

Abstract
In situ hybridization with 3DNA™ dendrimers is a novel tool for detecting low levels of mRNA in tissue sections and whole embryos. Fluorescently labeled dendrimers were used to identify cells that express mRNA for the skeletal muscle transcription factor MyoD in the early chick embryo. A small population of MyoD mRNA positive cells was found in the epiblast prior to the initiation of gastrulation, two days earlier than previously detected using enzymatic or radiolabeled probes for mRNA. When isolated from the epiblast and placed in culture, the MyoD mRNA positive cells were able to differentiate into skeletal muscle cells. These results demonstrate that DNA dendrimers are sensitive and precise tools for identifying low levels of mRNA in single cells and tissues.
Keywords: MyoD protein, In situ hybridization
Introduction
In situ hybridization is a powerful tool for analyzing gene expression. Although less quantitative than Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR), in situ hybridization has the advantage of being able to localize messenger RNA (mRNA) to individual cells and to reveal qualitative differences in the level of gene expression within a heterogeneous tissue. However, in situ hybridization does have its limitations. A low level of gene expression in a small number of cells can go undetected. Such was the case in studies of the onset of expression of the MyoD family of skeletal muscle specific transcription factors. RT-PCR demonstrated the presence of MyoD mRNA in tissues of the chick embryo that give rise to the muscle forming somites (1), whereas in situ hybridizations using enzymatic or radiolableled oligonucleotide probes marked the onset of MyoD expression after somite formation (2-4). Clearly, a more sensitive probe was required to detect the low level of MyoD mRNA in tissue sections of the early embryo.
Fluorescently labeled 3DNA™ dendrimers (Genisphere, Inc.) have proven to be sensitive and precise reagents for detecting mRNA in single cells in both tissue sections and early embryos (5). DNA dendrimers are branched, multilayered structures synthesized by sequential hybridizations of partially complementary DNA monomers (6-8). Antisense oligonucleotide sequences are ligated to the ends of the outer arms via the use of T4 DNA ligase. Arms remaining single stranded are available for hybridization and cross-linking of complementary oligonucleotides previously conjugated with a choice of fluorochromes, 32P, digoxigenin, biotin or enzymes capable of generating signal producing reactions.
The sensitivity and accuracy of fluorescent dendrimer probes were analyzed by mapping the expression of MyoD mRNA in the early chick embryo. MyoD mRNA was detected in the somites, the tissue that gives rise to nearly all the skeletal muscle in the body, as well as the presomitic mesoderm and epiblast of gastrulating and pregastrulating embryos (5). Additional confirmation of the validity of dendrimer labeling was carried out by sorting cells that express MyoD, and testing their potential to differentiate into skeletal muscle in vitro (9).
Materials and Methods
3DNA™ dendrimers
3DNA™ dendrimers labeled with the fluorochrome CyTM3 were obtained from Genisphere, Inc. (Hatfield, PA). The following cDNA sequences for antisense mRNA were used: MyoD, 5’-TTC TCA AGA GCA AAT ACT CAC CAT TTG GTG ATT CCG TGT AGT AGC TGC TG 3’(10), chicken glyceraldehyde-3-dehydrogenase (GAPDH), 5’-ATC AAG TCC ACA ACA CGG TTG CTG TAT CCA AAC TCA TTG TCA TAC CAG GAA 3’ (11), and embryonic fast myosin, 5’-CAG GAG GTG CTG CAG GTC CTT CAC CGT CTG GTC CAG GTT CTT CTT CAT CCT CTC TCC AGG 3’ (12). Dendrimers lacking a specific recognition sequence were used as a negative control for background fluoresence.
