

Abstract
Hepatocyte growth factor activator inhibitor-2 (HAI-2) is an inhibitor of many proteases in vitro, including the membrane-bound serine protease, matriptase. Studies of knock-out mice have shown that HAI-2 is essential for placental development only in mice expressing matriptase, suggesting that HAI-2 is important for regulation of matriptase. Previous studies have shown that recombinant expression of matriptase was unsuccessful unless co-expressed with another HAI, HAI-1. In the present study we show that when human matriptase is recombinantly expressed alone in the canine cell line MDCK, then human matriptase mRNA can be detected and the human matriptase ectodomain is shed to the media, suggesting that matriptase expressed alone is rapidly transported through the secretory pathway and shed. Whereas matriptase expressed together with HAI-1 or HAI-2 accumulates on the plasma membrane where it is activated, as judged by cleavage at Arg614 and increased peptidolytic activity of the cell extracts. Mutagenesis of Kunitz domain 1 but not Kunitz domain 2 abolished this function of HAI-2. HAI-2 seems to carry out its function intracellularly as this is where the vast majority of HAI-2 is located and since HAI-2 could not be detected on the basolateral plasma membrane where matriptase resides. However, minor amounts of HAI-2 not undergoing endocytosis could be detected on the apical plasma membrane. Our results suggest that Kunitz domain 1 of HAI-2 cause matriptase to accumulate in a membrane-bound form on the basolateral plasma membrane.
Keywords: Matriptase, HAI-2, HAI-1, Shedding
lntroduction
Hepatocyte growth factor activator inhibitor-2 (HAI-2), also known as placental bikunin [1,2], has emerged as a potential inhibitor of the type II transmembrane serine protease, matriptase [3,4]. HAI-2 is encoded by the SPINT2 gene and its function is essential for life as HAI-2 knock-out mice die during mid-gestation with signs of dysfunctional placental development and missing neural tube closure [5]. HAI-2 and matriptase double knock-out mice survive until birth with normal placental development showing that HAI-2 is unnecessary for placental development if matriptase is absent [3]. In contrast, the neural tube closure defects seem to be the result of lack of a different function of HAI-2, most likely inhibition of an unknown protease, as matriptase and HAI-2 double knock-out mice still carry this feature [3]. A recent publication clearly shows that matriptase and the serine protease, prostasin, are both components of a proteolytic cascade regulated by HAI-1 and HAI-2 important for development of the placenta [6].
Humans homozygous or compound heterozygous for an assortment of SPINT2 autosomal recessive mutations, suffer from congenital sodium diarrhea [7]. These individuals also display a broad range of developmental abnormalities that include duplication of internal organs, duplication and abnormal location of digits, craniofacial dysmorphisms, anal and choanal atresia, fistulas and hamartoma [7]. Together this suggests that HAI-2 plays an important role during tissue morphogenesis.
Matriptase plays a crucial role in maintaining functional epithelial barriers as matriptase knock-out mice have lethal epithelial barrier defects of the skin [8] and conditional knock-out of the protease in adult mice leads to lethal barrier defects in the intestine [9]. In humans, dysfunctional matriptase has been linked to an autosomal recessive form of ichthyosis characterized by a missense mutation (G827R) in the catalytic domain of matriptase [10]. Matriptase has also been coupled to cancer development as transgenic mice with a modest overexpression of the protease in the skin all developed dysplasia of which 70% progressed into carcinomas, whereas control littermates were unaffected [11].
The enzymatic activity of matriptase is not controlled by HAI-2 alone, as hepatocyte growth factor activator inhibitor-1 (HAI-1) encoded by the SPINT1 gene, also acts as an inhibitor of matriptase [11–14]. In the above mentioned study of matriptase-overexpressing mice, co-expression of matriptase together with HAI-1 completely abolished tumor formation suggesting that the enzymatic activity of matriptase can cause cancer [11]. Lack of HAI-1 is also lethal as the knock-out mice die during gestation [13].
Both HAI-2 and HAI-1 are type I transmembrane serine protease inhibitors belonging to the Kunitz family with a short intracellular tail and a larger extracellular part containing two inhibitory Kunitz domains (KD). HAI-2 consists of 252 amino acids adding up to a calculated molecular weight of approximately 28 kDa containing two potential N-glycosylation sites [1,2].
Matriptase, HAI-2 and HAI-1 are expressed in most epithelial cells [4,15–17]. Under normal physiological conditions many epithelial cells maintain a polarized state, in which the plasma membrane of differentiated cells is divided into an apical and a basolateral domain separated by tight-junctions, controlling paracellular flow and preventing diffusion of outer leaflet proteins between the apical and the basolateral plasma membrane domains. Matriptase is synthesized as a single-chain inactive protein, which is initially cleaved at Gly149 in its SEA domain [18]. The N-terminal fragment (1–149) remains non-covalently associated with the rest of the protein but dissociates away during western blotting sample preparation. Matriptase is subsequently activated by a proteolytic cleavage following Arg614 in the linker to the C-terminal serine protease domain. The serine protease domain remains covalently linked to the rest of the protein by a disulfide bridge [18]. Matriptase can be detected as a 70 kDa form under non-reducing conditions by western blotting. This is referred to as total matriptase, and represents both the zymogen and fully activated matriptase that are indiscernible under these conditions. However, under reducing conditions, the catalytic domain of activated matriptase becomes detectable on western blots as a separate 30 kDa band.
It is at present unclear where and how HAI-2 interacts with matriptase. In contrast matriptase and HAI-1 form a complex on the plasma membrane after activation of matriptase has taken place [19,20]. HAI-1 thus acts as a bonafide inhibitor of matriptase. However, the role of HAI-1 is not fully understood as it appears that matriptase cannot be recombinantly expressed in the absence of co-expression with HAI-1 [21,22]. In the present study we have investigated the subcellular localization and function of HAI-2. HAI-2 is smaller and less complex than HAI-1 and may therefore shed light on the function of HAIs during expression of matriptase. Our results suggest that matriptase recombinantly expressed without HAI-2 or HAI-1 is rapidly transported through the secretory pathway and shed, whereas matriptase expressed together with HAI-2 accumulate on the plasma membrane as a result of a process dependent on Kunitz domain 1 of HAI-2. Matriptase accumulated at the plasma membrane is activated, as judged by peptidolytic activity of the cell lysate and cleavage at Arg614.
Materials and methods
Cell culture
The human colon epithelial cell line Caco-2 was grown in minimal essential medium supplemented with 2 mM 1-glutamine, 10% fetal bovine serum, 1 × non-essential amino acids, 100 units/ml penicillin and 100 μg/ml streptomycin at 37 °C in an atmosphere of 5% CO2. Madin-Darby Canine Kidney (MDCK) epithelial cells were grown in minimal essential medium supplemented with 2 mM l-glutamine, 10% fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin at 37 °C in an atmosphere of 5% CO2. Medium for MDCK cells stably expressing human wild-type HAI-2 or HAI-2-EYFP had an addition of 0.65 mg/ml geneticin (G418) selection antibiotic. Chinese Hamster Ovary (CHO) cells were grown in HAM-F12 medium supplemented with 2 mM L-glutamine, 10% fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin at 37 °C in an atmosphere of 5% CO2.
For experiments 2 × 106 Caco-2 or MDCK cells were seeded into 0.4 μm-pore-size 24 mm or 5 × 105 cells into 0.4 μm-pore-size 12 mm Transwell® filter chambers (cat. no. 3412 and cat. no. 3401, Corning). Cells were grown to confluence forming a tight polarized monolayer allowing separate access to the apical and basolateral plasma membrane. Caco-2 cells plated on transwell filters were used for experiments 11 days post-confluence. The tightness of filter-grown cells was assayed by filling the inner chamber to the brim and allowing it to equilibrate overnight. The medium was changed every day.
Biotinylation of cell surface exposed proteins
Cells seeded on transwell filters were washed twice with ice-cold PBS╫ (PBS supplemented with 0.7 mM CaCl2 and 0.25 mM MgCl2) and biotin-labeled at either the apical or the basolateral side for 30 min at 4 °C, using 1 mg/ml EZ-Link® Sulfo-NHS-SS-Biotin (cat. no. 21331, Pierce) dissolved in PBS╫. Cells were then washed three times with ice-cold PBS╫ and excess biotin quenched for 5 min at 4 °C with 50 mM glycine in PBS╫, followed by an additional three times wash with ice-cold PBS╫. Next, cells were lysed in lysis buffer (PBS containing 1% Triton X-100, 0.5% deoxycholate and protease inhibitors 10 mg/l benzamidine, 2 mg/l pepstatin A, 2 mg/l leupeptin, 2 mg/l antipain and 2 mg/l chymostatin) and the lysates were spun at 4 °C for 10 min at 20,000g to pellet insoluble material. The supernatants were transferred to clean test tubes and were subsequently exposed to streptavidin or monomeric avidin pull-down of biotinylated proteins.
