

Abstract
A series of 5-(2′-indolyl)thiazoles were synthesized and evaluated for their cytotoxicity against selected human cancer cell lines. The reaction of thioamides 3 with 3-tosyloxypentane-2,4-dione 4 led to in situ formation of 5-acetylthiazole 5 which upon treatment with arylhydrazines 6 in polyphosphoric acid resulted in the formation of 5-(2′-indolyl)thiazoles 2. Among the synthesized 5-(2′-indolyl)thiazoles, compounds 2d–f, and 2h exhibited encouraging anticancer activity and also selectivity towards particular cell lines (IC50 = 10–30 μM). Further studies on the SAR of compound 2e may result in good anticancer activity.
Multicomponent reactions (MCRs) are one-pot multi-step reactions comprising three or more substrates. They are popular in drug discovery for being important tools in rapidly generating a library of novel compounds with diverse substitutions and molecular complexity1. The advantages associated with MCRs include atom economy, simple operation, and high yields of the products along with eliminating the need for isolation of intermediates. Additionally, the application of microwaves in organic synthesis has enabled the chemists to accelerate reactions, improve yields, and easily achieve desired products2,3,4.
Indole alkaloids extracted from plants and marine sources are well known for their chemistry and biology. The biological significance of these naturally available alkaloids secured much attention resulting in exploring the structural novelty of these molecules. 5-(2′-Indolyl)azoles 2 possess an indole ring at the C-5 position of azole ring as in 5-(3′-indolyl)azoles 1, but is connected at the second position of indole unlike the third position in natural derivatives (Fig. 1).
Figure 1. 5-(3′-Indolyl)azoles 1 and 5-(2′-Indolyl)azoles 2.
Thiazole derivatives are one of the most revered compounds with many applications and biological activities attributed to them. Indolylthiazole and its related heterocyclic systems are found in many natural products. The anticancer compounds, Camalexin5,6 and the naturally occurring BE 10988, are thiazole-substituted indole derivatives7. Additionally, the thiazole and benzothiazole derivatives of indole have been found to exhibit broad therapeutic activities including antimicrobial and antitumor8,9,10,11,12,13,14,15.
The naturally occurring bis(indolyl) alkaloids and their analogues, in general, have also exhibited significant anticancer activities16. For example, Nortopsentins and Topsentins represent a class of deep-sea sponge metabolites which display potent biological activities such as antitumor17, antiviral18, and antiinflammation19. Nortopsentin analogues, such as 2,4-bis(indolyl)thiazoles, also have exhibited good cytotoxicity against a panel of human tumor cell lines with GI50 values as low as 0.888 μM13. The closely related thiazolylbenzofuran derivatives20 have been shown to possess leukotriene and SRS-A antagonist or inhibitor activities. Our research group has prepared a series of indolylazoles such as 5-(3-indolyl)-1,3,4-oxadiazoles21, 4-(3-indolyl)oxazoles22, 5-(3-indolyl)-1,3,4-thiadiazoles23, and indolyl-1,2,4-triazoles24 as potential anticancer agents against human cancer cell lines.
Instigated by the role of indolylthiazoles and their bioisosteres in a variety of therapeutic activities as well as paucity of their one-pot preparation, we have contrived the synthesis of 5-(2′-indolyl)-2-substituted thiazoles 2 (Fig. 2). This one-pot synthesis was carried out by in situ generation of 5-acetylthiazole 5 from the reaction of thioamide 3 with 3-tosyloxypentane-2,4-dione 4 in ethanol with subsequent treatment of 5 with arylhydrazines 6 in polyphosphoric acid (PPA).
Figure 2. One-pot synthesis of 5-(2′-indolyl)thiazoles 2.

