ALS disease. The identification of C9ORF72 repeat expansion as the major factor responsible for ALS onset (DeJesus-Hernandez et al., 2011; Renton et al., 2011) in the familial forms has focused attention on the causative role of alterations in RNA metabolism, a line of research supported also by the involvement of mutations in TARDBP and FUS genes that encode DNA/RNA binding proteins in ALS development (Arai et al., 2006; Lagier-Tourenne et al., 2012; Neumann et al., 2006).
Although much work needs to be done with ALS iPSCs, this particular disease is probably the most heavily investigated neurological disorder with human iPSC modeling. The establishment of human cell platforms has allowed to test in vitro some of these pathogenic hypotheses and to model and investigate early disease mechanisms. Neural stem cells or MNs derived from ALS patients or bearing ALS mutations could be used in long-term cell cultures or co-culture with glial cells to examine pathophysiological hallmarks of the disease, including analysis of protein aggregates, altered pathways in the neurodegenerative process, in particular the imbalance of mitochondrial dynamics, electrophysiological properties, gene expression analyses, and examination of patterns of alternative pre-mRNA splicing. These studies might provide important insights into ALS, and provide cell-based assays for drug screening (Bilican et al., 2012; de Boer et al., 2014; Di Giorgio et al., 2008; Dimos et al., 2008; Donnelly et al., 2013; Kiskinis et al., 2014; Li et al., 2015b; Marchetto et al., 2008; Sareen et al., 2013; Wainger et al., 2014).
2.1. Modeling ALS with human PSCs
As a proof of principle for generating disease models using human ESCs, well characterized common human ESC lines were used to mimic the genetic changes in familial ALS patients by overexpressing mutant SOD1 (Karumbayaram et al., 2009). Alternatively, one could introduce mutations of known genes into normal human ESC lines. For instance, creation of SOD1 mutation at the original genomic SOD1 locus would recapitulate the genetic defects in a wide range of familial ALS patients (with A4V being the most lethal form, Figure 1). This approach could be extended to additional SOD1 mutants and other gene mutations identified in ALS. Another strategy makes use of safe-harbor or pre-engineered docking platform human ESC lines. These human ESC (or iPSC) lines usually harbor Cre-lox cassette or R4 integrase binding site at constitutive (such as the human ROSA, HPRT1, and the chromosome 19 adeno-associated virus AAV integration) loci, or lineage specific genes (such as the neural specific transcription factor Olig2) loci (Cerbini et al., 2015; Irion et al., 2007; Liu et al., 2011; Macarthur et al., 2012; Pei et al., 2015; Xue et al., 2009). This strategy ensures the re-targeting of mutation at predetermined and validated genomic loci, facilitating the subsequent cell differentiation and purification, as well as phenotype identification and comparison.
Figure 1.

An example for using human ESC and genetic engineering tools to study ALS. (A) Work flow. (B) Vector design for creating SOD1 A4V mutation in human ESC.
Using genetic modification to generate disease models with human ESCs is limited to known gene mutations, which only cover a small percentage of ALS patients (Boillee et al., 2006a; Brown, 1997; Cole and Siddique, 1999). In order to model a wider range of ALS cases, and to provide the opportunity to examine disease complexity, researchers have generated disease models using ALS patient-derived iPSCs, which open a new avenue to the generation of human neurons and glia that carry the genomes of individuals with familial or sporadic ALS. Differentiation of such stem cells to human MNs and glia is already offering new insights into ALS and molecular pathways that might provide new targets for effective therapy (Bilican et al., 2012; Donnelly et al., 2013; Haeusler et al., 2014; Kiskinis et al., 2014; Sareen et al., 2013; Wainger et al., 2014). There is a need for standardization of models so that isogenic lines differing only in the familial ALS mutation could be compared. There are exciting opportunities for more complex culture systems in either 3 dimensions, organoids culture (Lancaster et al., 2013), or with the addition of other defined cell types, such as astrocytes, oligodendrocytes, and microglia. This system opens many new doors for testing mechanisms and pathways, as well as for discovery of therapeutic targets and agents.
A unique opportunity of the human iPSC system is the ability to probe how complex human genomic architectures, i.e, the complexity of the genome content (single nucleotide polymorphism or SNPs, repeat sequences, duplications, copy number variations or CNVs, etc.), epigenetic composition, as well as the organization of the genome (Alkan et al., 2009; Dixon et al., 2015; Guo et al., 2015), predispose patients to ALS and how they influence the behavior of the various participating cell types. There is enormous promise in the utility of a human cellular system to predict how individual genetic and cellular phenotypic variation contributes to response to pharmacological intervention at clinically relevant levels. Moreover, the genetic technologies available with human iPSCs might allow the deciphering of how complex genomic architectures found in individual humans act together to generate susceptibility and variation in response to the environmental factors that might also contribute to or pharmacologically modify ALS phenotypes in patients.
Because non-cell autonomous effects have been observed in SOD1G93A mice (Boillee et al., 2006b; Nagai et al., 2007; Wichterle et al., 2002), the co-culturing of human iPSC-derived MNs with glial cells would prove very useful for the establishment of a robust in vitro system that could serve as the basis for detailed mechanistic studies of the interactions between SOD1 mutant MNs and glia; these studies also provide an assay for diffusible factors toxic to MNs. They might also provide a cell based assay for small molecules that promote survival of mutant SOD1 MNs. In addition, such studies validate the use of stem cells carrying disease-causing genes to study disease mechanisms. Thus, it is necessary and important to carry out such an approach using fibroblasts from both familial and sporadic ALS patients, converting them to iPSCs and then to MNs and glia for detailed mechanistic studies. The goal is to understand the mechanisms that underlie familial and sporadic forms of ALS, and to ultimately identify approaches for the development of new therapies. This is clearly of great importance, considering the devastating effects of the disease, the lack of mechanistic understanding, and the fact that neither a cure nor treatment is available. A robust in vitro model system in which the pathological events leading to ALS could be studied could lead to the identification of the factors that influence MN survival. The capacity of pluripotent stem cells to self-renew in culture, while retaining their pluripotent potential, provides the opportunity to produce virtually unlimited numbers of differentiated cell types to replenish those lost as a consequence of disease processes. An alternative, but equally important potential of iPSCs is to provide insights into disease mechanisms. In this case, iPSCs carrying the genes responsible for a particular disease could be induced to differentiate into the cell types affected in that disease. Studies of the differentiated cells in culture could provide important information regarding the molecular and cellular nature of events leading to pathology.
