

Abstract
Purpose
Perivascular epithelioid cell tumors (PEComas) represent a family of mesenchymal neoplasms, mechanistically linked through activation of the mTOR signaling pathway. There is no known effective therapy for PEComa, and the molecular pathophysiology of aberrant mTOR signaling provided us with a scientific rationale to target this pathway therapeutically. On this mechanistic basis, we treated three consecutive patients with metastatic PEComa with an oral mTOR inhibitor, sirolimus.
Patients and Methods
Patients with advanced PEComa were treated with sirolimus and consented to retrospective collection of data from their medical records and analysis of archival tumor specimens. Tumor response was determined by computed tomography scans obtained at the clinical discretion of the treating physicians. Tumors were assessed for immunohistochemical evidence of mTORC1 activation and genetic evidence of alterations in TSC1 and TSC2.
Results
Radiographic responses to sirolimus were observed in all patients. PEComas demonstrated loss of TSC2 protein expression and evidence of baseline mTORC1 activation. Homozygous loss of TSC1 was identified in one PEComa.
Conclusion
Inhibition of mTORC1, pathologically activated by loss of the TSC1/TSC2 tumor suppressor complex, is a rational mechanistic target for therapy in PEComas. The clinical activity of sirolimus in PEComa additionally strengthens the pathobiologic similarities linking PEComas to other neoplasms related to the tuberous sclerosis complex.
INTRODUCTION
Sarcomas are a heterogeneous collection of tumors sharing a common mesenchymal origin. Historically, treatment studies of pooled subtypes of sarcoma have shown overall poor responses to conventional chemotherapeutic agents.1 However, in recent years, identification of molecular subtypes of sarcoma has led to the application of effective targeted therapies, such as imatinib mesylate for the treatment of gastrointestinal stromal tumors that usually harbor activating mutations in the KIT receptor tyrosine kinase. Identification of molecular alterations in other sarcoma subtypes may lead to more effective therapies for this otherwise difficult-to-treat group of diseases.
The perivascular epithelioid cell tumor (PEComa) family of tumors consists of related mesenchymal neoplasms that exhibit myomelanocytic differentiation and share a distinctive cell type, the perivascular epithelioid cell, or PEC.2–4 The major members of this family include lymphangioleiomyomatosis (LAM), a disease predominantly presenting as numerous nodular and interstitial pulmonary lesions in premenopausal women; angiomyolipoma (AML), commonly identified as an asymptomatic renal lesion with evidence of vascular, muscle, and adipocytic differentiation; and PEComa, an epithelioid malignancy with clear-to-granular eosinophilic cytoplasm typically arising in the gastrointestinal tract, retroperitoneum, uterus, or somatic soft tissues, composed of nests and sheets of epithelioid or occasionally spindled cells, intimately related to blood vessel walls.
Tumors of the PEComa family are rare and usually occur sporadically. LAM and AML also are seen at high frequency in patients with tuberous sclerosis complex (TSC), a disorder caused by mutation of TSC1 or TSC2, for which the gene products negatively regulate mTORC1 through inhibition of the mTOR kinase activator RHEB. Recently Bissler et al5 described modest, transient improvement in lung function and reduction in size of AML in a trial of 25 patients with LAM and AML treated with sirolimus, an inhibitor of mTORC1. PEComas have rarely been reported in association with TSC.6
Most PEComas are benign tumors and do not recur after complete surgical resection. However, a subset of PEComas exhibit malignant behavior, with either locally invasive recurrences or development of distant metastases, most commonly in the lung. No effective therapy for malignant PEComa has been described. Because PEComas share activation of the mTOR pathway with LAM and AML in many instances,6,7 we treated three consecutive patients at two institutions with sirolimus.
PATIENTS AND METHODS
Patient Selection, Treatment, and Clinical Assessments
Three consecutive patients with PEComa who presented to Dana-Farber Cancer Institute or Memorial Sloan-Kettering Cancer Center were offered off-label treatment with sirolimus by their treating physicians. None of the patients exhibited clinical signs or a history of TSC. There was no significant family history of other cancers in any of the index patients. All patients provided informed consent for treatment with sirolimus as well as retrospective review of medical records and evaluation of archival tumor specimens according to institutional review board–approved protocols. The dosage of sirolimus (range, 1 mg every other day to 8 mg daily) was determined by the treating physician and was adjusted on the basis of trough sirolimus levels or patient tolerance. Disease status was assessed by CT scans at baseline and at intervals recommended by the treating physician. [18F]fluorodeoxyglucose–positron emission tomography scans were not performed as part of tumor assessment.