In situ hybridization
The in situ hybridization procedure (5) was modified from that of Sassoon and Rosenthal (13) and Raap et al. (14). Briefly, White Leghorn chick embryos were staged according to the method of Hamburger and Hamilton (15). Embryos were fixed in 4% formaldehyde in phosphate buffered saline (PBS), embedded in paraffin, sectioned at 10μm, and applied to 0.2% gelatin coated, 3-well Teflon printed slides (EMS). After paraffin removal, the tissue was permeabilized with Triton X-100 and pepsin, then incubated with dendrimers overnight at 37°C. Nuclei were labeled with bis-benzamide and the sections were mounted in Gelmount (Fisher). Sections were observed with a Nikon Eclipse 800 epifluorescence microscope (Optical Apparatus), and images were captured with the Optronics DEI 750 video camera and the Image Pro Plus image analysis software program (Phase 3 Imaging Systems).
In situ hybridization also was performed on whole embryos. Embryos were rinsed in PBS and transferred to gelatin coated, 1-well Teflon printed slides. The embryos were fixed, permeabilized, and labeled with dendrimers and bis-benzamide as described above.
Magnetic cell sorting
The epiblast was isolated from stages 3-5 embryos, dissociated in 0.25% trypsin and EDTA (Invitrogen), centrifuged, and resuspended in Dulbecco’s Minimal Essential Medium (DMEM) containing 5% fetal bovine serum, 5% horse serum, 100 U of penicillin and streptomycin (Invitrogen), and 5% chick embryo extract (Myo medium). The G8 monoclonal antibody that specifically labels cells expressing MyoD (9, 16) was added at a concentration of 2μg/ml for 45 minutes at 37°C. After centrifugation, IgM microbeads tagged with iron (Miltenyi Biotech) were added to the cells. Cell sorting was carried out with the MiniMacs column separation unit (Miltenyi Biotech).
Culturing of sorted cells
MyoD positive/G8 positive cells were cultured under conditions that promote skeletal myogenesis (1, 17). Twenty thousand cells in 15 μl of Myo medium were plated on a tissue culture dish coated with gelatin and fibronectin. After 90 minutes, dishes were flooded with 1 ml of serum and hormone free DMEM/F12 (Invitrogen).
Immunofluorescence localization
Cultures were fixed at 5 days with 2% formaldehyde, permeabilized with Triton X-100, and labeled with the skeletal muscle specific 12101 monoclonal antibody (18), followed by affinity purified goat anti-mouse Fab′-2 fragments conjugated with rhodamine (Jackson ImmunoResearch). Nuclei were stained with bis-benzamide.
Results
Localization of MyoD mRNA in the somites and segmental plate mesoderm with fluorescent dendrimers
The ability of DNA dendrimers to correctly localize MyoD mRNA was tested in sections through the somites of stage 14 embryos. In situ hybridization with enzymatic and radiolabeled probes had demonstrated strong MyoD labeling in the dorsal medial lip of the dermomyotome (2-4, 19). Labeling with MyoD dendrimers also was abundant in the same region of the somite (Fig. 1A). A few cells in the sclerotome and the neural tube were labeled with dendrimers, a finding consistent with the fact that cells from the neural tube of the mouse embryo and spinal cord of the chick fetus differentiate into skeletal muscle in vitro (16, 20). Dendrimers lacking a specific recognition sequence produced very little background, yielding only one to three randomly scattered dendrimers per section. By contrast, labeling with dendrimers to GAPDH was observed in all tissues (Fig. 1D).
Fig. 1. Localization of MyoD mRNA in the somite and segmental plate with fluorescent dendrimers.
