

Abstract
The unrestricted use of antibiotics has resulted in rapid acquisition of antibiotic resistance (AR) and spread of multidrug-resistant (MDR) bacterial pathogens. With the advent of next-generation sequencing technologies and their application in understanding MDR pathogen dynamics, it has become imperative to unify AR gene data resources for easy accessibility for researchers. However, due to the absence of a centralized platform for AR gene resources, availability, consistency, and accuracy of information vary considerably across different databases. In this article, we explore existing AR gene data resources in order to make them more visible to the clinical microbiology community, to identify their limitations, and to propose potential solutions.
INTRODUCTION
Over the years, antibiotics have vastly benefitted human and animal health in combatting bacterial infections. Apart from being widely used in clinical practice, antibiotics are also employed in agriculture, aquaculture, and intensive animal farming either as prophylactic agents or for therapeutic purposes (1–3). The unrestrained use of antibiotics has, however, resulted in a higher frequency of resistant human pathogens (4). Acquisition of antibiotic resistance (AR) can result from a variety of genomic alterations, for instance, single nucleotide mutations, large genomic changes such as insertions or deletions, chromosomal rearrangements, gene duplications, and, importantly, factors that have facilitated their rampant spread, i.e., carriage on plasmids and other mobile genetic elements (MGEs), including integrons and transposons (5, 6). A fitting example is the recently reported mcr-1 gene that has been linked to colistin resistance in humans and animals (7–10) and has been found to be associated with at least three different plasmids to date (7, 8, 10).
In the last decade, the emergence of multidrug-resistant (MDR) bacteria that harbor multiple antibiotic resistance mechanisms or genes severely limited therapeutic options (4). Common examples are some of the most important Gram-negative human pathogens such as Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Escherichia coli, which harbor MGEs carrying genes encoding enzymes such as extended-spectrum beta-lactamases (ESBLs) that can hydrolyze penicillins, cephalosporins, and monobactams, along with aminoglycoside-modifying enzymes and the Qnr protection proteins that confer resistance to the fluoroquinolones (11, 12). Thus, a single conjugation event involving such MGEs is enough to transform an antibiotic-sensitive pathogen into a MDR organism that can potentially cause infections that are nontreatable by the current antibiotic arsenal (13). Extremely worrisome are the rising rates of resistance to carbapenems, which are among the most important last-line antibiotics available to us (14). Carbapenemases such as those encoded by blaVIM, blaIMP, blaKPC, and blaNDM are also primarily encoded by MGEs, and, as a result of the high and differing levels of antibiotic selection pressure in hospitals, there has been a rapid evolution and spread of these enzymes with various substrate specificities (15). Currently, more than 40 blaVIM and 10 blaNDM variants are known (16). On the other hand, in important Gram-positive pathogens such as Staphylococcus aureus, resistance to beta-lactam antibiotics is mediated by the “staphylococcal cassette chromosome mec” (SCCmec) MGEs that integrate in a site-specific manner into the Staphylococcus genome (17). Interestingly, the marked differences in the antibiotic resistance profiles of community-associated and hospital-associated strains of methicillin-resistant S. aureus (CA-MRSA and HA-MRSA, respectively) can be largely attributed to the kind of SCCmec elements harbored by these strains. CA-MRSA bacteria harbor the smaller SCCmec type IV, V, or VII elements, whereas HA-MRSA typically contain the larger SCCmec type I, II, III, VI, or VIII elements that encode multiple resistance determinants in addition to the gene encoding beta-lactam resistance, mecA (17). While these examples highlight the complex combinations of emerging AR mechanisms and genes, they represent only the proverbial tip of the iceberg. The application of next-generation sequencing (NGS) technology to the study of pathogen genomes as well as to soil-, marine-, and human-associated metagenomes has given us unprecedented insights into unknown reservoirs and novel AR genes (18–21). Currently, a wealth of information with respect to AR genes is available online in AR gene databases (Table 1 and Fig. 1). As costs of sequencing are steadily decreasing and response times are shortening, the utility of NGS technology as a tool for tracking MDR pathogens in real time for routine hospital epidemiology or as an early warning system for outbreak detection is steadily increasing. Application of NGS to routine clinical microbiology and diagnostics will be especially useful in simplifying the technical algorithms utilized for typing and for antibiotic resistance detection. Currently, the majority of the routine microbiology laboratories still screen for MDR based on phenotypic susceptibility testing, which not only is subject to guidelines and breakpoints but also is time-consuming as it depends on pathogen growth. Previous NGS-based studies have demonstrated high concordance between in silico-predicted and phenotypic antimicrobial susceptibility (24, 36). Nonetheless, sequence-based predictions of phenotypic resistance for clinical purposes need to be made with caution. First, in contrast to phenotypic testing, sequencing data yield information only on resistance to antibiotics but not on susceptibility. Also, the absence of a resistance gene does not preclude the possibility of sensitivity to that antibiotic, as any new resistances that are not in the utilized AR gene database might have been missed. On the other hand, sequence-based predictions might potentially identify a gene whose presence leads to resistance during treatment even when it is not expressed during sensitivity testing under specific growth conditions. Thus, while application of NGS to antimicrobial susceptibility testing can result in a more efficient workflow, the large data sets generated here will be heavily reliant on the available AR gene data resources for quality reference data and interpretation, making it imperative that the latter are well curated, up to date, and comprehensive.
TABLE 1.
