

Abstract
Phthiocerol dimycocerosates (PDIMs) and phenolic glycolipids (PGLs) contribute to the pathogenicity of several mycobacteria. Biosynthesis of these virulence factors requires polyketide synthases and other enzymes that represent potential targets for the development of adjuvant antivirulence drugs. We used six isogenic Mycobacterium marinum mutants, each with a different gene knockout in the PDIM/PGL biosynthetic pathway, to probe the pleiotropy of mutations leading to PDIM− PGL−, PDIM+ PGL− or PDIM− PGL+ phenotypes. We evaluated the M. marinum mutants for changes in antibiotic susceptibility, cell envelope permeability, biofilm formation, surface properties, sliding motility and virulence in an amoeba model. The analysis also permitted us to begin exploring the hypothesis that different gene knockouts rendering the same PDIM and/or PGL deficiency phenotypes lead to M. marinum mutants with equivalent pleiotropic profiles. Overall, the results of our study revealed a complex picture of pleiotropic patterns emerging from different gene knockouts, uncovered unexpected phenotypic inequalities between mutants, and provided new insight into the phenotypic consequences of gene knockouts in the PDIM/PGL biosynthetic pathway.
Keywords: Mycobacterium, cell wall biosynthesis, polyketide, amoeba, biofilm, outer membrane lipids
Analysis of mycobacterial mutants with knockouts in the biosynthetic pathway of PDIM/PGL cell wall (glyco)lipids revealed a complex picture of pleiotropic patterns and uncovered unexpected phenotypic inequalities.
Graphical Abstract Figure.
Analysis of mycobacterial mutants with knockouts in the biosynthetic pathway of PDIM/PGL cell wall (glyco)lipids revealed a complex picture of pleiotropic patterns and uncovered unexpected phenotypic inequalities.
INTRODUCTION
The hallmark of the Mycobacterium genus is a unique cell wall that includes an outer membrane-like structure with unusual (glyco)lipids, some of which have been implicated in pathogenesis (Guenin-Mace, Simeone and Demangel 2009; Neyrolles and Guilhot 2011). Phthiocerol dimycocerosates (PDIMs) and phenolic glycolipids (PGLs) are two related families of (glyco)lipids thought to localize to the outer membrane of M. tuberculosis, M. leprae, and several opportunistic mycobacterial pathogens, including M. marinum (Mmar) (for a review, see Onwueme et al. 2005) (Fig. 1A). These unique (glyco)lipids are involved in pathogenicity through intricate and incompletely understood mechanisms (Arbues et al. 2014), and recent studies suggest complementary roles of PDIMs and PGLs in a complex immune evasion mechanism (Cambier et al. 2014). There is also evidence that, at least in some species, these lipids strengthen the cell envelope permeability barrier and increase the bacterium's intrinsic antibiotic resistance (Camacho et al. 2001; Alibaud et al. 2011; Chavadi et al. 2011; Yu et al. 2012; Soetaert et al. 2015).
Figure 1.

PDIMs, PGLs and chromosomal locus involved in their production. The structures are representative of those produced by M. marinum (A). The genes deleted in the M. marinum isogenic mutants analyzed in this study are highlighted with black-filled arrows (B). The roles of the gene products and the previously determined PDIM/PGL phenotype of each deletion mutant and corresponding complemented strain are outlined. The position of the enzymes missing in the mutants (*) in the biosynthetic pathway is schematically depicted (C).
The relevance of PDIMs and PGLs in mycobacterial biology highlights the enzymes needed for biosynthesis of these virulence factors as attractive targets for exploring the development of adjuvant antivirulence drugs. This consideration has provided thrust for numerous studies that led to the identification of several conserved genes required for PDIM and/or PGL production and the elucidation of the specific roles that many of these genes play in the pathway (for a review, see Quadri 2014). Interestingly, the knowledge gained from some of these studies guided the design of the first PGL biosynthesis inhibitor (Ferreras et al. 2008). The inhibitor targets a p-hydroxybenzoic acid adenylating enzyme (FadD22) essential for PGL synthesis and has potent activity in M. tuberculosis complex members and nontuberculous mycobacterial pathogens, such as Mmar (Ferreras et al. 2008; He et al. 2009). Mmar, a close genetic relative of the M. tuberculosis complex, causes tuberculosis-like disease in poikilotherms and peripheral granulomatous disease in humans (Cambau and Aubry 2011). Recent studies have shown both PDIMs and PGLs are required for Mmar virulence in the zebrafish model (Alibaud et al. 2011; Yu et al. 2012; Cambier et al. 2014).