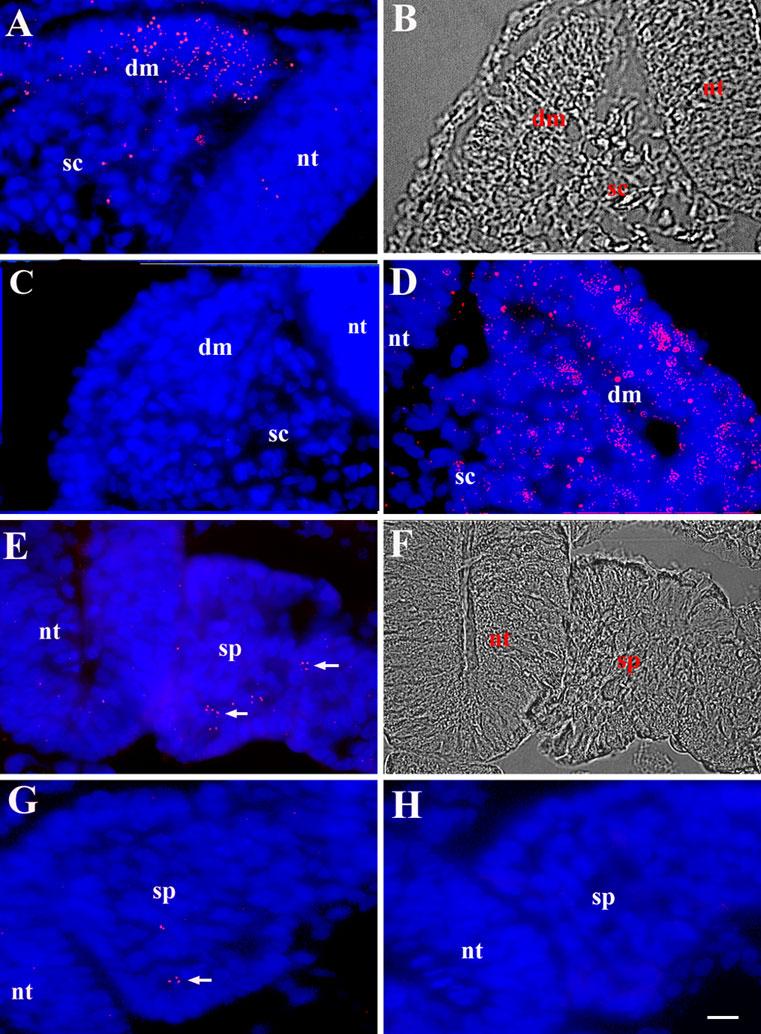
Sections through the stage 16 embryo were hybridized with Cy3 labeled dendrimers containing a recognition sequence for MyoD (A and E), GAPDH (D), embryonic fast myosin (G), or dendrimers lacking a specific recognition sequence (C). Photomicrographs are the merged images of Cy3 dendrimers (red) and bis-benzamide labeled nuclei (blue). Representative DIC images of A and E are shown in B and F. MyoD dendrimers were concentrated in the dermomyotome (dm) of the somite (A). A few dendrimers also were found in the sclerotome (sc) and neural tube (nt). GAPDH dendrimers labeled all tissues (D). Dendrimers lacking a specific recognition sequence did not bind to the somite or the segmental plate (sp) (C,H). MyoD mRNA was found in a small population of cells in the segmental plate (arrows) (E). Only a few dendrimers for myosin were found in the segmental plate (G). Bar, 9 μm.
We next examined the expression of MyoD in the segmental plate mesoderm, the tissue that gives rise to the somites. Although conventional probes for in situ hybridization did not detect MyoD mRNA in the segmental plate, fluorescent dendrimers revealed the presence of a subpopulation of MyoD mRNA positive cells throughout the tissue (Fig. 1E). Only a few dendrimers to embryonic fast myosin, a marker for differentiated skeletal muscle cells (Fig. 1G), were found in the segmental plate. One to three dendrimers lacking a specific recognition sequence were found per section (Fig. 1C and H).
Localization of MyoD mRNA in gastrulating and pregastrulating embryos
RT-PCR demonstrated that MyoD was expressed in the embryo prior to the formation of the segmental plate mesoderm (1). In situ hybridizations were performed on paraffin sections through embryos in the early stages of gastrulation, as well as non-embedded, whole embryos. In the gastrulating stage 4 embryo, MyoD mRNA was found in a small number of cells throughout the epiblast, mesoderm and endoderm (5) (Fig. 2A-D). The strongest staining was observed in the rostral end of the primitive streak near Hensen’s node (Fig. 2A).
Fig. 2. Localization of MyoD mRNA in the stage 4 and stage 1 embryo with fluorescent dendrimers.