Characteristics of available AR gene data resources
| No. | Database/repository | AR gene spectrum | Functionality and feature(s) | Last updatea | Reference |
|---|---|---|---|---|---|
| 1. | ARDB | All AR genes | Webtool, BLASTp, BLASTn | July 2009 | 22 |
| 2. | CARD | All AR genes | Webtool, BLASTp, BLASTn, gene ontology, gene identifier V2, annotation | April 2014 | 23 |
| 3. | ResFinder | All AR genes (except chromosome-specific genes) | Webtool, BLASTn | June 2015 | 24 |
| 4. | LacED | β-Lactamases | Webtool, BLASTp, ClustalW | 25 | |
| 5. | ResFams | All AR genes | BLASTp, Local BLAST, HMM profile | January 2015 | 26 |
| 6. | Patric | All AR genes | Webtool link to CARD and ARDB | December 2015 | 27 |
| 7. | HMP | Human body site-specific study resources | Webtool, BLASTp, BLASTn | November 2011 | 28 |
| 8. | RED-DB | All AR genes | BLASTn, BLASTp | ||
| 9. | U-CARE | Organism specific (E. coli) | BLASTp | 29 | |
| 10. | ARG-ANNOT | All AR genes | BLAST, BioEdit V7.25, annotation | 30 | |
| 11. | BLAD | β-Lactamases | Webtool | 31 | |
| 12. | CBMAR | β-Lactamases | Webtool, BLASTn, BLASTp, ClustalW, MEME/MAST | September 2014 | 32 |
| 13. | Lahey Clinic | β-Lactamases | β-lactamase classification and assigning allelic no.b | March 2015 | |
| 14. | Institut Pasteur | OKP, LEN, OXY | MLST database with additional information on specific β-lactamases | August 2015 | |
| 15. | Tetracycline + MLS nomenclature | Tetracycline and macrolide AR genes | Information on resistance mechanisms and nomenclature | June 2015 | |
| 16. | ABRES Finder | All AR genes | Links to external databases | ||
| 17. | INTEGRALL | Integron types and genetic context of AR genes | Webtool, BLASTn | August 2015 | 33 |
| 18. | RAC | Genetic context of AR genes | Webtool, resistance gene cassette annotation | 34 | |
| 19. | MvirDB | Virulence and toxin factors | BLASTn, BLASTp, link to ARGODB for AR genes | April 2014 | 35 |
Data are based on information available on the respective websites and/or in the respective publications.
Data have been moved to http://www.ncbi.nlm.nih.gov/pathogens/submit_beta_lactamase/.
FIG 1.
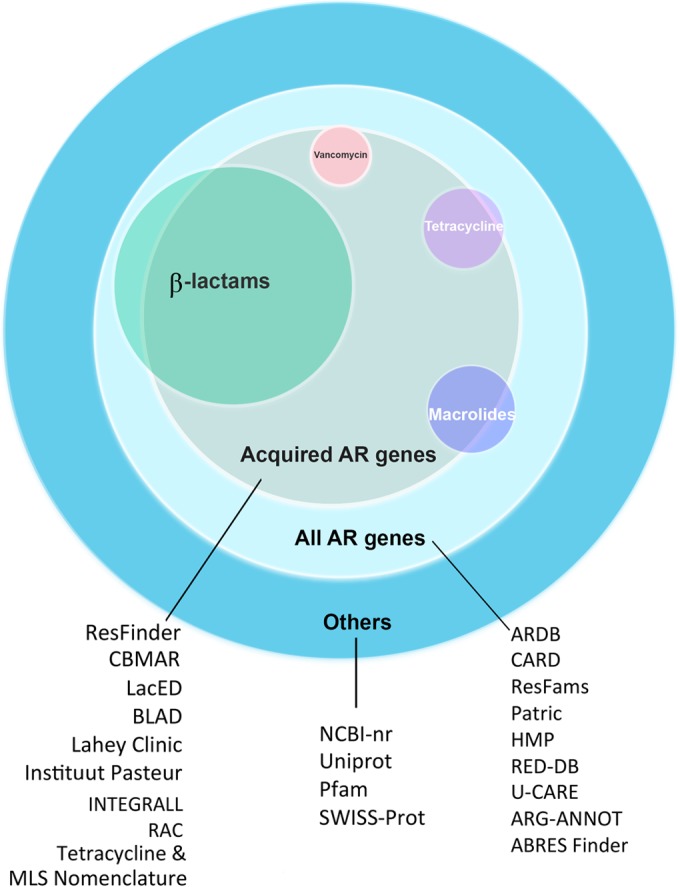
Nonexhaustive overview of available data resources in light of the functional classifications of resistance genes targeting different antibiotic classes. For instance, “β-lactams” refers to all beta-lactamase genes, including ESBLs and carbapenemases. Colors indicate the subsets of genes represented in the databases. The illustration is not drawn to scale.
We reviewed the currently available AR gene data resources with the aim of making them more visible to the clinical microbiology community, particularly emphasizing regular updates and easy accessibility to resources that include AR metadata from published literature. Additionally, we also demonstrated test runs on 4 available databases using in-house and publicly available data. This exercise revealed inconsistent search results, which we discuss in detail, and we propose two complementary approaches that call for combined efforts in addressing this issue.
ANTIBIOTIC RESISTANCE GENE DATABASES
AR gene data resources are online platforms that offer AR-related reference data in support of prediction of resistome- and gene-based antibiograms along with online bioinformatics tools for sequence comparisons, alignment, and annotation. These resources accept user nucleotide or protein sequences as queries and return predictions of their AR gene content, often with confidence-related statistics, annotation, and links to external related resources. We consider first several generalist resources and then move on to AR-focused resources, providing an anecdotal commentary and a tabulated summary of each open access data resource's key characteristics.
The NCBI nonredundant (NCBI-nr) data set (http://www.ncbi.nlm.nih.gov/nuccore) represents one of the largest of the publicly available generalist data resources that include AR genes and associated information. In some cases, however, search results obtained with NCBI-nr might not be specific in terms of gene subtypes, resulting in multiple hits with similar levels of identity and query coverage. It is important that results may vary depending on whether the query sequence is a part of the gene or also includes regions flanking the gene. Thus, additional manual verifications may be required for accurate predictions.
Popular further generalist options that relate to protein-level similarity are the UniProt Knowledgebase (UniProtKB; http://www.uniprot.org/) and the Protein Families Database (Pfam; http://pfam.xfam.org/), which together provide information on protein sequences, functional annotations, and conserved protein families (37, 38). UniProtKB offers an exhaustive collection of protein annotation, cross-references, and literature-derived annotations, while Pfam offers conserved protein families. Pfam uses profile hidden Markov model (HMM) software, HMMER3 (http://hmmer.janelia.org/), in order to identify and build HMMs of protein families. These generalist resources are of value not only because they are comprehensive for publicly available data but also because they serve to feed data to more-specialized AR-gene data resources (Table 1).