We have recently reported mutational studies that interrogated the involvement of the Mmar genes tesA, papA5, fadD22, fadD26, fadD28 and fadD29 in the production of PDIMs and PGLs (Chavadi et al. 2011, 2012; Vergnolle et al. 2015) (Fig. 1B). These six genes are conserved in M. tuberculosis and other mycobacteria (Onwueme et al. 2005) and their proposed functions in PDIM and/or PGL synthesis are outlined in Fig. 1B (for a review, see Quadri 2014). These studies were based on gene-specific, unmarked deletion mutants and each PDIM and/or PGL deficiency was complemented by episomal expression of a wild-type (WT) copy of the deleted gene, thus permitting unambiguous genotype–phenotype assignments (Fig. 1B). Overall, these findings are in line with those from other reports (Alibaud et al. 2011; Yu et al. 2012).
In this study, we utilized the Mmar isogenic mutants noted above to further interrogate the phenotypic consequences of each of the six gene knockouts. We evaluated the mutants in in vitro assays that probed for changes in antibiotic susceptibility, cell envelope permeability, biofilm formation, surface properties and sliding motility. We also explored the virulence of the mutants using the Dictyostelium discoideum model. The parallel analysis of these isogenic mutants permitted us to begin exploring the hypothesis that different knockouts in the pathway rendering the same PDIM and/or PGL deficiency lead to mutants with equivalent pleiotropic profiles. Exploration of this hypothesis is relevant in the context of considering specific enzymes of the pathway as potential targets for adjuvant drug development.
MATERIALS AND METHODS
Strains and culturing conditions
Mycobacteria (Table 1) were cultured in Middlebrook 7H9 medium supplemented with 10% ADN (5% BSA, 2% dextrose, 0.85% NaCl), 0.05% Tween-80 and 0.2% glycerol (supplemented 7H9) or Middlebrook 7H11 medium supplemented with 10% ADN and 0.5% glycerol (supplemented 7H11) (Parish and Stoker 1998). Kanamycin (30 μg mL−1) was added to the media for maintenance of pCP0 plasmids. Mmar WT and the Mmar mutants carried pCP0 (Ferreras et al. 2008), the vector used for episomal complementation. Dictyostelium discoideum DH1-10 was axenically cultured under standard conditions in HL5 medium (Eichinger and Rivero 2013).
Table 1.
Mycobacterial strains.
| Strain | Characteristics | Reference |
|---|---|---|
| M. marinum M (ATCC BAA-535) | Human clinical isolate, wild-type, PDIM+ PGL+ | Snapper et al. (1990) |
| M. marinum ΔfadD22 | fadD22 (MMAR_1761 / MMAR_RS08725)* deletion, PDIM+ PGL− | Vergnolle et al. (2015) |
| M. marinum ΔfadD22-c | fadD22 deletion, carries a pCP0 vector derivative expressing fadD22, PDIM+ PGL+ (complemented strain) | Vergnolle et al. (2015) |
| M. marinum ΔfadD26 | fadD26 (MMAR_1777 / MMAR_RS08805) deletion, PDIM− PGL+ | Vergnolle et al. (2015) |
| M. marinum ΔfadD26-c | fadD26 deletion, carries a pCP0 derivative expressing fadD26, PDIM+ PGL+ (complemented strain) | Vergnolle et al. (2015) |
| M. marinum ΔfadD28 | fadD28 (MMAR_1765 / MMAR_RS08745) deletion, PDIM− PGL− | Vergnolle et al. (2015) |
| M. marinum ΔfadD28-c | fadD28 deletion, carries a pCP0 derivative expressing fadD28, PDIM+ PGL+ (complemented strain) | Vergnolle et al. (2015) |
| M. marinum ΔfadD29 | fadD29 (MMAR_1759 / MMAR_RS08715) deletion, PDIM+ PGL− | Vergnolle et al. (2015) |
| M. marinum ΔfadD29-c | fadD29 deletion, carries a pCP0 derivative expressing fadD29, PDIM+ PGL+ (complemented strain) | Vergnolle et al. (2015) |
| M. marinum ΔtesA | tesA (MMAR_1778 / MMAR_RS08810) deletion, PDIM− PGL− | Chavadi et al. (2011) |
| M. marinum ΔtesA-c | tesA deletion, carries a pCP0 derivative expressing tesA, PDIM+ PGL+ (complemented strain) | Chavadi et al. (2011) |
| M. marinum ΔpapA5 | papA5 (MMAR_1768 / MMAR_RS08760) deletion, PDIM− PGL− | Chavadi et al. (2012) |
| M. marinum ΔpapA5-c | papA5 deletion, carries a pCP0 derivative expressing papA5, PDIM+ PGL+ (complemented strain) | Chavadi et al. (2012) |
| M. smegmatis MC2155 (ATCC 700084) | Nonpathogenic species, lacks the PDIM/PGL biosynthetic pathway | Snapper et al. (1990) |
Original and re-annotated locus tag designations as per National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/) are provided for each gene.