Streptavidin/monomeric avidin pull-down
Supernatants from lysed biotin-labeled cells were transferred to clean test tubes containing either Pierce® streptavidin agarose resin (cat. no. 20349, Pierce) or Pierce® immobilized monomeric avidin agarose resin (cat. no. 20267, Pierce). The agarose was prepared by washing the streptavidin and monomeric avidin agarose three times in lysis buffer or H2O respectively. After overnight incubation at 4 °C with end-over-end rotation, the samples were spun for 1 min at 1200g at 4 °C and the flow through fraction (FT), representing non-biotinylated proteins, were collected. The streptavidin and monomeric avidin agarose were then washed tree times with lysis buffer or 50 mM Tris–HCl pH 6.25, respectively. Biotinylated proteins were eluted from the streptavidin agarose resin by boiling the samples for 5 min at 95 °C in SDS sample buffer containing 100 mM DTT. For the monomeric avidin agarose resin biotinylated proteins were eluted with 4 mM biotin (cat. no. 29129, Pierce) in PBS╫ for 30 min at room temperature (RT), followed by addition of non-reducing SDS sample buffer and another 5 min incubation at RT. After elution of biotinylated proteins the samples were spun for 1 min at 1200g and the supernatants analyzed by SDS-PAGE and western blotting. Sample buffer were added to the collected FTs, with or without DTT for samples precipitated with streptavidin and monomeric avidin, respectively.
SDS-PAGE and western blotting
Proteins were separated on 10% SDS poly-acrylamide gels and transferred to Immobilon-P PVDF membranes (Millipore). The blots were blocked with 10% non-fat dry milk in PBS containing 0.1% Tween-20 (PBS-T) for 1 h at RT. The individual PVDF membranes were probed with primary antibodies diluted in 1% non-fat dry milk in PBS-T at 4 °C overnight, followed by 3 × 5 min wash in PBS-T and 1 h incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (Pierce). After 3 × 5 min wash with PBS-T the signal was developed using ECL® (enhanced chemiluminescence) reagent Super Signal West Femto Maximum Sensitivity Substrate (Pierce) according to the protocol supplied by the manufacturer, and visualized with a Fuji LAS-1000 camera and Intelligent DarkBox II (FujiFilm Sweden AB), using the program LAS1000 Lite v1.5.
Antibodies
Blots were probed with monoclonal mouse anti-human matriptase antibodies M24 [12] and M32 [23] (1:1000) recognizing total matriptase, i.e. both zymogen and active matriptase under non-reducing conditions, polyclonal rabbit anti-human matriptase/MT-SP1 (cat. no. IM1014, Calbiochem®) (1:1000) recognizing the serine protease domain of matriptase under reducing conditions, monoclonal mouse anti-human HAI-2 ectodomain (cat. no. MAB1106, R&D) (1:400) recognizing the extracellular part of HAI-2 under reducing conditions, monoclonal mouse anti-EYFP (JL-8) (cat. no. 632380, Clontech) (1:2000) recognizing EYFP under reducing conditions, mouse anti-human transferrin receptor (cat. no. 13-6800, Zymed) recognizing the transferrin receptor and antibodies against glyceraldehydes 3-phosphate dehydrogenase (GAPDH) (cat. no #MAB374, zymed laboratories inc.) as a loading control.
Endocytosis assay
Caco-2 cells seeded on 0.4 μm-pore-size 12 mm transwell filters were biotinylated at the apical side as described above. For the endocytosis experiment, internalization media (MEM Eagle Medium containing Earle's balanced salts, 0.02 M HEPES, 20 mM NaHCO3, 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin) preheated to 37 °C was added, and the cells incubated at 37 °C for the times indicated in the experiment. After incubation the following was done at 4 °C: Cells were washed twice with PBS╫ and biotin-labeled cells were stripped of any remaining cell-surface bound Sulfo-NHS-SS-Biotin by exposing the cells twice to a reducing buffer (50 mM glutathione, 75 mM NaCl, 75 mM NaOH, 1 mM EDTA and 0.5% BSA in H2O) for 20 min at gentle agitation. Cells were then washed twice with PBS╫ and excess glutathione quenched for 5 min, using 5 mg/ml iodoacetamide dissolved in PBS╫. Control samples representing total cell-surface protein biotinylation were not treated with reducing glutathione buffer. Next, cells were washed twice with PBS╫ and lysed as described above and samples were subsequently exposed to streptavidin pull-down.
Transfections and DNA constructs
For transient expression MDCK cells were transfected in suspension and CHO cells as adherent cells using Lipofectamine™ 2000 (Invitrogen), according to the manufacturers recommendations. Cell extracts of transiently transfected cells were obtained 48 h post-transfection. For co-transfections the same overall quantity of plasmid was used. Stable transfections of MDCK cells were performed using the calcium phosphate transfection procedure [24]. The cDNA coding for full-length human wild-type matriptase, HAI-1 and HAI-2 were incorporated into pcDNA3.1 plasmid vectors. For Mock transfections λ-DNA was used. The HAI-2 cDNA used in the present study contains a natural occurring SNP resulting in the amino acid substitution (V200L).
The cDNA coding for full-length human wild-type HAI-2, incorporated into the eukaryotic expression vector pLenti-EF1-SfiI-shuttle-IRES-PuroR (HAI-2-pLEF) [3], was subcloned into the eukaryotic expression vector pEYFP-N1 (cat. no. 6006-1, Clontech). The construct pEYFP-N1-HAI-2 vector was verified by sequencing (data not shown). Mutations in the cDNA coding for HAI-2 or matriptase were introduced by site-directed mutagenesis using the Gene Tailor™ Site-Directed Mutagenesis System (cat. no. 12397-014, Invitrogen) according the manufacturers recommendations.
Immunocytochemistry
Transfected MDCK cells were plated on transwell filters and grown to either 2 days post-transfection or 3 days post-confluence as indicated in the separate experiments. The following procedures were done at 4 °C. Cells were washed 2 × with PBS╫ and fixed using 4% (w/v) paraformaldehyde in 0.1 M phosphate buffer pH 7.4 for 20 min. After 2 × wash in PBS╫ the cells were permeabilized in 0.05% TritonX-100 in PBS╫ for 20 min and blocked for 30 min using PBS╫ containing 3% BSA (PBS/BSA). Primary antibody (1:100 in PBS/BSA) was added to the cells and allowed to incubate for 2 h at gentle agitation, then washed 3 × 5 min in PBS╫ and after that exposed to relevant AlexaFluor-conjugated secondary antibodies diluted in PBS/BSA for 1 h at gentle agitation. After a final 3 × 5 min wash in PBS╫ the cells were mounted in ProLong Gold antifade reagent (cat. no. P36930, Invitrogen). For cell surface staining Alexa Flour® 647 Phalloidin (cat. no. A22287, Invitrogen) (1:200 in PBS/BSA) was used and for endoplasmic reticulum detection the vector pDsRed2-ER (cat. no. 632409, Clontech) was used. Laser scanning confocal microscopy was performed using the Leica TCS SP2 system equipped with argon and helium-neon lasers. Images were acquired using a 63 × water immersion objective, NA 1.2 with a pinhole size of 1 and a pixel format of 1024 × 1024. Line averaging was used to reduce noise. For double- and triple labeling experiments sequential scanning was employed to allow the separation of signals from the individual channels. Acquired images were treated using Adobe Photoshop CS2 and Adobe Illustrator CS2.
Enzymatic activity assay
Cell extracts of transiently transfected MDCK-cells were prepared using the same procedure as described above for the biotinylation protocol, but no protease inhibitors was applied during the extraction procedure. Twenty microliters of cell extract was diluted with 20 mM HEPES pH 7.4, 140 mM NaCl supplemented with 0.1% BSA (Sigma) to a final volume of 180 μl in a 96 well plate and heated to 37 °C for 15 min. At time equal zero, 20 μl chromogenic substrate H-d-Isoleucyl-l-prolyl-l-arginine-p-nitroaniline (cat. no. S2288, Chromogenix) was added to a final concentration of 1 mM. Substrate conversion was followed in a standard plate reader by consecutive measurements of the absorbance at 405 nm at time points between 0 and 400 min while incubating the plate at 37 °C. The rate of substrate turnover was determined from the color development resulting from the pseudo-first order reaction due to a substrate concentration far greater than the expected pM range of protease content as judged by semi-quantitative western blot analysis. When testing the effect of protease inhibitors, 400 μM PMSF, 10 μM TLCK (Sigma), 10 μM Soy bean trypsin inhibitor and 1 μM aprotinin (Bovine trypsin inhibitor (Sigma)) were added to the wells prior to temperature equilibration to allow efficient inhibition before assaying the enzymatic activity by adding the chromogenic substrate.