The Antibiotic Resistance Genes Database (ARDB; http://ardb.cbcb.umd.edu/index.html), a manually curated specialist AR gene database, appeared very promising at the time of its introduction, combining information from several existing resources and offering AR gene (sub)type and ontology information. At launch, it comprised 13,293 genes, 377 types, 257 antibiotics, 632 genomes, 933 species, and 124 genera to which were applied a two-step filtering of vector sequences, synthetic constructs, and redundant genes and then removal of incomplete sequences, yielding 4,545 antibiotic resistance gene sequences (22). The resource features various tools for annotation and comparison of genes and genomes. Furthermore, a tool for mutation detection is also provided (22). The site allows upload of data as a single gene or in a batch mode for multiple genes or protein sequences. Though the site is functional and user-friendly, the major concern is with the updates of the database as, according to the database statistics, the last update was in July 2009. Following the last update, the database reported 23,137 genes, 380 types, 249 antibiotics, 632 genomes, 1,737 species, and 267 genera and included information on 2,881 vectors (vehicles for transmitting genetic material/genes from one organism to another), including plasmids.
The Comprehensive Antibiotic Resistance Database (CARD; http://arpcard.mcmaster.ca/) was first introduced with a β-lactamase ontology feature. Since its introduction in 2009, regular updates have been announced, with the most recent one in April 2014. The database facilitates access to exhaustive knowledge resources regarding antibiotic resistance genes and their associated proteins that additionally include antibiotics and corresponding targets. CARD data presents a well-developed AR Ontology (ARO) platform that has been expanded from the initial efforts of the ARDB. The ARO allows efficient investigation of molecular data by inclusion of classification of AR genes, functional ontology information, single nucleotide polymorphism (SNP) details for resistance genes, extensive microarray targets, Gene Ontology (GO), Sequence Ontology (SO), and Infectious Disease Ontology (IDO) (23, 37–39). Additionally, CARD also features a graphical Web tool called Resistance Gene Identifier (RGI), Version 2, that was introduced in October 2011 for annotation of query sequences. As of the latest update, CARD includes 3,008 genes tagged specifically for antibiotic resistance and 4,120 genes with AR-related functions. It permits query sequence upload in both batch mode (limited to 20 Mb) and single-sequence mode. The graphical interface was found to be user friendly and highly descriptive, with function-based classification of AR genes.
ResFinder version 2.1 (https://cge.cbs.dtu.dk//services/ResFinder/), most recently updated in July 2015, is a database that provides exhaustive information on AR genes from sequenced or partially sequenced bacterial isolates. Information on resistance genes acquired through horizontal gene transfer can be obtained here. ResFinder not only provides up-to-date information on AR genes but also offers enhanced flexibility in the user interface, which helps minimize unspecific hits. The current version of ResFinder allows a user to set the identity and length coverage thresholds to as low as 30% and 20%, respectively (40). One of the major advantages of ResFinder over other tools is that it accepts NGS raw reads, including de novo assembled contigs, without any limitations on size or sequence length. However, one of the limitations of ResFinder is that the information currently contained in the database is specific for acquired genes and therefore does not include AR mechanisms mediated by chromosomal mutations. Furthermore, the database accepts only nucleotide (and not protein) sequence queries for comparison.
The Lactamase Engineering Database (LacED; http://www.laced.uni-stuttgart.de/) provides systematic analysis and annotation of sequences that helps compare new entries to already existing ones. Furthermore, the database provides integrated tools for sequence comparison and multiple-sequence alignments such as the Basic Local Alignment Search Tool (BLAST) and ClustalW, respectively. The LacED database, however, specializes in information related to mutations, sequences, and structures of TEM and SHV β-lactamases (25).
ResFams (http://www.dantaslab.org/resfams) is a recently established resource for protein families, which are linked to their HMMs associated with AR function. It aims at providing accurate identification and annotation of AR genes. With the information provided, one can also get an overview of the ecology and evolution of the resistant pathogens. The ResFams platform is specifically targeted toward AR gene families and their HMMs, which are further associated with functional metagenomic data sets acquired from various sources, such as soil and human feces, as well as from 6,000 sequenced microbial-isolate genomes across diverse phylogenies and habitats. These data were then utilized to derive 166 HMM profiles comprising the major AR gene classes (26). The authors of the article emphasize that, for resistome analysis, this HMM-based approach is superior to that of the BLAST-based pairwise sequence alignment used by AR-specific databases that are biased toward human-associated organisms and that vastly underestimate the potential impact of environmental resistance reservoirs on AR in pathogens (26). The authors of the article demonstrated this by comparing ResFams HMMs with the BLAST-based ARDB and CARD databases for their ability to predict AR function and showed that 64% of the AR proteins identified using ResFams in both the soil and the human gut microbiota were not detected by BLAST. This increase in sensitivity over that of other AR data resources is expected with HMM-based analysis. HMMs are specific models that are constructed based on observed sequence variation sampled across gene or protein families and capture nuanced positional variability for the family. Searches of query sequences using these models return resulting matches that can be distant and not detectable using sequence-based matching such as that used in BLAST but that can represent valid homologous genes or proteins. However, HMM approaches come at a computational cost, in particular, where models are used in scans of high-volume whole-genome shotgun data. The implication of this cost for ResFams is that the user must provide local computational resources in order to run HMM-based searches. These HMMER tools need to be installed locally using LINUX/UNIX platforms, and the results appear in a tabular form without a graphical interface.
Antibiotic Resistance Gene Annotation (ARG-ANNOT; http://en.mediterranee-infection.com/article.php?laref=283%26titre=arg-annot-) is a rapid bioinformatics tool that is used in identifying putative new AR genes in bacterial genomes. ARG-ANNOT also provides data relating to point mutations. In another study, the tool was tested for its enhanced sensitivity and specificity for both complete and partial gene sequences (30).