An objective of this line of research is to characterize the molecular and physiological nature of the events leading to MN degeneration in ALS. This objective could be approached by detailed comparisons of the mRNAs and proteins produced in normal and patient derived MNs, and by physiological studies. An important goal is to identify gene products that track with progressive MN degeneration. Since patient derived MNs likely behave indistinguishably from primary MNs and the patients’ iPSCs could be readily expanded, this work would neither be limited by availability of material, nor are the observations likely to be confounded by possible heterogeneity in tissue samples. To validate results obtained using the in vitro system, one could employ microarray and RNA sequencing (RNAseq) analyses to profile patterns of gene expression in patient-derived as well as normal MNs obtained from healthy control individuals. These studies would also include detailed studies of the changes in pattern of alternative pre-mRNA splicing.
2.2. Limitations and potential mitigation strategies
Human ESCs are not always available in substantial amount for large scale in vitro studies. Furthermore, a great advantage of iPSCs is that they carry the genetic combination of an individual, thus permitting the study of patient-specific mutations. Issues concerning the heterogeneity of iPSC lines, which could provide misleading results, might be overcome by a careful experimental setting. This should be based on rigorous statistical analysis and standardized protocols. Moreover, the use of cutting-edge molecular methods allows correcting the mutation in patient-derived iPSCs, thus obtaining isogenic controls. Genome editing of human cells aims to repair or eliminate a mutation that could cause disease. The premise is that corrective changes to a sufficient number of cells carrying the mutation, in which the genetic fixes would last the lifetimes of the modified cells and their progeny, could provide isogenic controls for rigorous research or even a “one and done” curative treatment for patients. The newest addition to the genome-editing arsenal is CRISPR/Cas9, a bacteria-derived system that uses RNA molecules that recognize specific human DNA sequences. The RNAs act as guides, matching the nuclease to corresponding locations in the human genome. CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated) system is the simplest genome-editing tool to work with because it relies on RNA-DNA base pairing, rather than the engineering of proteins that bind particular DNA sequences. The CRISPR technique has dramatically expanded research on genome editing in human stem cells (Cong et al., 2013; Fu et al., 2014; Jinek et al., 2012; Li et al., 2015a; Xue et al., 2014; Yang et al., 2013a).
Another important advancement is that reliable results could be obtained with the use of non-integrative reprogramming methods. Advances in reprogramming methods have also led to the possibility of directly reprogramming mature cells (i.e., fibroblasts or astrocytes) into relevant cell types (i.e., induced MNs). These trans-differentiation methods could be assessed for ALS disease modeling. In addition to the well-studied role of MNs in ALS pathology, attention has recently been paid on glial cells in ALS pathogenesis. Indeed, astrocytes, oligodendrocytes and microglia have all proved to contribute to MN dysfunction and play key roles in neurodegeneration in ALS. It is crucial to pay attention to the cellular subtypes obtained in culture with varying differentiation protocols in order to avoid misleading results. Well-established cellular markers need to be used to assess both the cell phenotype and the maturation state. In addition, several studies have highlighted the possibility that reprogrammed cells might maintain an epigenetic memory of the cell of origin, which could interfere with the expression of membrane markers. It also needs to be pointed out that pathogenesis studies benefit from long-term observation, from the cell pluripotent state to the mature phenotype, in order to speculate on the time-dependent disease alteration. Nonetheless, patient-derived iPSCs have truly proven as invaluable tools for disease modeling, as they provide a direct way to test the possibility that a key molecule or pathway or a specific defect is responsible for a particular neurodegenerative disorder in humans. This approach involves the production of iPSCs from adult cells derived from individuals with the disorder and then inducing these iPSCs to form neurons and glial cells. Such an approach using patient-derived iPSCs has been applied to model ALS, revealing key pathogenic events in this disease.
3. Investigating key pathogenic events in ALS using PSCs
The development and optimization of iPSC-based platforms has allowed elucidating disease-specific mechanisms, which are exquisitely human. Further advances might eventually lead to the path to a full understanding of key pathogenic events, leading to the development of effective treatments for ALS. The majority of ALS cases are sporadic with only 5–10% of patients having a family history of the disease. Studies of familial ALS patients have identified a number of genes, mutation of which likely accounts for the onset of the disease. However, twin studies have estimated ALS heritability at greater than 60%, strongly suggesting that a majority of sporadic ALS cases also have a genetic basis. Genomic sequencing studies of sporadic ALS patients and their parents demonstrate that patients often carry compound heterozygous mutations that potentially inactivate genes whose loss leads to ALS onset. Examining the functions of these genes in iPSC-based models and systems to assess their likely association with ALS would provide a format for tailoring treatment options on the basis of the genetic underpinning of the disease.
3.1. Identification of genetic defects
While mutations of the SOD1 gene in an autosomal dominant trait have been identified in a small percentage of familial patients, no consistent genetic alterations have been found in sporadic subjects, who are the majority of ALS cases. Currently, transgenic rodents are the disease models available for pathogenesis study and drug testing. As no sporadic ALS disease models exist, the majority of preclinical trials are based on animal models of familial ALS that do not recapitulate sporadic ALS. In addition, rodent models, although do mimic disease phenotypes, often are not satisfying tools for studying pathogenesis in humans. These needs prompt us to model ALS using a reverse engineering approach, allowing for a developmental perspective for the neurodegenerative disease and providing a platform to determine key pathogenic events in disease development. ALS iPSCs could be used to determine how developmental dysfunction and pathogenic signaling might critically contribute to the development of MN and glial pathology in human ALS.