Patients, study material, treatment
This study includes sections from colonic biopsies taken by colonoscopy. The sections originate from a study in which the patients were referred for elective diagnostic lower endoscopy and asked for permission to take extra biopsies for scientific purposes. The study was approved by the local ethical committee (HA-2008-131) and informed consent was obtained from each patient. Biopsy specimens weighing approximately 5 mg were promptly fixed using 10 ml of pure acetone (cat no. L.00014.2500, Merck) and allowed to incubate for 48 h before treatment with a 25% sucrose solution (cat. no. A2211.100, Applichem) for 24 h. The biopsies were embedded in Tissue-Tek® (cat. no. 4583, Sakura Finetek) using dry-ice cooled isopentane (−70 °C) and then cryo-sectioned in 5 μm thick sections which were subsequently stored at −80 °C.
Immunohistochemistry
Sections were acclimatized to RT for 1 h, then rinsed with TBS-T (50 mM Tris, 150 mM NaCl, 0.5% Triton X-100, pH 7.6) until all of the Tissue-Tek® was removed and subsequently washed for 5 min with H2O. Endogenous peroxidases were then blocked by incubating the sections with 1% H2O2 for 2 × 10 min followed by another 5 min wash in H2O. Sections were next washed 2 × with TBS-T, then blocked with goat serum (1:100 in TBS-T) for 1 h and washed 2 × with TBS-T. Afterwards the monoclonal antibody mouse anti-human HAI-2 ectodomain diluted in Antibody Diluent (cat. no. S3022, Dako) to a concentration of 2.5 μg/ml was applied overnight at 4 °C. The following day the sections were allowed to acclimatize at RT for 30 min prior to a 2 × wash with TBS-T and secondary EnVision anti-mouse antibody (cat. no K4001, Dako) was applied for 45 min. Afterwards sections were washed 2 × with TBS-T and then allowed to incubate for 12 min with NovaRED (cat. no. SK-4800, Vector Laboratories) according to the protocol supplied by the manufacturer. Sections were subsequently washed 2 × with H2O and counterstained with Mayer's Haematoxylin for 30 s. Finally the sections were rinsed in H2O for 5 min before dehydration and mounting in Pertex™ Mounting Medium (cat. no. 20080, Sakura Finetek). In parallel sections the primary antibody was omitted and showed no staining. As a positive control M19, a monoclonal antibody against HAI-1, was included and showed staining along the basolateral membranes.
Results
Specific detection of HAI-2 and HAI-2-EYFP in mammalian cells
Using real time RT-PCR, we found significant amounts of mRNA coding for HAI-2 relative to the expression of beta-actin in Caco-2 cells (data not shown), suggesting that HAI-2 is expressed in these cells. To determine whether the HAI-2 antibody used in this study was specific, we compared the HAI-2 immunoreactive material of Caco-2 cells and the immunoreactive material of CHO and MDCK cells recombinantly expressing human HAI-2. A vector coding for full-length human HAI-2 cDNA was either transiently transfected into CHO cells or stably transfected into MDCK cells. Cell extracts of the transfected CHO and MDCK cells were then analyzed by SDS-PAGE and western blotting and compared to non-transfected cells and a Caco-2 cell extract, using the mouse monoclonal antibody “anti-human HAI-2 ectodomain” for detection (Fig. 1A). In both transfected cell lines, two bands of approximately 28 and 32 kDa could be detected, corresponding to full-length HAI-2. Additionally, a double band of 18-20 kDa was detected (Fig. 1A). These most likely represents proteolytically cleaved forms of HAI-2. Non-transfected CHO and MDCK cells showed no immunoreactivity, whereas the Caco-2 cell extract showed bands of equivalent molecular weight as CHO and MDCK cells recombinantly expressing HAI-2. This demonstrates that the “anti-human HAI-2 ectodomain” antibody specifically detects HAI-2 at least in these cell lines. It should however be noted that the apparent rate of HAI-2 cleavage vary between the three cell lines. Previous experiments showed that Caco-2 cells also express HAI-1 [19]. HAI-1 is detected as a 55 kDa protein when expressed in Caco-2 cells and is not detected by the anti-human HAI-2 ectodomain antibody (data not shown).
Fig. 1.
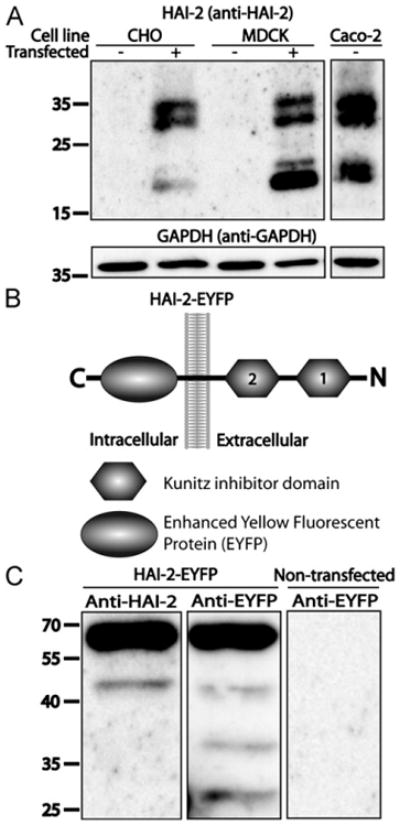
Specific detection of HAI-2 and HAI-2-EYFP: (A) Cell extracts of CHO cells transiently transfected with a vector coding for human HAI-2 and MDCK cells recombinantly expressing human HAI-2 were compared to non-transfected cells by SDS-PAGE and western blotting. For further comparison a cell extract of 11-days post-confluent human Caco-2 cells was included in the analysis. The blots were probed using monoclonal mouse anti-human HAI-2 ectodomain antibody targeting the extracellular part of HAI-2. Blots were stripped and reprobed with antibodies against GAPDH. Transfected cells are indicated by (+) and non-transfected controls by (−). The molecular weights are in kDa. Schematic representation of the fusion protein HAI-2-EYFP. HAI-2-EYFP is synthesized as a 57 kDa protein with EYFP located on the intracellular side of the plasma membrane and the two Kunitz domains (1 and 2) located on the extracellular side. Cell extracts of MDCK cells with a transient expression of HAI-2-EYFP were compared to non-transfected cells by SDS-PAGE and western blotting. The blots were probed using the antibody anti-human HAI-2 ectodomain or antibody against EYFP (anti-EYFP). The molecular weights are in kilodalton. Results shown are representative of three independent experiments.
To ease the detection of HAI-2 we constructed an expression vector encoding a fusion protein consisting of Enhanced Yellow Fluorescent Protein (EYFP) fused to the C-terminus of HAI-2 (Fig. 1B). The constructed vector, pEYFP-N1-HAI-2, was transiently transfected into MDCK cells and the cell extracts were analyzed by SDS-PAGE and western blotting (Fig. 1C). Both the anti-human HAI-2 ectodomain antibody and the antibody JL-8 (targeting EYFP) detected proteins of approximately 50 kDa and 60 kDa, which are the expected molecular weights for full-length HAI-2-EYFP and a HAI-2-EYFP cleaved form respectively, corresponding to the same difference in molecular weight as observed for endogenous HAI-2 in Caco-2 cells. Two additional bands appear when detecting HAI-2-EYFP using the JL-8 antibody, which most likely represents additional cleavage or degradation products of HAI-2-EYFP. No immunoreactivity could be detected in non-transfected MDCK cells using the JL-8 antibody.
In all, these results suggest that the anti-human HAI-2 ectodomain antibody specifically detects HAI-2 and that the fusion protein HAI-2-EYFP was successfully expressed.