Results and Discussion
The preparation of indolylthiazoles 2 was initiated with a trial reaction of 4-methoxybenzothioamide 3a with 3-tosyloxypentane-2,4-dione 4 in ethanol. The reaction mixture was then refluxed at 80 °C for 4 hours in ethanol without any catalyst to afford 5-acetylthiazole 5a in 90% yield. Upon confirming the synthesis of the 5-acetylthiazole 5a by comparing its melting point with that of literature report25, it was further reacted with an equimolar quantity of phenylhydrazine (6a) in ethanol at 80 °C, to produce the corresponding hydrazone 7a (cf. Fig. 3). The solid hydrazone 7a obtained was then subjected to a Fischer indole cyclization under varying acidic conditions (Table 1). In most of the cases attempted, either the reaction failed to initiate or led to a complex mixture with trace amounts of the desired product. The cyclization was then attempted by refluxing the hydrazone 7a in ethanol in the presence of HCl or p-toluenesulfonic acid (p-TsOH) or phosphotungstic acid (PTA). No desired product was formed under these reaction conditions even after extended heating (12 h).
Figure 3. Plausible mechanistic pathway for the formation of indolylthiazoles 2.

Table 1. Cyclization of hydrazone 7a.
| S.No. | Reagent | Condition | Solvent | Yield (%) |
|---|---|---|---|---|
| 1 | HCl | Reflux | Ethanol | – |
| 2 | H3PO4 (6 M) | Heating (80 °C) | – | – |
| 3 | H3PO4 (18 M) | Heating (80 °C) | – | – |
| 4 | p-TsOH | Microwave | Neat | – |
| 5 | p-TsOH | Reflux | Ethanol | – |
| 6 | p-TsOH | Reflux | Acetonitrile | – |
| 7 | p-TsOH | Grinding | Neat | – |
| 8 | Conc. H2SO4 | Heating (80 °C) | – | – |
| 9 | PPA | Room temp. | – | – |
| 10 | PPA | Heating (80 °C) | – | 30 |
| 11 | PPA | Reflux (110 °C) | Toluene | – |
| 12 | PPA | Microwave (80 °C) | Ethanol | 65 |
| 13 | ZnCl2 | Grinding | Neat | – |
| 14 | ZnCl2 | Microwave | Neat | – |
| 15 | ZnCl2 | Reflux | Ethanol | – |
| 16 | AcOH | Heating (116 °C) | – | – |
| 17 | PTA | Reflux (80 °C) | Ethanol | – |
| 18 | PTA | Grinding | – | – |
| 19 | HCOOH | Heating (80 °C) | – | – |
The cyclization was also attempted by directly heating the hydrazone 7a with phosphoric acid (H3PO4) or zinc chloride (ZnCl2) or conc. sulfuric acid (H2SO4) or formic acid (HCOOH) or PTA. Finally, the hydrazone 7a was heated in PPA at 80 °C for 15 min to obtain the desired indolylthiazole 2a in 30% yield. The structure of 2a was confirmed by 1H NMR and mass spectral data. 1H NMR spectrum of 2a showed characteristic singlets at δ 6.68 and δ 2.67 due to indole C3-H and C4 methyl of thiazole, respectively. The mass spectrum of 2a showed the expected molecular ion peak [M + H]+ at m/z 321.1 which was found to be in agreement with the calculated value.
The reaction was further simplified by the direct reaction of phenylhydrazine 6a with 5-acetyl-thiazole 5a in presence of PPA. Encouraged by the outcome, a one-pot trial was attempted by subjecting equimolar quantities of 4-methoxybenzothioamide 3a with 3-tosyloxypentane-2,4-dione 4 under microwave (MW) irradiation in ethanol at 80 °C for 10 min. Upon completion of the reaction, phenylhydrazine 6a was added to the reaction vessel and exposed to MW irradiation at the same temperature for an additional 10 min. After the addition of PPA (two drops) the contents were further exposed to MW for 15 min to afford the desired indolylthiazole 2a in 65% yield.