3.2. Investigation of mitochondria dysfunction
Mitochondrial dysfunction has been implicated in many human neurodegenerative diseases including ALS. Understanding the molecular basis of mitochondrial function and dysfunction in human cells would transform our understanding of the role of this critical organelle in human health and disease. It would be interesting to investigate mitochondrial function in the human ESC/iPSC-based models, i.e. to study how mitochondrial function dictates the fate of ALS human ESC/iPSCs and their differentiation toward MNs and glia, in hope to gain new insights into pathogenesis and treatment of the neurodegenerative disease. For example, it would be interesting to examine mitochondrial dysfunction in human ESCs/iPSCs with ALS SOD1 mutations. In addition, iPSC technology could be used as an innovative tool for human mitochondrial research since we could generate iPSCs with normal or dysfunctional mitochondria from healthy people and ALS patients, and then force the cells to differentiate into various cell lineages to evaluate how the state of mitochondrial function dictates the ability of the iPSCs to provide distinct lineages. Elucidating the complexity of human mitochondrial function through the use of ALS patient-derived iPSCs that are reprogrammed from a disease state allows for investigation into mechanisms that regulate mitochondrial mechanisms in ALS-specific iPSCs. Comparison of mitochondrial morphology and function in a variety of human ESCs and iPSCs would allow evaluation of basic mitochondrial function and structure to gain an appreciation for the variability in these human ESC/iPSCs and for the global effects on human ESC/iPSC function. It would also be interesting to direct human ESC/iPSCs with normal or ALS mitochondria to differentiate into various cell types of brain cells (MNs vs. astrocytes vs. oligodendrocytes), to evaluate how the state of mitochondrial function would dictate the ability for human ESC/iPSCs to provide distinct lineages. This approach might shed new light on mitochondrial regulation of cell behavior and to provide information for future therapeutic applications. Combined with mitochondrial imaging techniques, the iPSC technology would likely enhance our ability to monitor and measure human mitochondrial function or dysfunction in healthy and diseased cells. In addition, these studies should be complemented and strengthened by electron microscopy to evaluate the progress of the disease in WT and mutant stem cell-derived MNs, in the presence or absence of WT and mutant glia. Mitochondrial dysfunction has been implicated in ALS pathology as a modulator of MN apoptosis, calcium buffering, SOD1 inclusions, and other morphological alterations (Boillee et al., 2006a). Electron microscopy experiments could be carried out to evaluate the number, distribution and morphological changes known to be associated with mitochondrial dysfunction in ALS. In addition, physiological studies in this model using electrophysiology and calcium imaging would help determine the role of neuronal activity in the progression of ALS. The primary goal is to identify the mechanisms that trigger neuronal death at early stages, before the death of MNs. Electrophysiological studies and analysis of calcium activity could provide important clues with regards to pre- and post-synaptic changes associated with ALS. These experiments would help understand how ALS develops and how synaptic connectivity is established and regulated in the course of the disease.
3.3. RNA toxicity in ALS pathogenesis
Recent studies from a number of labs have identified a nucleic acid binding protein called TDP43 in high molecular weight or detergent insoluble fractions in spinal cords from sporadic ALS patients (Arai et al., 2006; Bilican et al., 2012; Lagier-Tourenne et al., 2012; Neumann et al., 2006). Additional biochemical studies identify both ubiquitinated and partially degraded TDP43 protein in ALS patients, and double-label immunofluorescence studies showed that ubiquitin and TDP43 co-localize in spinal cord MNs of ALS patients. As neither the abnormally sized TDP43 nor ubiquitin-/TDP43-immunoreactive inclusions were seen in either control patients or patients diagnosed with other neurodegenerative diseases, it was proposed that TDP43 pathology might either cause or result from a common underlying cause of ALS. Another surprising result was that TDP43 pathology was not restricted to neurons: TDP43-ir cytoplasmic inclusions were also found in glia. If TDP43 pathology indeed causes ALS, then it might do so either by gain of a toxic function or loss of a vital function. At this stage, because there is no experimental system in which to study cytoplasmic TDP43-ir inclusions, exploratory experiments on a possible gain of function would be very difficult. On the other hand, it might be that a loss of normal TDP43-regulated splicing leads to MN death. In that case using biochemical techniques to analyze normal TDP43 physiology in cell culture and mice models might elucidate the vital functions disturbed in ALS. TDP-43 has previously been implicated in alternative splicing and transcription (Acharya et al., 2006; Ayala et al., 2006; Buratti et al., 2001; Buratti et al., 2004). It is known that RNA processing is exquisitely regulated in the mammalian nervous system; the brain exhibits more alternative splicing than any other organ and many neuronal diseases are pathologies of splicing factors (Licatalosi and Darnell, 2006). If ALS is caused by misregulation of TDP43-dependent splicing, then a genome-wide analysis of its targets could provide major advances in our understanding of the disease (Bilican et al., 2012).
Given the technological feasibility in genomic sequencing-based technologies (RNASeq, ChIPSeq and CLIPSeq), it would be interesting to investigate the role of TDP43 in regulating alternative pre-mRNA splicing in MNs (Johnson et al., 2007; Ule et al., 2003; Ule et al., 2005a; Ule et al., 2005b; Ule et al., 2006), and to sequence RNA from stem cell-derived MNs treated with control siRNA or siRNA targeting TDP43 for knock down. Analysis of these data could identify genes dependent on TDP43 not only for their transcriptional regulation but also for different stages of the processing of their encoded RNAs such as alternative promoter usage, splicing, poly-adenylation and editing. It would also be interesting to then sequence biochemically purified DNA (ChIPSeq) or RNA (CLIPSeq) cross-linked to TDP43 using a TDP43 antibody. Comparison of these sequence tags with the RNASeq data might lead to testable hypotheses about the implications of TDP43 binding such as its preferred binding sites and the effect of TDP43 binding on transcription and RNA processing. Sequence analysis of the tags could also identify potential binding partners of TDP43. These data could be examined for genes and RNA processing events already known to be perturbed in ALS to uncover the role TDP43 pathology might play in these deleterious processes.