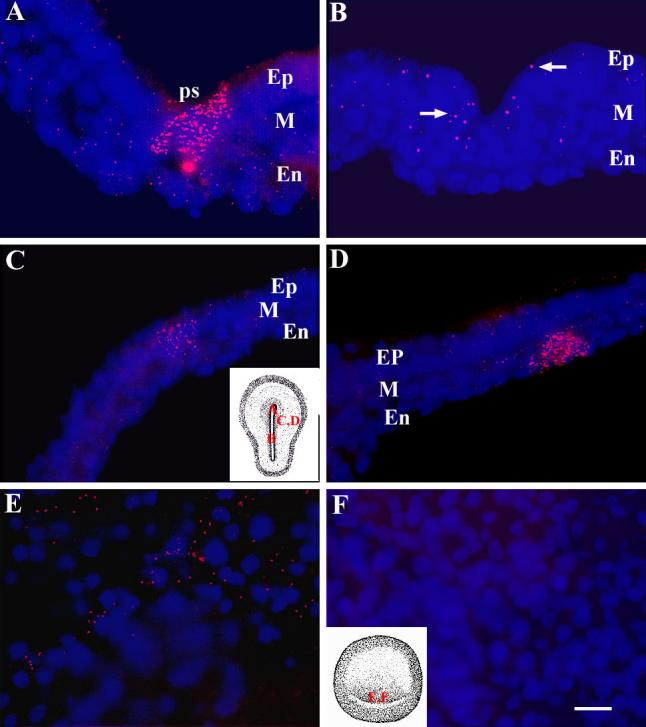
In situ hybridizations with dendrimers to MyoD (A-E) and embryonic fast myosin (F) were performed on sections through the stage 4 embryo (A-D) and whole stage 1 embryos (E-F). Photomicrographs are the merged images of Cy3 dendrimers (red) and bis-benzamide labeled nuclei (blue). C and F contain insets of drawings of the stage 4 and stage 1 embryo, respectively. The letters indicate the area shown at high magnification in the photomicrographs. Strong labeling with MyoD dendrimers was seen in cells ingressing through the primitive streak (ps) at the level of Hensen’s node (A). MyoD mRNA was less abundant in the posterior portion of the primitive streak (B). In the lateral region of the embryo, MyoD was found in adjacent cells of the epiblast (Ep) and mesoderm (M) and in the mesoderm and endoderm (En) (C and D). MyoD mRNA was localized to a subpopulation of cells in the posterior epiblast of the stage 1 embryo (E). Myosin mRNA was not detected in the stage 1 embryo (F). Bar, 9 μm.
Directly after the egg is laid, the stage 1 embryo consists of only 2 layers, the epiblast and hypoblast. Approximately 20 MyoD mRNA positive cells were identified in the posterior region of the epiblast (Fig. 2E). GAPDH dendrimers labeled most cells throughout the embryo. The lack of detection of GAPDH in every cell in paraffin sections could reflect an absence of ubiquitous expression at this stage of development or limited accessibility of dendrimers to mRNA in some cells. Only 1-3 dendrimers to myosin (Fig. 2F) or those lacking a specific recognition sequence bound to the entire embryo.
Confirmation of myogenic potential of cells labeled with MyoD dendrimers
To confirm that MyoD dendrimers were labeling cells with myogenic potential, MyoD mRNA positive cells were isolated from gastrulating embryos and placed in culture. To isolate the cells expressing MyoD mRNA, epiblasts were labeled with the G8 monoclonal antibody that recognizes a cell surface antigen present on the same cells that bind MyoD dendrimers (9, 16) (Fig. 3A). G8/MyoD positive and negative populations were separated by magnetic cell sorting (9). After 5 days in culture, cells were stained with monoclonal antibodies to sarcomeric myosin (21) and the skeletal muscle specific protein 12101 (18). In G8/MyoD positive cultures, 95% of the cells differentiated into skeletal muscle (Fig. 3B), where as only 4% of the G8/MyoD negative cells formed muscle (Fig. 3C) (9). These results demonstrate that dendrimers with a recognition sequence for MyoD mRNA accurately identify cells with myogenic potential.