Antibiotic susceptibility
The studies were conducted using a microdilution method (Chavadi et al. 2011). Plates were incubated at 30°C with orbital shaking (170 rpm) for 8 days and growth was assessed by measuring OD595 using a plate reader. MIC90 values for fold-change determinations were calculated from sigmoidal curves fitted to dose-response datasets using GraphPad Prism (GraphPad Software, Inc., Monroe, North Carolina, United States of America) as reported (Ollinger et al. 2013).
Ethidium bromide accumulation
Assay conditions were guided by those published (Yu et al. 2012). Briefly, mycobacteria resuspended in 50 mM phosphate-buffered solution, pH 7 (PBS) were loaded into black, flat-bottom, 96-well plates (Greiner Bio-One Co., La Jolla, California, United States of America) at 152 μL per well. Following loading, 8 μL of a 10% glucose solution in PBS and 40 μL of an ethidium bromide (EtBr) solution in PBS (25 mg L−1) were added to the wells and fluorescence intensity (535 nm excitation, 595 nm emission) was kinetically measured for 30 min using a plate reader. EtBr accumulation rates were determined as the slope of the linear regression fitted to datasets of relative fluorescence units versus time.
Biofilm and pellicle formation
MBEC™ plates (Innovotech Inc., Edmonton, Alberta, Canada) were loaded (150 μL per well) with bacterial cultures (106 CFU per mL; 7H9 medium with 0.2% glycerol) and incubated (30°C, 100 rpm, 4 weeks) for biofilm formation. Biofilm formation was quantified using a crystal violet colorimetric method (O'Toole 2011) and imaged using an Olympus BX41 microscope (Olympus Corp., Center Valley, Pennsylvania, United States of America). Pellicle formation was investigated using a 24-well plate assay similar to those reported (Pang et al. 2012; Tatham et al. 2012). Wells preloaded with Sauton's medium (1 mL per well) were surface inoculated with a saturated culture (10 μL) and pellicles were imaged after plate incubation (30°C, 5 weeks).
Sliding motility
The assay was conducted as reported (Tatham et al. 2012). Briefly, mycobacterial cultures were spotted (5 μL) on the center of sliding motility plates (7H9 medium, 0.3% or 0.5% agarose, 6% glycerol). Inoculated plates were incubated at 30°C (12 days and 14 days for 0.3% and 0.5% agarose plates, respectively). Sliding motility was determined as the diameter of the spreading colony.
Congo red binding
Plates of supplemented or nonsupplemented 7H11 medium without or with 100 μg mL−1 of Congo red (CR) (Tatham et al. 2012) were spot-inoculated with 2 μL of cultures grown to saturation. Inoculated plates were incubated at 30°C for 2 weeks (supplemented) or 4 weeks (nonsupplemented). Mycobacterial colonies were imaged using an Olympus SZX7 stereo microscope (Olympus Corp., Center Valley, Pennsylvania, United States of America).
Dictyostelium discoideum inhibition
The experiments were performed using a 24-well plate platform and adapted from a recently published assay (Alibaud et al. 2011). The inoculum of amoeba cells was expanded from the 100-to-10 000 reported range to a 10-to-150 000 range and escalated using 2-fold increments, as opposed to the 10-fold increments used previously. Plates were incubated for 14 days at 25°C. Starting on the third day, wells were visually inspected for formation of phagocytic plaques daily for 10 days. Amoebal growth was defined for any given set of duplicate wells (same amoeba inoculum) that exhibited at least two plaques in each well.