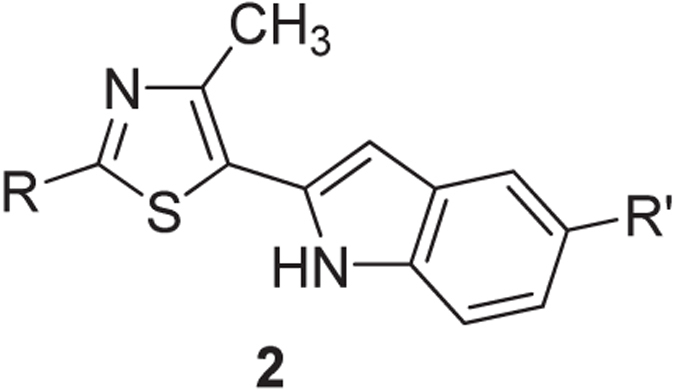
The developed protocol was extended to prepare various 5-(2′-indolyl)thiazoles 2b–j using thioamides 3 and arylhydrazines 6 in good yields (65–85%). The 4-methoxythiobenzamide, 3,4,5-trimethoxythiobenzamide and 4-chlorothiobenzamide exhibited almost similar reactivity towards 3-tosyloxypentane-2,4-dione. However, thioacetamide and indolylthioamide were more reactive and afforded the corresponding products in relatively higher yields.
The thioacetamide and phenylhydrazine reacted to afford 2d in 65% yield, whereas reaction involving indole-3-thioamide and phenylhydrazine afforded 2e in 80% yield. The yields of the compounds 2f–j were in the range of 70–80% demonstrating that substitutions on arylhydrazine have little effect on the formation of final products 2. The reaction of 4-methoxythiobenzamide with phenylhydrazine and 4-chlorophenylhydrazine yielded 2a and 2b in 65% and 68% yields, respectively. Arylhydrazines 6 bearing an electronegative atom (Cl, Br and F) were equally reactive with 5-acetylthiazole 5, whereas arylhydrazines 6 bearing an electron-donating group (4-OMe) were relatively more reactive to undergo the Fischer indole cyclization.
Formation of 5-(2′-indolyl)thiazoles 2 involves an initial nucleophilic displacement of a tosyloxy group in 4 by thioamide 3 to generate intermediate species A which undergoes cyclization with concomitant loss of water molecule to afford acetylthiazole 5. Subsequent reaction of 5 with arylhydrazines 6 leads to hydrazones 7 which undergoes a [3,3]-sigmatropic rearrangement as depicted in Fig. 3 to produce 5-(2′-indolyl)thiazoles 2.
Synthesized 5-(2′-indolyl)thiazoles compounds 2a–j were then screened for their in vitro cytotoxicity against five human cancer cell lines: breast (BT-474, MCF-7 and MDA-MB-231, MDA-MB-157) and colon (HTC-116). Anticancer activity of 5-(2′-indolyl)thiazoles 2a–j against cancer cell lines are expressed in terms of IC50 values (Table 2). Some of the compounds have shown moderate activity. Structure–activity relationship (SAR) study revealed that substitution at C-2 position of the thiazole ring is crucial for inducing cytotoxicity and selectivity against particular cancer cell line.
Table 2. Synthesis and anticancer activity (IC50 in μM) of 5-(2′-indolyl)thiazoles 2.