Shed matriptase can be detected in the media from all cells expressing matriptase whereas membrane-bound matriptase can only be detected when co-expressed with HAI-2, HAI-2-EYFP or HAI-1 — It has previously been reported that matriptase levels are low in the cells when exogenously expressed alone, a phenomenon that can be counteracted by co-expression of HAI-1 [21]. We therefore investigated whether HAI-2 and/or HAI-2-EYFP have similar abilities. Cell extracts of MDCK cells transiently expressing matriptase alone or expressing matriptase in combination with either wild-type HAI-2 or HAI-2-EYFP were analyzed by SDS-PAGE and western blotting (Fig. 2). Transient transfections of matriptase together with HAI-1, as well as λ-DNA (Mock) and non-transfected cells were included as controls. The protein levels of wild-type HAI-2 and HAI-2-EYFP, detected using the anti-human HAI-2 ectodomain antibody, are shown on Fig. 2A. HAI-2 immunoreactivity is clearly only present in cells co-expressing matriptase and HAI-2 (Fig. 2A, lane 5) and matriptase and HAI-2-EYFP (Fig. 2A, lane 6). The cell lysates were also analyzed for matriptase immunoreactivity using antibodies M24 and M32 detecting the non-reduced 70 kDa form of matriptase and IM1014 detecting the 30 kDa peptide indicating that activation has occurred. Cells transfected with the matriptase vector alone showed low levels of total and activated matriptase (Fig. 2B and C, lane 3), whereas no matriptase could be detected in non-transfected and Mock transfected cells, as assessed using the antibodies M24 and M32 to detect total matriptase and IM1014 to detect the catalytic domain (Fig. 2B and C, lanes 1 and 2). Cells co-transfected with a vector coding for wild-type HAI-1 resulted in detection of a higher matriptase protein level in the cell lysates when compared to cells expressing matriptase alone (Fig. 2 B and C, lane 3 and 4). Co-expression of matriptase with wild-type HAI-2 or HAI-2-EYFP resulted in almost identical matriptase protein levels as that observed for the matriptase and HAI-1 co-expression (Fig. 2 B and C, lanes 4–6). No matriptase–HAI-2 complexes could be detected when analyzing extracts by western blotting under non-reducing conditions (Fig. 2B, and data not shown). All blots were stripped and reprobed using antibodies against GAPDH to ensure that equal amounts of samples were loaded. We also analyzed the transfected canine MDCK cells for the presence of mRNA encoding human matriptase, in order to verify that the cells transfected with matriptase alone were indeed transfected. RNA was extracted from transfected cells and the content of matriptase mRNA was measured by real time RT–PCR as previously described [25]. Non-transfected and Mock transfected cells contained no or very low levels of matriptase mRNA/18S, whereas cells transfected with matriptase alone or matriptase in combination with HAI-1 or HAI-2 contained 105 fold more matriptase mRNA/18S (data not shown). No signal could be detected when reverse transcriptase was omitted. In summary, these results show that HAI-2 and HAI-2-EYFP function in a manner similar to HAI-1, resulting in a higher level of matriptase in the cells.
Fig. 2.
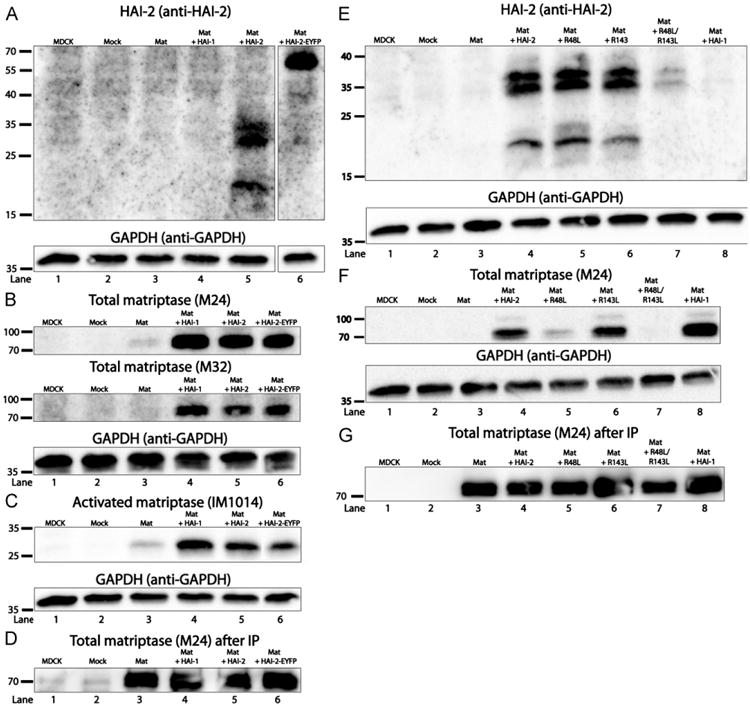
HAI-2 and HAI-2-EYFP are able to retain matriptase in a membrane-bound form. Cell extracts of MDCK cells transiently expressing matriptase alone or in combination with HAI-1, HAI-2 or HAI-2-EYFP were compared to non-transfected (MDCK) and λ-DNA (Mock) transfected cells by SDS-PAGE and western blotting. The blots were probed using the antibodies anti-human HAI-2 ectodomain, recognizing HAI-2 under reducing conditions (A), M24 and M32 detecting total matriptase under non-reducing conditions (B) and IM1014 detecting the catalytic domain of matriptase (here activated matriptase) under reducing conditions (C). The corresponding media were also collected, immunoprecipitated using antibodies against matriptase coupled to sepharose-beads and analyzed on western blots (D). Cell extracts of MDCK cells transiently expressing matriptase alone or in combination with HAI-2, HAI-2 R48L, HAI-2 R143L, and HAI-2 R48L/R143L or HAI-1 were analyzed on western blots using antibodies against HAI-2 (E) or matriptase (F). The corresponding media were also collected, immunoprecipitated and analyzed as described above (G). All blots of cell extracts were stripped and reprobed using antibodies against GAPDH. The molecular weights are in kilodalton. Results shown are representative of three independent experiments.
When matriptase is expressed alone it can either be degraded intracellularly or shed to the surroundings. To investigate whether matriptase is shed, media from MDCK cells expressing matriptase in various combinations with HAI-1, HAI-2, and HAI-2-EYFP were collected and immunoprecipitated using the antibodies against matriptase coupled to sepharose. Immunoprecipitates were analyzed using western blotting. Our analysis showed that matriptase could be detected in media from cells expressing matriptase, matriptase and HAI-1, matriptase and HAI-2, and matriptase and HAI-2-EYFP (Fig. 2D). No matriptase could be detected in media from un-transfected MDCK cells or Mock transfected MDCK cells (Fig. 2D). Similar results were obtained using the antibody M24 and M32. No HAI-1 or HAI-2 immunoreactivity could be detected in any of the immunoprecipitates (data not shown).
Kunitz domain 1 of HAI-2 is important for prevention of matriptase shedding
HAI-2 contains two inhibitory Kunitz domains. Using site-directed mutagenesis the ability of both Kunitz domains of HAI-2 to form an inhibitory complex with a serine protease was removed either individually (HAI-2 R48L and HAI-2 R143L) or simultaneously (HAI-2 R48L/R143L). In order to investigate the ability of the HAI-2 mutants to retain matriptase in the membrane-bound form, we analyzed extracts of MDCK cells transiently expressing matriptase alone or in combination with either wild-type HAI-2 or the HAI-2 mutants by SDS-PAGE and western blotting (Fig. 2E). The results showed that wild-type HAI-2, HAI-2 R48L and HAI-2 R143L was present at the same level as detected by antibodies against HAI-2 whereas HAI-2 R48L/R143L consistently was present at a lower level. The blot was re-probed with antibodies against GAPDH in order to show that equal amounts of cell lysates were loaded onto the gel (Fig. 2E). All cell extracts were also analyzed for the presence of matriptase using western blot (Fig. 2F). This analysis showed that HAI-2 R143L, wild-type HAI-2 and wild-type HAI-1 all, to a certain degree, retained matriptase in a membrane-bound form whereas HAI-2 containing the R48L mutation either alone or together with the mutation R143L showed a significantly reduced ability to prevent release of matriptase. The blot was reprobed with antibodies against GAPDH (Fig. 2F). Media were also collected and immunoprecipitated as described above. The immunopreciptates were analyzed using western blot (Fig. 2G) and showed that matriptase could be detected in all media from cells expressing matriptase, both when matriptase was expressed alone and when expressed together with HAI-2, HAI-2 R48L, HAI-2 R143L, and HAI-2 R48L/R143L, and HAI-1. Together, our results indicate that matriptase is fully shed except when co-expressed with HAI-2 carrying an intact Kunitz domain 1 or HAI-1.
