

Abstract
Molecular surveillance is essential to monitor HIV diversity and track emerging strains. We have developed a universal library preparation method (HIV-SMART [i.e., switching mechanism at 5′ end of RNA transcript]) for next-generation sequencing that harnesses the specificity of HIV-directed priming to enable full genome characterization of all HIV-1 groups (M, N, O, and P) and HIV-2. Broad application of the HIV-SMART approach was demonstrated using a panel of diverse cell-cultured virus isolates. HIV-1 non-subtype B-infected clinical specimens from Cameroon were then used to optimize the protocol to sequence directly from plasma. When multiplexing 8 or more libraries per MiSeq run, full genome coverage at a median ∼2,000× depth was routinely obtained for either sample type. The method reproducibly generated the same consensus sequence, consistently identified viral sequence heterogeneity present in specimens, and at viral loads of ≤4.5 log copies/ml yielded sufficient coverage to permit strain classification. HIV-SMART provides an unparalleled opportunity to identify diverse HIV strains in patient specimens and to determine phylogenetic classification based on the entire viral genome. Easily adapted to sequence any RNA virus, this technology illustrates the utility of next-generation sequencing (NGS) for viral characterization and surveillance.
Molecular characterization of human immunodeficiency virus type 1 (HIV-1) has revealed an exceptional level of sequence diversity. Inherent factors such as an error-prone reverse transcriptase (RT) along with high rates of replication and recombination all contribute to this variability, reaching as high as 10% within a single individual (1, 2). While a limited number of subtypes and circulating recombinant forms (CRFs) predominate in a particular geographical region, global diversification of HIV is continually being driven by the movement of people around the world and social changes (3–5). Surveillance is essential to monitor global HIV diversity and to identify newly emerging strains, since this will have significant implications from the perspective of screening, diagnostic testing, patient monitoring (e.g., viral load assays), drug resistance, and vaccine development.
The utility of next-generation sequencing (NGS) has been demonstrated for a variety of applications related to HIV patient monitoring, including identification of drug resistance mutations (6–10) and prediction of coreceptor tropism (11, 12). Examples generally entail sequencing only of pol or env amplicons, respectively, to a depth necessary to identify low-level variants. However, full genomes are also of interest for detection of quasispecies and tracking immune evasion through variation in CD8 T lymphocyte epitopes with linkage elsewhere in the genome (13, 14). We recently implemented a randomly primed NGS approach to conduct HIV surveillance and characterize clinical specimens for coinfecting viruses (15). For specimens with viral loads of >4.8 log10 copies/ml, full genomes were obtained 70% of the time; however, the percentage of HIV reads varied significantly among specimens (0.002 to 4.11%).
cDNA synthesis with primers specific for HIV could potentially yield greater overall coverage and sequencing depth. In an article by Henn et al. (16), for example, subtype B-specific primers were used to generate 4 overlapping amplicons (2 to 3 kb in length) that cover the entire genome and were then sequenced by 454 pyrosequencing. A similar strategy for simian immunodeficiency virus (SIV) has been applied using Nextera prior to pyrosequencing (14, 17). Notably, in each approach the strain was known and primers were designed accordingly. From the standpoint of conducting surveillance and characterization of specimens infected with unknown strains, this is not possible. Indeed, HIV-1 is classified into four highly divergent groups: M (major), O (outlier), N (non-M, non-O), and P. The group M pandemic branch is divided into nine subtypes (A to D, F to H, J, and K) and more than 70 CRFs from intersubtype and inter-CRF recombination (Los Alamos National Lab HIV Sequence Database [www.hiv.lanl.gov]); CRFs account for nearly 20% of infections worldwide (18). A large-scale analysis of 2,996 genomic full-length sequences revealed the average nucleotide diversities are 37.5% between groups, 14.7% between subtypes, and 8.2% within subtypes (19).
A universal method for full genome characterization that reliably captures HIV sequence heterogeneity is needed. Gall et al. recently reported a strategy that permits amplification and genome assembly of a wide variety of HIV-1 strains (20). It is similar to the strain-specific approaches in that long individual amplicons must be generated and purified before NGS libraries are generated. Here, we describe a much simpler approach that can be used directly on patient samples and demonstrate its applicability for HIV-1 group M, including numerous subtypes, CRFs, and unique recombinant forms (URFs), as well as for group O, N, and P strains and HIV-2.
MATERIALS AND METHODS
Clinical specimens.
HIV-infected blood donations were obtained from blood banks in Cameroon, South Africa, and Thailand. Collection of discarded, deidentified blood donations for research purposes was approved by local ethics committees, and informed consent was obtained. Preliminary strain classification was based on reverse transcriptase PCR (RT-PCR) amplification of RNA extracted from plasma followed by population sequencing by Sanger and phylogenetic analysis of subgenome regions of gag, pol, and env as previously described (21). HIV-1 viral loads were determined using the RealTime HIV-1 assay on the m2000 system following the manufacturer's instructions (Abbott Molecular, Des Plaines, IL).
Virus isolates.
HIV strains representing a wide range of HIV-1 group M subtypes and CRFs, groups N, O, and P, and HIV-2 were isolated from patient specimens and propagated in cell culture; patient information and GenBank accession numbers for previously reported viral sequences are provided in Table S1 in the supplemental material. For the group M viruses, preliminary HIV-1 strain classification was based primarily on either gag or pol (PR/RT) sequences. For the group N and P strains, complete genome sequences had been determined, and for the group O strains, partial genome sequences were available. For cell culture propagation, patient plasma or peripheral blood mononuclear cells (PBMCs) were incubated with human PBMCs that were previously activated with phytohemagglutinin (PHA) for 3 days in RPMI buffer containing 10% complement-inactivated fetal bovine serum (FBS). Twice per week, cultured cells were resuspended in fresh RPMI medium supplemented with interleukin-2 (IL-2) (10 μl/liter), gentamicin (50 μg/ml), Polybrene (2 μg/ml), and 10% FBS. Peak viral replication was monitored by measuring reverse transcriptase activity (Cavidi, Uppsala, Sweden). Cell-free culture supernatant was harvested and stored at −80°C. To determine HIV-1 virus titer, culture supernatant was diluted into HIV-uninfected human plasma (normal human plasma [NHP]), and viral loads were measured using the RealTime HIV-1 assay. For the HIV-2 isolate, the viral load was measured on the m2000 system using a research assay as previously described, except that a pumpkin-derived RNA was used as an internal control and RNA extracted from electron microscopy-quantified virus particles of the HIV-2 NIH-Z strain was used as the HIV-2 calibrator (22).
RNA extractions.
For virus isolate library construction, culture supernatants were diluted in normal human plasma (NHP) and automated RNA extractions were performed either with the QIAamp DNA blood minikit on the QiaCube (Qiagen, Carlsbad, CA) for total nucleic acid or on an m2000sp instrument using the Abbott RNA sample preparation system (Abbott Molecular; Des Plaines, IL) according to the manufacturer's instructions. Sample volumes were 200 μl and 500 or 600 μl with elution volumes of 100 μl and 70 μl, respectively. The resulting RNA inputs ranged from 8,019 to 385,592 copies for each cDNA reaction and are listed in Table 1.
TABLE 1.
Summary of virus isolate NGS resultsa
| Specimen ID | Control expt no. or HIV group | Subtype | Viral RNA input (no. of copies) | No. of reads: |
% of: |
Sequence depth (×) | Depth SD (×) | ||
|---|---|---|---|---|---|---|---|---|---|
| Total | HIV | HIV reads | Genome coverage | ||||||
| LA09 | 1 | CRF01 | 33,583 | 2,445,293 | 1 | 0 | 0.5 | ND | ND |
| LA09 | 2 | CRF01 | 33,583 | 1,374,471 | 10,615 | 0.77 | 99 | 231 | 392 |
| LA09 | 3 | CRF01 | 33,583 | 1,204,284 | 43,305 | 3.6 | 99 | 853 | 510 |
| LA09 | 4 | CRF01 | 33,583 | 1,105,601 | 120,937 | 10.9 | 99 | 2,541 | 1,851 |
| LA09 | 5 | CRF01 | 33,583 | 118,422 | 32 | 0.03 | 24 | 2.4 | 1.6 |
| LA09 | 6 | CRF01 | 33,583 | 723,407 | 1,837 | 0.25 | 84 | 40 | 43 |
| LA51 | 1 | O | 42,076 | 1,698,542 | 14 | 0 | 14 | 0.2 | 0.5 |
| LA51 | 2 | O | 42,076 | 1,556,269 | 181,343 | 11.6 | 100 | 3,812 | 7,714 |
| LA51 | 3 | O | 42,076 | 1,177,271 | 129,330 | 11.0 | 100 | 2,502 | 1,727 |
| LA51 | 4 | O | 42,076 | 1,669,639 | 670,727 | 40.2 | 100 | 12,934 | 10,999 |
| LA51 | 5 | O | 42,076 | 192,466 | 69 | 0.04 | 34 | 1.1 | 2.3 |