Histologic and Immunohistochemical Evaluations
Hemotoxylin and eosin staining and immunohistochemical stains of sections of archival formalin-fixed paraffin-embedded tissue were performed according to standard techniques for routine clinical pathologic evaluation. For additional immunohistochemical analyses, slides were processed through a xylene and ethanol series, subjected to Dako Antigen Retrieval procedure (Dako, Carpinteria, CA), and stained with a monoclonal mouse antiphospho-S6 antibody (S240/244, clone DAK-S6-240; Dako, Carpinteria, CA) or a polyclonal rabbit anti-TSC2 antibody (gift of Vijaya Ramesh, Massachusetts General Hospital, Boston, MA) followed by horseradish peroxidase detection.
Genetic Analyses
Genomic DNA was extracted from paraffin-embedded tumor tissue or peripheral-blood mononuclear cells and was assessed for copy number changes by mulitplex ligation-dependent probe amplification (MLPA) with nine exonic probes for TSC1 and TSC2, respectively, and five control probes from other genomic locations.8 Signals were normalized to values obtained with DNA extracted from control tissues. A value of 1 represented two copies of the assayed gene, 0.5 represented one copy, and 0 represented loss of both copies.
Fluorescence in situ hybridization (FISH) was performed by using interphase nuclei isolated from archival paraffin-embedded tissue and fosmid DNA probes corresponding to TSC1 (G248P87270H4 and G248P87503E5) and TSC2 (G248P89119C12 and G248P84443G9) with 4′,6-diamidino-2-phenylindole counterstaining, according to standard methods.9 Genomic DNA from PEComas was amplified for PIK3CA (exons 10 and 21), AKT1 (exon 3), and KRAS (exons 2 and 3), and then was sequenced by standard dideoxy sequencing.
RESULTS
Patient outcomes with systemic therapy are summarized in narrative format in this Results section. Appendix Table A1 indicates the outcomes of systemic therapy for index patients as well.
Patient 1
Patient 1 is a 65-year-old man who underwent resection of a 20-cm retroperitoneal PEComa in 2005, complicated by preoperative tumor rupture. Multifocal retroperitoneal recurrences were noted on surveillance imaging, and all sites of disease were resected in 2007. Additional sites of disease were noted on surveillance scans 3 months later.
He participated in a phase I study of an oral inhibitor of MET and developed rapid disease progression. In 2008, he began treatment with sirolimus 8 mg daily and achieved a serum trough level of 36 ng/mL; sirolimus has been well tolerated except for mild fatigue. Significant reduction in size of all tumors was noted on serial CT scans (Fig 1A), with near complete disappearance at 1 year. Both treatment and response are ongoing currently at 16 months of follow-up.
Fig 1.