Hexanucleotide (GGGGCC)n repeats, located at the noncoding exon of the C9orf72 (C9) gene, has been identified to be the most common causative genetic changes in amyotrophic lateral sclerosis (ALS) patients with or without frontotemporal dementia (FTD) (Almeida et al., 2013; Donnelly et al., 2013; Haeusler et al., 2014; Sareen et al., 2013). ALS research has therefore focused more on possible RNA toxicity caused neurodegeneration. Aberrant expansion of GGGGCC repeats might act through forming RNA foci of G-quadruplexes and RNA-DNA hybrids, binding and sequestering RNA-biding proteins, and subsequently disrupting the normal process of transcription, promoter methylation/demethylation, or translation, resulting in dipeptide formation, cytoplasmic inclusion, and RAN (repeat associated non-ATG) translation (Almeida et al., 2013; Donnelly et al., 2013; Gijselinck et al., 2012; Haeusler et al., 2014; Sareen et al., 2013; Todd and Paulson, 2013; Xi et al., 2013; Zu et al., 2011). However, the size of repeats and their exact function in disease initiation and progression have yet to be determined. The relationship of the size of the GGGGCC repeat expansion across different neural cell types or non-neural cell types that might correlate disease pathogenesis, disease onset and progress has not been fully understood (Nordin et al., 2015). One of the limitations is the lack of availability of matched patient isogenic samples. iPSCs provide a renewable source of patient autologous cells that not only retain identical genetic information but also give rise to all neural cell types. Meanwhile, the rapid advancement of genome editing tools including the CRISPR/Cas9 system would allow for the generation of a collection of isogenic iPSCs with desired (GGGGCC)n length from patient iPSCs. These advancements combined have provided unprecedented opportunities to whittle down the versatile symptoms in ALS/FTD (Figure 2). They may also facilitate in the research on other diseases involving GGGGCC or trinucleotide repeat expansion including Parkinson’s disease, Huntington’s disease, Fragile X syndrome, and myotonic dystrophy.
Figure 2.
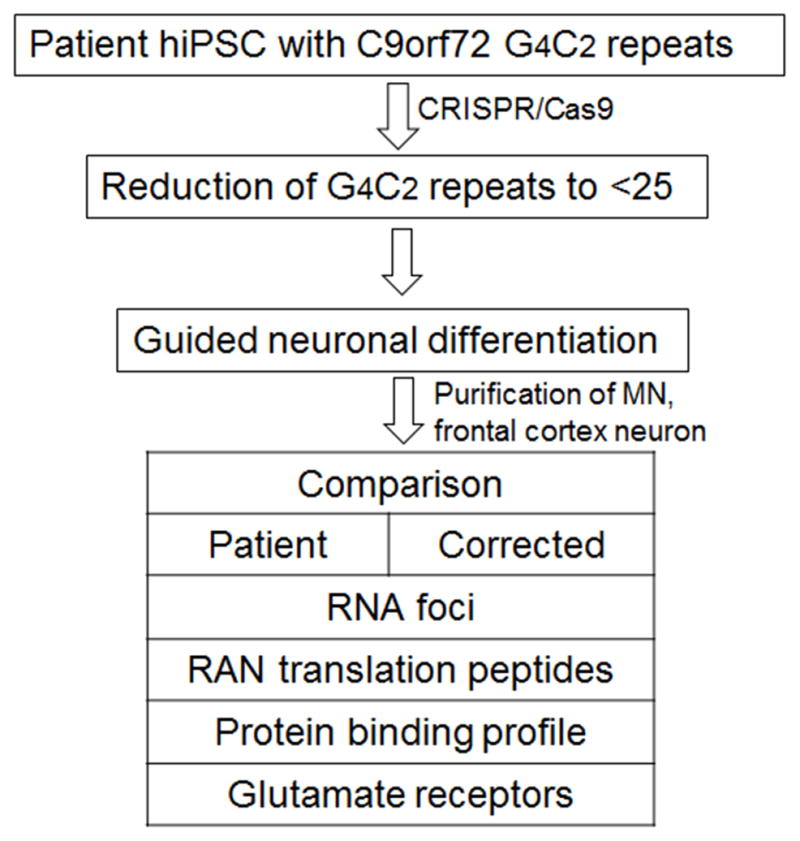
An example for using patient derived human iPSC and CRISPR/Cas9 system for genetic correction in ALS. RAN foci: repeat-associated non-ATG translation.
4. Drug screening for ALS
Cellular reprogramming offers a unique approach to modeling human disorders, as it captures a patient’s genome in relevant cell types that could be propagated in vitro. This kind of “disease-in-a-dish“ approach allows progressive time course analyses of target cells, offering a window of opportunity to reveal molecular or pathway alterations before symptomatic onset. It would be interesting to integrate neurotransmitter/calcium imaging probes to patient-specific iPSC-derived neurons and glial cells to create a platform for modeling “brain disease in a dish”. Such cultured neural networks would enable us to visualize how the precise, guided communication between neurons and glia develops, and how it breaks down in disease. With this system, we could test a library of drugs to identify ones that could correct communication defects in a patient- and disease-specific manner (Bilican et al., 2012; Di Giorgio et al., 2008; Drawnel et al., 2014; Egawa et al., 2012; Mercola et al., 2013; Yang et al., 2013b). Such drug screening would not be possible in living patients. Working with large patient cohorts is important to understand how brain cells derived from the diverse human genetic background respond to specific drugs. As many neurodegenerative disorders share similar neurological symptoms, comparison of in-vitro-derived patient-specific neural cells provides insights into common trends and unique aspects of each neurological disease.