Matriptase co-expressed with HAI-2 is mainly located on the plasma membrane
Recombinant matriptase expressed alone was previously shown to be located mainly intracellularly partly co-localized with the Golgi marker GM130 [21] whereas matriptase co-expressed with HAI-1 is located mainly at the plasma membrane [21,26]. To investigate whether HAI-2 has a similar effect, MDCK cells were transiently transfected with a matriptase vector alone or in combination with a vector coding for either HAI-1, HAI-2 or HAI-2-EYFP. Cells were grown for 48 h on transwell filters before fixation and immunocytochemical staining with M32 to detect matriptase and phalloidin targeting F-actin as a surface marker. Confocal microscopy showed that low levels of matriptase, located in intracellular structures, could be detected in cells transfected with the matriptase vector alone (Fig. 3A–C). It should be noted that with HAI-1, HAI-2 or HAI-2-EYFP omitted very few cells appeared to contain matriptase, which is in agreement with our previous findings (Fig. 2B, lane 3). Matriptase could be detected at the plasma membrane co-localizing with phalloidin on cells co-expressing matriptase with either HAI-1, HAI-2, or HAI-2-EYFP. Inspecting 100 cells at random expressing matriptase and HAI-1 and 100 cells expressing matriptase and HAI-2 showed in 100% of the cases a clear co-localization of matriptase and phalloidin. When inspecting cells expressing matriptase alone the intensity of staining was clearly lower and in a similar area less than 10 cells clearly expressing matriptase could be detected, of these none exhibited co-localization between phalloidin staining and matriptase staining. HAI-2-EYFP surprisingly seemed to locate mainly intracellularly (Fig. 3I–J) in cells co-expressing matriptase and HAI-2-EYFP. The indication of intracellular localization of HAI-2 was investigated further (see below). These results suggest that matriptase locates to the plasma membrane when co-expressed with HAI-2.
Fig. 3.
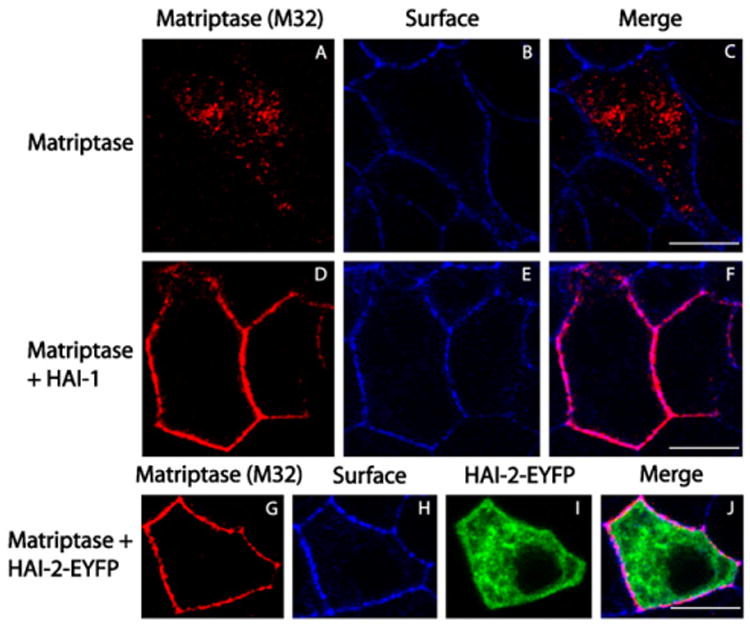
Matriptase accumulate on the plasma membrane when co-expressed with HAI-2-EYFP. MDCK cells were transiently transfected with a matriptase vector alone or in combination with a vector coding for either HAI-1 or HAI-2-EYFP, and grown for 48 h on transwell filters before fixation and immunocytochemical staining with M32 (targeting total matriptase) and the cell surface-marker phalloidin (targeting F-actin). The cells were visualized using confocal microscopy showing granular-like intracellular structures of matriptase when expressed alone (A–C) and plasma membrane localization when co-expressed with HAI-1 (D–F) or HAI-2-EYFP (G–J). (A, D and G) total matriptase (red), (B, E and H) cell surface F-actin (blue), (I) HAI-2-EYFP (green) and (C, F and J) merge. Scale bars equals 10 μm. Results shown are representative of three independent experiments.
Co-expression of matriptase with either HAI-1 or HAI-2 increases the peptidolytic activity of cell extracts
We have previously shown that the activating proteolytic cleavage of matriptase occurs when the protein is located on the basolateral plasma membrane [19]. In order to obtain some information on the effect of retaining matriptase in a membrane-bound form by the co-expression with either HAI-1 or HAI-2, we decided to test the enzymatic activity of cell extracts and media towards a peptide based chromogenic substrate, isoleucil-prolyl-arginine-p-nitroaniline. This substrate is known to be readily cleaved by a broad spectrum of serine proteases. MDCK cells were transfected with matriptase alone, matriptase and HAI-1, or matriptase and HAI-2. Transfections were also made with matriptaseS805A alone, matriptaseS805A and HAI-1, or matriptaseS805A and HAI-2 along with Mock-transfected cells in order to determine the background level of peptidolytic hydrolysis.
Strikingly, a significant increase in peptidolytic activity was observed in the cell extracts when matriptase was expressed together with HAI-1 or HAI-2 as compared to the basal level observed for HAI-1 or HAI-2 expressed alone (Fig. 4). Importantly, no substrate conversion above the Mock background was observed when matriptase was transiently expressed alone, in agreement with western blot analysis showing that almost no matriptase is present in the absence of HAI-1 or HAI-2 (Fig. 2B and C, lane 3). The addition of the trypsin inhibitor aprotinin, previously shown to be an efficient inhibitor of matriptase [27], completely reduced the activity of the extracts from matriptase expressed with HAI-1 or HAI-2 down to the Mock background (Fig. 4, the three outmost columns to the right). To support the direct involvement of the active site of matriptase in the observed activities of the extracts we expressed the catalytic inactive mutant, matriptaseS805A. Assay of matriptaseS805A, matriptaseS805A and HAI-1, or matriptaseS805A and HAI-2 all showed a rate of substrate conversion in the order of 0.2–0.4 mAU/min similar to the Mock extracts (Fig. 4). The basal turnover of substrate in the Mock extract is for the vast majority unlikely to stem from serine proteases, as addition of neither of the serine protease inhibitors PMSF, TLCK, or Soy bean trypsin inhibitor caused significant reduction (data not shown). These results indicate that the peptidolytic activity in the cell extracts increases with increasing amounts of activated matriptase, as detected by IM1014 (compare Fig. 2C, lanes 3–5 and Fig. 4). We were unable to detect peptidolytic activity in the media or immunoprecipitates of media generated as described above even after prolonged incubation for up to 30 h.
Fig. 4.
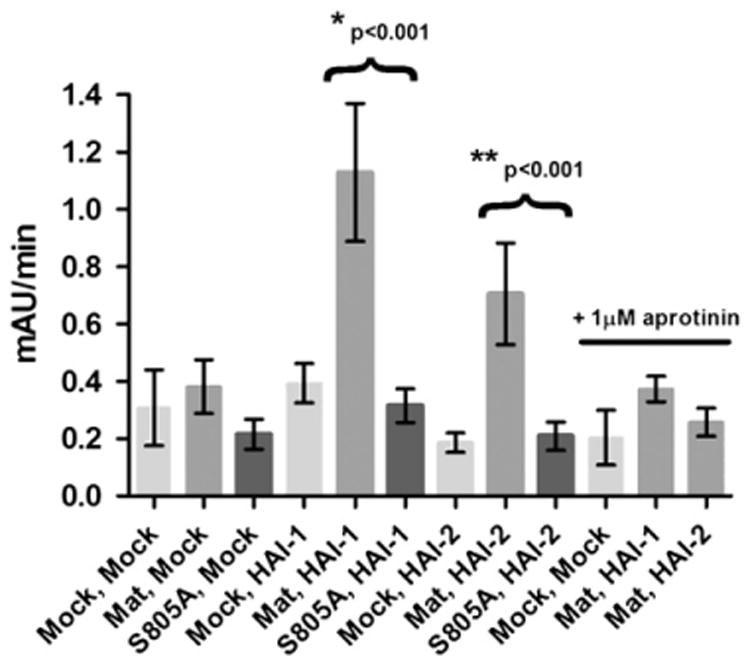
Enzymatic activity in cell extracts. Each column represents the rate of turnover of a chromogenic substrate in the extracts from the indicated double transfected cells (x-axis). From left to right, columns 1–3, columns 4–6 and columns 7–9 represent Mock, HAI-1 and HAI-2, respectively, co-transfected with Mock (light gray columns), native matriptase (Mat, medium gray columns) and as a control matriptase S805A (S805A, dark gray columns). The last three columns to the right represent Mock and matriptase with HAI-1 or HAI-2 in the presence of 1 μM of the matriptase inhibitor aprotinin. Each column represents the mean value for the rate of turnover of a chromogenic substrate from at least three independent determinations from three independent transfections with the corresponding standard deviation. Significant increases in rate above the respective Mock control are indicated with the corresponding p values by Student's t-test. N=3.