| LA51 | 6 | O | 42,076 | 649,493 | 1,114 | 0.17 | 93 | 20 | 17 |
| LA01 | HIV-M | A | 160,741 | 2,570,888 | 268,802 | 10.5 | 100 | 5,684 | 2,901 |
| LA02 | HIV-M | B | 86,324 | 2,475,214 | 238,949 | 9.7 | 100 | 4,950 | 2,284 |
| LA03 | HIV-M | B | 211,897 | 2,302,602 | 96,464 | 4.2 | 100 | 1,449 | 1,272 |
| LA04 | HIV-M | B | 116,445 | 2,682,766 | 396,688 | 14.8 | 100 | 8,401 | 4,750 |
| LA05 | HIV-M | B | 153,507 | 2,411,778 | 345,127 | 14.3 | 100 | 7,095 | 5,210 |
| LA06 | HIV-M | B | 150,014 | 2,459,618 | 475,929 | 19.3 | 100 | 10,060 | 5,207 |
| LA07 | HIV-M | C | 12,194 | 2,278,578 | 544,430 | 23.9 | 100 | 11,351 | 5,916 |
| LA08 | HIV-M | C | 70,168 | 2,357,246 | 96,126 | 4.1 | 100 | 2,011 | 915 |
| LA17 | HIV-M | D | 188,853 | 329,806 | 110,622 | 33.5 | 99 | 2,319 | 1,628 |
| LA18 | HIV-M | D | 385,592 | 1,343,884 | 73,603 | 5.5 | 100 | 1,340 | 979 |
| LA19 | HIV-M | H | 29,789 | 755,902 | 82,593 | 10.9 | 100 | 1,742 | 1,770 |
| LA20 | HIV-M | F | 46,358 | 571,138 | 350,627 | 61.4 | 100 | 7,338 | 4,644 |
| LA21 | HIV-M | F1 | 67,008 | 983,916 | 306,748 | 31.2 | 100 | 6,359 | 4,454 |
| LA22 | HIV-M | F1 | 92,498 | 967,966 | 50,825 | 5.3 | 100 | 1,020 | 518 |
| LA23 | HIV-M | G | 127,680 | 1,420,142 | 84,297 | 5.9 | 100 | 1,623 | 1,231 |
| LA24 | HIV-M | G | 90,391 | 1,606,488 | 111,036 | 6.9 | 100 | 2,063 | 1,409 |
| LA57 | HIV-M | G | 80,174 | 1,057,838 | 115,153 | 10.9 | 100 | 2,171 | 1,850 |
| LA25 | HIV-M | H | 24,213 | 1,357,150 | 24,151 | 1.8 | 100 | 474 | 304 |
| LA26 | HIV-M | J | 237,755 | 1,400,214 | 101,588 | 7.3 | 100 | 1,952 | 1,189 |
| LA09 | HIV-M | CRF01 | 33,583 | 2,123,166 | 41,644 | 2.0 | 100 | 898 | 436 |
| LA10 | HIV-M | CRF02 | 153,507 | 699,406 | 477,506 | 68.3 | 100 | 10,240 | 6,315 |
| LA11 | HIV-M | CRF02 | 42,280 | 809,298 | 363,898 | 45.0 | 100 | 7,738 | 4,818 |
| LA12 | HIV-M | CRF02 | 12,135 | 1,449,450 | 164,538 | 11.4 | 100 | 3,447 | 2,761 |
| LA13 | HIV-M | CRF06 | 130,655 | 1,129,112 | 22,376 | 2.0 | 100 | 444 | 656 |
| LA14 | HIV-M | CRF06 | 197,757 | 1,196,210 | 47,768 | 4.0 | 100 | 935 | 583 |
| LA58 | HIV-M | CRF06 | 383,735 | 1,013,130 | 546,977 | 54.0 | 100 | 10,025 | 6,402 |
| LA15 | HIV-M | CRF11 | 292,502 | 910,784 | 145,379 | 16.0 | 100 | 2,848 | 1,484 |
| LA56 | HIV-M | URF | 149,290 | 1,201,436 | 474,347 | 39.5 | 100 | 8,568 | 5,668 |
| LA27 | HIV-N | 38,800 | 1,912,588 | 578,940 | 30.3 | 100 | 14,376 | 7,049 | |
| LA28 | HIV-N | NT | 1,564,286 | 155,741 | 10.0 | 100 | 3,422 | 1,604 | |
| LA29 | HIV-O | NT | 1,166,164 | 22,650 | 1.9 | 100 | 467 | 282 | |
| LA30 | HIV-O | NT | 1,508,470 | 105,088 | 7.0 | 100 | 2,124 | 1,206 | |
| LA31 | HIV-O | NT | 1,108,592 | 1,604 | 0.1 | 100 | 33 | 22 | |
| LA32 | HIV-O | 40,750 | 3,916,336 | 325,129 | 8.3 | 100 | 7,354 | 5,601 | |
| LA33 | HIV-O | 18,456 | 940,134 | 23,171 | 2.5 | 100 | 485 | 429 | |
| LA34 | HIV-O | 8,019 | 1,336,488 | 50,865 | 3.8 | 100 | 986 | 589 | |
| LA45 | HIV-O | 65,168 | 142,776 | 50,083 | 35.1 | 100 | 1,088 | 1,563 | |
| LA46 | HIV-O | 167,506 | 129,252 | 70,585 | 54.6 | 100 | 1,452 | 1,820 | |
| LA47 | HIV-O | 73,119 | 1,208,542 | 576,658 | 47.7 | 100 | 11,189 | 9,229 | |
| LA48 | HIV-O | 105,689 | 201,200 | 118,043 | 58.7 | 100 | 2,527 | 3,410 | |
| LA49 | HIV-O | 136,154 | 398,948 | 123,981 | 31.1 | 100 | 2,461 | 3,002 | |
| LA50 | HIV-O | 225,960 | 118,364 | 32,685 | 27.6 | 100 | 638 | 850 | |
| LA51 | HIV-O | 42,076 | 1,700,174 | 362,061 | 21.3 | 100 | 6,500 | 6,260 | |
| LA52 | HIV-O | 87,909 | 2,270,530 | 608,102 | 26.8 | 100 | 10,625 | 8,983 | |
| LA53 | HIV-O | 54,204 | 2,124,180 | 644,939 | 30.4 | 100 | 11,960 | 8,888 | |
| LA54 | HIV-O | 136,154 | 1,719,776 | 790,881 | 46.0 | 100 | 14,826 | 13,213 | |
| LA55 | HIV-O | 13,306 | 1,824,796 | 1,013,709 | 55.6 | 100 | 18,650 | 13,996 | |
| LA35 | HIV-P | 40,000 | 1,979,032 | 350,522 | 17.7 | 100 | 8,228 | 4,928 | |
| LA36 | HIV-2 | A | 28,469 | 1,035,972 | 165,620 | 16.0 | 100 | 3,489 | 2,658 |
Listed for each specimen are the HIV group, the subtype determined by full genome sequences, and the calculated viral RNA input (copies/reaction) for each HIV-SMART cDNA reaction. Control experiments numbered 1 to 6 are listed in the second column. Reported for NGS are the total unprocessed reads, reads aligning to the HIV genome, percentage of HIV reads of the total, percentage of genome coverage, average sequence depth, and the standard deviation of the sequence depth. ND, not determined; NT, not tested. Additional information for isolates containing demographic data and references is found in Table S1 in the supplemental material.
For clinical specimens, plasma was pretreated with benzonase as described below. The m2000sp extraction used 500 or 600 μl of sample with RNA elution in 70 μl. Where noted, manual extractions were also performed using the MagMax kit (Life Technologies, Grand Island, NY), Qiagen viral minikit, Ultra Sens virus kit (Qiagen), and PureLink kit (Life Technologies) or automated extractions as described above according to the manufacturer's instructions. To assess RNA recovery from manual RNA extraction kits, extracted material was substituted for m2000sp eluate prior to RT-PCR master mix addition, and real-time fluorescence detection on the m2000rt instrument with the RealTime HIV-1 assay was performed.
Clinical specimen pretreatment.
Clinical plasma specimens were spun at 5,000 rpm (2,650 × g) for 5 min at room temperature, and supernatants were transferred to fresh tubes. One-tenth volume of 10× benzonase buffer (200 mM Tris-Cl [pH 7.5], 100 mM NaCl, 20 mM MgCl2) and 250 U/ml ultrapure benzonase (Sigma, St. Louis, MO) were added to 0.9 volume of plasma to degrade free DNA and RNA (23, 24). Samples were incubated at 37°C for 3 h and as noted were then filtered by centrifugation (5,000 rpm) through 0.22-μm-pore spin filters (Millipore, Billerica, MA) before extraction. Where also noted, postextraction treatments were performed with Turbo DNase (Life Technologies) by adding 6.5 μl of 10× Turbo buffer and 1 μl of DNase to 60 μl of RNA. Following a 25-min incubation at 37°C, reaction mixtures were inactivated with 6.5 μl of inactivation bead solution for 5 min at room temperature and then centrifuged at 10,000 rpm for 1.5 min before the supernatant was transferred to a fresh tube.
HIV-SMART library preparation.
Libraries were prepared using the SMARTer PCR cDNA synthesis kit essentially as described by the manufacturer (Clontech, Mountain View, CA), with the following modifications. The HIV-SMART (i.e., switching mechanism at 5′ end of RNA transcript) fusion primers, which have the SMART sequence (5′-AAGCAGTGGTATCAACGCAGAGTAC-3′) added to the 5′ end of each HIV-specific reverse primer listed in Table 2, were used for cDNA synthesis. First-strand synthesis reaction mixtures consisting of 3.5 μl of RNA, 0.5 μl of 24 μM SMART primer mix (1.2 μM final concentration for each [described below]), and 0.5 μl of 12 μM 3′SMART CDS primer II A (0.6 μM final concentration) were heated at 72°C for 3 min, and then the temperature was lowered to 47°C for 2 min before the addition of 5.5 μl of master mix (2 μl of 5× first-strand buffer, 0.25 μl of 100 mM dithiothreitol [DTT], 1 μl of 10 mM deoxynucleoside triphosphate [dNTP] mixture, 1 μl of 12 μM SMARTer II A oligonucleotide, 0.25 μl of 40 U/μl RNase inhibitor, and 1 μl of 100 U/μl SMARTScribe reverse transcriptase). cDNA synthesis reaction mixtures of clinical specimens were incubated at 47°C for a total of 90 min (42°C for virus isolates), terminated at 70°C for 10 min, and brought to 4°C before the addition of 0.25 μl RNAse H (2 U/μl [Life Technologies]). Reaction mixtures were incubated at 37°C for 20 min, returned to 4°C, and diluted to 50 μl with water.
TABLE 2.
HIV-SMART primer names along with positions relative to the HXB2 reference genomea
| Primer name | Position | Primer included in: |
Primer sequence | ||
|---|---|---|---|---|---|
| N4 | N6 | N10 | |||
| HS10 | 682–703 | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACAGCCGAGTCCTGCGTCGAGAGA-3′ | ||
| HS15 | 1825–1847 | √ | √ | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACACTCCCTGRCAKGCTGTCATCAT-3′ |
| HS20 | 2077–2096 | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACTTCCCTAAAAAATTAGCCTG-3′ | ||
| HS25 | 2376–2395 | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACCCTATCATTTTTGGTYTCCA-3′ | ||
| HS30 | 3303–3325 | √ | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACTTYTGTATRTCATTGACAGTCCA-3′ | |
| HS45 | 4395–4420 | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACTCTARTTGCCATATYCCTGGACTRCA-3′ | ||
| HS55 | 5195–5218 | √ | √ | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACCTARTGGGATRTGTACTTCTGAAC-3′ |
| HS65 | 6203–6229 | √ | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACCTCRTTDCCACTGTCTTCTBCTCTTTC-3′ | |
| HS85 | 8346–8365 | √ | √ | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACGGTGARTATCCCTKCCTAAC-3′ |
| HS99 | 9607–9628 | √ | √ | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACTCAAGGCAAGCTTTATTGAGGC-3′ |
| 3′SMART CDS IIA | Poly(A) | √ | √ | √ | 5′-AAGCAGTGGTATCAACGCAGAGTACT(30)N-1N-3′ |
The HXB2 reference genome accession number is K03455. In each reverse primer, the SMART adaptor precedes the sequence complementary to HIV. Checks indicate which primers are included in the N4, N6, and N10 mixtures.