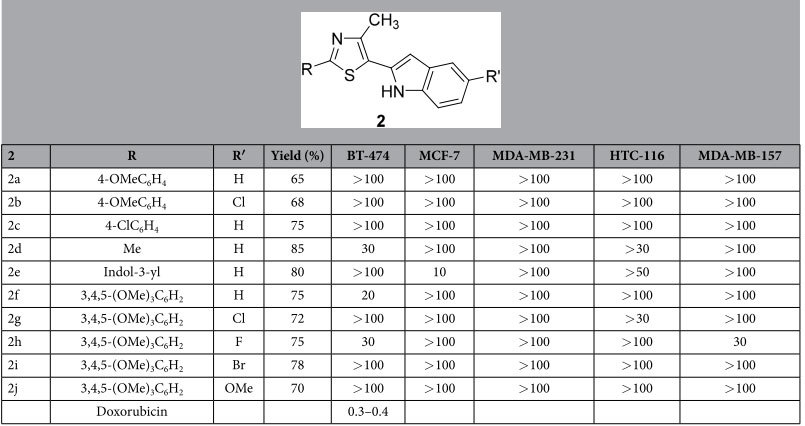
The presence of a 4-methoxyphenyl group at C-2 position and absence of any substituent on the C-5 indole ring led to compound 2a not having any significant activity against the tested cancer cell lines (IC50 > 100 μM). Upon placing a chloro substituent on the indole ring of 2a led to compound 2b also without any improvement in activity (IC50 > 100 μM). The anticancer activity was further studied by replacing the 4-methoxyphenyl in 2a with 4-chlorophenyl resulting in compound 2c without any improvement in activity (IC50 > 100 μM). The presence of a methyl substituent at the C-2 position of thiazole 2d exhibited selectivity towards breast cancer cell line, BT-474, with an improved activity (IC50 = 30 μM), but relatively moderate towards colon cancer cell line, HTC-116 (IC50 > 30 μM). The presence of a heterocyclic indole ring at the C-2 position resulted in bis(indolyl)thiazole 2e, which closely resembles the naturally occurring bisindolyl alkaloids such as Nortopsentins. Compound 2e exhibited very good selectivity and potency (IC50 = 10 μM) when compared to other compounds of the series.
It was earlier shown that one of the important structural units is a trimethoxyphenyl moiety in several known natural antimitotic agents (Combretastatin A-4, Colchicine, Podophyllotoxin and Steganacin) that binds at the Colchicine site of tubulin26,27. Incorporation of such a crucial structural feature of lead anticancer compounds into 5-(2′-indolyl)thiazoles 2 may results in a potent anticancer compound. Compounds 2f–j were synthesized with a 3,4,5-trimethoxy phenyl substitution at C-2 position of the thiazole ring anticipating improved activity and selectivity towards a particular cell line. The compound 2f with no substituents on the indole ring at C-5 of thiazole exhibited very good selectivity towards BT-474 breast cancer cell line (IC50 = 20 μM) whereas, the presence of a fluoro substituent on the indole ring (compound 2h) could inhibit both BT-474 and MDA-MB-157 (IC50 = 30 μM). The other compounds exhibited moderate to insignificant activity. Further improvement in activity may be achieved to a great extent by investigating the compound 2e with further substitutions on both indole rings.
Conclusions
The synthesis of prominent 5-(2′-indolyl)thiazoles was achieved in one-pot using a sequential reaction of thioamides, 3-tosyloxypentane-2,4-dione and arylhydrazines. All the 5-(2′-indolyl)thiazoles were obtained in good yields. Overall, the protocol is appreciable in terms of the high exploratory power, short duration and ease of availability of the starting materials. The generality of this protocol is demonstrated by preparing a diverse library of 5-(2′-indolyl)thiazoles from various thioamides and arylhydrazines. The similitude of the 5-(2′-indolyl)thiazoles to naturally occurring indole-based alkaloids envisage their biological importance. The anticancer activity studied against breast and colon cancer cell lines show that the compounds 2d–f, and 2h exhibit encouraging anticancer activity and also selectivity towards particular cell lines (IC50 = 10–30 μM). Further studies on the SAR of 2e may result in compounds with greatly improved anticancer activity.