The majority of HAI-2 and HAI-2-EYFP are located intracellularly
Matriptase is activated on the basolateral plasma membrane [19]. We therefore expected HAI-2, presuming it works as a matriptase inhibitor, to be located there as well. The genetic engineered fusion protein HAI-2-EYFP is located intracellularly, but may behave as a misfolded protein retained in the endoplasmatic reticulum (ER), we therefore investigate the subcellular localization of wild-type HAI-2. For this purpose Caco-2 cells, expressing HAI-2 endogenously, were grown to 11-days post confluence, at which time a tight polarized monolayer of cells have formed. Membrane-bound proteins were labeled from either the apical or the basolateral side using a non-membrane permeable biotinylation reagent, and the biotinylated proteins were isolated using streptavidin pull-down. The isolated proteins were then compared to non-biotinylated proteins found in the corresponding flow through fractions and to a Caco-2 cell extract by SDS-PAGE and western blotting, using the anti-human HAI-2 ectodomain antibody (Fig. 5A). The results show that small amounts of HAI-2 can be detected amongst the proteins biotinylated from the apical side of the Caco-2 cells (Fig. 5A, lane 1). However HAI-2 could mainly be detected in the flow through fractions. As the biotin used in these experiments is non-permeable to the plasma membrane this indicates that the majority of HAI-2 is located intracellularly. Overexposure of the apical and basolateral lanes confirmed the result (Fig. 5A, lanes 6 and 7).
Fig. 5.
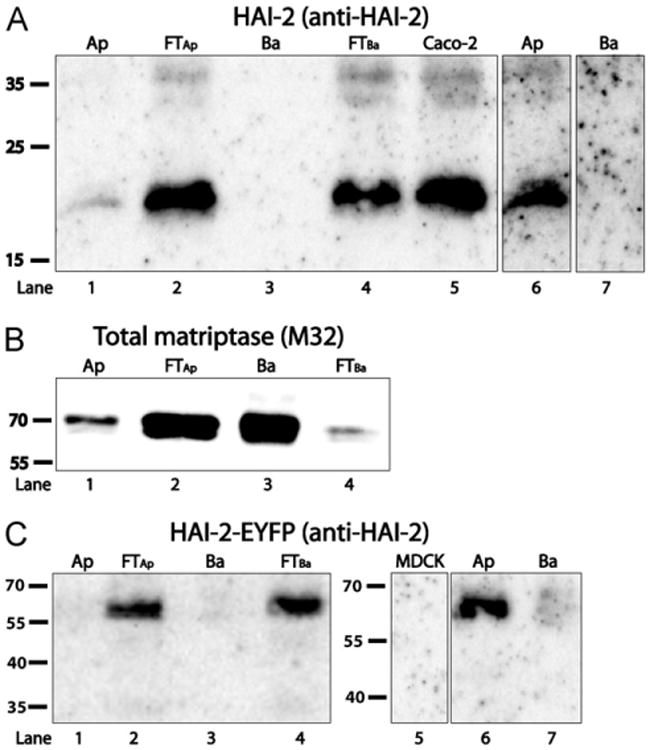
HAI-2 and HAI-2-EYFP can be detected on the apical plasma membrane, but the majority is located intracellularly. Caco-2 cells were grown on transwell filters to 11-days post-confluence and biotinylated on either the apical (Ap) or the basolateral (Ba) side. The apical and basolateral protein pull-downs were then compared with their respective flow through fractions (FTs) and to an extract of Caco-2 cells by SDS-PAGE and western blotting, using the anti-HAI-2 ectodomain antibody for detection (A). An over-exposure of the apical and basolateral lanes is also shown (A, lane 6 and 7). Lane 1 and 6 thus show the same blot, with different exposure times and similarly lane 3 and 7 shows the same blot with different exposure times (B) For comparison, the pull-downs as well as the FTs were analyzed by SDS-PAGE and western blotting using the M32 antibody for detection of total matriptase. (C) A similar biotinylation experiment performed on MDCK cells stably expressing HAI-2-EYFP was analyzed by SDS–PAGE and western blotting, using the anti-HAI-2 ectodomain antibody for detection. Overexposure of the apical and basolateral pull-downs compared to a cell extract of non-transfected MDCK cells is shown on the right (C, lanes 5–7). Lanes 5–7 are from the same gel. All molecular weights are in kilodalton. Results shown are representative of three independent experiments.
Monomeric avidin pull-down was performed on a parallel set of samples and the blots probed with the M32 monoclonal antibody, detecting total matriptase (Fig. 5B). Monomeric avidin was used since bound material can be released with excess biotin, as M32 only recognize matriptase under non-reducing conditions. The experiment shows that matriptase mainly locates to the basolateral plasma membrane, confirming that basolateral plasma membrane proteins were efficiently biotinylated under these conditions (Fig. 5B, lane 3).
To further investigate if the fusion protein HAI-2-EYFP behaved in a manner similar to wild-type HAI-2, MDCK cells stably expressing HAI-2-EYFP were grown to 3-days post confluence on transwell filters and a biotinylation procedure identical to the one described above was repeated. As for endogenous HAI-2 this experiment showed that HAI-2-EYFP was predominantly found in the flow through fractions (Fig. 5C, lane 1–4). An over exposure revealed a minor amount of HAI-2-EYFP on the apical plasma membrane (Fig. 5C, lane 6).
These results suggest that HAI-2 is located intracellularly, except for a minor fraction located on the apical plasma membrane.
The majority of intracellular HAI-2-EYFP is found in the endoplasmic reticulum
To investigate the subcellular localization of HAI-2 in more detail, MDCK cells stably expressing HAI-2-EYFP were seeded on transwell filters and transiently transfected with the plasmid vector pDsRed2-ER, expressing a fluorescence fusion protein fused to both the ER retention sequence KDEL and to the ER targeting sequence of calreticulin. When reaching three days of post-confluence the cells were fixed and stained with the surface-marker phalloidin. Confocal microscopy of the cells showed that the majority of HAI-2-EYFP co-localized with the ER marker (Fig. 6). In this setup, we were unable to detect HAI-2-EYFP at the apical plasma membrane.
Fig. 6.
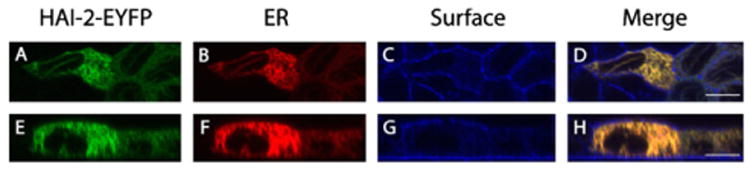
The majority of intracellular HAI-2-EYFP is found in the ER. MDCK cells with a stable expression of HAI-2-EYFP were transiently transfected with the vector pDsRed2-ER, expressing an ER marker, and grown on transwell filters to three days post-confluence. The cells were then fixed, stained with the surface-marker phalloidin (targeting F-actin) and visualized using confocal microscopy. Top view (A–D) and side view (E–H) of fixed cells showing HAI-2-EYFP localization to the ER. (A and E) HAI-2-EYFP (green), (B and F) ER (red), (C and G) surface F-actin (blue) and (D and H) merge. Scale bars equals 10 μm. Results shown are representative of two independent experiments.
These results strongly indicate that the majority of intracellular HAI-2-EYFP is found in the ER.
Endogenously expressed HAI-2 in the large intestines is also located intracellularly
As the fusion protein HAI-2-EYFP may be mislocated to the ER due to misfolding we wanted to investigate the intracellular localization of HAI-2 in a more physiological setting. Therefore human colon tissue samples were immunohistochemically stained using the antibody anti human HAI-2 ectodomain. Both epithelial colonocytes (data not shown) and crypt cells showed strong staining for HAI-2, which was clearly located intracellularly (Fig. 7A, arrows). Also a colon carcinoma tissue sample was immunostained and cells with cancerous morphology showed clear intracellular staining (Fig. 7B, arrows). These results show that HAI-2 localizes intracellularly when endogenously expressed, in both normal and malignant tissues. This is in agreement with our other results using biotinylation techniques and the HAI-2-EYFP fusion protein.
Fig. 7.