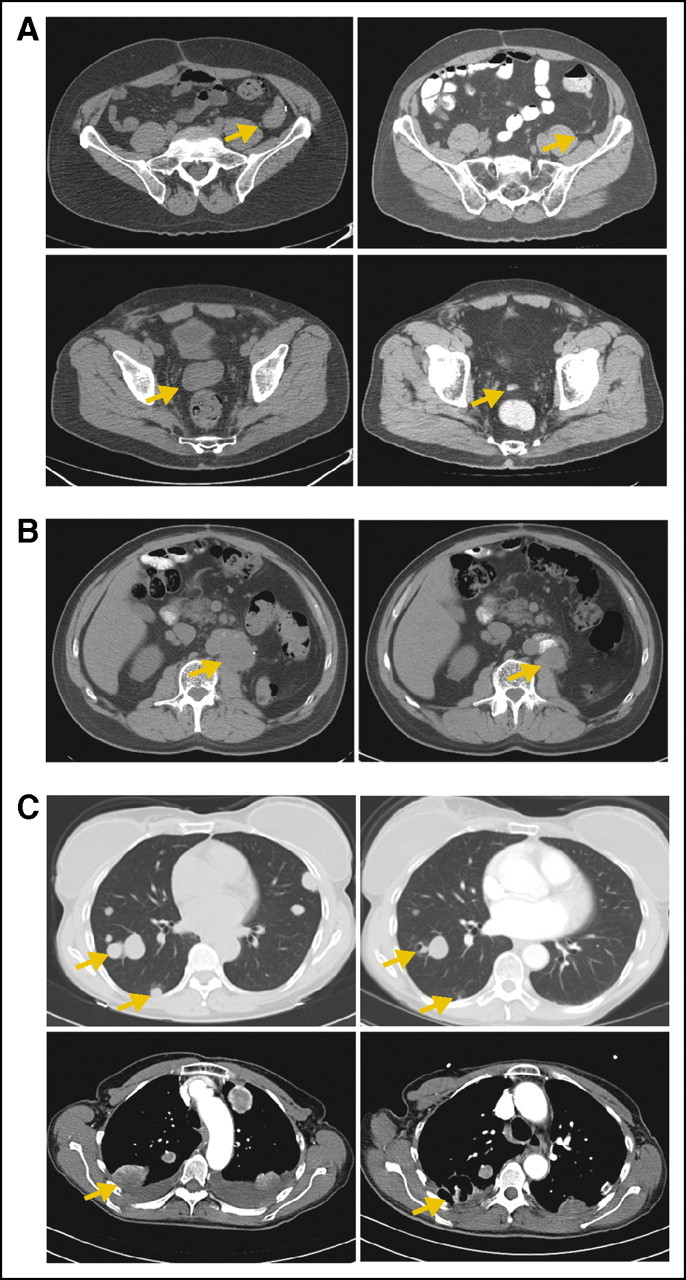
Computed tomography images of perivascular epithelioid cell tumors (PEComas; left panels) at baseline and (right panels) after treatment with sirolimus. All images are chosen to show the maximal size of the lesions indicated by arrows; other lesions may be out of phase in the two studies. (A) Retroperitoneal PEComa before and after 1 year of treatment. (B) Renal PEComa before and after 9 months of treatment. (C, top) Metastatic uterine PEComa in lung before and after 6 weeks of treatment; some lesions are improved, whereas others remain stable. (C, bottom, left) Before and (right) 3 weeks after treatment with sorafenib.
Patient 2
Patient 2 is a 70-year-old man who presented with hematuria in 2001. A radical nephrectomy was performed for removal of a 9-cm renal mass, and the pathology was initially interpreted as a poorly differentiated sarcomatoid variant of clear cell carcinoma. Five years later, a local recurrence was detected and resected. One year later, the patient developed flank pain and again was noted to have developed a locoregional recurrence. Treatment with sunitinib was initiated but was stopped with evidence of disease progression after 6 weeks.
In 2008, the patient presented to one of our institutions, and the pathologic diagnosis was reclassified as PEComa. Sirolimus 4 mg daily was initiated but was complicated by diarrhea and fatigue. The dose was reduced to 1 mg every other day, and improvement in the adverse effect profile and reduction in flank pain were noted. Restaging studies demonstrated reduction in size of the tumor (Fig 1B), with 40% reduction in the longest diameter and continued disease control for 10 months before evidence of progression despite a sirolimus level of 9.4 ng/mL. He subsequently had an additional surgical resection and remains alive with disease.
Patient 3
Patient 3 was a 61-year-old woman who presented with uterine bleeding in 2007. A total abdominal hysterectomy was performed, which identified a 9-cm PEComa with malignant features arising from the cervix. Staging studies demonstrated numerous bilateral pulmonary metastases. Sirolimus 4 mg orally each day was initiated. Restaging studies performed after 6 weeks of treatment revealed interval reduction in size and central cavitation of most of the pulmonary nodules (Fig 1C, top). Repeat evaluation at 3 months demonstrated significant progression of disease; a serum trough sirolimus level was 5 ng/mL. The dose of sirolimus was increased to 8 mg daily, but restaging studies 1 month later showed additional progression of disease, and the serum trough sirolimus level was 7 ng/mL. The sirolimus dose was decreased to 2 mg daily, and clarithromycin 500 mg daily was added to inhibit CYP3A4, the major enzyme responsible for metabolism of sirolimus. The serum trough sirolimus level increased to 20 ng/mL, and CT scans 1 month later demonstrated stabilization of the majority of lesions and interval reduction in the size of some nodules. Additional progression of disease was noted 1 month later, and treatment was empirically changed to sorafenib 200 mg twice daily and sirolimus 4 mg daily. Significant cavitation of lung masses developed within 2 weeks (Fig 1C, bottom) with improvement in symptoms and obviation of the need for supplemental oxygen, although bilateral pneumothoraces also developed. After 2 additional months of disease control, symptomatic worsening of pulmonary parenchymal disease ensued, and the patient died.