Experimental
Representative procedure for the synthesis of 5-(2′-indolyl)thiazole (2a)
A mixture of 4-methoxybenzothioamide (2.0 mmol) and 3-tosyloxypentane-2,4-dione (2.0 mmol) in ethanol was subjected to MW irradiation (100 watt power) for 10 min at 80 °C temperature. Upon consumption of the thioamide as indicated by TLC, an appropriate phenylhydrazine (2.1 mmol) was added to reaction mixture and exposed to MW for 10 min at same temperature. Polyphosphoric acid (two-drops) was added to the reaction mixture and irradiated in MW for 15 min at 80 °C. The reaction contents were diluted with water and extracted with ethyl acetate (2 × 10 mL). The combined organic phase was dried over anhydrous sodium sulphate, concentrated in vacuo, and purified by passing through a column of silica-gel using ethyl acetate-hexane as eluent to afford the pure 5-(2′-indolyl)thiazole 2a in 65% yield.
Analytical data of the synthesised 5-(2′-indolyl)thiazoles 2
5-(1H-Indol-2-yl)-2-(4-methoxyphenyl)-4-methylthiazole (2a). Yield 45%. Rf = 0.51 (hexane/EtOAc, 6:4). m.p. 182–185 °C. 1H NMR (400 MHz, CDCl3) δ: 8.28 (s, br, 1H), 7.91–7.78 (m, 2H), 7.63 (d, J = 7.80 Hz, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.23 (dt, J = 7.08 and 1.12 Hz, 1H), 7.15 (dt, J = 7.88 and 0.92 Hz, 1H), 6.97–6.94 (m, 2H), 6.69–6.68 (m, 1H), 3.86 (s, 3H), 2.67 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C19H16N2OS: 321.1; found: 321.1.
5-(5-Chloro-1H-indol-2-yl)-2-(4-methoxyphenyl)-4-methylthiazole (2b). Yield 48%. Rf = 0.59 (hexane/EtOAc, 6:4). m.p. 197–198 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.05 (s, 1H), 7.86 (d, J = 8.76 Hz, 2H), 7.51 (s, 1H), 7.37 (d, J = 8.52 Hz, 1H), 7.07 (dd, J = 8.48 and 1.84 Hz, 1H), 6.97 (d, J = 8.80 Hz, 2H), 6.56 (s, 1H), 3.86 (s, 3H), 2.65 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C19H15ClN2OS: 355.1; found: 355.1.
2-(4-Chlorophenyl)-5-(1H-indol-2-yl)-4-methylthiazole (2c). Yield 55%. Rf = 0.61 (hexane/EtOAc, 6:4). m.p. 172–175 °C. 1H NMR (400 MHz, CDCl3) δ: 8.51 (s, br, 1H), 7.98–7.92 (m, 2H), 7.64 (d, J = 7.84 Hz, 1H), 7.46–7.42 (m, 3H), 7.17 (dt, J = 7.08 and 1.12 Hz, 1H), 7.15 (dt, J = 8.04 and 1.04 Hz, 1H), 6.73 (s, 1H), 2.74 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C18H13ClN2S: 325.1; found: 325.2.
5-(1H-Indol-2-yl)-2,4-dimethylthiazole (2d). Yield 65%. Rf = 0.56 (hexane/EtOAc, 6:4). m.p. 238 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.38 (s, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.42 (dd, J = 8.1, 0.8 Hz, 1H), 7.17–7.11 (m, 1H), 7.04 (ddd, J = 7.9, 7.1 and 1.0 Hz, 1H), 6.65–6.62 (m, 1H), 2.70 (s, 3H), 2.53 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C13H12N2S: 229.1; found: 229.2.
5-(1H-Indol-2-yl)-2-(1H-indol-3-yl)-4-methylthiazole (2e). Yield 60%. Rf = 0.32 (hexane/EtOAc, 6:4). m.p. 146–149 °C. 1H NMR (400 MHz, CDCl3) δ: 8.73 (s, 1H), 8.33 (s, 1H), 8.01 (s, 1H), 7.92 (s, 1H), 7.55 (s, 1H), 7.45–7.38 (m, 4H), 7.24–7.21 (m, 3H), 2.17 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C20H15N3S: 330.1; found: 330.2.