The HIV-SMART primer mixtures were combined as follows: for N4, HS15, HS55, HS85, and HS99; for N6, HS15, HS30, HS55, HS65, HS85, and HS99. The N10 mixture included all primers shown in Table 2. The N6 mixture was used for all virus isolates and clinical specimen libraries unless indicated. SMART cDNA was amplified by long-distance PCR on a thermocycler as follows using Advantage II reagents (Clontech): 7.5 μl SMART cDNA, 7.5 μl 10× Advantage 2 PCR buffer, 1.5 μl 50× dNTP mix (10 mM), 1.5 μl 5′ PCR primer IIA (12 μM), 1.5 μl 50× Advantage 2 polymerase mix, and 55.5 μl water (total of 75 μl). Reaction mixtures were cycled as follows: step 1, 95°C for 1 min; step 2, 95°C for 15 s, 65°C for 30 s, and 68°C for 3 min followed by hold at 4°C. Unless indicated, 35 cycles of step 2 were performed. Amplified HIV-SMART cDNA was purified with AMP-Pure magnetic beads (Beckman Coulter, Indianapolis, IN) using a ratio of 1.8× beads to sample (e.g., 135 μl). Incubation times and washes followed the manufacturer's recommendations. Libraries were eluted in 30 μl of 10 mM Tris-Cl (pH 7.5) and then quantified on a Qubit instrument (Invitrogen) using double-stranded DNA (dsDNA) broad-range detection kit reagents (Life Technologies).
NGS library preparation.
The purified cDNA SMART libraries were diluted to 0.2 ng/μl with water, and 5 μl (e.g., 1-ng input) was added to Nextera XT reaction mixtures (Illumina, San Diego, CA). Compatible barcodes were selected, and the manufacturer's protocol was followed, except that 16 cycles of PCR were performed instead of 12. Libraries were purified once more with 0.7× AMP-Pure beads (35 μl) and eluted in 30 μl of Illumina resuspension buffer (RSB). Library concentrations were measured on a BioAnalyzer 2200 TapeStation using a D1K screentape (Agilent Technologies, Santa Clara, CA) based on integration of peaks from 150 to 700 nucleotides (nt) and then adjusted to a 1 nM final concentration before multiplexing. Libraries were combined in equal volumes, denatured with 0.1 N (final concentration) NaOH for 5 min, and diluted to 20 pM with HT1 buffer. The multiplex library was diluted once more with HT1 to 12 pM, and 1% PhiX loading control was added. The multiplex library was denatured at 96°C for 2 min, snap-chilled on ice, and then run on a MiSeq instrument using a 500-cycle MiSeq reagent kit v2 (Illumina).
NGS analysis.
Barcodes were parsed on the MiSeq instrument, and reads were filtered for Q-scores above 30. Fastq files were imported into CLC Genomics Workbench 8.0 software (CLC bio/Qiagen, Aarhus, Denmark), Illumina paired-end reads 1 and 2 were merged (paired-end distance of 100 to 250 nt), and duplicate reads were removed. Reads were trimmed for quality (limit = 0.05) and ambiguity (2-nt maximum), and the SMART adaptor sequence was removed. Reads below 50 nt were discarded; paired-end reads and broken pairs were aligned to a selected HIV reference sequence. The following alignment settings were applied: mismatch = 2, insertion = 3, deletion = 3, length fraction = 0.7, and similarity fraction = 0.8. The preliminary strain classification using RT-PCR and population sequencing by the Sanger method performed in advance of NGS studies guided the selection of the reference genome used for the initial alignment of NGS data; however, HXB2 can substitute for another HIV-1 group M strain when no information is available. The consensus sequence obtained from the first alignment then served as a reference to refine the consensus in a second round of alignment, which was typically sufficient to arrive at the final sequence when no gaps in coverage were present. When gaps in coverage were observed, the data were aligned to one or more additional reference genomes. HIV-mapped reads were then extracted from each alignment, combined, and de novo assembled. Should contigs be formed that bridged these gaps, these sequences were merged with NGS data in Sequencher 5.2.3 software to create a final consensus sequence. The raw NGS data were realigned to the final consensus sequence to generate the NGS statistics. Open reading frames were verified and annotated using SeqBuilder (DNASTAR Lasergene v11.2).
Minor variant analysis.
Values for the conservation of each consensus base call were determined in CLC-Bio for each nucleotide position in the N05-5 genome and averaged, and the standard deviation (SD) was calculated for 14 experiments having an average sequence depth of ≥400×. The Low Frequency Variant Detection tool in CLC Bio was used to estimate a sequencing error rate and identify significant alleles and their frequencies. The statistical model for error is based on quality scores at each position, and no assumption is made about the ploidy of the sample when predicting variants. The cutoff for significance was set at 1%, and the minimum frequency for variants was set at 5.0% for global detection (see Table S3 in the supplemental material) and at 2% for the region shown in Fig. 6C. Default settings for error rate calculations were chosen.
FIG 6.

(A) The reproducibility of consensus base call percentages in 14 N05-5 experiments. Histograms report the proportions of nucleotides in the genome (9,603 bp) by their ranges of base call frequency standard deviations. (B) The N05-5 pol integrase sequence was analyzed. Histograms (above) represent consensus base call frequencies (±SD) at each position accumulated over 14 experiments (also represented in the consensus sequence). Scatter plots (below) show the frequency versus position of minor variants present at ≥2% for each experiment.
Phylogenetic analysis.
Final genomic sequences were aligned with a subset of the Los Alamos HIV Database full genome alignment (www.hiv.lanl.gov). The alignments were gap-stripped and converted to PHYLIP format using BioEdit Sequence Alignment Editor (version 5.0.9; Tom Hall, North Carolina State University, Raleigh, NC). Phylogenetic analysis was performed with the PHYLIP software package (version 3.5c; J. Felsenstein, University of Washington, Seattle, WA). Evolutionary distances were estimated with DNADIST (Kimura two-parameter method), and phylogenetic relationships were determined by NEIGHBOR (neighbor-joining method). Branch reproducibility of trees was evaluated using SEQBOOT (100 replicates) and CONSENSE. Programs were run with default parameters. Trees were constructed using TreeExplorer (version 2.12; Koichiro Tamura of Tokyo Metropolitan University, Tokyo, Japan). Viral sequences were individually analyzed for evidence of recombination using SimPlot (version 3.5.1; S. Ray, Johns Hopkins University, Baltimore, MD) (25, 26). SimPlot calculates and plots the percentage of identity of the query sequence to a panel of reference sequences in a sliding window, which is moved across the alignment in steps, to identify intersubtype mosaicism. If recombination was indicated, BOOTSCAN and FINDSITE were performed, and breakpoints were confirmed by constructing phylogenetic trees for each subfragment.
Nucleotide sequence accession numbers.
Full-genome consensus sequences have been deposited into GenBank under the following accession numbers: KU168256 (LA01), KU168257 (LA02), KU168258 (LA03), KU168259 (LA04), KU168260 (LA05), KU168261 (LA06), KU168262 (LA07), KU168263 (LA08), KU168264 (LA09), KU168265 (LA10), KU168266 (LA11), KU168267 (LA12), KU168268 (LA13), KU168269 (LA14), KU168270 (LA15), KU168271 (LA17), KU168272 (LA18), KU168273 (LA19), KU168274 (LA20), KU168275 (LA21), KU168276 (LA22), KU168277 (LA23), KU168278 (LA24), KU168279 (LA25), KU168280 (LA26), KU168281 (LA29), KU168282 (LA30), KU168283 (LA31), KU168284 (LA32), KU168285 (LA33), KU168286 (LA34), KU168287 (LA36), KU168288 (LA45), KU168289 (LA46), KU168290 (LA47), KU168291 (LA48), KU168292 (LA49), KU168293 (LA50), KU168294 (LA51), KU168295 (LA52), KU168296 (LA53), KU168297 (LA54), KU168298 (LA55), KU168299 (LA56), KU168300 (LA57), KU168301 (LA58), KU168302 (44-10), KU168303 (10-10), KU168304 (1193-8), KU168305 (54-7), KU168306 (62-15), KU168307 (1031-19), KU168308 (8119636), KU168309 (10047107229), KU168310 (100-17), and KU168311 (N05-5).
RESULTS
Design and construction of HIV-SMART libraries for NGS.
Highly conserved stretches of HIV-1 sequence were identified through an alignment of selected reference genomes containing HIV-1 groups M, N, and O. Sites spaced approximately 1.5 to 2 kb from one another were selected to design degenerate primers for initiation of virus-specific cDNA synthesis (Fig. 1; Table 2). In these primers, viral sequences (20 to 30 nt) were fused to a common adaptor sequence (SMART [25 nt]) to make use of the SMARTer PCR kit for cDNA synthesis (red-blue arrows). In place of the oligo(dT)-SMART fusion primer, mixtures of HIV-SMART oligonucleotides are substituted that hybridize to viral RNA to initiate reverse transcription of HIV genomes. The terminal transferase activity of reverse transcriptase (RT) adds nontemplated bases (5′-CCCAU-3′; shown as XX) to the 3′ end of nascent cDNA, to which anneals an oligonucleotide containing the complement of these bases fused to the SMART sequence. The polymerase switches strands to fill in the adaptor so that the SMART sequence is now present at both ends of cDNA, allowing for PCR amplification of libraries with a single SMART primer (Fig. 1). Following bead purification and fluorometric quantification, libraries are then “tagmented” with Nextera XT reagents, whereby topoisomerases cleave double-stranded cDNA at ∼300-bp intervals to covalently link Illumina sequencing primer binding sites, sample indices, and flow cell adaptors (Fig. 1). A low number of PCR cycles generates highly concentrated libraries (>50 nM) with a size range of 200 to 700 bp (peak at ∼330 bp) and average inserts size of ∼190 bp, which are then multiplex sequenced in a 2- by 250-bp paired-end MiSeq run.