RESULTS AND DISCUSSION
Different gene knockouts in the PDIM/PGL biosynthetic pathway increase antibiotic susceptibility and weaken the cell envelope permeability barrier by different magnitudes
Recent studies in Mmar indicated that disruption of tesA, fadD28 or fadD26 produces antibiotic hypersusceptibility (Alibaud et al. 2011; Chavadi et al. 2011; Yu et al. 2012) and that disruption of fadD28 or fadD26 increases cell envelope permeability, as judged by dye accumulation assays (Yu et al. 2012). These studies support the notion that loss of PDIMs leads to antibiotic hypersusceptibility and increased cell envelope permeability, but provide no information as to whether selective loss of PGLs affects these phenotypic traits as well. To address this knowledge gap and expand our understanding of the phenotypic consequences of inactivating different genes required for PDIM and/or PGL production, we tested the susceptibility of our six Mmar mutants to antibiotics of different classes. In addition, we investigated the EtBr accumulation rates in the mutants to probe for changes in cell envelope permeability.
In the susceptibility experiments, the group of antibiotics tested included rifampicin, doxycycline, ciprofloxacin and streptomycin, which are used in the treatment of Mmar infections (Rallis and Koumantaki-Mathioudaki 2007). A strain was defined as having altered susceptibility when it displayed >2-fold change in MIC90 value relative to WT. As per this criterion, each mutant showed hypersusceptibility to at least one of the six antibiotics tested, while none of the mutants displayed reduced susceptibility to any of these antibiotics (Fig. 2A). The tesA and papA5 mutants showed qualitatively and quantitatively comparable hypersusceptibility profiles (Fig. 2A). The profiles of the remaining mutants were, for the most part, qualitatively comparable, yet quantitatively attenuated relative to that of the tesA and papA5 mutants (Fig. 2A). The susceptibility changes appears to be antibiotic-specific and uncorrelated with antibiotic lipophilicity (e.g. log P (Kerns and Di 2008)). Episomal expression in the mutants of a WT copy of the deleted gene flattened the hypersusceptibility profiles (Fig. 2B). Notably, the attenuated hypersusceptibility profile of the fadD28 mutant is unexpected because the mutant shares the PDIM− PGL− phenotype with the tesA and papA5 mutants and has an increase in envelope permeability comparable to that of these mutants (See below). The reason for this phenotypic inequality is unclear. Perhaps production of anomalous lipid variants with surrogate acyl chains in the absence of mycocerosic acids due to fadD28 deletion in Mmar ΔfadD28 is responsible for the unexpected phenotypic profile. Appearance of anomalous PDIM-like variants with abnormal acyl chains has been documented in the mycocerosic acid-deficient mutant of M. bovis BCG (Azad et al. 1996; Fitzmaurice and Kolattukudy 1998).
Figure 2.

Antibiotic susceptibility and ethidium bromide accumulation rate of M. marinum strains. Fold-change in MIC90 values between the WT strain and the mutants (A) or the mutants constitutively expressing an episomal WT copy of the deleted gene (B) are shown. A strain was defined as having an altered antibiotic susceptibility when it displayed >2-fold change (dotted line) relative to WT. The antimicrobial susceptibility results represent means and SEM of three independent experiments. The ethidium bromide (EtBr) accumulation data (C) correspond to means and SEM of five independent experiments.
The six gene knockouts also correlated with an increase in EtBr accumulation rate, a reporter of cell envelope permeability barrier integrity (Fig. 2C). Knockouts leading to concurrent loss of PDIMs and PGLs or selective loss of PDIMs roughly doubled the rate of EtBr accumulation, whereas knockouts selectively eliminating PGLs produced only a 25% increase. Many factors are likely to influence the antibiotic susceptibility of each mutant to individual antibiotics, including differences in drug uptake and/or efflux. The weakening of the permeability barrier might also affect the antibiotic susceptibility of the mutants by permitting better antibiotic penetration. However, the pattern of changes of EtBr accumulation rate (Fig. 2C) and antibiotic susceptibility (Fig. 2A) do not fully correlate across mutants. Taken together, the results indicate that only two out of the three knockouts producing concurrent loss of PDIMs and PGLs lead to a marked increase in susceptibility to specific antibiotics and that mutations resulting in selective loss of PDIMs or PGLs affect antibiotic susceptibility to a lesser degree. The results also suggest that PDIMs have a more significant role than PGLs in the integrity of the cell envelope permeability barrier.