5-(1H-Indol-2-yl)-4-methyl-2-(3,4,5-trimethoxyphenyl)thiazole (2f). Yield 55%. Rf = 0.30 (hexane/EtOAc, 6:4). m.p. 126–129 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.25 (s, 1H), 7.65 (d, J = 8.02 Hz, 1H), 7.38 (dd, J = 8.1 and 0.8 Hz, 1H), 7.17–7.13 (m, 1H), 7.05–7.03 (m, 1H), 6.95 (s, 2H), 6.64 (s, 1H), 4.02 (s, 6H), 3.93 (s, 3H), 2.80 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C21H20N2O3S: 381.1; found: 381.1.
5-(5-Chloro-1H-indol-2-yl)-4-methyl-2-(3,4,5-trimethoxyphenyl)thiazole (2g). Yield 52%. Rf = 0.32 (hexane/EtOAc, 6:4). m.p. 126–127 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.25 (s, 1H), 7.51 (s, 1H), 7.37 (d, J = 8.52 Hz, 1H), 7.12 (s, 2H), 7.07 (dd, J = 8.48 and 1.84 Hz, 1H), 6.56 (s, 1H), 4.00 (s, 6H), 3.95 (s, 3H), 2.84 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C21H19ClN2O3S: 415.1; found: 415.1.
5-(5-Fluoro-1H-indol-2-yl)-4-methyl-2-(3,4,5-trimethoxyphenyl)thiazole (2h). Yield 55%. Rf = 0.40 (hexane/EtOAc, 6:4). m.p. 128–129 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.38 (s, 1H), 7.62–7.61 (m, 1H), 7.48–7.51 (m, 1H), 6.90 (s, 2H), 7.11–7.09 (m, 1H), 6.59 (s, 1H), 4.02 (s, 6H), 3.97 (s, 3H), 2.87 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C21H19FN2O3S: 399.1; found: 399.1.
5-(5-Bromo-1H-indol-2-yl)-4-methyl-2-(3,4,5-trimethoxyphenyl)thiazole (2i). Yield 58%. Rf = 0.41 (hexane/EtOAc, 6:4). m.p. 128–131 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.39 (s, 1H), 7.98–7.97 (m, 1H), 7.66–7.64 (m, 1H), 7.05 (s, 2H), 7.43–7.41 (m, 1H), 6.76 (s, 1H), 3.97 (s, 6H), 3.93 (s, 3H), 2.82 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C21H19BrN2O3S: 458.0; found: 458.1 [M + H]+ and 460.1 [M + H + 2]+.
5-(5-Methoxy-1H-indol-2-yl)-4-methyl-2-(3,4,5-trimethoxyphenyl)thiazole (2j). Yield 50%. Rf = 0.43 (hexane/EtOAc, 6:4). m.p. 129–132 °C. 1H NMR (400 MHz, DMSO-d6) δ: 11.34 (s, 1H), 7.57–7.56 (m, 1H), 7.38–7.36 (m, 1H), 7.01 (s, 2H), 6.90–6.94 (m, 1H), 6.72 (s, 1H), 3.96 (s, 6H), 3.92 (s, 3H), 3.79 (s, 3H), 2.68 (s, 3H). MS (ESI): m/z [M + H]+ calcd for C22H22N2O4S: 411.1; found: 411.2.
In vitro anticancer screening
Five human cancer cell lines (BT-474, MCF-7, MDA-MB-231, HTC-116, and MDA-MB-157) were cultured in RPMI 1640 media supplemented with 10% heat inactivated fetal bovine serum and 1% penicillin/streptomycin. They were seeded in 96-well plates at a density of 4 × 103 cells per well for 12 h. Cells were incubated with various concentrations of the compounds ranging from 10 nM to 1 mM. After 48 h, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to the final concentration of 0.2 mg/mL and incubated for 30 min. The cells were washed twice with PBS and lysed in 100 μL dimethylsulfoxide, and the absorbance was measured at 570 nm using Tecan Spectrafluor Plus.