FIG 1.
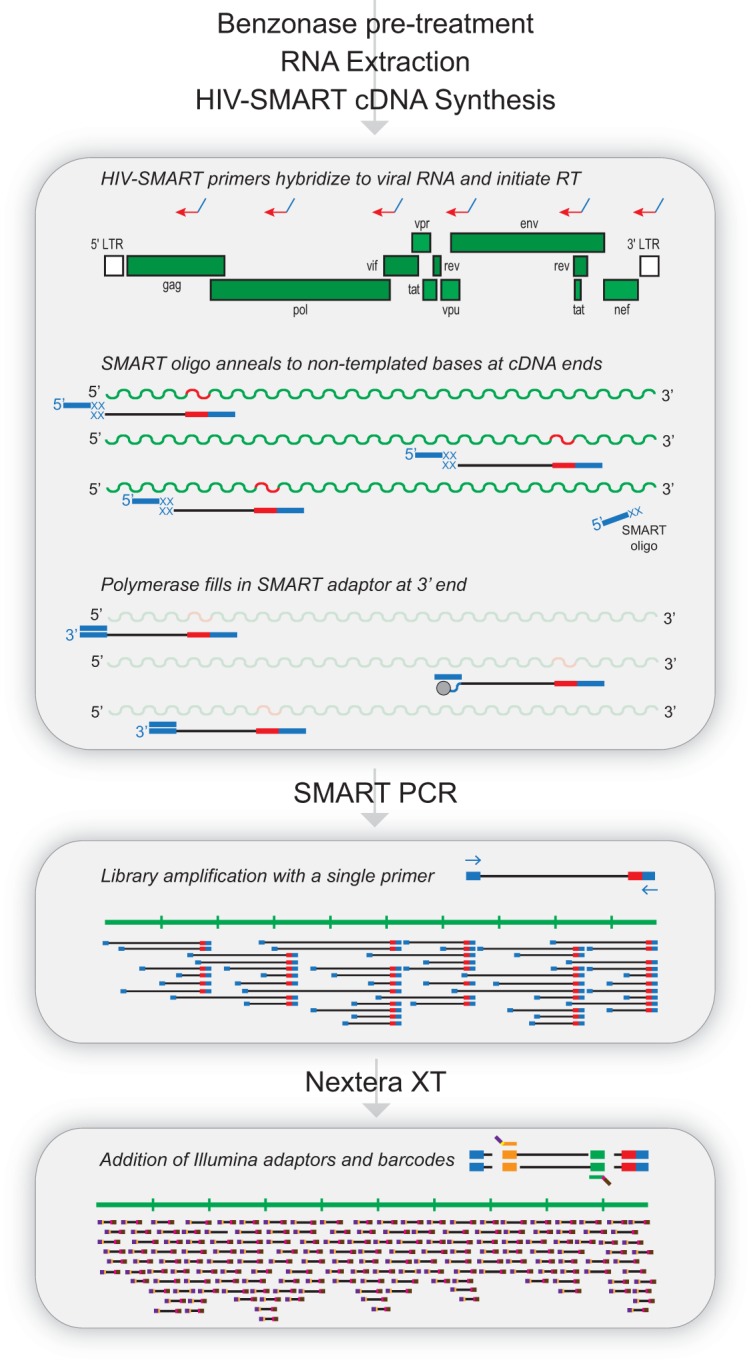
The HIV-SMART protocol workflow. Benzonase treatment is required for clinical specimens prior to RNA extraction. The HIV-1 genome is shown with the locations of six HIV-SMART fusion primers that initiate reverse transcription of viral RNA. In the cDNA reaction, nontemplated bases (XX) are added to the ends of nascent cDNA, to which the SMART oligonucleotide anneals. The RT polymerase switches strands to transcribe the complement of the oligonucleotide, leaving the SMART adaptor at both ends of cDNA. Long-distance PCR with a single SMART primer amplifies libraries and is followed by fragmentation and addition of Illumina primer and barcode sequences with Nextera XT reagents.
Full genome sequencing and characterization of virus isolates by HIV-SMART.
To demonstrate the principle and specificity of the method, control libraries were made from virus culture supernatants infected with either a CRF01_AE recombinant (LA09) or a group O strain (LA51). NGS statistics are reported in Table 1, and coverage maps are shown in Fig. 2A. HIV packages 2 copies of (+) single-stranded RNA (ssRNA) genomes that are 5′ capped, unspliced, and 3′ polyadenylated, and therefore we reasoned that priming with oligo(dT) should facilitate cDNA synthesis (16). This indeed yielded complete sequences for both isolates; however, there was an obvious 3′ bias in coverage, with multiple regions having ≤10× depth. The average depths for nt 1 to 8400 compared to the 8400 end were 99× versus 1,158× for LA09 and 1,315× versus 19,132× for LA51. In comparison, use of the N6 mixture of HIV-SMART primers produced more uniform coverage and the same or greater average depth: e.g., LA09, 853× ± 510× for HIV-SMART versus 231× ± 391× for oligo(dT). Addition of these primer sets together had a highly synergistic effect, yielding 14- and 3-fold greater percentages of HIV reads for LA09 and 3.4- and 3.6-fold greater percentages for LA51 compared to oligo(dT) and the HIV-SMART mixture alone, respectively. When no RT primers were used (not shown; experiment 1) or the SMART amplification step was omitted, both control libraries were extremely dilute and had <70 HIV reads. The use of cDNA directly in the Nextera step yielded <0.04% HIV reads for both isolates, with 24 to 34% genome coverage at 1.1× to 2.4× depth. In contrast, including the HIV SMART amplification step produced full genomes with a 365- to 1,004-fold increase in percentage of HIV reads and a 1,058- to 11,246-fold increase in coverage depth. Finally, reverse transcription with six SMART primers designed against an unrelated RNA virus, while still achieving ≥84% coverage for each isolate, had percentage of HIV reads and coverage depths that were 14- to 65- and 21- to 124-fold less than those of HIV-SMART primers alone, respectively, suggesting the method tolerates mismatches but requires sequence specificity for optimal performance.
FIG 2.

(A) Coverage maps of control libraries for LA09-CRF01_AE and LA51-group O. cDNA synthesis (with the corresponding control experiment number in Table 1 given in parentheses) was with oligo(dT)-SMART only (experiment 2), N6 HIV-SMART primers only (experiment 3), N6 HIV-SMART+oligo(dT)-SMART (experiment 4), the same as experiment 4 without the SMART-PCR amplification step (experiment 5), and non-HIV-SMART primers (experiment 6). (B) Neighbor-joining phylogenetic tree of HIV-1 isolate sequences (indicated in red by LA number only); alignment was 8,001 nt in length after gap stripping, and bootstrap values of 100% are shown.
To assess the broad utility of this method, a panel of highly diverse virus isolates was selected, including 28 group M (different subtypes and CRFs), 2 group N, and 17 group O strains, 1 group P strain, and 1 HIV-2 strain, all diluted into NHP to viral loads ranging from 5.61 to 7.74 log10 copies/ml. Three consecutive MiSeq runs were performed, multiplexing 12, 14, or 23 libraries, and all 49 libraries achieved 99 to 100% genome coverage in a single attempt. The percentage of HIV reads (median, 14.8%; 145,379 reads) and the sequence depth (median, 2,848×) for each isolate are reported in Table 1. Sequence coverage was broadly uniform across the genome, reflected by the low standard deviation values relative to the mean (Table 1). Equivalent results were obtained irrespective of HIV-1 group (see Fig. S1 in the supplemental material), with median percentages of HIV-1 reads and sequence depths of 10.9% and 2,583× for group M, 20.1% and 8,899× for group N, and 27.6% and 2,460× for group O (Table 1). Despite the high sequence divergence from HIV-1 group M, both the group P and HIV-2 samples performed exceedingly well with this approach, each obtaining full genome coverage from 17.7% and 16.0% of the total reads, respectively.
Focusing on the multiplexed run of 23 libraries, a median of only1.1 million total reads was obtained per barcode, yet this was more than sufficient to yield full genome sequences for each strain. The percentage of reads aligning to HIV varied from 0.14% to 68.3%, with a median of 6.97% (101,588 reads), corresponding to a median sequence depth of 1,952× (range of 33.3 to 10,240×) reads per nucleotide.
We compared all of the NGS consensus sequences to the available subgenomic sequences (not shown) as well as to published sequences for YBF30 (group N, LA27) and RBF168 (group P, LA35) and observed 99.5% and 98.3% concordance, respectively, between the NGS- and Sanger-generated sequences (see Table S1 in the supplemental material). The majority of differences involved degenerate nucleotides (e.g., Y or R) obtained by population sequencing, which the depth of the NGS data now afforded the ability to confirm or resolve.
A neighbor-joining phylogenetic tree derived from virus isolate sequences is shown in Fig. 2B. The majority of full-length sequences clustered tightly with reference sequences corresponding to their preliminary classification based on subgenome sequences. Notable among these new genomes are LA19 (subtype H), LA25 (subtype H), and LA26 (subtype J), rare pure subtypes for which few database entries are available. While LA19 is basal on the H branch and was preliminarily classified as subtype H in gag and F in pol RT, SimPlot shows it is subtype H across the entire genome (see Fig. S2A in the supplemental material) and BOOTSCAN analysis failed to show evidence of recombination (data not shown). Similarly, LA08 branches basally with subtype C, but no recombination was observed (see Fig. S2B). For isolates LA03 and LA06, in which classification was initially uncertain based on subgenomic pol PR/RT sequence, the aid of full genome sequences now demonstrated they both cluster tightly with subtype B and show no evidence of recombination (Fig. 2B; see Fig. S2C and S2D). LA57 was classified as CRF14_BG based on pol PR/RT sequence but is subtype G throughout the genome (see Fig. S2E); the isolate sequence is most closely related to the subtype G parent of CRF14, but there is no subtype B sequence in env. Only LA56 was determined to be a URF comprised of alternating CRF02_AG and CRF06_cpx sequence. SimPlots, bootscanning, and individual trees describing its genomic structure are found in Fig. S3 in the supplemental material.
Optimization of HIV-SMART protocol for clinical specimens.