Fig. 3. Isolation and differentiation of cells that express MyoD mRNA in the stage 4 embryo.
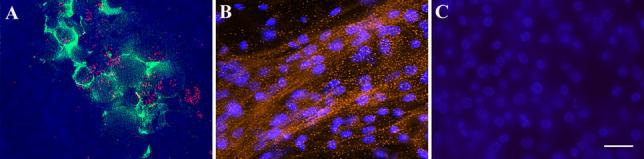
The stage 4 embryo was double labeled with MyoD dendrimers (red) and the G8 monoclonal antibody followed by a secondary antibody conjugated with Alexa 488 (green). Nuclei were labeled with bis-benzamide (blue). Epiblast cells co-expressed the G8 antigen and MyoD mRNA (A). The G8 and MyoD positive and negative populations were isolated by magnetic cell sorting. On the fifth day in culture, cells were stained with the skeletal muscle specific antibody 12101 and a secondary antibody conjugated with rhodamine (red). Most G8 positive cells differentiated into skeletal muscle cells (B), whereas few cells in G8 negative cultures were labeled with the 12101 antibody (C). Bar, 9 μm.
Discussion
The sensitivity and resolution of the in situ hybridization procedure has been enhanced with the use of fluorescent dendrimers as probes for mRNA. Dendrimers revealed the presence of MyoD mRNA two days earlier than previously detected with enzymatic or radiolabeled probes (2-4, 18). Single cells containing a low level of MyoD mRNA were resolved due to the preciseness of the fluorescent signal and the minimal level of background from nonspecific binding.
Messenger RNA can be detected at low levels in whole embryos at early stages of development and in tissue sections. Dendrimers also can be used to localize mRNA in cultured cells. The ability to label dendrimers with a variety of fluorochromes permits the localization of different mRNAs in the same tissue section. Dendrimers also can be tagged with 32P, biotin, and digoxigenin. Furthermore, in situ hybridizations with dendrimers can be combined with antibody labeling to localize mRNA and protein within the same cell. This capability led to the discovery of a cell surface antibody for cells expressing MyoD mRNA that was then used to isolate MyoD mRNA positive cells from the embryo and fetal organs (9, 16). The specificity of MyoD dendrimers for cells with myogenic potential was confirmed by demonstrating that the isolated population differentiated into skeletal muscle in culture (9, 16).
The protocol for using dendrimers is straightforward, particularly for those experienced with conventional methods of in situ hybridization. The usual considerations of avoiding RNAase and DNAase contamination, selecting anti-sense oligonucleotide sequences that remain linear, and optimization of hybridization temperature also apply when using dendrimer probes. A unique feature of the dendrimer procedure is the use of pepsin instead of Proteinase K. The appropriate temperature and length of incubation with pepsin must be carefully determined. Exposure to pepsin for 10 minutes at room temperature results in adequate digestion of most tissues for dendrimer penetration and preserves morphology. However, dense tissues, such as liver, require a 12-minute treatment with pepsin at room temperature, while the more fragile tissues of the early embryo are treated for only 5 minutes at 4°C.
Enzymatically labeled oligonucleotide probes that are smaller than dendrimers are particularly useful when performing whole mount in situ hybridization with older embryos and thicker tissues. While enzymatic probes are capable of amplifying the signal, resolution can be compromised due to the diffusion of the reaction product and accumulation of background. Neither dendrimers nor enzymatically labeled oligonucleotides are quantitative in tissue sections; however, relative comparisons of signal intensity can be made with both types of probes. Although the sensitivity of dendrimers needs to be determined, conceivably, a single mRNA could be detected with dendrimers due to the number of fluors present on each molecule.
The most significant advantages of using fluorescent dendrimers for in situ hybridization are their low level of background from nonspecific binding and pinpoint emission from the fluorescent tag. A single labeled cell can be detected among thousands of non-expressing cells. Observing the “needle in the haystack” is not only critical for determining the onset of gene expression in a subpopulation of cells within the embryo, but for detecting stem cells in adult tissues and early metastasis of cancer cells.