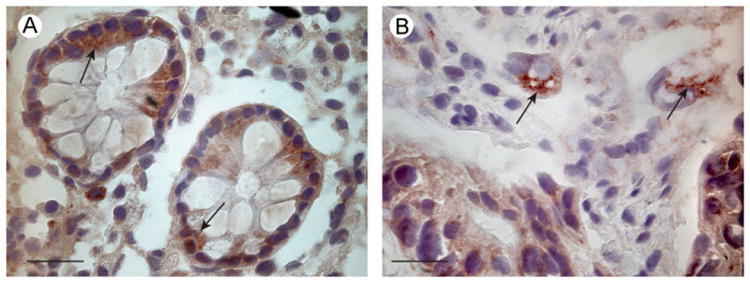
Immunohistochemical staining of human colon tissue sections show intracellular distribution of HAI-2. Colon tissue sections were stained using the anti-HAI-2 ectodomain antibody. (A) Staining of normal tissue showed a distinct intracellular signal in epithelial cells, as indicated by arrows. (B) Colorectal cancer tissue from the same individual also showed an intracellular distribution of HAI-2. Arrows indicate examples of cells with cancerous morphologs profiles. Scale bars equals 20 μm.
HAI-2 is not endocytosed from the apical membrane
Proteins primarily located in the ER often contain retrieval signals, implying that they are constantly routed back to the ER from locations further down the secretory pathway. We therefore investigated whether HAI-2 on the apical plasma membrane is a transient pool which recycles back to the ER using surface protein biotinylation. The biotinylation reagent, sulfo-NHS-SS-biotin, used in this experiment consists of a protein binding NHS group, linked via a reducible disulfide bond to a biotin group, and is consequently susceptible to reduction. The non-membrane permeable reducing agent, glutathione was used for reduction. To assay for possible endocytosis biotinylated Caco-2 cells were chased for a given amount of time, at which point they were exposed to reducing conditions, cleaving the linker and releasing the biotin group. Biotinylated proteins pulled down after reduction are indicative of endocytosis, as endocytosed proteins avoid reduction. In this experiment cells were incubated over a period of 0–180 min at 37 °C and thereafter exposed twice to glutathione, followed by streptavidin pull-down and analysis using SDS-PAGE and western blotting. A parallel set of samples not exposed to glutathione, representing the total amount of biotinylated proteins was included as a control.
Within the time frame used in this experiment no endocytosis of HAI-2 could be detected, as is clearly seen by the complete removal of all biotinylated HAI-2 following glutathione reduction (Fig. 8). The lower molecular weight form of HAI-2 was the predominant one on the apical plasma membrane (Fig. 8). Full-length HAI-2 was only seen on overexposed blots and no endocytosis could be detected (Fig. 8 and data not shown). As a control the blot was stripped and re-probed using antibodies against the transferrin receptor which is known to be endocytosed from the plasma membrane. The blot showed that the transferrin receptor is slowly endocytosed from the apical plasma membrane (Fig. 8). These results indicate that neither of the two forms of HAI-2 were endocytosed within the time frame of the experiment.
Fig. 8.
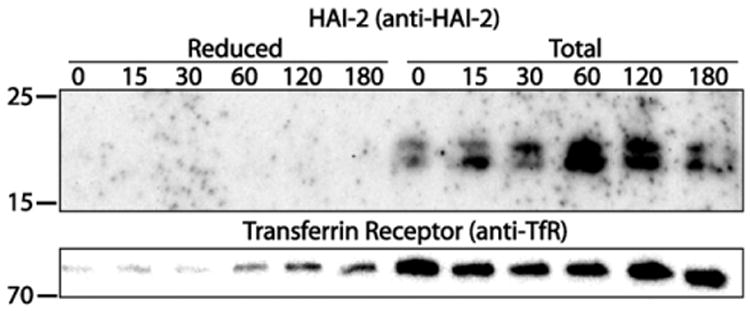
HAI-2 is not endocytosed from the apical membrane. Caco-2 cells were grown on transwell filters to 11-days post-confluence and biotinylated at the apical side, before incubation at 37°C to the time points (min) indicated on the figure. The apical surfaces were next either treated or not treated with reducing glutathione, resulting in samples that were stripped (reduced) and non-stripped (total) respectively of the surface bound S-NHS-SS biotin. Biotinylated proteins were then pulled down and analyzed by SDS-PAGE and western blotting, using the anti-HAI-2 ectodomain antibody for detection (upper panel). The blot was subsequently stripped and reprobed using antibodies against the transferrin receptor (lower panel). HAI-2 was not detected in the reduced samples, whereas HAI-2, predominantly in the cleaved form, was detected in the total non-reduced samples. No endocytosis of HAI-2 can thus be detected whereas the transferrin receptor is endocytosed. The molecular weights are in kDa. Results shown are representative of three independent experiments.
Discussion
When human matriptase was recombinantly expressed alone in canine MDCK cells the presence of human matriptase mRNA could be readily detected whereas we could hardly detect matriptase in the cell extracts. However, shed matriptase could be detected in the media. Matriptase was detected intracellularly in a few of these cells where matriptase levels were highest (Fig. 3A). This suggests that matriptase expressed alone is rapidly transported though the secretory pathway and shed to the surroundings—resulting in a steady state level in the cells below the detection limit of the antibodies.
In contrast to that, co-expression of matriptase with HAI-1 or HAI-2 causes matriptase to accumulate on the plasma membrane co-localizing with the surface marker phalloidin. Our results indicate that Kunitz domain 1 of HAI-2 is responsible for this activity of HAI-2 and that HAI-1 has the same ability. It has previously been shown that mutagenesis of Kunitz domain 1 of HAI-1 in a similar manner renders it unable to make matriptase accumulate on the plasma membrane [21].
The level of shed matriptase appeared to be the same in all scenarios despite a significant amount being retained in the cell extracts when the inhibitors were co-expressed with functional Kunitz domains (Fig. 2B and D, F and G). This is unlikely to be the result of an increased expression level of matriptase in cells co-transfected with HAI-2 or HAI-1 since matriptase mRNA levels are similar. Another possibility is that the HAI's serves as a checkpoint or gateway to alter the turnover into the media and retain a certain level of the protease. In this way the cellularly retained fraction might have accumulated over time with a slower turnover rate, while the shed matriptase could undergo breakdown on a relatively rapid scale masking the difference.
Protease inhibitors like HAI-1 and HAI-2 are thus via their Kunitz domain 1, able to reduce release of the ectodomain of matriptase. The most likely explanation for this is that both HAI-1 and HAI-2 are able to inhibit proteolytic release of matriptase. This proteolysis may takes place in the secretory pathway as no HAI-2 could be detected on the basolateral plasma membrane co-locating with matriptase. Matriptase may resemble corin in this respect as corin is also shed from the cells as a consequence of proteolytic cleavage [28]. Addition of protease inhibitors was able to abolish proteolytic release of corin [28]. We performed a similar experiment but in our system we were unable to prevent release of matriptase using a variety of protease inhibitors added to the media (A. Nonboe and L.K. Vogel, unpublished results) which may speak in favor of intracellular shedding of matriptase. Shed matriptase could also be detected when matriptase S805A was expressed alone and when matriptase S805A was co-expressed with HAI-1 and HAI-2 (A. Nonboe and L.K. Vogel, data not shown) suggesting that the shedding of matriptase is not caused by the proteolytic activity of matriptase. More investigations are needed to elucidate the mechanism resulting in the shedding of matriptase.
Alternatively, the shedding of matriptase may be due to the cleavage in the SEA domain taking place in the secretory pathway. As a consequence hereof, matriptase is only attached to the membrane by non-covalent forces that may not be strong. The SEA domain may thus spontaneously dissociate in the secretory pathway except when matriptase has interacted with a membrane-bound HAI. We find this unlikely as no HAI-2 could be detected on the plasma membrane co-locating with matriptase which one would expect if HAIs serve to tether matriptase to the membrane.
It has been shown, that matriptase and HAI-2 interact directly in vitro [4]. They are therefore likely to also interact directly when co-localized in vivo. However, as HAI-2 seems to be absent from the basolateral plasma membrane where active matriptase resides we hypothesize that interaction between matriptase and HAI-2 takes place intracellularly most likely in the ER.
To examine the effect of increasing level of membrane-bound matriptase, we decided to measure the enzymatic activity in cell extracts from cells transiently expressing matriptase and its inhibitors. The increased enzymatic activity in the cell extract where matriptase is co-transfected with either HAI-1 or HAI-2 seems to stem from a serine protease as the activity is blocked by the presence of a range of serine proteases inhibitors including aprotinin. That the increase in enzymatic activity in the cell extracts seems to stem directly from or at least requires the presence of activated matriptase is supported by the observation that no increase in activity was observed when expressing the catalytic inactive matriptaseS805A with or without the co-expression of HAI-1 and/or HAI-2. As no specific matriptase inhibitor is yet commonly available, we cannot rule out that other proteases activated by matriptase, and not matriptase itself, is responsible for the observed enzymatic activity. The preparation of cell extracts could cause matriptase-inhibitor complexes to dissociate resulting in increasing enzymatic activity. However, we have previously purified matriptase-HAI-1 complexes through several steps of affinity purification but not observed any dissociation [19]. It is safe to conclude that the observed increase in enzymatic activity in the extracts follows the increasing amount of matriptase protein and results, either directly or indirectly, from the catalytic activity of matriptase. It is highly likely that the biological activity of matriptase is inherent in its catalytic activity. It is therefore interesting that it seems to be the membrane-bound form of matriptase and not the shed form that is catalytically active.