Correlative Studies
Hematoxylin and eosin–stained tumor sections showed sheets of epithelioid cells with granular eosinophilic cytoplasm, intimately associated with blood vessels, and with frequent mitoses, pleomorphism, and necrosis suggestive of malignant behavior (Fig 2A). Immunohistochemical stains were positive for melanocytic markers HMB45 and Melan-A, variably positive for smooth muscle actin and desmin, and negative for cytokeratins and S100 (not shown). Specimens from all three patients demonstrated strong, diffuse, cytoplasmic staining for phosphorylated S6 protein (Fig 2B) consistent with activation of mTORC1. Lesional cells showed loss of expression of tuberin, the gene product of TSC2, whereas expression generally was maintained in normal vessel wall vascular smooth muscle cells (Fig 2C). The specificity of the anti-TSC2 antibody was confirmed in TSC2-null and -expressing cell lines (Appendix Fig A1).
Fig 2.
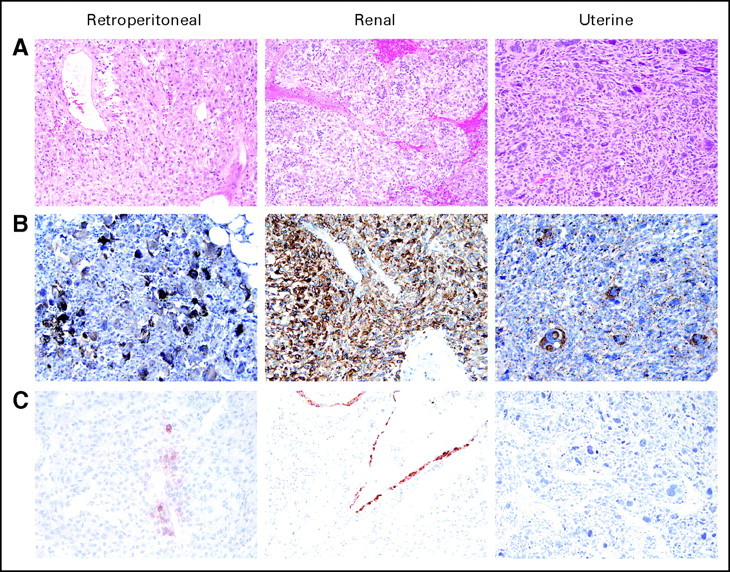
Perivascular epithelioid cell tumor (PEComa) histology and immunohistochemical stains. Formalin-fixed paraffin-embedded sections stained with (A) hematoxylin and eosin, (B) antiphospho-S6 antibody, and (C) anti-TSC2 antibody.
The integrity of the TSC1 and TSC2 genomic loci was examined by MLPA8 (Fig 3). Genomic DNA was extracted from paraffin-embedded tumor tissue (Figs 3A, 3B, and 3C) or peripheral blood (Fig 3D) and was assessed for copy number changes by MLPA by using nine exonic probes for TSC1 and TSC2, respectively, and five control probes from other genomic locations.
Fig 3.