Acknowledgments
We thank Steven Deluca, Robert Strony and David Beckmann for their technical assistance. These studies were supported by the National Institute of Health HD043157, and HD36650-01 to M. George-Weinstein.
Appendix
Protocols
Solutions
Hybridization buffer
30ml Deionized Formamide (Sigma)
5ml 20X SSC Buffer (Sigma)
345mg Sodium Phosphate (Sigma)
2.5g Dextran Sulfate (Sigma)
pH to 7.4 then add Nuclease Free Water (Ambion) to a final volume of 50ml
Store at 4°C in the dark
DNA stock solution
10mg Sonicated Salmon Sperm (SSS) DNA (Sigma) in 1ml Nuclease Free Water
Warm slightly or sonicate to get DNA into solution
Store at -20°C
Dilute 1:20 in Hybridization Buffer before use
Final concentration when diluted is 0.5mg/ml
RNA stock solution
10mg Yeast RNA (Boehringer Mannheim) in 1ml Nuclease Free Water
Warm slightly or sonicate to get RNA into solution
Store at -20°C
Dilute 1:20 in Hybridization Buffer before use
Final concentration when diluted is 0.5mg/ml
60% formamide in 2X SSC buffer
60mls Deionized Formamide
10mls 20X SSC
30mls Nuclease Free Water
Store at room temperature in the dark.
Myo medium
84mls DMEM (Invitrogen)
5mls Fetal Bovine Serum (Invitrogen)
5mls Horse Serum (Invitrogen)
5mls Chick Embryo Extract (22)
1ml Penicillin and Streptomycin 10,000 IU (Cellgro)
Equipment list
37°C Incubator
80°C Oven
Microtome
Procedure for in situ hybridization with 3DNA™ dendrimers
A. In situ hybridization with paraffin sections
1. Sectioning paraffin embedded tissues
All solutions are prepared with nuclease free water (Ambion).
Tissue embedded in paraffin is sectioned at 10μm and placed in 3-well Teflon printed slides (Electron Microscopy Science) coated with 0.2% gelatin in nuclease free water.
Place one section in each well.
2. Deparaffinizing, hydrating, and dehydrating sections
Xylene and ethanol rinses are carried out in glass staining dishes.
All solutions are prepared with nuclease free water.
Warm slides in a 80°C oven for 1 minute to melt the wax. This will help the tissue adhere to the slides.
Place slides in xylene for 10 minutes.
Place slides in fresh xylene for an additional 10 minutes.
Rinse in 100% ethanol for 2 minutes.
Hydrate tissue by placing slides in a graded series of ethanol: 90%, 80%, 70%, 50%, and 30%, 5 minutes in each solution.
Rinse slides with 0.85% NaCl, 100μl per well, for 10 minutes. To remove solutions, tap base of slide on a paper towel.
Rinse in 100μl PBS for 1 minute.
Fix tissue with 100μl 4% formaldehyde in PBS for 20 minutes.
Dehydrate tissue in 70%, 90% and 100% ethanols, 5 minutes in each solution.
3. Prehybridization
All solutions are prepared with nuclease free water.
Treat slides with 100μl 0.1%Triton X-100 for 10 minutes.
Rinse slides with 100μl PBS for 1 minute.
Incubate with 0.1% Pepsin (Boehringer Manheim) in 0.01M HCl, 33μl per well, for 5 to 10 minutes at room temperature or 4°C, depending on the tissue.
Rinse with 33μl PBS for 1 minute.
Postfix with 33μl 1% formaldehyde in PBS for 10 minutes
Dehydrate tissue in 70%, 90%, and 100% ethanols, 5 minutes in each solution.
Air dry slides.
4. Hybridization
All solutions are prepared with nuclease free water
Place yeast RNA into 80°C oven for 10 minutes.
-
To 500μl of Hybridization buffer add:
25μl of SSS DNA stock solution
25μl of Yeast RNA stock solution
Dilute dendrimers in hybridization buffer containing DNA and RNA to a final concentration of 3pg/μl.