Additional Information
How to cite this article: Vaddula, B. R. et al. One-pot synthesis and in-vitro anticancer evaluation of 5-(2′-indolyl)thiazoles. Sci. Rep. 6, 23401; doi: 10.1038/srep23401 (2016).
Acknowledgments
The authors thank the University Grants Commission (UGC), New Delhi for the financial support. B.R.V. was supported in part by an appointment to the Research Participation Program for the U.S. Environmental Protection Agency, Office of Research and Development, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the EPA.
Footnotes
Author Contributions D.K. and B.R.V. designed the work; B.R.V. carried out the research work, data analysis and compiled the manuscript. M.P.T. supplemented the research work with additional experiments and data analysis. R.S. carried out anticancer activity evaluation of the reported compounds. M.A.G. edited and revised the manuscript. D.K. supervised the overall research work progress and revised the manuscript. All authors reviewed the manuscript.
References
- Zhu J. & Bienaymé H. Multicomponent reactions. 1st Ed. edn, (Weinheim, 2005). [Google Scholar]
- Polshettiwar V. & Varma R. S. Microwave-Assisted Organic Synthesis and Transformations using Benign Reaction Media. Acc. Chem. Res. 41, 629–639 (2008). [DOI] [PubMed] [Google Scholar]
- Gawande M. B., Bonifacio V. D. B., Luque R., Branco P. S. & Varma R. S. Benign by design: catalyst-free in-water, on-water green chemical methodologies in organic synthesis. Chem. Soc. Rev. 42, 5522–5551 (2013). [DOI] [PubMed] [Google Scholar]
- Gawande M. B., Shelke S. N., Zboril R. & Varma R. S. Microwave-Assisted Chemistry: Synthetic Applications for Rapid Assembly of Nanomaterials and Organics. Acc. Chem. Res. 47, 1338–1348 (2014). [DOI] [PubMed] [Google Scholar]
- Browne L. M., Conn K. L., Ayert W. A. & Tewari J. P. The camalexins: new phytoalexins produced in the leaves of camelina sativa (cruciferae). Tetrahedron 47, 3909–3914 (1991). [Google Scholar]
- Pedras M. S. C., Okanga F. I., Zaharia I. L. & Khan A. Q. Phytoalexins from crucifers: synthesis, biosynthesis, and biotransformation. Phytochemistry 53, 161–176 (2000). [DOI] [PubMed] [Google Scholar]
- Oka H. et al. A new topoisomerase-II inhibitor, BE-10988, produced by a streptomycete. J. Antibiot. 44, 486–491 (1991). [DOI] [PubMed] [Google Scholar]
- Ayer W. A., Craw P. A., Ma Y.-t. & Miao S. Synthesis of camalexin and related phytoalexins. Tetrahedron 48, 2919–2924 (1992). [Google Scholar]
- Dzurilla M., Ružinský M., Kutschy P., Tewari J. & Ková ik V. Application of 2-substituted ethyl isothiocyanates and 2-aminothiols in the synthesis of the analogs of indole phytoalexin camalexin. Collect. Czech. Chem. Commun. 64, 1448–1456 (1999). [Google Scholar]
- Moody C. & Swann E. Synthesis of the naturally occurring indolequinone BE 10988, an inhibitor of topoisomerase II. J. Chem. Soc., Perkin Trans. 1, 2561–2565 (1993). [Google Scholar]
- Moody C. J., Swann E., Houlbrook S., Stephens M. A. & Stratford I. J. Synthesis and biological activity of thiazolylindolequinones, analogs of the natural product BE 10988. J. Med. Chem. 38, 1039–1043 (1995). [DOI] [PubMed] [Google Scholar]
- Moody C. J., Roffey J. R., Stephens M. A. & Stratford I. J. Synthesis and cytotoxic activity of indolyl thiazoles. Anti-Cancer Drugs 8, 489–499 (1997). [DOI] [PubMed] [Google Scholar]
- Jiang B. & Gu X.-H. Syntheses and cytotoxicity evaluation of bis(indolyl)thiazole, bis(indolyl)pyrazinone and bis(indolyl)pyrazine: analogues of cytotoxic marine bis(indole) alkaloid. Bioorg. Med. Chem. 8, 363–371 (2000). [DOI] [PubMed] [Google Scholar]
- Cheng X. M., Filzen G. F., Geyer A. G., Lee C. & Trivedi B. K. Substituted thiazoles and oxazoles that modulate PPAR activity. WIPO patent WO2003074051 (2003).