Application of this technique directly to patient specimens will be of the greatest utility. Surprisingly, despite the use of virus-specific primers and HIV-infected plasma specimens with viral loads of ≥5 log10 copies/ml, the same protocol used for the virus isolates yielded poor genome coverage and <0.1% HIV-1 reads on several clinical specimens tested (data not shown). For example, following RNA extraction (Ultra Sens virus kit) of specimen CHU1756, a CRF02_AG infection, and cDNA synthesis with either 100 nM or 1 μM SMART primers (final concentration of each primer in the reaction mixture), only percentages of coverage of 9% and 55% were achieved, respectively (Fig. 3A, first and third panels). Therefore, efforts were undertaken to optimize the method for clinical specimens using high-titer, non-subtype B HIV-1-infected plasma specimens collected in Cameroon.
FIG 3.

(A) NGS coverage plots of CHU1756 with (+) and without (−) benzonase pretreatment, from libraries reverse transcribed with 100 nM or 1 μM N6 mixtures of HIV-SMART primers. For coverage plots, the x axis shows the nucleotide position and the y axis shows sequence depth. (B) HIV-1 RNA recovery. RNA was extracted manually with the Qiagen Ultra-Sens kit and spiked into quantitative PCR (qPCR) mixtures, with recovery measured using the HIV-1 viral load assay (shown in copies per milliliter). CHU1756 (CRF02) was untreated (lane 1) or benzonase pretreated with addition of carrier RNA (lane 2), pretreated without carrier RNA (lane 3), pretreated with linear acrylamide (lane 4), or DNase treated postextraction with carrier (lane 5), without carrier (lane 6), or with linear acrylamide (lane 7).
To enrich for viral reads and reduce the contribution of human background, nuclease treatments were evaluated before and after RNA extraction of CHU1756, and NGS was performed. Pretreatment for 3 h with benzonase, which cleaves both free RNA and DNA, improved sequence coverage dramatically, increasing it from 9% to 63% and from 55% to 83% with 100 nM and 1 μM N6 primer mixtures, respectively. Measurement of HIV-1 viral loads showed that benzonase did not alter recovery of viral nucleic acid (Fig. 3B, lanes 1 and 2). The addition of carrier RNA [e.g., yeast tRNA or oligo(A)] helped the recovery of HIV-1 RNA for the Ultra Sens virus kit and other manual extraction methods but was not necessary for the m2000 extraction method (see Fig. S4A in the supplemental material); linear acrylamide did not substitute for carrier RNA (Fig. 3B, lanes 2 to 4). With a postextraction Turbo DNase step, HIV-1 recovery was either poor or highly variable (Fig. 3B, lanes 5 to 7; see Fig. S4), and specimen filtration (0.22-μm pore) had a neutral effect (data not shown). The pretreatment regimen therefore included an initial clarifying spin and a 3-h preincubation with benzonase.
Two additional high-titer clinical specimens (N05-5 and CHU2727) were selected to determine the most suitable extraction method(s). Automated extraction performed on the m2000sp or manually with MagMax or Qiagen viral minikits (using yeast tRNA carrier) produced equivalent results, recovering ≥100% HIV-1 RNA compared to the starting specimen viral load (see Fig. S4A in the supplemental material). Using the HIV-SMART cDNA synthesis conditions described below, N05-5 RNA from the m2000, MagMax, and Qiagen viral minikit extractions all yielded 100% genome coverage, 1.6 to 2.6% HIV-1 reads, and >800× depth (Fig. 4B; see Fig. S4B). The percentages of RNA recovery from the Ultra Sens virus kit [with poly(A) carrier] used in the initial experiments shown in Fig. 3 were only 2.5% for N05-5 and 0.98% for CHU2727 and were approximately 7% and 24% of the starting amount for each virus with the DNA blood (on QiaCube) and PureLink (manual) kits, respectively. Libraries prepared from these extractions all suffered from gaps in coverage (68 to 99%) and low depth (39× to 139×). Thus, NGS results correlated directly with extraction recovery data (see Fig. S4B).
FIG 4.

(A) NGS coverage plots of N05-5 are shown for increasing numbers of HIV-SMART primers (top) and temperature for RT (bottom). (B) Histograms summarizing N05-5 optimization experiments. Shown are the percentages of genome coverage (top) and HIV reads of total (middle) and median (±SD) (bottom) sequence depth. Red indicates the condition(s) chosen for the optimized protocol. (C and D) NGS coverage plots for 8 clinical specimens prepared with the optimized protocol (C) and histograms of NGS results (D) (as in panel B). The specimen ID and strain classification are shown.
The remainder of the HIV-SMART cDNA synthesis protocol was dissected using N05-5 RNA extracted by the MagMax or m2000 methods (Fig. 4). The number, and therefore the distance, between HIV-SMART primers proved an important consideration (Fig. 4A). Mixtures of N4 and N6 primers yielded 100% genome coverage, 1.1 to 2.5% HIV-1 reads, and an average depth ranging from 842× to 1,806×. Counterintuitively, the use of additional primers (N10) produced numerous gaps in genome coverage (84%) and led to a dramatic loss in HIV-1 reads (0.01%) and sequence depth (12×). Using the N10 primers, primer extension was poor, resulting in 83% of HIV-containing reads lost to trimming (e.g., <50 nt). Due to the more uniform coverage produced, the N6 primer mixture was used for the remaining optimization experiments (Fig. 4A and B; see Table S2 in the supplemental material).
Full genome coverage at an average depth of 712× to 1,598× was obtained over a range of primer concentrations (100 nM to 1 μM), but at 3 μM, genome coverage decreased to 91%, with a substantial decrease in sequence depth (11×) (Fig. 4B; see Fig. S5A in the supplemental material). A 100-fold dilution of the N05-5 RNA yielded similar trends at these primer concentrations (see Fig. S5B and Table S2 in the supplemental material), and thus a 1 μM primer mixture was selected as optimal.
An increase in reverse transcription temperature from 42°C to 47°C was the decisive factor that led to a 4-fold increase in HIV-1 reads from 1.6% to 6.6% and increased coverage depth by over 7-fold to 6,119× (Fig. 4A and B). Lowering the temperature to 37°C provided no advantage. Raising it to 52°C elevated the percentage of HIV-1 dramatically to 28% and sequence depth to 43,639×, but genome coverage dropped to 47% due to cDNA synthesis initiating from only 3 of 6 primers and reduced read lengths (Fig. 4A and B).
Other parameters tested that showed a modest impact on results are summarized in Fig. 4B and Table S2 in the supplemental material. The addition of the oligo(dT)-SMART primer in a clinical sample setting had only a minimally positive effect (∼2-fold), and incorporation of an RNase H cleavage step after cDNA synthesis also proved beneficial (data not shown). As few as 8 cycles of SMART PCR yielded 100% genome coverage at 200× depth. However, the initial choice of between 25 and 35 cycles proved ideal: both obtained ∼1% HIV-1 reads and ∼800× depth, while % HIV-1 reads and depth plateaued beyond 35 cycles (Fig. 4B). The resulting optimized protocol for clinical specimens is described in detail in Materials and Methods.
Sequencing clinical specimens with HIV-SMART.
To evaluate the robustness of the HIV-SMART clinical protocol, we selected a diverse set of high-titer HIV-1 group M specimens (>5 log10 copies/ml) for which we had preliminary strain classification based on subgenomic sequences. The 8 specimens included CRF02_AG, subtype G and URFs from Cameroon, a subtype C specimen from South Africa, and a CRF01_AE specimen from Thailand. From a single MiSeq run of the eight libraries, a median of 4.7 million reads was obtained for each specimen, and between 0.63 and 6.74% of reads aligned to an HIV-1 reference sequence (see Table S2 in the supplemental material). Importantly, all achieved between 95 and 100% genome coverage, with average sequence depths ranging from 494× to 6,481× (Fig. 4C and D). The relatively low standard deviation for each sample (Fig. 4D) is indicative of the uniformity of coverage achieved and the tolerance of the method for sequence diversity, seen initially with virus isolates and now also with clinical specimens.
Phylogenetic trees of full-length genomes were constructed for strain classification (see Fig. S6 in the supplemental material). Sequences for 10-10 and 1031-19, which are basal on the CRF02 branch, and 62-15, which is basal on the subtype G branch, were analyzed further by SimPlot and bootscanning. Specimen 1031-19 did not exhibit recombination; however, 10-10 is a URF consisting of subtype D and CRF02 (see Fig. S7 in the supplemental material). Specimen 62-15 appeared to be a recombinant of subtype G and CRF02, but bootscanning and subregion trees show it is subtype G throughout (see Fig. S8 in the supplemental material). Analysis of the N05-5 specimen classified it as a URF, consisting of subtype A, D, and unclassified sequence (see Fig. S9 in the supplemental material).
HIV-SMART sensitivity.
For clinical specimens, the HIV-SMART approach must work over a range of viral loads. The sensitivity of the method was ascertained by taking RNA extracted from a CRF02-infected plasma specimen (100-17), serially diluting it into RNA extracted from an HIV-negative Cameroonian specimen to maintain a consistent level of host nucleic acid background, and then performing the HIV-SMART protocol. The starting viral load of specimen 100-17 was 5.79 log10 copies/ml, for an estimated cDNA synthesis input of 13,100 RNA copies. For the neat sample, a full genome sequence with an average depth of 3,427× was generated from the 2.08% of the total reads that mapped to HIV (Fig. 5A). At the 1:10 dilution, an input of 1,310 RNA copies yielded 0.34% HIV-1 reads to achieve 93% genome coverage at 570× depth. At the 1:100 dilution, 130 copies was sufficient to obtain 47% genome coverage, and at the 1:1,000 dilution (13 copies), 27% of the genome was obtained. The dilution series of 100-17 demonstrated a linear correlation between input viral RNA copy number and genome coverage for the HIV-SMART assay (see Fig. S10A in the supplemental material).
FIG 5.

Coverage plots of HIV-SMART NGS data for CRF02-infected clinical specimens. (A) Serial dilution of 100-17 neat and at 1:10, 1:100, and 1:1,000 with the calculated viral load shown; coverage is based on reads aligned to the 100-17 consensus genome sequence. (B) Patient specimens (ID indicated) with viral loads varying from 3 to 4.5 log10 copies/ml. Coverage is based on reads aligning to the CRF02 reference, GHNJ188. (C) SimPlot analysis was performed with gap-stripped alignments of partial genomes using a 400-bp window, 20-bp step, and Kimura 2-parameter test, with a transition/transversion (T/t) value of 2.0 for specimens 1454-09 (4.08 log10; 2,104-bp alignment) and 339-31 (3.04 log10; 4,923-bp alignment).