Add 33μl of dendrimer solution to each well.
Cover with 22mm square, siliconized coverslips (Hampton Research), one per well. Do not use nonsiliconized coverslips.
Place slides in a 100mm petri dish on filter paper soaked with 20X SSC buffer to create a humid chamber.
Place dish in 80°C oven for 10 minutes.
Place dish in a 37°C incubator and incubate overnight.
Place slides in 100mm petri dish with 15mls PBS for 15 minutes to float coverslips off the well and then gently remove them from slides.
Incubate slides in 60% formamide in 2X SSC buffer for 10 minutes.
Counterstain with Bis-Benzamide, 1μg/ml deionized water, for 5 minutes.
Place 1 drop of Gelmount medium (Fisher) on each slide well and apply one 22 x 50mm coverslip over all three wells.
After one hour, seal edges of coverslip with nail polish. Store in dark. For best results let slide sit overnight before viewing. View within one week of preparation.
B. In situ hybridization with whole embryos
All solutions are prepared with nuclease free water.
Rinse embryo in PBS.
Place embryo on a 1mm nucleopore filter (Costar).
Transfer embryo to a 1 well Teflon printed slide coated with 0.2% gelatin.
Fix embryo in 100μl 4% formaldehyde for 20 minutes.
Dehydrate embryo in 100μl 70%, 90%, and 100% ethanols, 5 minutes in each solution.
Follow the same procedure as above for the remainder of the protocol, beginning with the Triton X-100 step during prehybridization. Use 100μl of solution for each step.
Remove the embryo from the nucleopore filter just prior to placing in 100% ethanol during the prehybridization step. Position the embryo in the center of the slide for air drying. DO NOT coverslip the embryo during the overnight hybridization step.
References
- George-Weinstein M, Gerhart JV, Reed R, Flynn J, Callihan B, Mattiacci M, Miehle M, Foti G, Lash JW, Weintraub H. Skeletal myogenesis: the preferred pathway of chick embryo epiblast cells in vitro. Dev Biol. 1996;173:279–291. doi: 10.1006/dbio.1996.0023. [DOI] [PubMed] [Google Scholar]
- Sassoon D, Lyon G, Wright WE, Lin V, Lassar A, Weintraub H, Buckingham M. Expression of two myogenic regulatory factors myogenin and MyoD1 during mouse embryogenesis. Nature. 1989;341:303–307. doi: 10.1038/341303a0. [DOI] [PubMed] [Google Scholar]
- Pownall ME, Emerson CP. Sequential activation of three myogenic regulatory genes during somite morphogenesis in quail embryos. Dev Biol. 1992;151:67–79. doi: 10.1016/0012-1606(92)90214-2. [DOI] [PubMed] [Google Scholar]
- Charles de la Brousse F, Emerson CP. Localized expression of a myogenic regulatory gene, qmf1 in the somite dermatome of avian embryos. Genes Dev. 1990;4:567–581. doi: 10.1101/gad.4.4.567. [DOI] [PubMed] [Google Scholar]
- Gerhart JV, Baytion M, DeLuca S, Getts R, Lopez C, Niewenhuis R, Nilsen TW, Olex S, Weintraub H, George-Weinstein M. DNA dendrimers localize MyoD mRNA in presomitic tissues of the chick embryo. J Cell Biol. 2000;149:825–833. doi: 10.1083/jcb.149.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen T, Grayzel J, Prensky W. Dendritic nucleic acid structures. J Theor Biol. 1997;187:273–284. doi: 10.1006/jtbi.1997.0446. [DOI] [PubMed] [Google Scholar]
- Volgelbacker HH, Getts RC, Tian N, Labaczewski R, Nilsen T. DNA dendrimers: assembly and signal amplification. Proc Am Chem Soc. 1997;76:458–460. [Google Scholar]
- Wang J, Jiang M, Nilsen TW, Getts RC. Dendritic nucleic acid probes for DNA biosensors. J Am Chem Soc. 1998;120:8281–8282. doi: 10.1021/ja980619p. [DOI] [Google Scholar]