Gene knockouts in the PDIM/PGL biosynthetic pathway are detrimental to biofilm formation and affect cell surface properties but not sliding motility
Recent studies have implicated the production of Mmar cell wall glycolipids known as lipooligosaccharides in biofilm formation and sliding motility (Ren et al. 2007). Loss of lipooligosaccharide production also leads to increased binding of the hydrophobic dye CR, a marker of cell envelope hydrophobicity (Etienne et al. 2009). These observations raise the possibility that other cell wall (glyco)lipids of Mmar might be relevant for these phenotypic traits. To our knowledge, the effect of PDIM or PGL production on biofilm formation, sliding motility and CR binding has not been reported. Thus, we explored these phenotypic traits in the Mmar mutants and expanded the study to assess the effect of the gene knockouts on pellicle formation (biofilm-type growth on a liquid surface) as well.
As expected, the WT formed biofilms on the pegs of the MBEC device (Fig. 3A) and its biofilm microcolonies displayed the characteristic serpentine cords (Fig. 3B) (Ceri et al. 1999; Hall-Stoodley et al. 2006). The mutants exhibited smaller and/or less abundant microcolonies (Fig. 3A) and less biofilm formation overall (32%–80% reduction relative to WT) (Fig. 3C). The tesA, papA5 and fadD26 mutants (all sharing loss of PDIMs) had more compact cell aggregates and less cording than WT (Fig. 3B). On the other hand, the microcolonies of the fadD28 mutant retained the WT phenotype (not shown). Thus, as seen in the antibiotic susceptibility experiments, the phenotype of the fadD28 mutant unexpectedly deviated from that of the other two PDIM− PGL− mutants (tesA and papA5). Episomal expression in the mutants of a WT copy of the deleted gene tended to increase biofilm formation and corrected the cording deficiency.
Figure 3.

Biofilm formation by M. marinum strains. Representative peg (A) and biofilm microcolony (B) images of the WT, two mutants (one PDIM− PGL− and one PDIM+ PGL−), and corresponding genetically complemented strains are shown. The results with these mutants are comparable to those seen with the rest of the mutants. Biofilm formation on the pegs was quantified using a standard colorimetric method (C). The results represent means and SEM of a minimum of six independent experiments. Student's t-test p values versus WT: *, <0.05; **, <0.01. All strains formed comparable pellicles on the surface of the liquid growth medium (D). The representative pellicle shown is for the WT.
The WT and the mutants had an equally robust pellicle formation capacity and their pellicles showed a comparably rich surface topology with folds, ridges and creases (Fig. 3D). There was no significant difference in sliding motility between the WT and the mutants either (Student's t-test P values > 0.05, n = 3; data not shown). On supplemented 7H11 plates, the WT and the mutants developed the same reddish coloration in the presence of CR (Fig. 4A). In contrast, the mutants displayed a drastic increase in CR staining relative to WT on nonsupplemented 7H11 plates (Fig. 4B). Episomal expression in the mutants of a WT copy of the deleted gene led to reversion to WT CR staining only in the tesA and fadD28 mutants, despite the fact that all the genetically complemented mutants regained PDIM and/or PGL production capacity (Fig. 1C) (Chavadi et al. 2011, 2012; Vergnolle et al. 2015). Taken together, our findings indicate that loss of PDIMs and/or PGLs affect biofilm formation and CR binding (in a medium composition-dependent manner), but do not compromise pellicle formation or sliding motility. To our knowledge, this is the first study indicating that loss of PDIMs and/or PGLs affects biofilm formation.
Figure 4.

CR binding properties of M. marinum strains. Plates of supplemented or nonsupplemented medium without or with CR were spot-inoculated with M. marinum cultures, incubated for growth, and imaged. Images of the WT, two mutants (one PDIM− PGL− and one PDIM+ PGL−), and corresponding genetically complemented strains are shown. The results shown for these mutants are comparable to those seen with the rest of the mutants, except by the complementation differences noted in the text. The data is representative of three independent experiments.