To evaluate method sensitivity on several distinct specimens, eight CRF02-infected specimens were selected with viral loads of approximately 3.0 (339-31 and 612-22), 3.5 (ADB1004-42 and 915-58), 4.0 (409-18 and 1454-09), and 4.5 (421-10 and 560-22) log10 copies/ml. These corresponded to approximately 21, 67, 212, and 672 copies of input RNA per cDNA reaction, respectively. A representative coverage plot for each viral load tested is shown in Fig. 5B, with all results detailed in Table S2 in the supplemental material. Within the range of 3.0 to 4.5 log10 copies/ml, genome coverage varied between 13 and 61%, regardless of the viral load, although the sequence depth was markedly improved for the higher-titer specimens (e.g., ∼4.3× at 3 log versus >1,200× at 4.0 to 4.5 log). In contrast to the serial dilution of 100-17, the relationship between viral load and genome coverage was nonlinear (see Fig. S10B in the supplemental material). Nevertheless, despite the lack of full coverage, we were still able to determine subtype classification. SimPlot analysis was performed on gap-stripped alignments of two strains with as little as 2,104 nt (1454-09) or 4,923 nt (339-31) of sequence, and each was determined to be CRF02_AG (Fig. 5C).
HIV-SMART reproducibility and variant detection.
To assess HIV-SMART reproducibility, we examined a total of 14 distinct NGS data sets derived from specimen N05-5 RNA in which we obtained 100% coverage and ≥400× sequence depth (Fig. 4A and B; see Table S2 in the supplemental material). With rare exceptions, all base calls were 100% identical and generated the same consensus sequence. At each nucleotide position in the genome, a percentage of frequency of the consensus base call was determined in CLC-Bio for each experiment. The standard deviation of this frequency was calculated based on all 14 libraries (Fig. 6A). For 83% of consensus base calls, the standard deviation was less than 1%, and for only 1.4% of calls it was ≥5%, indicating positions that are generally polymorphic.
An overall depth of 400× allowed minor variants present at ≥5% to be detected at a minimum of 20× depth (27). Using a CLC-Bio algorithm that does not assume organism ploidy and estimates a probabilistic error rate, we report in Table S3 in the supplemental material the frequency (occurrence/depth) of statistically significant (P > 0.01) minor variants present globally at >5% in the 14 N05-5 sequencing experiments. Focusing on a region of Pol integrase with a median of >650× depth as an example: nt 4768 is an A 82.2% ± 6.9% of the time, nt 4773 is a C 93.6% ± 4.3% of the time, and nt 4787 is an A 98.0% ± 1.8% of the time; the remainder of the base call frequencies shown approach 100% and therefore do not change (Fig. 6B, histogram). While the relative frequency varied (median, 19.3% ± 7.0%) between replicates (Fig. 6B, scatter plot), the same A4768G polymorphism was consistently detected at a >2% cutoff in 14/14 experiments. The less abundant C4773T polymorphism was also detected at the >2% cutoff in 10/14 (median, 6.1% ± 2.9%) experiments. The four samples in which C4773T was not detected were all sequenced on the same run, each of which also had a correspondingly lower frequency for A4768G. The A4787G polymorphism was present in only 5/14 experiments at a frequency of 4.1% ± 1.1%. Therefore, the HIV-SMART method generates a reproducible consensus sequence from which the same minor variants can be repeatedly detected depending on their relative abundance.
Sequence variation was also examined at lower titers, for which results generally exhibited less depth as well as overall genome coverage. The partial genome consensus sequences (69 to 75% coverage) derived from N05-5 diluted at 1:100 (3.79 log10; see Fig. S5B in the supplemental material) were aligned to the full-length genome and gap-stripped, and the percentage of identity was calculated. The sample obtained at 100 nM primer, with 69% coverage (6,612-nt consensus sequence) and an average 33× depth, was 97.5% identical to the N05-5 consensus. Removal of regions with a depth of 5 or fewer reads (4,629 nt) increased the identity to 99.9%. Similarly, the sample obtained at 300 nM primer, with 75% coverage (7,199 nt) and 22× depth, had 99.1% identity, and removal of regions with <5 reads (5,812 nt) increased the identity to 99.9%. Even at low coverage, the consensus sequence generated by HIV-SMART is still highly reliable.
DISCUSSION
We describe here a novel method to obtain full genome sequences of HIV. The use of conserved HIV-SMART fusion primers to generate NGS libraries amplified all HIV-1 groups, including several M subtypes, CRFs and URFs, and groups N, O, and P. Unexpectedly, HIV-2, which is highly divergent from HIV-1 (∼50% at the nucleotide level) and was not considered in the primer design, was also successfully sequenced (19). This universal approach does not require prior knowledge of viral classification and therefore obviates the need to design subtype/group-specific reagents. The method is rapid (2 days) and reliably obtained full coverage at substantial depths for a diversity of HIV-1 genomes, whether from cultured virus or clinical specimens. Even at the lower range of sensitivity, enough genome coverage of reliable sequence was obtained to permit strain classification. Therefore, beyond its demonstrated utility in characterization of HIV-1 sequence diversity, the HIV-SMART approach offers a viable new alternative for leveraging NGS to conduct viral surveillance.
The SMART approach was originally conceived with amplification of mRNA in mind, initiating reverse transcription via priming from the 3′ poly(A) tail of transcripts (28). Here we demonstrated that fusion primers targeting multiple sites of RT initiation throughout the same HIV transcript could generate complete genomic sequences and selectively enrich for viral sequences, normally present at far lower levels in clinical samples compared to host background nucleic acid. Cultured virus isolates were used as a model system to demonstrate the HIV-SMART principle and the breadth of diversity it could accommodate. A preponderance of metagenomics papers describing new methods have also relied on cultured isolates or transfected plasmids with lower host backgrounds and higher virus titers (13, 29–33). However, sequencing directly from plasma is the most expedient workflow and reveals which replication-competent viruses are circulating in the patient. We optimized the protocol for this purpose and found that the addition of benzonase prior to extraction increased HIV-1 reads (∼10- to 50-fold) and genome coverage (from 28% to 54%), presumably by decreasing human background nucleic acid, while a postextraction DNase treatment was often detrimental (Fig. 3; see Table S2 in the supplemental material) (24, 30). The selection of nucleic acid extraction method also drove performance, with higher HIV-1 RNA recovery correlating with higher genome coverage. Despite a wide variation in extraction efficiencies, the HIV-SMART protocol still achieved >90% genome coverage from all but one method for high-titer specimens. While the range of primer concentrations appears flexible, the number of HIV-SMART primers is not. Up to 6 primers spaced at least 1.5 to 2 kb apart provided optimal results, while adding more led to inhibition of cDNA synthesis, possibly due to steric hindrance on the same transcript or from unanticipated primer-primer interactions. An increase in cDNA synthesis temperature from the recommended 42°C to 47°C resulted in a 4-fold increase in % HIV-1 reads and 7-fold increase in sequence depth. At higher temperatures (e.g., 52°C), we saw a dramatic increase in HIV-1 reads coupled to a precipitous (>50%) drop in genome coverage. It remains to be seen whether increasing primer melting temperatures—for example, by chemical modification of the backbone (e.g., locked nucleic acids)—can restore annealing and still yield complete and accurate sequences at elevated temperatures.
The NGS-generated consensus genomes agreed with our prior population sequencing data and published sequences, were found to be reproducible between runs (e.g., N05-5), and were reliable over a range of sequence depths (see Fig. S5B in the supplemental material). This fidelity and the coverage depths typically achieved by HIV-SMART allowed us to identify the same polymorphisms in pol and elsewhere in the genome across multiple experiments, despite being done at different times and under slightly different conditions (Fig. 6B; see Table S3 in the supplemental material). Although the relative abundance of minor variants fluctuated between experiments, overall the results consistently identified the same polymorphism, which suggests that full-genome HIV-SMART data sets are suitable for qualitative analysis of drug resistance (e.g., protease, RT, and integrase [IN]), immune escape mutations (gp41), and CCR5/CXCR4 tropism. Given that cDNAs were not assigned an individual primer identification (ID) (34), the issue of whether reads were resampled was not addressed; however, variants were filtered using a statistical error rate model. The current HIV-SMART approach is not amenable to the addition of primer IDs since the Nextera “tagmentation” step would cleave these tags off the ends of inserts (Fig. 1) and is therefore not quantitative.
In a study by Gall et al., an analogous NGS-based method using conserved primer sets was described, and it has been used recently to obtain complete genomes for 85% of subtype C strains from South African specimens with viral loads of >log4 copies/ml (20, 35). A key advantage that the HIV-SMART protocol provides is the simplicity of one cDNA reaction per sample and amplification with a single primer, rather than 4 separate RT-PCRs followed by amplicon purification, quantification, and pooling. Another study required 2 to 6 cDNA reactions and 5 to 35 separate PCRs to generate libraries for full-genome sequencing on three different NGS platforms (13). The speed of Nextera XT, the tremendous sequence depth, an error rate of 0.1%, and the current read lengths (v3 = 600 nt) obtained on the ubiquitous Illumina MiSeq are also preferable to the time-consuming library amplification steps, lower throughput, and higher error rates associated with other platforms (11, 36). We estimate the cost of the m2000 extraction, HIV-SMART library prep, and MiSeq sequencing reagent was approximately $165 per genome for the multiplex run of 23 libraries.
There is still considerable off-target (non-HIV) amplification occurring in clinical specimens despite gene-specific priming. As to whether the HIV-SMART approach represents an improvement over random priming for direct sequencing from patient samples, a comprehensive side-by-side evaluation has not been attempted. However, libraries made from comparable non-subtype B Cameroonian specimens with viral loads of ≥4.5 log10 prepared by RdA/RdB random priming (37–39) yielded a median of 0.14% ± 0.8% HIV-1 reads, whereas those prepared by HIV-SMART virus-specific priming yielded a median of 2.48% ± 2.33% HIV-1 reads, for a >17-fold increase in viral reads (15). The HIV-SMART approach is also an improvement relative to published methods; one reported ≤311 viral reads for numerous high-titer (5 to 6 log10 copies/ml) HIV specimens, and another which started with a larger volume of specimen (4 to 8 ml of plasma) yielded incomplete coverage and 0.007 to 1.44% viral reads (24, 40). Our results with the optimized HIV-SMART protocol (Fig. 4C) are comparable to those obtained for HIV clinical samples using the Ovation transcriptome sequencing (RNA-seq) method described by Malboeuf et al. (41). In this study, specimens with ≥4.5 log10 copies/ml yielded approximately 5 million reads with 1.1% to 7.1% HIV-1 and full coverage at >600× sequence depth (41). A noted advantage of the SMART technology is the ability to consistently sequence the 5′ and 3′ ends of viruses, challenging regions often missed by random-primed methods due to secondary structure (42–44). Therefore, the HIV-SMART approach is an attractive, simple alternative that generally performs better than current unbiased priming methods.