A distinctive new approach to CNS drug discovery is multi-scale network analysis based systems drug screening on the human iPSC-based technology platform of “brain in a dish” in which neural network or brain connectome is derived from neural differentiation of patient-derived iPSCs. This network neuropharmacology (Csermely et al., 2005; Hopkins, 2008) approach involves application of network analysis to determine the set of proteins within the brain network or the wiring of neural circuits that are most critical in the disease pathway, and then chemical biology to identify molecules capable of targeting that set of proteins. By addressing the complexity of disease and by seeking to harness the ability of drugs to influence many different proteins, network pharmacology differs from conventional drug discovery approaches, which have generally been based on highly specific targeting of a single protein. Network pharmacology has the potential to provide new treatments for complex diseases such as ALS where conventional approaches have failed to deliver satisfactory therapies.
In all of life that is relevant to human diseases, there are hundreds of thousands of proteins in the dysfunctional brain network. Every drug compound binds in varying degrees to many different proteins, and could affect very many proteins in other ways, such as through changing their expression, or phosphorylation state. The huge variety of possible protein targets and the large variety of patterns of possible effects on proteins create an enormous range of possible interactions between a drug molecule and the underlying biological system, i.e. the dysfunctional brain network. For this fundamental reason, the biological effects of a molecule’s interactions with pathogenic, pathological and normal cells have been difficult to predict. Existing pharmacotherapies are limited and carry substantial risk of adverse effects. The iPSC-based technology platform of network pharmacology enables analysis of networks associated with the dysfunctional neural network underlying a particular CNS disease. In each case it identifies a disease signature, a set of points in the network at which intervention would have maximum impact. Then the goal is to seek drug candidates whose footprint, the set of proteins impacted by the presence of the drug, matches the relevant disease signature. Drug candidates discovered from this platform of iPSC-based systems pharmacology for disorders of neuro-connectome would more rapidly progress to development pipelines. Widespread integration of iPSC platform with genome-wide association studies and -omics would ultimately allow implementation of this technology in a personalized medicine context. In the case of ALS, iPSC technology provides unique advantages for the discovery of factors that cause ALS and how these factors respond to genetic or pharmacologic manipulation. The generation of human neurons and glia carrying various familial ALS mutations or sporadic ALS genomes would provide a uniquely human cellular system for the evaluation of potential therapeutic interventions. Probing the human genetic and biochemical pathways in MNs and glial cells provides unique advantages owing to the likely differences between humans and other model organisms such as mice. Future work along this direction is warranted.
5. Cell therapy for ALS
Studies on the treatment of ALS are roughly divided into two strategies. One is to reproduce the pathology of ALS and search for a therapeutic drug to improve it. The other is the complementation and regeneration of MNs with ALS, which have lost their function to appropriately stimulate muscle, by cell transplantation. Recently, much attention has been placed on stem cell-based regeneration strategies as a promising new treatment for ALS (Lepore et al., 2008; Papadeas et al., 2011). Cell therapy for ALS aims to regenerate MNs and additionally provide a favorable microenvironment by replacing astrocytes and microglia. MNs have been generated in vitro from several stem cell sources. However, to be clinically successful in ALS, transplanted MN precursors should extend axons across long distances and integrate with the existing neural circuitries and innervate target muscle fibers. Preclinical studies in rat models showed that spinal transplantation of MN progenitors could extend axons to ventral roots and innervate host muscles resulting in partial recovery from paralysis. Although this sounds promising, several finer characteristics need to be defined before clinical translation of this technology. For example, MN progenitors need to be delivered at multiple sites along the spinal cord and they should be directed to cervical, thoracic or lumbar phenotypes. It should also be evaluated whether corticospinal neurons that degenerate in ALS could be effectively replaced. Perhaps the most important hurdle in MN regeneration would be to ensure that the hostile microenvironment for this cell type in the spinal cord is also favorably modified.
Replacing lost MNs using human ESCs or iPSCs remains a potential therapy for ALS, but relies on transplanting MNs expected to survive and form long-distance projections (from brain to spinal cord, and/or spinal cord to muscle) with functional connections. This is a very challenging proposition given the long distances required for axonal outgrowth in the adult compared to development (when the MNs originally make contact with the muscle). In addition, the new MNs are placed in a very toxic environment where all those around them are dying. Alternatively, transplanting cells that have the ability to support the survival of existing MNs or potentially “detoxify” the environment is a far more practical idea. Support cell types could be generated from other tissues such as mesenchymal stem cells (MSCs) isolated from the bone marrow and neural stem cells (NSCs) isolated from fetal brain. These cells are not as likely to make primitive tumors as those derived from pluripotent cells. They are a safer potential product and have provided a much faster path to the clinic, although pluripotent stem cell research is developing rapidly and clearly this field would expand enormously in the coming years.
There have been several advancements using MSCs and NSCs, all of which rely on using the stem cells to stimulate the survival of existing MNs rather than MN replacement. MSCs and NSCs have been used to generate immunomodulatory cells, growth factor-releasing cells, functional support cells such as glia, or GABAergic interneurons to modify MN survival and activity. Several clinical studies (Choi et al., 2010; Connick et al., 2011; Karussis et al., 2010; Mazzini et al., 2012; Xiao et al., 2015) have examined the effect of transplanting MSCs to alter the inflammatory microenvironment in the spinal cord of ALS patients. Intravenous allo-transplantation of MSCs showed migration of the cells to the pathologic lesions. Intra-spinal injections of autologous MSCs reduced inflammation and MN loss and led to significant improvement in ALS patients. Although these studies show that ALS patients could benefit from MSC transplantation, continued fundamental research to understand the specific effects mediated by these transplanted cells would be necessary to improve therapeutic directions. This understanding would also facilitate optimizing human ESCs or patient specific iPSCs protocols for ALS treatment.