The Pathosystems Resource Integrated System (Patric; https://www.Patricbrc.org/portal/portal/patric/AntibioticResistance) provides a platform for genome assembly, protein family comparisons, genome annotations, metadata information such as AR, and pathway comparisons. Patric collects public genome data and currently provides AR data from ARDB and CARD (27).
The Human Microbiome Project (http://hmpdacc.org/HMGOI/), in efforts to characterize the human microbiome, developed a reference set of 3,000 microbial-isolate genomes. The HMP also provides a large collection of AR genes (28).
The Resistance Determinants Database (RED-DB; http://www.fibim.unisi.it/REDDB/) is a nonredundant collection of resistance genes from various nucleotide sequence databases. One can easily look up the database based on the cluster or reference gene names.
The User-friendly Comprehensive Antibiotic Resistance Repository of Escherichia coli (U-CARE; http://www.e-bioinformatics.net/ucare/) is a manually curated resource that provides E. coli-related AR information, including information from 52 antibiotics and 107 resistance genes and associated information of transcription factors and SNPs (29).
The β-lactamase Database (BLAD; http://www.blad.co.in) includes resistance patterns of all classes of β-lactamases collected from published data and NCBI and the crystal structures of proteins from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do). BLAD allows sequence comparison using the BLAST search tool. Apart from facilitating access to information regarding the three-dimensional (3D) structures and physiochemical properties of bound ligands, BLAD also provides links to the popular nucleotide and protein databases (31). The resource specializes in β-lactamase-related information.
The Comprehensive β-lactamase Molecular Annotation Resource (CBMAR; http://14.139.227.92/mkumar/lactamasedb) is a recently established AR gene database which provides a fully interactive environment for data access to an exhaustive range of β-lactamase resources (32). It provides extensive metadata along with detailed molecular and biochemical information which could reveal further insights into novel β-lactamases. CBMAR also features tools such as BLAST and searches for family-specific fingerprints employing MAST (Motif Alignment and Search Tool; http://meme-suite.org/tools/mast). Information related to proteins, nucleotides, protein structures, alignments, mutation profiles, and phylogenetic trees can be downloaded from the database. According to site statistics, the most recent update was performed on 9 September 2014.
Lahey (http://www.lahey.org/studies/) is a conventional database/repository for β-lactamase classification and amino acid sequences for blaTEM, blaSHV, and blaOXA extended-spectrum and other inhibitor-resistant enzymes. Information on plasmid-borne quinolone resistance genes (qnr genes) and qnr allele designations can also be accessed from http://www.lahey.org/qnrStudies/.
At the time of this review, due to the transitioning of the database to a new location, it was not assessed.
The Institut Pasteur database (http://bigsdb.web.pasteur.fr/) provides multilocus sequence type (MLST) data for Klebsiella pneumoniae, including information specific to β-lactamases OKP, LEN, and OXY.
The latest resources regarding tetracycline and macrolide-lincosamide-streptogramin (MLS) AR genes can be obtained from the Tetracycline and MLS Nomenclature database (http://faculty.washington.edu/marilynr/). The latest update for the tetracycline and MLS resources was in August 2015.
The Antibiotic Resistance Gene Finder (ABRESFinder; http://www.bioindians.org/ABRES/) is an AR gene resource that includes information on the gene subfamilies and on the mechanisms of resistance from various sources. ABRESFinder provides links to tools such as BLAST, ClustalW, and Primer3Plus and is mainly focused on AR-related information from India.
Additionally, we would also like to shed light on some of the databases that provide information on integrons, AR-related gene cassettes, and virulence factors. INTEGRALL, the integron database (http://integrall.bio.ua.pt), is a freely available Web tool that provides information on integron sequences and their genetic arrangements with respect to AR genes (33). Furthermore, annotation and information of gene cassettes in mobile integrons can also be accessed using the Repository of Antibiotic Resistance Cassettes (RAC; http://rac.aihi.mq.edu.au/rac/) (34).
The MvirDB (http://mvirdb.llnl.gov/) is a database that targets genes for signature discovery, mainly in identification and characterization of both functional and genetic signatures. MvirDB has a collection of virulence factors and toxins, including AR gene-related resources from several other databases. Apart from aiding medical researchers, the database also aims at centralizing information for biodefense purposes concerning virulence factors and toxins, especially for tracking genetically engineered organisms. The Web interface includes two tools—Virulence browser and Virulence BLAST Interface—for sequence identification and comparison. MvirDB also compares entries to data in a high-throughput microbial annotation database (MannDB; http://manndb.llnl.gov/) (35). Although the Web tools were found to be functional and easy to work with, we observed some broken links in the documentation section.
Besides the specific characteristics discussed above, one of the common issues that arises with AR gene databases is that of false-positive predictions due to certain housekeeping genes that are ubiquitously present in bacterial and, sometimes, in mammalian genomes. For instance, dihydrofolate reductases (encoded by dhfr) are important enzymes catalyzing the folic acid pathway in bacteria and are targeted by the antibiotic trimethoprim. Resistance arises either by overproduction of chromosomal DHFR due to a promoter mutation in E. coli or by production of an altered chromosomally encoded DHFR due to a single amino acid substitution in the dhfr gene in S. aureus (41). However, naturally insensitive enzymes have also been reported in some organisms (41, 42) and the mammalian dhfr genes are also highly similar to their bacterial counterparts. Expectedly, this gene is highly represented in metadata from various communities, including fecal and soil sources (42), and it may be challenging for most AR gene databases, which do not include information from soil and ecological microbiome studies, to single out hits as false-positive predictions. ResFams is one database that includes soil resistome metadata, which aids accurate predictions. Furthermore, terminologies or gene names might differ; for example, dhfr is often referred to as dfrA in certain databases. This ambiguity can be counteracted by conducting a parallel protein-domain-based search using related databases such as Pfam. Thus, although a number of databases are available at our disposal, working with large amounts of data still requires fine tuning of parameters, such as identity levels, E values, and bit scores, to predict the right AR genes and obtain better sensitivity.