Different gene knockouts in the PDIM/PGL biosynthetic pathway lead to different attenuation levels in an amoeba model
The phagocytic amoeba D. discoideum is a commonly used model to study virulence of mycobacteria and other pathogens (Tosetti, Croxatto and Greub 2014). Mycobacterial pathogens such as Mmar multiply intracellularly in permissive endosomal compartments and produce amoebal growth inhibition (Hagedorn and Soldati 2007; Kennedy, Morisaki and Champion 2012). We probed the virulence of the Mmar mutants (Fig. 5) using a D. discoideum inhibition assay in which a Mmar tesA transposon mutant previously showed attenuation (Alibaud et al. 2011). We found a drastic reduction in virulence (∼300-fold–∼500-fold relative to WT) for the tesA, papA5, fadD28 and fadD26 mutants. On the other hand, the fadD22 and fadD29 mutants exhibited a modest virulence reduction (3-fold). Episomal expression of a WT copy of the deleted gene increased the virulence of the mutants (Fig. 5). Our results indicate that concurrent loss of PDIMs and PGLs or selective loss of PDIMs leads to severe attenuation in the D. discoideum model. In contrast, selective loss of PGLs is relatively inconsequential in this model. These findings support the view that PDIMs have a more critical role than PGLs in the pathogenicity of Mmar against D. discoideum. In M. tuberculosis, disruption of fadD28 impairs the ability of the pathogen to arrest phagolysosome maturation (Brodin et al. 2010), possibly by the lack of PDIMs to elicit pathogen-directed reorganization of host membrane lipids (Astarie-Dequeker et al. 2009). This precedent highlights the possibility that the attenuation of the Mmar mutants might emerge from a reduced capacity to arrest the amoebal phagolysosome maturation pathway and create protective niches. Interestingly, even the most attenuated mutant remained significantly more virulent than the nonpathogenic M. smegmatis. This suggests that Mmar has multiple virulence factors contributing to its interplay with the amoeba. This notion is not surprising considering that Mmar and amoebae share environmental niches and that amoebae are thought to act as evolutionary training grounds for intracellular bacterial pathogens (Molmeret et al. 2005).
Figure 5.
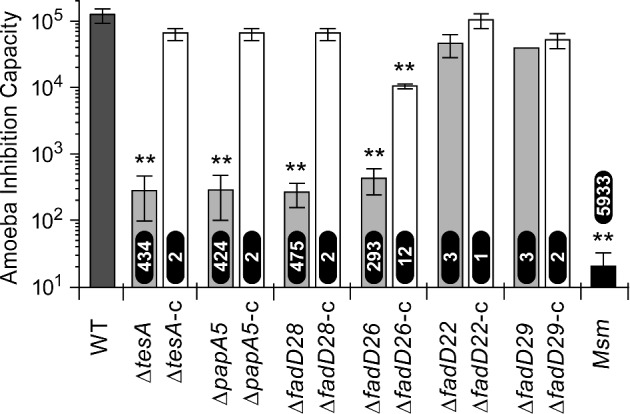
Inhibition of D. discoideum by M. marinum strains. The virulence of the M. marinum strains was comparatively assessed using a D. discoideum inhibition assay. In this assay, the highest amoeba inoculum inhibited by each strain is considered a measure of its virulence. The fold reduction in virulence of each strain relative to M. marinum WT is shown at the base of each bar for the mutants and above the bar for M. smegmatis (Msm). The results correspond to means and SEM of three experiments. **, Student's t-test P < 0.01 (versus WT).
CONCLUSIONS
Our findings provide new insight into the consequences (or lack thereof) of different knockouts in the PDIM/PGL biosynthetic pathway on various phenotypic traits. The complex pleiotropic patterns arising from the different knockouts might be influenced by changes in metabolic flux redirecting different biosynthetic precursor surpluses to production of other (glyco)lipids (Jain et al. 2007; Ferreras et al. 2008; Lee et al. 2013). Accumulation of different biosynthetic intermediates, some of which might be repurposed for the production of unusual (glyco)lipids with potential phenotypic impacts (Azad et al. 1996; Fitzmaurice and Kolattukudy 1998), might also contribute to define the pleiotropic patterns of the mutants seen here. (Glyco)lipidomic profiling will be needed to investigate these possibilities and assist the unraveling of the molecular mechanisms underlying the phenotypic profiles of the mutants. The parallel analysis of isogenic mutants reported herein permitted us to uncover unexpected phenotypic inequalities. Most noticeably, the differences in antibiotic susceptibility and microcolony morphology between the tesA (or papA5) and fadD28 knockouts (both leading to a PDIM− PGL− phenotype) suggest that the enzymes encoded by these genes might not be equivalent targets for adjuvant drug development. In more general terms, our findings advocate that the notion that different enzymes of a metabolic pathway for which elimination equally leads to pathway shutdown are equivalent potential targets for drug development might not always hold true.
Acknowledgments
We are grateful for the endowment support from Carol and Larry Zicklin. We thank Milan Patel, Pawandeep Singh, Kang Li, Tanmai Shah and Claudia Zmijewski for assistance with the mutant characterization experiments and Prof. Derrick Brazill (Hunter College) for advice with the amoeba experiments.