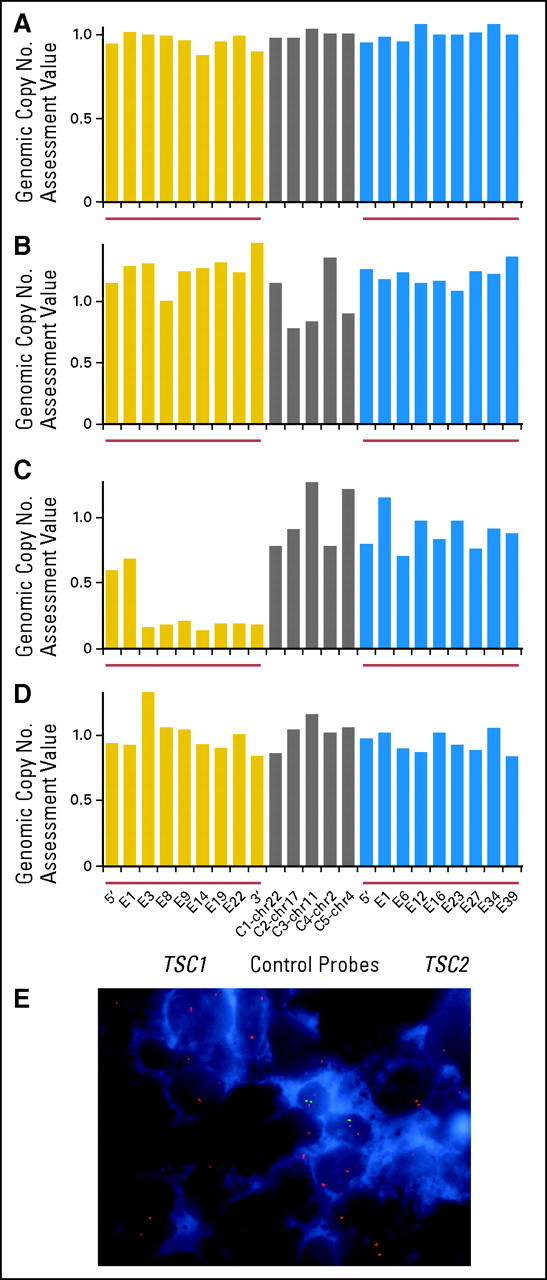
Mulitplex ligation-dependent probe amplification (MLPA) and fluorescent in situ hybridization (FISH) assays of TSC1 and TSC2 in perivascular epithelioid cell tumor (PEComa) samples. Genomic DNA from (A-C) tumor or (D) peripheral blood assessed for copy number by mulitplex ligation-dependent probe amplification (MLPA). Signals were normalized to values from control tissues. (E) FISH using (green) TSC1-specific DNA probes, (red) chromosome 16 centromeric probes, and nuclei isolated from uterine PEComa tissue.
This technique allowed genomic copy number assessment; a normalized value of 1 indicated two intact alleles, and values of 0.5 and 0 indicated a single allele and complete genomic loss, respectively. Marked reduction in signal from the TSC1 gene was observed in the uterine tumor specimen from patient 3 (Figs 3C to D), consistent with somatic biallelic deletion of TSC1 (Fig 3A). MLPA demonstrated no alterations in tumor samples from the retroperitoneal PEComa (patient 1; Fig 3A) or the renal PEComa (patient 2; Fig 3B), which suggests that mechanisms other than gross alteration of the TSC1 or TSC2 genomic loci were important in loss of TSC2 expression in those two tumors.
FISH, performed by using interphase nuclei isolated from archival paraffin-embedded tissue and probes corresponding to TSC1 and TSC2, confirmed homozygous loss of TSC1 (Fig 3E, green signal) in greater than 60% of nuclei from the uterine PEComa sample, whereas the centromeric region of chromosome 9 (Fig 3E, red signal) was maintained.
Karyotype analysis of the retroperitoneal PEComa (patient 2), previously performed for clinical purposes, showed two copies of chromosome 16 as well as an add([16p]; not shown). The add(16p13) involved chromosome breakage within band p13, loss of material distal to the break, and addition of unidentified chromosomal material in its place. Because TSC2 maps to 16p13, interphase FISH was performed to determine the TSC2 copy number. Three copies of TSC2 were detected (not shown), which suggests that the breakpoint of the add16 maps distal to the TSC2 probe. The possibility of small deletions of or point mutations in TSC2, which are undetectable by FISH, cannot be excluded. No other evidence of deletion or rearrangement of either TSC1 or TSC2 was observed.