ASSESSMENT OF THE PERFORMANCE OF AR GENE DATABASES USING GENE SEQUENCES, WHOLE-GENOME SEQUENCES, AND (FUNCTIONAL) METAGENOMICS DATA
Next, we carried out test runs on the selected databases, namely, ARDB, CARD, ResFinder, and CBMAR, using our in-house data and those of others. We selected those databases in particular because ARDB, CARD, and ResFinder are among the most popular AR-related reference data sources. CBMAR is a recently established database that offers a comprehensive collection of data resources and tools related to AR genes. The query sequences used to assess the databases consist of AR gene sequences, whole-genome sequences, and metagenomics data, including whole-genome shotgun and functional-metagenomics sequences.
To further verify the availability of the latest resources and the accuracy of AR gene predictions from the 4 databases, we selected two of the known carbapenemase genes, blaVIM and blaNDM, and their variants as query sequences. The entire sequences of blaVIM-1 (KT124311), blaVIM-2 (KR337992.1), blaVIM-4 (AJ585042.1), blaVIM-19 (KT124310), and blaVIM-35 (JX982634.1) and of blaNDM genes such as blaNDM-1 (KP770030.1), blaNDM-2 (JF703135.1), blaNDM-4 (KP772213), blaNDM-6 (KJ872581.1), and blaNDM-8 (NG_036906.1) were downloaded from the NCBI database and used in our analysis. Runs were performed with the BLAST parameters set to the default for each of the databases used. Of the 10 genes that we used for screening the 4 databases, 3 (blaVIM-1, blaVIM-2, and blaVIM-4) were predicted correctly by all 4 of the databases. blaVIM-19 and blaVIM-35 were incorrectly predicted by the ARDB and CBMAR databases. ARDB returned several nonspecific hits to the blaVIM gene type, with average similarity percentages of 94.12% and 96.43% for blaVIM-19 and blaVIM-35, respectively. The results shortlisted blaVIM genes but not the variants used as the queries. In the case of CBMAR, blaVIM-19 and blaVIM-35 yielded nonspecific hits; the top 10 hits pointed to the blaVIM-4 gene and to the blaVIM-1, blaVIM-4, and blaVIM-5 genes, respectively. The CARD and ResFinder databases produced correct results. In the case of the blaNDM genes, BLAST results from the ARDB and CBMAR databases returned no hits. However, the CARD and ResFinder databases were found to consistently return correct hits (Fig. 2a). We did not observe any differences in the results that corresponded to the use of the entire sequence or of the partial sequence as the query.
FIG 2.

Comparison of AR gene data resources ARDB, CARD, ResFinder, and CBMAR using single-gene sequences, whole-genome sequences, and metagenomics data sets as queries. (a) BLAST results obtained with blaVIM and blaNDM genes and their variants as queries against the four databases. (b and c) Results obtained using whole-genome sequences (H-EMRSA-15, JKD6008, and UAS391) (b) and metagenomic sequences (45) (c) as queries.
Next, we utilized whole-genome sequences of 3 MRSA strains, UAS391, H-EMRSA-15, and JKD6008, with GenBank accession numbers CP007690, CP007659 (43), and CP002120 (44), respectively, in order to assess the ability of the databases to predict AR genes and their variants from whole-genome sequence data. Results obtained using whole-genome sequences showed that CARD detected the maximum number of AR genes—6 for UAS391 and JKD6008 and 4 for the H-EMRSA-15 strains. ResFinder predicted 5 for JKD6008 and 1 each for UAS391 and H-EMRSA-15. CBMAR detected 1 each for JKD6008 and H-EMRSA-15 and no hits for UAS391 (Fig. 2b). We were unable to receive results from ARDB as our sequence files of 2.8 Mb were not accepted as queries.
Additionally, we screened the databases with a publicly available whole-genome shotgun metagenomics data set with primary accession number PRJEB3977. The data were obtained from a study considering the effects of a decolonization strategy on the gut resistome (45). Utilizing these data as the query, CARD predicted the maximum number of resistance genes—a total of 11, including 2 aminoglycoside resistance genes, 7 β-lactamases, and 2 either undefined genes or other genes. While ResFinder detected a total of 2, including 1 aminoglycoside and 1 β-lactamase, ARDB predicted a total of 4, 1 each from aminoglycoside, β-lactamase, tetracycline, and others (Fig. 2c). In this case, CBMAR detected no genes.
We screened the databases using functional metagenomics data that came from a recently concluded study of the naso-oro-pharyngeal resistome from 150 healthy individuals across five countries representing the northern (Sweden), southern (Spain), eastern (Poland and Slovakia), and western (Belgium) parts of Europe (J. Vervoort, B. B. Xavier, M. Joossens, Y. Darzi, A. Versporten, C. Lammens, J. Raes, H. Goossens, and S. Malhotra-Kumar, submitted for publication). Here, we utilized a functional metagenomic approach in order to identify differences in the presence of antibiotic resistance genes harbored by healthy individuals and attempted to correlate the results to the antibiotics consumed in each particular country. Samples were enriched overnight in the presence of different antibiotics, and the DNA was isolated, sheared, and cloned in E. coli. Plasmid DNA from the resistant clones was sequenced by Illumina (HiSeq), followed by filtering out of vector-specific sequence reads and de novo assembly of remaining reads using Velvet v1.2.10 (46). Derived contigs were used for a BLAST search for AR genes against ResFinder, ARDB, CARD, and CBMAR. We first utilized ResFinder and CARD-RGI for primary screening, and results were predicted by both tools. As ResFinder is restricted to acquired resistance genes, norA, a multidrug efflux transporter gene identified by CARD, was not identified by ResFinder. Similarly, trimethoprim resistance-conferring genes such as the dfrA (dhfr) gene were predicted only by ResFinder and not by the CARD (RGI), CBMAR, and ARDB databases. Apart from these predictions, we observed that HMM-based Resfams searches performed using our resistome data gave us additional/novel dfr variants and also predicted two (dfrA8 and dfrG) resistance genes previously identified by ResFinder. However, not all dfr genes identified by ResFams in our data were resistance related, which calls for caution while interpreting output from broader databases that include soil/environmental microbiome data.