All told, we deployed HIV-SMART to obtain 56 novel full-length genomes, including 3 URFs, 3 rare group M subtypes (H and J), and 17 group O strains. A diverse set of group M clinical specimens were fully sequenced as long as the viral loads were ≥5 log10 copies/ml (see Fig. S10 in the supplemental material). While we showed that shallow sequence depth obtained from lower-titer samples is still highly reliable, the challenge now is to improve upon sensitivity in patient specimens. The sensitivity is currently lower than that achieved by Gall et al.; however, the use of RNA concentrators after extraction/before cDNA synthesis has led to the ability to now consistently obtain full genome sequences from samples having ≥4.5 log10 copies/ml (M. A. Rodgers, A. Vallari, B. Harris, C. McArthur, L. Sthreshley, and C. A. Brennan, unpublished data). We observed that as viral loads decreased, the likelihood of individual primers failing increased, however, just which ones appeared largely stochastic, since coverage patterns varied randomly from one specimen to the next, irrespective of titer (Fig. 5B). Predictably, primer HS85, situated in a highly diverse region (env) and only 20 nt in length, failed most frequently. The lack of a direct correlation between titer and coverage has been seen with other methods and presumably is attributed to the variability in host backgrounds (41, 45). Indeed, despite benzonase pretreatment, there were still large percentages of host reads sequenced in these clinical specimens, suggesting this step generally helps but does not completely eliminate background (data not shown). In addition, at viral loads of ≤4.5 log10 copies/ml, the lengths of cDNAs were highly variable, from as short as 100 nt to >2 kb.
The SMART approach described here for HIV may be easily adapted to any RNA virus. In addition to the core or NS5B, genotyping of hepatitis C (HCV) typically involves interrogating the 5′ untranscribed region (UTR), a region with extensive secondary structure that this method reliably sequences through (46, 47). Treatment indications for new direct-acting antiviral (DAA) therapies vary by HCV viral genotype, so it may be important to consider the potential clinical impact of recombination and identify the specific genotype found at the drug target: i.e., NS3, NS5A, and NS5B versus the generic genotype identified by the 5′ UTR or core (48). The ability to readily sequence HCV genomes could also provide a comprehensive view of the presence of resistance-associated variants (RAVs), which may have implications for treatment choices—particularly in the retreatment of patients who have failed to achieve sustained virological response (SVR) on previous DAA regimens (49–52). To sequence influenza virus rapidly by NGS, methods for cloning and sequencing that rely on amplification from conserved elements at termini of viral subgenomic fragments or those requiring multiple individual PCRs as input could readily be adapted to the SMART approach (53, 54). As clinical and diagnostic virology in general comes to rely increasingly on NGS, it is necessary to develop reliable, streamlined, and interchangeable protocols for these and other RNA viruses of medical importance (36, 55, 56).
The complexity of HIV strains has increased significantly due to natural evolution and intersubtype recombination; recombinant strains are now prevalent worldwide (57). Thus, it is essential to monitor HIV diversity accurately within populations and to determine phylogenetic classification based on the entire viral genome. The HIV-SMART approach combines the specificity of HIV-directed priming with a built-in tolerance for extensive sequence diversity, illustrating its utility for viral characterization and potential for surveillance, particularly when a priori knowledge of the viral strain is absent.
Supplementary Material
ACKNOWLEDGMENTS
Clinical specimens from Cameroonian blood donors were obtained from Lazare Kaptué, Universite des Montagnes, Bangangte, Cameroon. We thank Mary Rodgers and Gavin Cloherty for critical reading of the manuscript.
Funding Statement
This research was funded by and conducted at Abbott Laboratories. M.G.B., J.Y., R.W.T., and C.A.B. are employees of Abbott Laboratories.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.02479-15.
REFERENCES
- 1.Hemelaar J. 2013. Implications of HIV diversity for the HIV-1 pandemic. J Infect 66:391–400. doi: 10.1016/j.jinf.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V. 2001. Evolutionary and immunological implications of contemporary HIV-1 variation. Br Med Bull 58:19–42. doi: 10.1093/bmb/58.1.19. [DOI] [PubMed] [Google Scholar]
- 3.Brennan CA, Yamaguchi J, Devare SG, Foster GA, Stramer SL. 2010. Expanded evaluation of blood donors in the United States for human immunodeficiency virus type 1 non-B subtypes and antiretroviral drug-resistant strains: 2005 through 2007. Transfusion 50:2707–2712. doi: 10.1111/j.1537-2995.2010.02767.x. [DOI] [PubMed] [Google Scholar]
- 4.Pyne MT, Hackett J Jr, Holzmayer V, Hillyard DR. 2013. Large-scale analysis of the prevalence and geographic distribution of HIV-1 non-B variants in the United States. J Clin Microbiol 51:2662–2669. doi: 10.1128/JCM.00880-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semaille C, Barin F, Cazein F, Pillonel J, Lot F, Brand D, Plantier JC, Bernillon P, Le Vu S, Pinget R, Desenclos JC. 2007. Monitoring the dynamics of the HIV epidemic using assays for recent infection and serotyping among new HIV diagnoses: experience after 2 years in France. J Infect Dis 196:377–383. doi: 10.1086/519387. [DOI] [PubMed] [Google Scholar]
- 6.Callegaro A, Di Filippo E, Astuti N, Ortega PA, Rizzi M, Farina C, Valenti D, Maggiolo F. 2014. Early clinical response and presence of viral resistant minority variants: a proof of concept study. J Int AIDS Soc 17:19759. doi: 10.7448/IAS.17.4.19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley DM, Chin EN, Bimber BN, Sanabani SS, Tarosso LF, Costa PR, Sauer MM, Kallas EG, O'Connor DH. 2012. Low-cost ultra-wide genotyping using Roche/454 pyrosequencing for surveillance of HIV drug resistance. PLoS One 7:e36494. doi: 10.1371/journal.pone.0036494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ekici H, Rao SD, Sonnerborg A, Ramprasad VL, Gupta R, Neogi U. 2014. Cost-efficient HIV-1 drug resistance surveillance using multiplexed high-throughput amplicon sequencing: implications for use in low- and middle-income countries. J Antimicrob Chemother 69:3349–3355. doi: 10.1093/jac/dku278. [DOI] [PubMed] [Google Scholar]
- 9.Fisher RG, Smith DM, Murrell B, Slabbert R, Kirby BM, Edson C, Cotton MF, Haubrich RH, Kosakovsky Pond SL, Van Zyl GU. 2015. Next generation sequencing improves detection of drug resistance mutations in infants after PMTCT failure. J Clin Virol 62:48–53. doi: 10.1016/j.jcv.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Diaz A, McCormick A, Booth C, Gonzalez D, Sayada C, Haque T, Johnson M, Webster D. 2014. Analysis of transmitted HIV-1 drug resistance using 454 ultra-deep-sequencing and the DeepChek-HIV system. J Int AIDS Soc 17:19752. doi: 10.7448/IAS.17.4.19752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Archer J, Weber J, Henry K, Winner D, Gibson R, Lee L, Paxinos E, Arts EJ, Robertson DL, Mimms L, Quinones-Mateu ME. 2012. Use of four next-generation sequencing platforms to determine HIV-1 coreceptor tropism. PLoS One 7:e49602. doi: 10.1371/journal.pone.0049602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swenson LC, Daumer M, Paredes R. 2012. Next-generation sequencing to assess HIV tropism. Curr Opin HIV AIDS 7:478–485. doi: 10.1097/COH.0b013e328356e9da. [DOI] [PubMed] [Google Scholar]
- 13.Giallonardo FD, Topfer A, Rey M, Prabhakaran S, Duport Y, Leemann C, Schmutz S, Campbell NK, Joos B, Lecca MR, Patrignani A, Daumer M, Beisel C, Rusert P, Trkola A, Gunthard HF, Roth V, Beerenwinkel N, Metzner KJ. 2014. Full-length haplotype reconstruction to infer the structure of heterogeneous virus populations. Nucleic Acids Res 42:e115. doi: 10.1093/nar/gku537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes AL, Becker EA, Lauck M, Karl JA, Braasch AT, O'Connor DH, O'Connor SL. 2012. SIV genome-wide pyrosequencing provides a comprehensive and unbiased view of variation within and outside CD8 T lymphocyte epitopes. PLoS One 7:e47818. doi: 10.1371/journal.pone.0047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luk KC, Berg MG, Naccache SN, Kabre B, Federman S, Mbanya D, Kaptue L, Chiu CY, Brennan CA, Hackett J Jr. 2015. Utility of metagenomic next-generation sequencing for characterization of HIV and human pegivirus diversity. PLoS One 10:e0141723. doi: 10.1371/journal.pone.0141723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bimber BN, Dudley DM, Lauck M, Becker EA, Chin EN, Lank SM, Grunenwald HL, Caruccio NC, Maffitt M, Wilson NA, Reed JS, Sosman JM, Tarosso LF, Sanabani S, Kallas EG, Hughes AL, O'Connor DH. 2010. Whole-genome characterization of human and simian immunodeficiency virus intrahost diversity by ultradeep pyrosequencing. J Virol 84:12087–12092. doi: 10.1128/JVI.01378-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemelaar J. 2012. The origin and diversity of the HIV-1 pandemic. Trends Mol Med 18:182–192. doi: 10.1016/j.molmed.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Li G, Piampongsant S, Faria NR, Voet A, Pineda-Pena AC, Khouri R, Lemey P, Vandamme AM, Theys K. 2015. An integrated map of HIV genome-wide variation from a population perspective. Retrovirology 12:18. doi: 10.1186/s12977-015-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gall A, Ferns B, Morris C, Watson S, Cotten M, Robinson M, Berry N, Pillay D, Kellam P. 2012. Universal amplification, next-generation sequencing, and assembly of HIV-1 genomes. J Clin Microbiol 50:3838–3844. doi: 10.1128/JCM.01516-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brennan CA, Bodelle P, Coffey R, Devare SG, Golden A, Hackett J Jr, Harris B, Holzmayer V, Luk KC, Schochetman G, Swanson P, Yamaguchi J, Vallari A, Ndembi N, Ngansop C, Makamche F, Mbanya D, Gurtler LG, Zekeng L, Kaptue L. 2008. The prevalence of diverse HIV-1 strains was stable in Cameroonian blood donors from 1996 to 2004. J Acquir Immune Defic Syndr 49:432–439. doi: 10.1097/QAI.0b013e31818a6561. [DOI] [PubMed] [Google Scholar]