FUNDING
This work was supported in part by National Institutes of Health grant R15AI105884 a grant from The Potts Memorial Foundation, both awarded to LENQ.
Conflict of interest. None declared.
REFERENCES
- Alibaud L, Rombouts Y, Trivelli X, et al. A Mycobacterium marinum TesA mutant defective for major cell wall-associated lipids is highly attenuated in Dictyostelium discoideum and zebrafish embryos. Mol Microbiol. 2011;80:919–34. doi: 10.1111/j.1365-2958.2011.07618.x. [DOI] [PubMed] [Google Scholar]
- Arbues A, Lugo-Villarino G, Neyrolles O, et al. Playing hide-and-seek with host macrophages through the use of mycobacterial cell envelope phthiocerol dimycocerosates and phenolic glycolipids. Front Cell Infect Microbiol. 2014;4:173. doi: 10.3389/fcimb.2014.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astarie-Dequeker C, Le Guyader L, Malaga W, et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog. 2009;5:e1000289. doi: 10.1371/journal.ppat.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad AK, Sirakova TD, Rogers LM, et al. Targeted replacement of the mycocerosic acid synthase gene in Mycobacterium bovis BCG produces a mutant that lacks mycosides. P Natl Acad Sci USA. 1996;93:4787–92. doi: 10.1073/pnas.93.10.4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, Poquet Y, Levillain F, et al. High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog. 2010;6:e1001100. doi: 10.1371/journal.ppat.1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho LR, Constant P, Raynaud C, et al. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem. 2001;276:19845–54. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- Cambau E, Aubry A. Mycobacterium marinum. In: Schlossberg D, editor. Tuberculosis and Nontuberculous Mycobacterial Infections. Herndon, VA: ASM Press; 2011. pp. 586–600. [Google Scholar]
- Cambier CJ, Takaki KK, Larson RP, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014;505:218–22. doi: 10.1038/nature12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceri H, Olson ME, Stremick C, et al. The calgary biofilm device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J Clin Microbiol. 1999;37:1771–6. doi: 10.1128/jcm.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavadi S, Onwueme K, Edupuganti U, et al. The mycobacterial acyltransferase PapA5 is required for biosynthesis of cell wall-associated phenolic glycolipids. Microbiology. 2012;158:1379–87. doi: 10.1099/mic.0.057869-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavadi SS, Edupuganti UR, Vergnolle O, et al. Inactivation of tesA reduces cell wall lipid production and increases drug susceptibility in mycobacteria. J Biol Chem. 2011;286:24616–25. doi: 10.1074/jbc.M111.247601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichinger L, Rivero F. Dictyostelium Discoideum Protocols. Springer Verlag, Heidelberg, Germany: Humana Press; 2013. [Google Scholar]
- Etienne G, Malaga W, Laval F, et al. Identification of the polyketide synthase involved in the biosynthesis of the surface-exposed lipooligosaccharides in mycobacteria. J Bacteriol. 2009;191:2613–21. doi: 10.1128/JB.01235-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras JA, Stirrett KL, Lu X, et al. Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation. Chem Biol. 2008;15:51–61. doi: 10.1016/j.chembiol.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice AM, Kolattukudy PE. An acyl-CoA synthase (acoas) gene adjacent to the mycocerosic acid synthase (mas) locus is necessary for mycocerosyl lipid synthesis in Mycobacterium tuberculosis var. bovis BCG. J Biol Chem. 1998;273:8033–9. doi: 10.1074/jbc.273.14.8033. [DOI] [PubMed] [Google Scholar]
- Guenin-Mace L, Simeone R, Demangel C. Lipids of pathogenic mycobacteria: contributions to virulence and host immune suppression. Transbound Emerg Dis. 2009;56:255–68. doi: 10.1111/j.1865-1682.2009.01072.x. [DOI] [PubMed] [Google Scholar]
- Hagedorn M, Soldati T. Flotillin and RacH modulate the intracellular immunity of Dictyostelium to Mycobacterium marinum infection. Cell Microbiol. 2007;9:2716–33. doi: 10.1111/j.1462-5822.2007.00993.x. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Brun OS, Polshyna G, et al. Mycobacterium marinum biofilm formation reveals cording morphology. FEMS Microbiol Lett. 2006;257:43–9. doi: 10.1111/j.1574-6968.2006.00143.x. [DOI] [PubMed] [Google Scholar]