Finally, in order to check for the availability of up-to-date reference information, we screened all of the selected databases for the recently reported mcr-1 gene, which has been linked to colistin resistance in bacteria. In our observation, as of 16 December 2015, ResFinder was the only database that correctly detected this gene.
In summary, of the 4 popular databases that we screened for latest information and accuracy, ARDB was found to provide information limited to the gene name but not the actual variant. Records of the blaNDM genes were also missing in ARDB. Although ARDB is considered one of the most popular databases in identification of novel AR genes, lack of regular updates has limited its scope. CARD was found to accurately predict the query gene variant and to provide several related hits in the BLAST results. We observed ResFinder to accurately predict all of the query genes. As for the CBMAR database, we found that predictions using the nucleotide sequence of the variant gene (blaVIM) provided correct hits to related blaVIM gene variants in 3 of 5 searches. Whereas a BLAST search performed with blaNDM variants returned no results, using a protein sequence query of these genes produced correct results.
On the basis of our results, we suggest that CARD and ResFinder are ideal when single gene sequences are used as the query. However, using whole-genome sequences and metagenomic sequencing data, CARD performs better than the rest. The ResFinder database, which was found to be up to date and accurate, currently detects only acquired genes and ignores chromosomal mutations. The ARDB is limited in its scope due to the lack of regular updates. As for the CBMAR database, on referring to the resources available for download, we found that information related to all the query variants was available and fully updated. This suggests that, while the sequence repository of the CBMAR database was found to be up to date, the search tools may need to be updated.
A FUTURE PATH
While we have noted the value of the data resources available to support AR-related work, we have also noted a number of limitations. These include gaps, inconsistent results of searches against different resources with the same query data, and a lack of up-to-date reference data. While it is beyond the scope of this minireview for us to formulate solutions to these issues, and it is certainly true that expertise beyond ours alone will be required for these solutions, we take the opportunity here to lay out some thinking that we hope will be useful in stimulating, and perhaps steering, community discussions as to the solutions. We trust that we, and others, will be able to take advantage of existing initiatives, such as the Horizon 2020 COMPARE [COllaborative Management Platform for detection and Analyses of (Re-)emerging and food-borne outbreaks in Europe; http://www.compare-europe.eu/] project, to facilitate and energize these community discussions.
Our proposal is to rise to the challenge with two complementary approaches, the first simpler to lay out in practical terms, the second requiring significant conceptual planning before the practical work. The first approach charges the community to develop and implement appropriate best practices and standards in the gathering of reference AR data, in the description, publication, and dissemination of these data, and in the presentation of methodologies and algorithms offered through the services of each data resource. Establishing best practices on the basis of the open sharing of richly and systematically described reference data (such as sequences, annotations, alignments, and models) is a step that will reduce redundant efforts in discovering source data for analysis and curation in specialist resources and will maximize opportunities to fill gaps. Systematic descriptions of computational methods and query services offered by specialist AR data resources will aid in users' selection of appropriate tools for their analyses and minimize the risk of misinterpretation. In this first approach, we do not seek to fill gaps where they exist in AR data resource services or to benchmark precision and reliability but rather seek to create a landscape of transparent and tractable elements that can contribute to many different current and future analytical infrastructures.
Our second approach calls for decisions as to how data resources, both generalist and AR specialist, should move forward to fill gaps in coverage, to provide consistency between query tools that are intended to serve the same function, to remove redundant data processing, curation, and software development steps to maximize overall productivity, and to guide consumers in making informed analyses using the most appropriate tools. Clearly, broad community engagement will be required to tackle these issues. While the AR data resource provider and consumer community will no doubt present very specific needs, a number of successful initiatives in other domains will be informative. RNAcentral, for example, is the product of a broad collaboration between 38 specialist data resources covering different families of noncoding RNA genes (47) in a model that centralizes database components for noncoding sequences and comprehensive search and discovery across, so far, 22 of the collaborating data resources, while maintaining expert curation and specialist Web access at the expert site. A second example, which differs in its model, comes from the Generic Model Organism Database (GMOD) project (http://gmod.org/), which provides software tools for the maintenance and presentation of model organism data across many community projects, including FlyBase (http://flybase.org/), WormBase (http://www.wormbase.org/#01-23-6), and DictyBase (http://dictybase.org/), for example.
SUMMARY AND CONCLUSIONS
In this minireview, we have compiled information on the available data resources that relate to AR function. In the fight against the spread of MDR pathogens, a collective effort is being made in establishing these resources to share knowledge in free and accessible ways. While we find substantial value in what is already available, we note a number of limitations, including those that relate to frequency of updates and to the functionality and comprehensiveness of the resources as a whole. Indeed, broader coverage, consistent gene terminologies, and up-to-date, centralized records (i.e., records that are unified with respect to soil, human, and other microbiome/resistome data) will be crucial in identifying and tracking the (novel) genomic alterations that are acquired by bacterial pathogens upon progression to antibiotic resistance.
At a time when NGS has become somewhat affordable and relatively rapid and MDR poses an ever-greater challenge to public and animal health, a greater need is created for comprehensive, up-to-date, and interoperable AR gene data resources. We hope that this evaluation will initiate a strategic response among data resource managers in which they come together to work out mutual solutions, which would make it easier for their operations to be sustained and kept more up to date. As coordination at the international level of pathogen-genomics efforts grows, we urge that attention be paid to sustaining and extending AR gene data resources as a critical component of our response to MDR.
ACKNOWLEDGMENTS
This work and A.J.D. are financially supported by the European Union Horizon 2020 Research and Innovation Programme COMPARE [COllaborative Management Platform for detection and Analyses of (Re-)emerging and food-borne outbreaks in Europe; grant number 643476). B.B.X. is supported by University of Antwerp research funds (BOF-DOCPRO 2012-27450).
We declare that we have no conflicts of interest.