- Nowakowski J. 5-Heteroyl indole derivatives. US5409941 (1995).
- Diana P. et al. Synthesis and antitumor properties of 2,5-bis(3′-indolyl)thiophenes: analogues of marine alkaloid nortopsentin. Bioorg. Med. Chem. Lett. 17, 2342–2346 (2007). [DOI] [PubMed] [Google Scholar]
- Shaaban M. R., Saleh T. S., Mayhoub A. S., Mansour A. & Farag A. M. Synthesis and analgesic/anti-inflammatory evaluation of fused heterocyclic ring systems incorporating phenylsulfonyl moiety. Bioorg. Med. Chem. 16, 6344–6352 (2008). [DOI] [PubMed] [Google Scholar]
- Schlecker R. & Thieme P. C. The synthesis of antihypertensive 3-(1,3,4-oxadiazol-2-yl)phenoxypropanolahines. Tetrahedron 44, 3289–3294 (1988). [Google Scholar]
- Liras S., Allen M. P. & Segelstein B. E. A mild method for the preparation of 1, 3, 4-oxadiazoles: Triflic anhydride promoted cyclization of diacylhydrazines. Synth. Commun. 30, 437–443 (2000). [Google Scholar]
- Masaaki M., Kazuo O., Shinji S., Hiroshi M. & Türk G. H. New thiazolylbenzofuran derivatives, processes for the preparation thereof and pharmaceutical composition comprising the same. EP0528337B1 (1999).
- Kumar D., Sundaree S., Johnson E. O. & Shah K. An efficient synthesis and biological study of novel indolyl-1,3,4-oxadiazoles as potent anticancer agents. Bioorg. Med. Chem. Lett. 19, 4492–4494 (2009). [DOI] [PubMed] [Google Scholar]
- Kumar D., Kumar N. M., Sundaree S., Johnson E. O. & Shah K. An expeditious synthesis and anticancer activity of novel 4-(3′-indolyl)oxazoles. Eur. J. Med. Chem. 45, 1244–1249 (2010). [DOI] [PubMed] [Google Scholar]
- Kumar D., Maruthi Kumar N., Chang K.-H. & Shah K. Synthesis and anticancer activity of 5-(3-indolyl)-1,3,4-thiadiazoles. Eur. J. Med. Chem. 45, 4664–4668 (2010). [DOI] [PubMed] [Google Scholar]
- Kumar D., Narayanam M. K., Chang K.-H. & Shah K. Synthesis of novel indolyl-1,2,4-triazoles as potent and selective anticancer agents. Chem. Biol. Drug Des. 77, 182–188 (2011). [DOI] [PubMed] [Google Scholar]
- Varma R., Kumar D. & Liesen P. Solid state synthesis of 2-aroylbenzo [b] furans, 1, 3-thiazoles and 3-aryl-5, 6-dihydroimidazo [2, 1-b][1, 3] thiazoles from -tosyloxyketones using microwave irradiation. J. Chem. Soc., Perkin Trans. 1, 4093–4096 (1998). [Google Scholar]
- Jordan A., Hadfield J. A., Lawrence N. J. & McGown A. T. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med. Res. Rev. 18, 259–296 (1998). [DOI] [PubMed] [Google Scholar]
- Hadfield J. A., Ducki S., Hirst N. & McGown A. T. Tubulin and microtubules as targets for anticancer drugs. Prog. Cell Cycle Res. 5, 309–325 (2003). [PubMed] [Google Scholar]