- Gerhart JV, Neely C, Stewart B, Perlman J, Beckman D, Wallon M, Knudsen K, George-Weinstein M. Epiblast cells that express MyoD recruit plutipotent cells to the skeletal muscle lineage. J Cell Biol. 2004;164:739–746. doi: 10.1083/jcb.200309152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechesne CA, Wei J, Eldridge L, Gannoun-Zaki P, Millasseau L, Bougueleret D, Caterina D, Paterson BM. E-box and MEF-2 independent muscle-specific expression, positive autoregulation, and cross-activation of the chicken MyoD (CMD1) promoter reveal an indirect regulatory pathway. Mol Cell Biol. 1994;14:869–877. doi: 10.1128/mcb.14.8.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugaiczyk A, Haron JR, Stone EM, Dennison OE, Rothblum KN, Schwartz RJ. Cloning and sequencing of a deoxyribonucleic acid copy of a glyceraldehydes-3-phosphate dehydrogenase messenger ribonucleic acid isolated from chicken muscle. Biochemistry. 1983;22:1605–1613. doi: 10.1021/bi00276a013. [DOI] [PubMed] [Google Scholar]
- Freyer GC, Robbins J. The analysis of a chicken myosin heavy chain cDNA clone. J Biol Chem. 1983;258:7149–7154. [PubMed] [Google Scholar]
- Sassoon D, Rosenthal N. Detection of messenger RNA by in situ hybridization. Methods Enzymol. 1993;225:384–404. doi: 10.1016/0076-6879(93)25027-y. [DOI] [PubMed] [Google Scholar]
- Raap KA, van de Rijke FM, Dirks RW. MRNA in situ hybridization to in vitro cultured cells. In: Choo KHA editor. Method in Molecular Biology . Vol 33. Totowa, NJ: Humana Press; 1994. p.293-300. [DOI] [PubMed]
- Hamburger V, Hamilton HL. A series of normal stages in development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Gerhart JV, Bast B, Neely C, Iem S, Amegbe P, Niewenhuis R, Miklasz S, Cheng PF, George-Weinstein M. MyoD-positive myoblast are present in mature fetal organs lacking skeletal muscle. J Cell Biol. 2001;155:381–391. doi: 10.1083/jcb.200105139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George-Weinstein M, Gerhart JV, Foti G, Lash JW. Maturation of myogenic and chondrogenic cells in the presomitic mesoderm of the chick embryo. Exp Cell Res. 1994;211:263–274. doi: 10.1006/excr.1994.1086. [DOI] [PubMed] [Google Scholar]
- Kitner CR, Brockes JP. Monoclonal antibodies identify blastemal cells derived from dedifferentiating muscle in newt limb regeneration. Nature. 1984;308:67–69. doi: 10.1038/308067a0. [DOI] [PubMed] [Google Scholar]
- Ott MO, Bober E, Lyons G, Arnold H, Buckingham M. Early expression of the myogenic regulatory gene, myf-5 in precursor cells of skeletal muscle in the mouse embryo. Development. 1991;111:1097–1107. doi: 10.1242/dev.111.4.1097. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Vivarelli E, Cusella-De Angelis G, Rocancourt D, Buckingham M, Cossu G. A population of myogenic cells derived from the mouse neural tube. Neuron. 1994;13:813–821. doi: 10.1016/0896-6273(94)90248-8. [DOI] [PubMed] [Google Scholar]
- Bader D, Masakki T, Fischman DA. Immunochemical analysis of myosin heavy chain during avian embryogenesis in vivo and in vitro. J Cell Biol. 1995;95:763–770. doi: 10.1083/jcb.95.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLullo C, Gerhart JV, George-Weinstein M. Preparation of chick-striated muscle cultures. In: Tuan R and Lo C, Editors, Methods in Molecular Biology, Humana Press, Totowa, NJ. 2000; 137: 337-349. [DOI] [PubMed]