- 22.Chang M, Gottlieb GS, Dragavon JA, Cherne SL, Kenney DL, Hawes SE, Smith RA, Kiviat NB, Sow PS, Coombs RW. 2012. Validation for clinical use of a novel HIV-2 plasma RNA viral load assay using the Abbott m2000 platform. J Clin Virol 55:128–133. doi: 10.1016/j.jcv.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merck. 9 February 2016, accession date Benzonase endonuclease: the smart solution for DNA removal. Merck, Darmstadt, Germany: https://www.bioneer.dk/files/Files/Benzonase1.pdf. [Google Scholar]
- 24.Law J, Jovel J, Patterson J, Ford G, O'Keefe S, Wang W, Meng B, Song D, Zhang Y, Tian Z, Wasilenko ST, Rahbari M, Mitchell T, Jordan T, Carpenter E, Mason AL, Wong GK. 2013. Identification of hepatotropic viruses from plasma using deep sequencing: a next generation diagnostic tool. PLoS One 8:e60595. doi: 10.1371/journal.pone.0060595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray S. 9 February 2016, accession date SCRoftware. http://sray.med.som.jhmi.edu/SCRoftware/. [Google Scholar]
- 26.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muzzey D, Evans EA, Lieber C. 2015. Understanding the basics of NGS: from mechanism to variant calling. Curr Genet Med Rep 3:158–165. doi: 10.1007/s40142-015-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chenchik A, Zhu Y, Diatchenko L, Li R, Hill J, Siebert P. 1998. Generation and use of high-quality cDNA from small amounts of total RNA by SMART PCR, p 305–319. In Siebert P, Larrick J (ed), Gene cloning and analysis by RT-PCR. Biotechniques Books, Natick, MA. [Google Scholar]
- 29.Djikeng A, Halpin R, Kuzmickas R, Depasse J, Feldblyum J, Sengamalay N, Afonso C, Zhang X, Anderson NG, Ghedin E, Spiro DJ. 2008. Viral genome sequencing by random priming methods. BMC Genomics 9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall RJ, Wang J, Todd AK, Bissielo AB, Yen S, Strydom H, Moore NE, Ren X, Huang QS, Carter PE, Peacey M. 2014. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J Virol Methods 195:194–204. doi: 10.1016/j.jviromet.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ragupathy V, Gao F, Sanchez A, Schito M, Denny T, Busch M, Zhao J, Mbondji C, Vemula S, Hewlett I. 2015. PCR-free full genome characterization of diverse HIV-1 strains by Nextgen sequencing. Webcast. CROI 2015: Conf Retrovir Opportunistic Infect, Seattle, WA, 23 to 26 February 2015 http://www.croiwebcasts.org/st/themed?link=nav&linkc=sesstype. [Google Scholar]
- 32.Svraka S, Rosario K, Duizer E, van der Avoort H, Breitbart M, Koopmans M. 2010. Metagenomic sequencing for virus identification in a public-health setting. J Gen Virol 91:2846–2856. doi: 10.1099/vir.0.024612-0. [DOI] [PubMed] [Google Scholar]
- 33.Willerth SM, Pedro HA, Pachter L, Humeau LM, Arkin AP, Schaffer DV. 2010. Development of a low bias method for characterizing viral populations using next generation sequencing technology. PLoS One 5:e13564. doi: 10.1371/journal.pone.0013564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. 2011. Accurate sampling and deep sequencing of the HIV-1 protease gene using a primer ID. Proc Natl Acad Sci U S A 108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danaviah S, Manasa J, Wilkinson E, Pillay S, Sibisi Z, Msweli S, Pillay D, de Oliveira T. 2015. Near full length HIV-1 sequencing to understand HIV phylodynamics in Africa in real time. Webcast. CROI 2015: Conf Retrovir Opportunistic Infect, Seattle, WA, 23 to 26 February 2015 http://www.croiwebcasts.org/st/themed?link=nav&linkc=sesstype. [Google Scholar]
- 36.Quinones-Mateu ME, Avila S, Reyes-Teran G, Martinez MA. 2014. Deep sequencing: becoming a critical tool in clinical virology. J Clin Virol 61:9–19. doi: 10.1016/j.jcv.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greninger AL, Chen EC, Sittler T, Scheinerman A, Roubinian N, Yu G, Kim E, Pillai DR, Guyard C, Mazzulli T, Isa P, Arias CF, Hackett J, Schochetman G, Miller S, Tang P, Chiu CY. 2010. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS One 5:e13381. doi: 10.1371/journal.pone.0013381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naccache SN, Federman S, Veeraraghavan N, Zaharia M, Lee D, Samayoa E, Bouquet J, Greninger AL, Luk KC, Enge B, Wadford DA, Messenger SL, Genrich GL, Pellegrino K, Grard G, Leroy E, Schneider BS, Fair JN, Martinez MA, Isa P, Crump JA, DeRisi JL, Sittler T, Hackett J Jr, Miller S, Chiu CY. 2014. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res 24:1180–1192. doi: 10.1101/gr.171934.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorber K, Chiu C, Webster D, Dimon M, Ruby JG, Hekele A, DeRisi JL. 2008. The long march: a sample preparation technique that enhances contig length and coverage by high-throughput short-read sequencing. PLoS One 3:e3495. doi: 10.1371/journal.pone.0003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li L, Deng X, Linsuwanon P, Bangsberg D, Bwana MB, Hunt P, Martin JN, Deeks SG, Delwart E. 2013. AIDS alters the commensal plasma virome. J Virol 87:10912–10915. doi: 10.1128/JVI.01839-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malboeuf CM, Yang X, Charlebois P, Qu J, Berlin AM, Casali M, Pesko KN, Boutwell CL, DeVincenzo JP, Ebel GD, Allen TM, Zody MC, Henn MR, Levin JZ. 2013. Complete viral RNA genome sequencing of ultra-low copy samples by sequence-independent amplification. Nucleic Acids Res 41:e13. doi: 10.1093/nar/gks794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alfson KJ, Beadles MW, Griffiths A. 2014. A new approach to determining whole viral genomic sequences including termini using a single deep sequencing run. J Virol Methods 208:1–5. doi: 10.1016/j.jviromet.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 43.Logan G, Freimanis GL, King DJ, Valdazo-Gonzalez B, Bachanek-Bankowska K, Sanderson ND, Knowles NJ, King DP, Cottam EM. 2014. A universal protocol to generate consensus level genome sequences for foot-and-mouth disease virus and other positive-sense polyadenylated RNA viruses using the Illumina MiSeq. BMC Genomics 15:828. doi: 10.1186/1471-2164-15-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wellenreuther R, Schupp I, Poustka A, Wiemann S. 2004. SMART amplification combined with cDNA size fractionation in order to obtain large full-length clones. BMC Genomics 5:36. doi: 10.1186/1471-2164-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartolini B, Giombini E, Abbate I, Selleri M, Rozera G, Biagini T, Visco-Comandini U, Taibi C, Capobianchi MR. 2015. Near full length hepatitis C virus genome reconstruction by next generation sequencing based on genotype-independent amplification. Dig Liver Dis 47:608–612. doi: 10.1016/j.dld.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Hara K, Rivera MM, Koh C, Sakiani S, Hoofnagle JH, Heller T. 2013. Important factors in reliable determination of hepatitis C virus genotype by use of the 5′ untranslated region. J Clin Microbiol 51:1485–1489. doi: 10.1128/JCM.03344-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. 2001. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 30:892–897. [DOI] [PubMed] [Google Scholar]
- 48.Hedskog C, Doehle B, Chodavarapu K, Gontcharova V, Crespo Garcia J, De Knegt R, Drenth JP, McHutchison JG, Brainard D, Stamm LM, Miller MD, Svarovskaia E, Mo H. 2015. Characterization of hepatitis C virus intergenotypic recombinant strains and associated virological response to sofosbuvir/ribavirin. Hepatology 61:471–480. doi: 10.1002/hep.27361. [DOI] [PubMed] [Google Scholar]
- 49.Donaldson EF, Harrington PR, O'Rear JJ, Naeger LK. 2015. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology 61:56–65. doi: 10.1002/hep.27375. [DOI] [PubMed] [Google Scholar]
- 50.Halfon P, Sarrazin C. 2012. Future treatment of chronic hepatitis C with direct acting antivirals: is resistance important? Liver Int; 32(Suppl 1):S79–S87. doi: 10.1111/j.1478-3231.2011.02716.x. [DOI] [PubMed] [Google Scholar]
- 51.Schneider MD, Sarrazin C. 2014. Antiviral therapy of hepatitis C in 2014: do we need resistance testing? Antiviral Res 105:64–71. doi: 10.1016/j.antiviral.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 52.Sullivan JC, De Meyer S, Bartels DJ, Dierynck I, Zhang EZ, Spanks J, Tigges AM, Ghys A, Dorrian J, Adda N, Martin EC, Beumont M, Jacobson IM, Sherman KE, Zeuzem S, Picchio G, Kieffer TL. 2013. Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin Infect Dis 57:221–229. doi: 10.1093/cid/cit226. [DOI] [PubMed] [Google Scholar]
- 53.Hoper D, Hoffmann B, Beer M. 2011. A comprehensive deep sequencing strategy for full-length genomes of influenza A. PLoS One 6:e19075. doi: 10.1371/journal.pone.0019075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibson RM, Schmotzer CL, Quinones-Mateu ME. 2014. Next-generation sequencing to help monitor patients infected with HIV: ready for clinical use? Curr Infect Dis Rep 16:401. doi: 10.1007/s11908-014-0401-5. [DOI] [PubMed] [Google Scholar]
- 56.Radford AD, Chapman D, Dixon L, Chantrey J, Darby AC, Hall N. 2012. Application of next-generation sequencing technologies in virology. J Gen Virol 93:1853–1868. doi: 10.1099/vir.0.043182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. 2008. The challenge of HIV-1 subtype diversity. N Engl J Med 358:1590–1602. doi: 10.1056/NEJMra0706737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.