- He W, Soll CE, Chavadi SS, et al. Cooperation between a coenzyme A-independent stand-alone initiation module and an iterative type I polyketide synthase during synthesis of mycobacterial phenolic glycolipids. J Am Chem Soc. 2009;131:16744–50. doi: 10.1021/ja904792q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Petzold CJ, Schelle MW, et al. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. P Natl Acad Sci USA. 2007;104:5133–8. doi: 10.1073/pnas.0610634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy GM, Morisaki JH, Champion PA. Conserved mechanisms of Mycobacterium marinum pathogenesis within the environmental amoeba Acanthamoeba castellanii. Appl Environ Microb. 2012;78:2049–52. doi: 10.1128/AEM.06965-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns EH, Di L. Lipophilicity. In: Kerns EH, Di L, editors. Drug-Like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization. San Diego: Academic Press; 2008. pp. 43–7. [Google Scholar]
- Lee W, VanderVen BC, Fahey RJ, et al. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem. 2013;288:6788–800. doi: 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molmeret M, Horn M, Wagner M, et al. Amoebae as training grounds for intracellular bacterial pathogens. Appl Environ Microb. 2005;71:20–8. doi: 10.1128/AEM.71.1.20-28.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyrolles O, Guilhot C. Recent advances in deciphering the contribution of Mycobacterium tuberculosis lipids to pathogenesis. Tuberculosis (Edinb) 2011;91:187–95. doi: 10.1016/j.tube.2011.01.002. [DOI] [PubMed] [Google Scholar]
- O'Toole GA. Microtiter dish biofilm formation assay. J Vis Exp. 2011;47:e2437. doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollinger J, Bailey MA, Moraski GC, et al. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS One. 2013;8:e60531. doi: 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onwueme KC, Vos CJ, Zurita J, et al. The dimycocerosate ester polyketide virulence factors of mycobacteria. Prog Lipid Res. 2005;44:259–302. doi: 10.1016/j.plipres.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Pang JM, Layre E, Sweet L, et al. The polyketide Pks1 contributes to biofilm formation in Mycobacterium tuberculosis. J Bacteriol. 2012;194:715–21. doi: 10.1128/JB.06304-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish T, Stoker NG, Walker JM, editors. Mycobacteria Protocols. Vol. 101. Totowa, New Jersey: Humana Press; 1998. General culture methodology and safety considerations; pp. 15–30. [Google Scholar]
- Quadri LE. Biosynthesis of mycobacterial lipids by polyketide synthases and beyond. Crit Rev Biochem Mol. 2014;49:179–211. doi: 10.3109/10409238.2014.896859. [DOI] [PubMed] [Google Scholar]
- Rallis E, Koumantaki-Mathioudaki E. Treatment of Mycobacterium marinum cutaneous infections. Expert Opin Pharmaco. 2007;8:2965–7298. doi: 10.1517/14656566.8.17.2965. [DOI] [PubMed] [Google Scholar]
- Ren H, Dover LG, Islam ST, et al. Identification of the lipooligosaccharide biosynthetic gene cluster from Mycobacterium marinum. Mol Microbiol. 2007;63:1345–59. doi: 10.1111/j.1365-2958.2007.05603.x. [DOI] [PubMed] [Google Scholar]
- Snapper SB, Melton RE, Mustafa S, et al. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–9. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- Soetaert K, Rens C, Wang XM, et al. Increased vancomycin susceptibility in Mycobacteria: a new approach to identify synergistic activity against multidrug-resistant Mycobacteria. Antimicrob Agents Ch. 2015;59:5057–60. doi: 10.1128/AAC.04856-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatham E, Chavadi S, Mohandas P, et al. Production of mycobacterial cell wall glycopeptidolipids requires a member of the MbtH-like protein family. BMC Microbiol. 2012;12:118. doi: 10.1186/1471-2180-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosetti N, Croxatto A, Greub G. Amoebae as a tool to isolate new bacterial species, to discover new virulence factors and to study the host-pathogen interactions. Microb Pathogenesis. 2014;77:125–30. doi: 10.1016/j.micpath.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Vergnolle O, Chavadi SS, Edupuganti UR, et al. Biosynthesis of cell-envelope-associated phenolic glycolipids in Mycobacterium marinum J Bacteriol 2015. 197 1040–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Tran V, Li M, et al. Both phthiocerol dimycocerosates and phenolic glycolipids are required for virulence of Mycobacterium marinum. Infect Immun. 2012;80:1381–9. doi: 10.1128/IAI.06370-11. [DOI] [PMC free article] [PubMed] [Google Scholar]