PEComa DNA samples were sequenced to search for known activating mutations in the PIK3CA and AKT1 genes as well as for the common mutations that occur in KRAS. No mutations were detected in any of the three PEComas in any of these three genes.
DISCUSSION
Metastatic PEComa is a rare form of sarcoma for which no effective therapy has been described previously and which has a uniformly fatal outcome. We have observed significant clinical responses in three patients treated with sirolimus, an inhibitor of mTORC1, including one patient with an ongoing near complete response of greater than 14 months duration. Tumors from all three patients showed evidence of mTORC1 activation, and somatic deletion of TSC1 was identified as the likely mechanism in patient 3. TSC1 acts to stabilize TSC2, so its genomic loss will lead to lack of TSC2 expression.10 The mechanisms of mTOR activation in patients 1 and 2 are unclear, but they potentially include small deletions or inactivating or missense mutations in TSC1 or TSC2 that would similarly account for loss of TSC2 expression in those tumors.
Other evidence to support activation of the mTOR pathway in PEComas also has recently been described. Kenerson et al7 reported immunohistochemical evidence of mTORC1 activity in 15 PEComas and absence of AKT phosphorylation in 14 tumors, which suggests the loss of TSC1 or TSC2 as potential mechanisms. Similarly, Pan et al6 described elevated phospho-p70S6K and reduced phospho-AKT in 11 of 12 PEComas.6 Seven of these tumors had loss of heterozygosity of the TSC2 region, and one additionally showed loss of heterozygosity of TSC1.
Our data are consistent with findings to date of the activity of mTOR inhibitors in tumors known to be biologically related to PEComas, specifically AML and LAM. After case reports of patients with renal AML and LAM responding to sirolimus were published,11–13 Bissler et al5 reported on treatment of 25 patients with AML or LAM with sirolimus for 12 months followed by 12 months of observation.5 After 12 months of therapy, AML volume decreased 53% but returned to 86% of baseline after the year of observation, which indicated the need for continued inhibition to maintain tumor shrinkage. Less-impressive improvements in respiratory function were observed in patients with LAM, which also reversed on observation alone. Interestingly, facial angiofibromas associated with tuberous sclerosis also have significantly improved on sirolimus in a case report.14 There are presently no reports of mTOR inhibitors as treatment of clear cell “sugar” tumors of the pancreas or lung, another neoplasm considered to be a PEComa, but it is expected that such tumors would respond to mTOR-directed therapy as well.
The efficacy of mTOR inhibitors has been explored in patients with a heterogenous mix of other metastatic sarcomas, in each case with only a modest response rate.15,16 However, the status of mTOR activation of these sarcomas is unknown, although in one study the presence of S6 phosphorylation correlated with a higher likelihood of disease control with an mTOR inhibitor.17 Taken together, these observations suggest that activation of mTOR through loss of the TSC1/TSC2 repressor complex, or potentially by other means, is likely a common and critically pathogenic event in PEComas.
Inhibition of mTOR has resulted in significant clinical activity in patients with PEComa and merits additional investigation in a prospective study. Absence of immunohistochemical evidence of TSC2 expression or the less-specific presence of S6 phosphorylation may be predictive markers for responsiveness to inhibitors of mTORC1. These findings additionally unify the concept of PEComa, AML, and LAM as closely related pathologic entities, from histology to genetic changes to evidence of therapeutic benefit from mTOR blockade.
Acknowledgment
We thank Vijaya Ramesh, MSc, PhD, for her kind gift of anti-TSC2 antibodies.
Appendix
Fig A1.