Transplantation of human VEGF-overexpressing NSCs conferred protection on MNs and delayed disease progression (Vande Velde and Cleveland, 2005). Based on these principles, NeuralStem Inc., a company based in the US has received FDA approval for ALS clinical trials by transplantation of human fetal-derived NSCs into the spinal cord (https://clinicaltrials.gov/ct2/show/NCT01348451?term=neuralstem&rank=2). At the present time, fetal NSC transplantation is considered safer than using of human ESC-derived NSCs due to risk of teratoma formation. NSCs differentiated from human iPSCs were administered by intravenous injections in the ALS mice which exhibited improved neuromuscular function and motor unit pathology and significantly increased life span (Boulis and Federici, 2011; Boulis et al., 2011; Glass, 2010; Glass et al., 2012; Lunn et al., 2011; Riley et al., 2012). The therapeutic effects of these cells were partially attributed to the production of neurotrophic factors (Krakora et al., 2013).
In addition, during early signs of ALS, counteracting MN loss by transplantation of supporting glial cell types to release neurotrophic molecules or by modifying the inflammatory milieu is a more realistic clinical goal for ALS treatment. Several lines of preclinical data have provided the rationale for this approach. For example, in a rodent model for ALS, glial restricted precursors (GRPs) extensively differentiated into mature astrocytes in grafts, and prevented MN loss and reduced microgliosis (Lepore et al., 2008; Papadeas et al., 2011).
A recent study showed that transplantation of human iPSC-derived glial-rich neural progenitor cells into ALS model mice extends their lifespans. The iPSC-derived glial-rich neural progenitor cells were transplanted them into the lumbar cord of the ALS model mice. The transplanted cells differentiated into astrocytes, and the survival time of mice with the graft was extended compared to that of those without the graft. It was also suggested that the transplanted cells improve the spinal cord environment by increasing neurotrophic factors. Neurotrophic factor is important in the pathology of ALS, and it is being investigated as a target of treatment. Cells of the glial lineage have a function to prepare the environment around MNs. In the future, it would be interesting to test whether the transplantation of healthy iPSC-derived MNs or combination of MNs and glia might more efficiently recover the function. This iPSC-based approach would help to realize the promise of autologous cell-based stem cell therapy as well as avoid many ethical concerns associated with human ESCs, but many issues remain to be overcome, and a lot of time is necessary before reaching the stage of investigating the therapeutic effects in humans.
6. Summary
We reviewed the current progress of disease modeling of ALS. A reverse engineering approach of using patient specific iPSCs allows for modeling the cellular degenerative phenotypes ALS. The iPSC technology together with advanced genome editing also provides an excellent platform for detailed investigation of ALS pathogenesis, as well as drug screening and testing. Such a platform combined with animal studies will offer useful clues for preclinical studies and clinical trials for the treatment of ALS.
Highlights.
Patient-derived iPSCs can be used to investigate the key pathogenic processes of ALS, using the reprogramming technology to “de-differentiate” patient-specific skin fibroblasts back to stem cells, and then “re-differentiate” them into specific neural lineages to create appropriate in vitro models of “disease in a dish”.
The ESC/iPSC-based platform can be used to determine the key pathogenic events in ALS disease progression and pathogenic development by a reverse engineering approach to identify key mechanisms in the neuronal versus glial causes of ALS.
The iPSC-based technology platform of network pharmacology enables analysis of networks associated with the dysfunctional neural network underlying ALS and other CNS diseases.
iPSCs provide promising cell source for cell therapy in ALS.
Acknowledgments
This work was supported by the Department of Neurosurgery, University of Texas Health Science Center at Houston, Memorial Hermann Foundation-Staman Ogilvie Fund (to Y.L.), the Bentsen Stroke Center Fund (to Y.L.), Mission Connect-TIRR Foundation (to Y.L.), NIH/NIAMS subcontract (to Y.L.), NIH R01NS061983 and R01ES015988 (to W.D.), the National Multiple Sclerosis Society (to W.D.), and Shriners Hospitals for Children (to W.D.).
Glossary
- ALS
amyotrophic lateral sclerosis
- C9ORF72
chromosome 9 open reading frame 72
- ChIPSeq
chromatin immunoprecipitation with high throughput sequencing
- CLIPSeq
cross-linking immunoprecipitation with high throughput sequencing
- CNV
copy number variation
- ESCs
embryonic stem cells
- FTD
frontotemporal dementia
- GRP
glial restricted precursors
- PSCs
pluripotent stem cells (ESCs and iPSCs are collectively called PSCs)
- MNs
motor neurons
- MSCs
mesenchymal stem cells
- NSCs
neural stem cells
- RNAseq
High throughput RNA sequencing
- SNP
single nucleotide polymorphism
- SOD1
superoxide dismutase-1
- VEGF
Vascular endothelial growth factor
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acharya KK, Govind CK, Shore AN, Stoler MH, Reddi PP. cis-requirement for the maintenance of round spermatid-specific transcription. Dev Biol. 2006;295:781–90. doi: 10.1016/j.ydbio.2006.04.443. [DOI] [PubMed] [Google Scholar]
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, Sahinalp SC, Gibbs RA, Eichler EE. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–7. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–99. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Pagani F, Baralle FE. TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett. 2006;580:1339–44. doi: 10.1016/j.febslet.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–6. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A. 2008;105:15558–63. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, Park IH, Friedman BA, Daley GQ, Wyllie DJ, Hardingham GE, Wilmut I, Finkbeiner S, Maniatis T, Shaw CE, Chandran S. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A. 2012;109:5803–8. doi: 10.1073/pnas.1202922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312:1389–92. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Boulis N, Federici T. Surgical approach and safety of spinal cord stem cell transplantation. Neurosurgery. 2011;68:E599–600. doi: 10.1227/NEU.0b013e3182095e2e. [DOI] [PubMed] [Google Scholar]