Biography

Surbhi Malhotra-Kumar has been an Associate Professor of Medical Microbiology at the University of Antwerp, Antwerp, Belgium, since 2011. Surbhi has a double M.S. in Medical Microbiology and Molecular Biology and a Ph.D. in Medical Microbiology from University of Antwerp. Her research is focused on overlapping areas highly relevant to human health, such as biofilm-related infections, the impact of antibiotic use on drug resistance and pathogenetic evolution of bacteria, and development of rapid diagnostic assays for hospital and community pathogens. She capitalizes on new technologies in bridging the gap between fundamental and clinical research and has more than 60 well-cited, peer-reviewed publications. Surbhi has received several awards for her work, including postdoctoral fellowships from Funds for Scientific Research—Flanders (FWO-F), the Young Investigator Award, 2011, instituted by the European Society of Clinical Microbiology and Infectious Diseases, and the Eugène Yourassowsky Award in Microbiology and Infectious Diseases, 2006, instituted by the Université Libre de Bruxelles, Brussels, Belgium.
REFERENCES
- 1.Cabello FC. 2006. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol 8:1137–1144. doi: 10.1111/j.1462-2920.2006.01054.x. [DOI] [PubMed] [Google Scholar]
- 2.Burns A, Shore AC, Brennan GI, Coleman DC, Egan J, Fanning S, Galligan MC, Gibbons JF, Gutierrez M, Malhotra-Kumar S, Markey BK, Sabirova JS, Wang J, Leonard FC. 2014. A longitudinal study of Staphylococcus aureus colonization in pigs in Ireland. Vet Microbiol 174:504–513. doi: 10.1016/j.vetmic.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Aarestrup FM. 2015. The livestock reservoir for antimicrobial resistance: a personal view on changing patterns of risks, effects of interventions and the way forward. Philos Trans R Soc Lond B Biol Sci 370:20140085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies J, Davies D. 2010. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mwangi MM, Wu SW, Zhou Y, Sieradzki K, de Lencastre H, Richardson P, Bruce D, Rubin E, Myers E, Siggia ED, Tomasz A. 2007. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A 104:9451–9456. doi: 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ploy MC, Lambert T, Couty JP, Denis F. 2000. Integrons: an antibiotic resistance gene capture and expression system. Clin Chem Lab Med 38:483–487. [DOI] [PubMed] [Google Scholar]
- 7.Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, Doi Y, Tian G, Dong B, Huang X, Yu LF, Gu D, Ren H, Chen X, Lv L, He D, Zhou H, Liang Z, Liu JH, Shen J. 18 November 2015. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 16:161–168 doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 8.Hasman H, Hammerum AM, Hansen F, Hendriksen RS, Olesen B, Agerso Y, Zankari E, Leekitcharoenphon P, Stegger M, Kaas RS, Cavaco LM, Hansen DS, Aarestrup FM, Skov RL. 2015. Detection of mcr-1 encoding plasmid-mediated colistin-resistant Escherichia coli isolates from human bloodstream infection and imported chicken meat, Denmark 2015. Euro Surveill 20:pii=30085. doi: 10.2807/1560-7917.ES.2015.20.49.30085. [DOI] [PubMed] [Google Scholar]
- 9.Malhotra-Kumar S, Xavier BB, Das AJ, Lammens C, Hoang HTT, Pham NT, Goossens H. 7 January 2016. Colistin-resistant Escherichia coli harbouring mcr-1 isolated from food animals in Hanoi, Vietnam. Lancet Infect Dis 16:286–287. doi: doi: 10.1016/S1473-3099(16)00014-1. [DOI] [PubMed] [Google Scholar]
- 10.Malhotra-Kumar S, Xavier BB, Das AJ, Lammens C, Butaye P, Goossens H. 2016. Colistin resistance gene mcr-1 harboured on a multidrug resistant plasmid. Lancet Infect Dis 16:283–284. [DOI] [PubMed] [Google Scholar]
- 11.Carattoli A. 2013. Plasmids and the spread of resistance. Int J Med Microbiol 303:298–304. doi: 10.1016/j.ijmm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Vervoort J, Gazin M, Kazma M, Kotlovsky T, Lammens C, Carmeli Y, Goossens H, Malhotra-Kumar S; SATURN WP1 and MOSAR WP2 study groups. 2014. High rates of intestinal colonisation with fluoroquinolone-resistant ESBL-harbouring Enterobacteriaceae in hospitalised patients with antibiotic-associated diarrhoea. Eur J Clin Microbiol Infect Dis 33:2215–2221. doi: 10.1007/s10096-014-2193-9. [DOI] [PubMed] [Google Scholar]
- 13.Alekshun MN, Levy SB. 2007. Molecular mechanisms of antibacterial multidrug resistance. Cell 128:1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Pournaras S, Poulou A, Voulgari E, Vrioni G, Kristo I, Tsakris A. 2010. Detection of the new metallo-beta-lactamase VIM-19 along with KPC-2, CMY-2 and CTX-M-15 in Klebsiella pneumoniae. J Antimicrob Chemother 65:1604–1607. doi: 10.1093/jac/dkq190. [DOI] [PubMed] [Google Scholar]
- 15.Queenan AM, Bush K. 2007. Carbapenemases: the versatile beta-lactamases. Clin Microbiol Rev 20:440–458, table of contents. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cantón R, Akóva M, Carmeli Y, Giske CG, Glupczynski Y, Gniadkowski M, Livermore DM, Miriagou V, Naas T, Rossolini GM, Samuelsen Ø, Seifert H, Woodford N, Nordmann P; European Network on Carbapenemases. 2012. Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin Microbiol Infect 18:413–431. doi: 10.1111/j.1469-0691.2012.03821.x. [DOI] [PubMed] [Google Scholar]
- 17.Malachowa N, DeLeo FR. 2010. Mobile genetic elements of Staphylococcus aureus. Cell Mol Life Sci 67:3057–3071. doi: 10.1007/s00018-010-0389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vervoort J, Xavier BB, Stewardson A, Coenen S, Godycki-Cwirko M, Adriaenssens N, Kowalczyk A, Lammens C, Harbarth S, Goossens H, Malhotra-Kumar S. 2015. Metagenomic analysis of the impact of nitrofurantoin treatment on the human faecal microbiota. J Antimicrob Chemother 70:1989–1992. [DOI] [PubMed] [Google Scholar]