The present study shows that matriptase is subject to several mechanisms controlling its subcellular localization and ensuring that active matriptase is co-expressed with a suitable inhibitor. The reason for the tight control of matriptase activity is emphasized by the fact that uncontrolled matriptase activity may lead to severe aberrant morphogenesis or carcinogenesis.
Conclusion
Our results suggest that Kunitz domain 1 of HAI-2 is necessary for accumulation of membrane-bound matriptase on the basolateral plasma membrane, where it is subsequently activated. Furthermore, matriptase seems to be rapidly transported down the secretory pathway and shed if expressed without HAI-2 or HAI-1.
Acknowledgments
Funding: This work was supported by The Harboe Foundation, The Brothers Hartmanns Foundation, The A.P. Møllers Foundation for the Advancement of Medical Science, The Lundbeck Foundation, Architect Holger Hjortenberg and Hustru Dagmar Hjortenbergs Foundation, Else and Mogens Wedell-Wedellsborg Foundation, Aage and Johanne Louis-Hansens Foundation, The NIDCR Intramural Programs and The Danish Cancer Society.
Abbreviations
- CHO
Chinese hamster ovary
- ECL®
enhanced chemiluminescence
- ER
endoplasmic reticulum
- EYFP
enhanced yellow fluorescent protein
- FT
flow through fraction
- HAI-1
hepatocyte growth factor activator inhibitor-1
- HAI-2
hepatocyte growth factor activator inhibitor-2
- MDCK
Madin-Darby canine kidney
- RT
room temperature
References
- 1.Kawaguchi T, Qin L, Shimomura T, Kondo J, Matsumoto K, Denda K, Kitamura N. J Biol Chem. 1997;272:27558–27564. doi: 10.1074/jbc.272.44.27558. [DOI] [PubMed] [Google Scholar]
- 2.Marlor CW, Delaria KA, Davis G, Muller DK, Greve JM, Tamburini PP. J Biol Chem. 1997;272:12202–12208. doi: 10.1074/jbc.272.18.12202. [DOI] [PubMed] [Google Scholar]
- 3.Szabo R, Hobson JP, Christoph K, Kosa P, List K, Bugge TH. Development. 2009;136:2653–2663. doi: 10.1242/dev.038430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szabo R, Hobson JP, List K, Molinolo A, Lin CY, Bugge TH. J Biol Chem. 2008;283:29495–29504. doi: 10.1074/jbc.M801970200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P, Leighton PA, Goodrich LV, Lu X, Avery BJ, Tate P, Dill K, Pangilinan E, Wakenight P, Tessier-Lavigne M, Skarnes WC. Nat Genet. 2001;28:241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- 6.Szabo R, Uzzun SK, Kosa P, Shylo NA, Godiksen S, Hansen KK, Friis S, Gutkind JS, Vogel LK, Hummler E, Camerer E, Bugge TH. PLOS Genet. 2012;8:e1002937. doi: 10.1371/journal.pgen.1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinz-Erian P, Muller T, Krabichler B, Schranz M, Becker C, Ruschendorf F, Nurnberg P, Rossier B, Vujic M, Booth IW, Holmberg C, Wijmenga C, Grigelioniene G, Kneepkens CM, Rosipal S, Mistrik M, Kappler M, Michaud L, Doczy LC, Siu VM, Krantz M, Zoller H, Utermann G, Janecke AR. Am J Hum Genet. 2009;84:188–196. doi: 10.1016/j.ajhg.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.List K, Haudenschild CC, Szabo R, Chen W, Wahl SM, Swaim W, Engelholm LH, Behrendt N, Bugge TH. Oncogene. 2002;21:3765–3779. doi: 10.1038/sj.onc.1205502. [DOI] [PubMed] [Google Scholar]
- 9.List K, Kosa P, Szabo R, Bey AL, Wang CB, Molinolo A, Bugge TH. Am J Pathol. 2009;175:1453–1463. doi: 10.2353/ajpath.2009.090240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basel-Vanagaite L, Attia R, Ishida-Yamamoto A, Rainshtein L, Ben AD, Lurie R, Pasmanik-Chor M, Indelman M, Zvulunov A, Saban S, Magal N, Sprecher E, Shohat M. Am J Hum Genet. 2007;80:467–477. doi: 10.1086/512487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.List K, Szabo R, Molinolo A, Sriuranpong V, Redeye V, Murdock T, Burke B, Nielsen BS, Gutkind JS, Bugge TH. Genes Dev. 2005;19:1934–1950. doi: 10.1101/gad.1300705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin CY, Anders J, Johnson M, Dickson RB. J Biol Chem. 1999;274:18237–18242. doi: 10.1074/jbc.274.26.18237. [DOI] [PubMed] [Google Scholar]
- 13.Szabo R, Molinolo A, List K, Bugge TH. Oncogene. 2007;26:1546–1556. doi: 10.1038/sj.onc.1209966. [DOI] [PubMed] [Google Scholar]
- 14.Wang JK, Lee MS, Tseng IC, Chou FP, Chen YW, Fulton A, Lee HS, Chen CJ, Johnson MD, Lin CY. Am J Physiol Cell Physiol. 2009;297:C459–C470. doi: 10.1152/ajpcell.00201.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oberst M, Anders J, Xie B, Singh B, Ossandon M, Johnson M, Dickson RB, Lin CY. Am J Pathol. 2001;158:1301–1311. doi: 10.1016/S0002-9440(10)64081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oberst MD, Singh B, Ozdemirli M, Dickson RB, Johnson MD, Lin CY. J Histochem Cytochem. 2003;51:1017–1025. doi: 10.1177/002215540305100805. [DOI] [PubMed] [Google Scholar]
- 17.List K, Szabo R, Molinolo A, Nielsen BS, Bugge TH. Am J Pathol. 2006;168:1513–1525. doi: 10.2353/ajpath.2006.051071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuzuki S, Murai N, Miyake Y, Inouye K, Hirayasu H, Iwanaga T, Fushiki T. Biochem J. 2005;388:679–687. doi: 10.1042/BJ20041639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friis S, Godiksen S, Bornholdt J, Selzer-Plon J, Rasmussen HB, Bugge TH, Lin CY, Vogel LK. J Biol Chem. 2011;286:5793–5802. doi: 10.1074/jbc.M110.186874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godiksen S, Selzer-Plon J, Pedersen ED, Abell K, Rasmussen HB, Szabo R, Bugge TH, Vogel LK. Biochem J. 2008;413:251–259. doi: 10.1042/BJ20071496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oberst MD, Chen LY, Kiyomiya K, Williams CA, Lee MS, Johnson MD, Dickson RB, Lin CY. Am J Physiol Cell Physiol. 2005;289:C462–C470. doi: 10.1152/ajpcell.00076.2005. [DOI] [PubMed] [Google Scholar]
- 22.Lin CY, Tseng IC, Chou FP, Su SF, Chen YW, Johnson MD, Dickson RB. Front Biosci. 2008;13:621–635. doi: 10.2741/2707. [DOI] [PubMed] [Google Scholar]
- 23.Lin CY, Wang JK, Torri J, Dou L, Sang QA, Dickson RB. J Biol Chem. 1997;272:9147–9152. [PubMed] [Google Scholar]
- 24.Vogel LK, Spiess M, Sjostrom H, Noren O. J Biol Chem. 1992;267:2794–2797. [PubMed] [Google Scholar]
- 25.Vogel LK, Saebo M, Skjelbred CF, Abell K, Pedersen EDK, Vogel U, Kure EH. BMC Cancer. 2006;6:176. doi: 10.1186/1471-2407-6-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogel LK, Godriksen S. Thromb Haemostasis. 2005;93:A17. [Google Scholar]
- 27.Yamasaki Y, Satomi S, Murai N, Tsuzuki S, Fushiki T. J Nutr Sci Vitaminol (Tokyo) 2003;49:27–32. doi: 10.3177/jnsv.49.27. [DOI] [PubMed] [Google Scholar]
- 28.Jiang J, Wu S, Wang W, Chen S, Peng J, Zhang X, Wu Q. J Biol Chem. 2011;286:10066–10072. doi: 10.1074/jbc.M110.185082. [DOI] [PMC free article] [PubMed] [Google Scholar]