- Boulis NM, Federici T, Glass JD, Lunn JS, Sakowski SA, Feldman EL. Translational stem cell therapy for amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;8:172–6. doi: 10.1038/nrneurol.2011.191. [DOI] [PubMed] [Google Scholar]
- Brown RH., Jr Amyotrophic lateral sclerosis. Insights from genetics. Arch Neurol. 1997;54:1246–50. doi: 10.1001/archneur.1997.00550220050013. [DOI] [PubMed] [Google Scholar]
- Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20:1774–84. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, Pagani F, Baralle FE. Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet. 2004;74:1322–5. doi: 10.1086/420978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerbini T, Funahashi R, Luo Y, Liu C, Park K, Rao M, Malik N, Zou J. Transcription activator-like effector nuclease (TALEN)-mediated CLYBL targeting enables enhanced transgene expression and one-step generation of dual reporter human induced pluripotent stem cell (iPSC) and neural stem cell (NSC) lines. PLoS One. 2015;10:e0116032. doi: 10.1371/journal.pone.0116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MR, Kim HY, Park JY, Lee TY, Baik CS, Chai YG, Jung KH, Park KS, Roh W, Kim KS, Kim SH. Selection of optimal passage of bone marrow-derived mesenchymal stem cells for stem cell therapy in patients with amyotrophic lateral sclerosis. Neurosci Lett. 2010;472:94–8. doi: 10.1016/j.neulet.2010.01.054. [DOI] [PubMed] [Google Scholar]
- Cole N, Siddique T. Genetic disorders of motor neurons. Semin Neurol. 1999;19:407–18. doi: 10.1055/s-2008-1040855. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connick P, Kolappan M, Patani R, Scott MA, Crawley C, He XL, Richardson K, Barber K, Webber DJ, Wheeler-Kingshott CA, Tozer DJ, Samson RS, Thomas DL, Du MQ, Luan SL, Michell AW, Altmann DR, Thompson AJ, Miller DH, Compston A, Chandran S. The mesenchymal stem cells in multiple sclerosis (MSCIMS) trial protocol and baseline cohort characteristics: an open-label pre-test: post-test study with blinded outcome assessments. Trials. 2011;12:62. doi: 10.1186/1745-6215-12-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M, Ferri A, Valle C, Carri MT. Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci. 2013;55:44–9. doi: 10.1016/j.mcn.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci. 2005;26:178–82. doi: 10.1016/j.tips.2005.02.007. [DOI] [PubMed] [Google Scholar]
- de Boer AS, Koszka K, Kiskinis E, Suzuki N, Davis-Dusenbery BN, Eggan K. Genetic validation of a therapeutic target in a mouse model of ALS. Sci Transl Med. 2014;6:248ra104. doi: 10.1126/scitranslmed.3009351. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–48. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–21. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, Diao Y, Liang J, Zhao H, Lobanenkov VV, Ecker JR, Thomson JA, Ren B. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–6. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–28. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, Gerard R, Badi L, Kam-Thong T, Bu L, Jiang X, Hoflack JC, Kiialainen A, Jeworutzki E, Aoyama N, Carlson C, Burcin M, Gromo G, Boehringer M, Stahlberg H, Hall BJ, Magnone MC, Kolaja K, Chien KR, Bailly J, Iacone R. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014;9:810–21. doi: 10.1016/j.celrep.2014.09.055. [DOI] [PubMed] [Google Scholar]
- Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, Aoi T, Watanabe A, Yamada Y, Morizane A, Takahashi J, Ayaki T, Ito H, Yoshikawa K, Yamawaki S, Suzuki S, Watanabe D, Hioki H, Kaneko T, Makioka K, Okamoto K, Takuma H, Tamaoka A, Hasegawa K, Nonaka T, Hasegawa M, Kawata A, Yoshida M, Nakahata T, Takahashi R, Marchetto MC, Gage FH, Yamanaka S, Inoue H. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med. 2012;4:145ra104. doi: 10.1126/scitranslmed.3004052. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Igoudjil A, Magrane J, Li Y, Hansen JM, Manfredi G, Glass JD. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain. 2011;134:196–209. doi: 10.1093/brain/awq314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–23. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Baumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- Glass JD. The promise and the reality of stem-cell therapies for neurodegenerative diseases. Cerebrum. 2010;2010:24. [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Boulis NM, Johe K, Rutkove SB, Federici T, Polak M, Kelly C, Feldman EL. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells. 2012;30:1144–51. doi: 10.1002/stem.1079. [DOI] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, Lu Y, Wu Y, Jia Z, Li W, Zhang MQ, Ren B, Krainer AR, Maniatis T, Wu Q. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell. 2015;162:900–10. doi: 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–90. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–72. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irion S, Luche H, Gadue P, Fehling HJ, Kennedy M, Keller G. Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat Biotechnol. 2007;25:1477–82. doi: 10.1038/nbt1362. [DOI] [PubMed] [Google Scholar]
- Israelson A, Ditsworth D, Sun S, Song S, Liang J, Hruska-Plochan M, McAlonis-Downes M, Abu-Hamad S, Zoltsman G, Shani T, Maldonado M, Bui A, Navarro M, Zhou H, Marsala M, Kaspar BK, Da Cruz S, Cleveland DW. Macrophage Migration Inhibitory Factor as a Chaperone Inhibiting Accumulation of Misfolded SOD1. Neuron. 2015 doi: 10.1016/j.neuron.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–81. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karumbayaram S, Kelly TK, Paucar AA, Roe AJ, Umbach JA, Charles A, Goldman SA, Kornblum HI, Wiedau-Pazos M. Human embryonic stem cell-derived motor neurons expressing SOD1 mutants exhibit typical signs of motor neuron degeneration linked to ALS. Dis Model Mech. 2009;2:189–95. doi: 10.1242/dmm.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, Bulte JW, Petrou P, Ben-Hur T, Abramsky O, Slavin S. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:1187–94. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, Mellin C, Merkle FT, Davis-Dusenbery BN, Ziller M, Oakley D, Ichida J, Di Costanzo S, Atwater N, Maeder ML, Goodwin MJ, Nemesh J, Handsaker RE, Paull D, Noggle S, McCarroll SA, Joung JK, Woolf CJ, Brown RH, Eggan K. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14:781–95. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakora D, Mulcrone P, Meyer M, Lewis C, Bernau K, Gowing G, Zimprich C, Aebischer P, Svendsen CN, Suzuki M. Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model. Mol Ther. 2013;21:1602–10. doi: 10.1038/mt.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, Wancewicz E, Kim AS, Watt A, Freier S, Hicks GG, Donohue JP, Shiue L, Bennett CF, Ravits J, Cleveland DW, Yeo GW. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–97. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–9. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, Maragakis NJ. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11:1294–301. doi: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xue H, Long B, Sun L, Truong T, Liu Y. Efficient generation of hiPSC neural lineage specific knockin reporters using the CRISPR/Cas9 and Cas9 double nickase system. J Vis Exp. 2015a:e52539. doi: 10.3791/52539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Balasubramanian U, Cohen D, Zhang PW, Mosmiller E, Sattler R, Maragakis NJ, Rothstein JD. A comprehensive library of familial human amyotrophic lateral sclerosis induced pluripotent stem cells. PLoS One. 2015b;10:e0118266. doi: 10.1371/journal.pone.0118266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. 2006;52:93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–38. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jiang P, Deng W. OLIG gene targeting in human pluripotent stem cells for motor neuron and oligodendrocyte differentiation. Nat Protoc. 2011;6:640–55. doi: 10.1038/nprot.2011.310. [DOI] [PubMed] [Google Scholar]
- Lunn JS, Sakowski SA, Federici T, Glass JD, Boulis NM, Feldman EL. Stem cell technology for the study and treatment of motor neuron diseases. Regen Med. 2011;6:201–13. doi: 10.2217/rme.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarthur CC, Xue H, Van Hoof D, Lieu PT, Dudas M, Fontes A, Swistowski A, Touboul T, Seerke R, Laurent LC, Loring JF, German MS, Zeng X, Rao MS, Lakshmipathy U, Chesnut JD, Liu Y. Chromatin insulator elements block transgene silencing in engineered human embryonic stem cell lines at a defined chromosome 13 locus. Stem Cells Dev. 2012;21:191–205. doi: 10.1089/scd.2011.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11:1615–26. doi: 10.1089/ars.2009.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–89. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–57. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Mazzini L, Vercelli A, Ferrero I, Boido M, Cantello R, Fagioli F. Transplantation of mesenchymal stem cells in ALS. Prog Brain Res. 2012;201:333–59. doi: 10.1016/B978-0-444-59544-7.00016-0. [DOI] [PubMed] [Google Scholar]
- Mercola M, Colas A, Willems E. Induced pluripotent stem cells in cardiovascular drug discovery. Circ Res. 2013;112:534–48. doi: 10.1161/CIRCRESAHA.111.250266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–22. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Nordin A, Akimoto C, Wuolikainen A, Alstermark H, Jonsson P, Birve A, Marklund SL, Graffmo KS, Forsberg K, Brannstrom T, Andersen PM. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv064. [DOI] [PubMed] [Google Scholar]
- Papadeas ST, Kraig SE, O’Banion C, Lepore AC, Maragakis NJ. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A. 2011;108:17803–8. doi: 10.1073/pnas.1103141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Sierra G, Sivapatham R, Swistowski A, Rao MS, Zeng X. A platform for rapid generation of single and multiplexed reporters in human iPSC lines. Sci Rep. 2015;5:9205. doi: 10.1038/srep09205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ Consortium I. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley J, Federici T, Polak M, Kelly C, Glass J, Raore B, Taub J, Kesner V, Feldman EL, Boulis NM. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: a phase I safety trial, technical note, and lumbar safety outcomes. Neurosurgery. 2012;71:405–16. doi: 10.1227/NEU.0b013e31825ca05f. discussion 416. [DOI] [PubMed] [Google Scholar]
- Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Todd PK, Paulson HL. C9orf72-associated FTD/ALS: when less is more. Neuron. 2013;80:257–8. doi: 10.1016/j.neuron.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–5. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen K, Mele A, Darnell RB. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005a;37:376–86. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, Darnell RB. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005b;37:844–52. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–6. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cleveland DW. VEGF: multitasking in ALS. Nat Neurosci. 2005;8:5–7. doi: 10.1038/nn0105-5. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–97. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–16. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J, Robertson J, Rogaeva E. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet. 2013;92:981–9. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Yang R, Biswas S, Qin X, Zhang M, Deng W. Mesenchymal stem cells and induced pluripotent stem cells as therapies for multiple sclerosis. Int J Mol Sci. 2015;16:9283–302. doi: 10.3390/ijms16059283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Wu S, Papadeas ST, Spusta S, Swistowska AM, MacArthur CC, Mattson MP, Maragakis NJ, Capecchi MR, Rao MS, Zeng X, Liu Y. A targeted neuroglial reporter line generated by homologous recombination in human embryonic stem cells. Stem Cells. 2009;27:1836–46. doi: 10.1002/stem.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Wu J, Li S, Rao MS, Liu Y. Genetic Modification in Human Pluripotent Stem Cells by Homologous Recombination and CRISPR/Cas9 System. Methods Mol Biol. 2014 Mar 11; doi: 10.1007/7651_2014_73. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013a;154:1370–9. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ, Ngo HD, Rosowski KA, Schein PA, Ackeifi CA, Arvanites AC, Davidow LS, Woolf CJ, Rubin LL. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 2013b;12:713–26. doi: 10.1016/j.stem.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108:260–5. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]