- 19.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Doré J; MetaHIT Consortium, Antolín M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, Delorme C, et al. 2011. Enterotypes of the human gut microbiome. Nature 473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caniça M, Manageiro V, Jones-Dias D, Clemente L, Gomes-Neves E, Poeta P, Dias E, Ferreira E. 2015. Current perspectives on the dynamics of antibiotic resistance in different reservoirs. Res Microbiol 166:594–600. doi: 10.1016/j.resmic.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. 2012. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu B, Pop M. 2009. ARDB–Antibiotic Resistance Genes Database. Nucleic Acids Res 37:D443–D447. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, Kalan L, King AM, Koteva K, Morar M, Mulvey MR, O'Brien JS, Pawlowski AC, Piddock LJ, Spanogiannopoulos P, Sutherland AD, Tang I, Taylor PL, Thaker M, Wang W, Yan M, Yu T, Wright GD. 2013. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thai QK, Bos F, Pleiss J. 2009. The Lactamase Engineering Database: a critical survey of TEM sequences in public databases. BMC Genomics 10:390. doi: 10.1186/1471-2164-10-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson MK, Forsberg KJ, Dantas G. 2015. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. The ISME J 9:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, Gillespie JJ, Gough R, Hix D, Kenyon R, Machi D, Mao C, Nordberg EK, Olson R, Overbeek R, Pusch GD, Shukla M, Schulman J, Stevens RL, Sullivan DE, Vonstein V, Warren A, Will R, Wilson MJ, Yoo HS, Zhang C, Zhang Y, Sobral BW. 2014. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Human Microbiome Project Consortium. 2012. A framework for human microbiome research. Nature 486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha SB, Uttam V, Verma V. 2 May 2015. u-CARE: user-friendly Comprehensive Antibiotic resistance Repository of Escherichia coli. J Clin Pathol doi: 10.1136/jclinpath-2015-202927:jclinpath-2015-202927. [DOI] [PubMed] [Google Scholar]
- 30.Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, Rolain JM. 2014. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother 58:212–220. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danishuddin M, Hassan Baig M, Kaushal L, Khan AU. 2013. BLAD: a comprehensive database of widely circulated beta-lactamases. Bioinformatics 29:2515–2516. doi: 10.1093/bioinformatics/btt417. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava A, Singhal N, Goel M, Virdi JS, Kumar M. 2014. CBMAR: a comprehensive beta-lactamase molecular annotation resource. Database (Oxford) 2014:bau111. doi: 10.1093/database/bau111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moura A, Soares M, Pereira C, Leitao N, Henriques I, Correia A. 2009. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics 25:1096–1098. doi: 10.1093/bioinformatics/btp105. [DOI] [PubMed] [Google Scholar]
- 34.Tsafnat G, Copty J, Partridge SR. 2011. RAC: Repository of Antibiotic resistance Cassettes. Database (Oxford) 2011:bar054. doi: 10.1093/database/bar054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou CE, Smith J, Lam M, Zemla A, Dyer MD, Slezak T. 2007. MvirDB—a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res 35(Database issue):D391–D394. doi: 10.1093/nar/gkl791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zankari E, Hasman H, Kaas RS, Seyfarth AM, Agerso Y, Lund O, Larsen MV, Aarestrup FM. 2013. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J Antimicrob Chemother 68:771–777. doi: 10.1093/jac/dks496. [DOI] [PubMed] [Google Scholar]
- 37.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M. 2005. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol 6:R44. doi: 10.1186/gb-2005-6-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldfain A, Smith B, Cowell LG. 2010. Dispositions and the infectious disease ontology. Formal ontology in information systems (Fois 2010). 209:400–413. [Google Scholar]
- 40.Zankari E. 2014. Comparison of the Web tools ARG-ANNOT and ResFinder for detection of resistance genes in bacteria. Antimicrob Agents Chemother 58:4986. doi: 10.1128/AAC.02620-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eliopoulos GM. 2001. Resistance to trimethoprim-sulfamethoxazole. Clin Infect Dis 32:1608–1614. http://cid.oxfordjournals.org/content/32/11/1608.long. [DOI] [PubMed] [Google Scholar]
- 42.Forsberg KJ, Patel S, Gibson MK, Lauber CL, Knight R, Fierer N, Dantas G. 2014. Bacterial phylogeny structures soil resistomes across habitats. Nature 509:612–616. doi: 10.1038/nature13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sabirova JS, Xavier BB, Hernalsteens JP, De Greve H, Ieven M, Goossens H, Malhotra-Kumar S. 2014. Complete genome sequences of two prolific biofilm-forming Staphylococcus aureus isolates belonging to USA300 and EMRSA-15 clonal lineages. Genome Announc 2:pii:e00610-14. doi: 10.1128/genomeA.00610-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howden BP, Seemann T, Harrison PF, McEvoy CR, Stanton JA, Rand CJ, Mason CW, Jensen SO, Firth N, Davies JK, Johnson PD, Stinear TP. 2010. Complete genome sequence of Staphylococcus aureus strain JKD6008, an ST239 clone of methicillin-resistant Staphylococcus aureus with intermediate-level vancomycin resistance. J Bacteriol 192:5848–5849. doi: 10.1128/JB.00951-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buelow E, Gonzalez TB, Versluis D, Oostdijk EA, Ogilvie LA, van Mourik MS, Oosterink E, van Passel MW, Smidt H, D'Andrea MM, de Been M, Jones BV, Willems RJ, Bonten MJ, van Schaik W. 2014. Effects of selective digestive decontamination (SDD) on the gut resistome. J Antimicrob Chemother 69:2215–2223. doi: 10.1093/jac/dku092. [DOI] [PubMed] [Google Scholar]
- 46.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.RNAcentral Consortium. 2015. RNAcentral: an international database of ncRNA sequences. Nucleic Acids Res 43(Database issue):D123–D129. doi: 10.1093/nar/gku991. [DOI] [PMC free article] [PubMed] [Google Scholar]