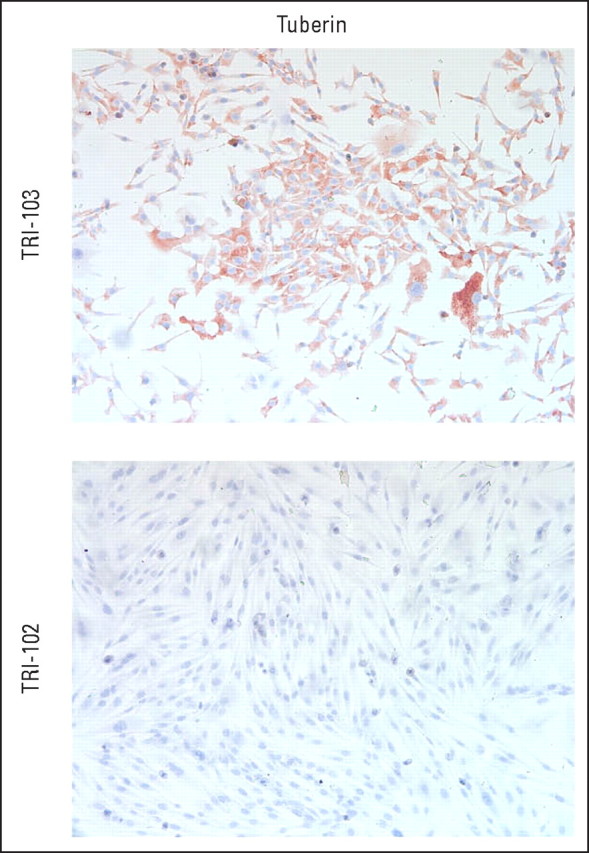
Validation of anti-TSC2 antibody. TSC2-null TRI-102 cells and TSC2-expressing TRI-103 cells were grown on coverslips, were fixed, and were stained with anti-TSC2 antibody followed by horseradish peroxidase detection.
Table A1.
Summary of Therapeutics Administered to Index Patients With PEComa
| Patient | Prior Therapy | Time to Progression (weeks) | Sirolimus Treatment |
Subsequent Therapy |
||
|---|---|---|---|---|---|---|
| Dosage | Outcome | Type | Outcome | |||
| 1 | Investigational MET inhibitor | 6 | 8 mg daily | Radiologic improvement; on sirolimus 16 months (without disease progression) | NA | Alive with disease |
| 2 | Sunitinib | 6 | 4→1 mg daily | Radiologic improvement; progression after 10 months | Surgery | Alive with disease |
| 3 | NA | NA | 4 mg | Radiologic improvement; Progression after 3 months; dose increased with stabilization for 2 months before progression | Sirolimus with sorafenib 200 mg orally twice daily | Tumor cavitation and stabilization; died as a result of disease after 3 months |
Abbreviations: PEComa, perivascular epithelioid cell tumors; NA, not applicable.
Footnotes
Supported in part by the sPECial Fund for PEComa Research and by the Virginia and D.K. Ludwig Fund for Cancer Research (to the Dana-Farber/Harvard Ludwig Center).
Presented in part at the 14th Annual Meeting of the Connective Tissue Oncologic Society,November 13-15, 2008, London, United Kingdom.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Andrew J. Wagner, Genentech (C); George D. Demetri, Ariad (C), Novartis (C), Pfizer (C), Genentech (C); Robert G. Maki, Novartis (C) Stock Ownership: None Honoraria: Robert G. Maki, Novartis Research Funding: Andrew J. Wagner, Genentech; Robert G. Maki, Novartis, Pfizer Expert Testimony: George D. Demetri, Ariad (U) Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Andrew J. Wagner, Jeffrey A. Morgan, Christopher D.M. Fletcher, Azra H. Ligon, George D. Demetri, David J. Kwiatkowski, Robert G. Maki
Financial support: Andrew J. Wagner, George D. Demetri
Administrative support: Andrew J. Wagner, George D. Demetri
Provision of study materials or patients: Andrew J. Wagner, Jeffrey A. Morgan, Christopher D.M. Fletcher, Cristina R. Antonescu, Robert G. Maki
Collection and assembly of data: Andrew J. Wagner, Izabela Malinowska-Kolodziej, Jeffrey A. Morgan, Wei Qin, Christopher D.M. Fletcher, Natalie Vena, Azra H. Ligon, Cristina R. Antonescu, Nikhil H. Ramaiya, David J. Kwiatkowski, Robert G. Maki
Data analysis and interpretation: Andrew J. Wagner, Izabela Malinowska-Kolodziej, Jeffrey A. Morgan, Christopher D.M. Fletcher, Natalie Vena, Azra H. Ligon, Nikhil H. Ramaiya, David J. Kwiatkowski, Robert G. Maki
Manuscript writing: Andrew J. Wagner, Izabela Malinowska-Kolodziej, Christopher D.M. Fletcher, Azra H. Ligon, George D. Demetri, David J. Kwiatkowski, Robert G. Maki
Final approval of manuscript: Andrew J. Wagner, Izabela Malinowska-Kolodziej, Jeffrey A. Morgan, Wei Qin, Christopher D.M. Fletcher, Natalie Vena, Azra H. Ligon, Cristina R. Antonescu, Nikhil H. Ramaiya, George D. Demetri, David J. Kwiatkowski, Robert G. Maki
REFERENCES
- 1.Wagner A. Treatment of advanced soft tissue sarcoma: Conventional agents and promising new drugs. J Natl Compr Canc Netw. 2007;5:401–410. doi: 10.6004/jnccn.2007.0035. [DOI] [PubMed] [Google Scholar]
- 2.Hornick JL, Fletcher CD. PEComa: What do we know so far? Histopathology. 2006;48:75–82. doi: 10.1111/j.1365-2559.2005.02316.x. [DOI] [PubMed] [Google Scholar]
- 3.Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: Pathology and pathogenesis. Hum Pathol. doi: 10.1016/j.humpath.2009.05.011. epub ahead of print on July 15, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Folpe AL, Mentzel T, Lehr HA, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: A clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol. 2005;29:1558–1575. doi: 10.1097/01.pas.0000173232.22117.37. [DOI] [PubMed] [Google Scholar]
- 5.Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan CC, Chung MY, Ng KF, et al. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): Genetic evidence for the relationship of PEComa with angiomyolipoma. J Pathol. 2008;214:387–393. doi: 10.1002/path.2289. [DOI] [PubMed] [Google Scholar]
- 7.Kenerson H, Folpe AL, Takayama TK, et al. Activation of the mTOR pathway in sporadic angiomyolipomas and other perivascular epithelioid cell neoplasms. Hum Pathol. 2007;38:1361–1371. doi: 10.1016/j.humpath.2007.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389–400. doi: 10.1007/s00439-006-0308-9. [DOI] [PubMed] [Google Scholar]
- 9.Firestein R, Bass AJ, Kim SY, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benvenuto G, Li S, Brown SJ, et al. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene. 2000;19:6306–6316. doi: 10.1038/sj.onc.1204009. [DOI] [PubMed] [Google Scholar]
- 11.Herry I, Neukirch C, Debray MP, et al. Dramatic effect of sirolimus on renal angiomyolipomas in a patient with tuberous sclerosis complex. Eur J Intern Med. 2007;18:76–77. doi: 10.1016/j.ejim.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Taillé C, Debray MP, Crestani B. Sirolimus treatment for pulmonary lymphangioleiomyomatosis. Ann Intern Med. 2007;146:687–688. doi: 10.7326/0003-4819-146-9-200705010-00022. [DOI] [PubMed] [Google Scholar]
- 13.Wienecke R, Fackler I, Linsenmaier U, et al. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis. 2006;48:e27–e29. doi: 10.1053/j.ajkd.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 14.Hofbauer GF, Marcollo-Pini A, Corsenca A, et al. The mTOR inhibitor rapamycin significantly improves facial angiofibroma lesions in a patient with tuberous sclerosis. Br J Dermatol. 2008;159:473–475. doi: 10.1111/j.1365-2133.2008.08677.x. [DOI] [PubMed] [Google Scholar]
- 15.Chawla SP, Tolcher AW, Staddon AP, et al. Updated results of a phase II trial of AP23573, a novel mTOR inhibitor, in patients with advanced soft tissue or bone sarcomas. J Clin Oncol. 2006;24(suppl):521s. abstr 9505. [Google Scholar]
- 16.Okuno SH, Mahoney MR, Bailey HH, et al. A multicenter phase II consortium study of the mTOR inhibitor CCI-779 in advanced soft tissue sarcomas. J Clin Oncol. 2006;24(suppl):521s. abstr 9504. [Google Scholar]
- 17.Iwenofu OH, Lackman RD, Staddon AP, et al. Phospho-S6 ribosomal protein: A potential new predictive sarcoma marker for targeted mTOR therapy. Mod Pathol. 2008;21:231–237. doi: 10.1038/modpathol.3800995. [DOI] [PubMed] [Google Scholar]