

Abstract
In eukaryotic ribosome biogenesis, U3 snoRNA base pairs with the pre-rRNA to promote its processing. However, U3 must be removed to allow folding of the central pseudoknot, a key feature of the small subunit. Previously, we showed that the DEAH/RHA RNA helicase Dhr1 dislodges U3 from the pre-rRNA. DHR1 can be linked to UTP14, encoding an essential protein of the preribosome, through genetic interactions with the rRNA methyltransferase Bud23. Here, we report that Utp14 regulates Dhr1. Mutations within a discrete region of Utp14 reduced interaction with Dhr1 that correlated with reduced function of Utp14. These mutants accumulated Dhr1 and U3 in a pre-40S particle, mimicking a helicase-inactive Dhr1 mutant. This similarity in the phenotypes led us to propose that Utp14 activates Dhr1. Indeed, Utp14 formed a complex with Dhr1 and stimulated its unwinding activity in vitro. Moreover, the utp14 mutants that mimicked a catalytically inactive dhr1 mutant in vivo showed reduced stimulation of unwinding activity in vitro. Dhr1 binding to the preribosome was substantially reduced only when both Utp14 and Bud23 were depleted. Thus, Utp14 is bifunctional; together with Bud23, it is needed for stable interaction of Dhr1 with the preribosome, and Utp14 activates Dhr1 to dislodge U3.
INTRODUCTION
Ribosomes are complex ribonucleoprotein (RNP) particles composed of an intricate assembly of folded rRNA interdigitated with ribosomal proteins (r-proteins). Faithful assembly of this RNP is critical for efficient and accurate cellular translation. Eukaryotic cells devote more than 200 trans-acting factors (1–3) to the assembly of ribosomes. This highly dynamic process (4) involves extensive structural rearrangements of RNA-RNA, RNA-protein, and protein-protein interactions (5, 6). To investigate how structural rearrangements are regulated to occur at the proper stage of assembly, our studies centered on the RNA helicase Dhr1, which dissociates a key RNA-RNA interaction, dislodging U3 snoRNA from the pre-rRNA of the preribosome (1, 3, 7).
U3 is instrumental in orchestrating early pre-rRNA processing. Ribosome assembly in Saccharomyces cerevisiae begins with the cotranscriptional assembly of the 90S preribosome, a large complex of r-proteins and trans-acting factors, including proteins and snoRNAs, on the growing 5′ end of the nascent 35S transcript (8–10). The pre-rRNA undergoes extensive modification and processing (11) to liberate the mature 18S, 5.8S, and 25S rRNAs that are embedded between transcribed spacer elements. U3 snoRNA is required for the early cleavages at sites A0 and A1 to generate the mature 5′ end of 18S and cleavage at A2 to separate the pre-40S (containing 20S pre-rRNA) from the pre-60S subunits (12, 13). Under conditions in which cleavage at A2 is blocked or reduced, cleavage at A3 can generate the pre-60S precursor. Thus, U3 is required for biogenesis of the small but not the large subunit.
U3 is also expected to promote proper rRNA folding. U3 hybridizes with the pre-rRNA at multiple sites within the 35S precursor: at sites in the 5′ external transcribed spacer, the 5′ end of 18S, and additional positions within 18S (12–17). The central pseudoknot (CPK) is a key architectural feature of the small subunit (SSU) that physically coordinates the four rRNA domains and sets up the decoding center. The CPK is formed when nucleotides in the loop of the first stem-loop structure of 18S (nucleotides [nt] 4 to 20) base pair with nt 1137 to 1144 of 18S. In the preribosome, U3 is expected to base pair with these elements of the CPK based on genetic and phylogenetic covariation analysis (14, 17). However, pre-rRNA hybridization with U3 blocks the folding of the CPK. Thus, removal of U3 from the preribosome is a prerequisite for, and perhaps promotes, CPK formation. We have recently shown that the essential DEAH/RHA RNA helicase Dhr1 is responsible for removing U3 from the pre-rRNA to allow folding of the CPK (18). How Dhr1 activity is regulated to ensure its activity at the correct point in folding and assembly of the SSU is addressed herein.
Most RNA helicases lack intrinsic regulation. Rather, they contain ancillary domains flanking the helicase core that provide binding sites for regulatory cofactors (19). DEAH/RHA helicases commonly use G-patch proteins as accessory factors for activation of enzymatic activity (20). For example, the yeast spliceosomal helicase Prp2 requires the G-patch protein Spp2, and Prp43, involved in ribosome biogenesis and spliceosome disassembly, is stimulated by at least three different G-patch proteins (Ntr1, PfaI, and Gno1) that recruit Prp43 to its respective substrates (reviewed in reference 20).
To identify regulators of Dhr1, we explored candidate proteins that interact with it. The methyltransferase Bud23 modifies the guanosine base at position 1575 in 18S rRNA (21, 22). Bud23 is recruited to the preribosome at a relatively late step in 40S biogenesis, before A2 cleavage, and remains associated with the pre-40S particle in the nucleus (21, 23). We took advantage of the slow-growth defect of bud23 mutants as a genetic entry point to dissect a constellation of functionally linked proteins. We found that mutations in DHR1, UTP2, or UTP14 partially suppressed the growth defect and 40S assembly defect of bud23Δ mutants (18, 21, 23). We also showed that Bud23 binds to the N-terminal extension of Dhr1, making Bud23 a candidate for recruiting or regulating Dhr1 (24). However, disruption of the interaction between Bud23 and Dhr1 had no observed effect on cell growth, leaving open the question of how Dhr1 is regulated.
In this study, we have turned our attention to a second functional interactor of bud23Δ, UTP14. Utp14 is an essential and highly conserved protein of uncharacterized function initially identified as a component of the 90S preribosome (8). Interestingly, dysfunction of one of the copies of the tissue-specific allele of Utp14 causes infertility in men and may contribute to scleroderma and ovarian cancer (reviewed in reference 25). We show that Utp14 interacts directly with Dhr1 to activate its unwinding activity. Our data suggest that Utp14 activates Dhr1 and together with Bud23 recruits this helicase to the assembling preribosome.
MATERIALS AND METHODS
Strains, plasmids, and media.
All yeast strains used in this study are listed in Table 1. Yeast were grown at 30°C in YEPD (2% peptone, 1% yeast extract, 2% dextrose) or YEPgal (2% peptone, 1% yeast extract, 1% galactose) or synthetic dropout (SD) medium containing 2% glucose, unless otherwise indicated. All plasmids and oligonucleotides used in this study are listed in Tables 2 and 3, respectively.
TABLE 1.
Strains used in this work
| Strain | Genotype | Source or reference |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Open Biosystems |
| PJ69–4α | MATα trp1-901 leu2-3,112 ura3-52 his3Δ200 gal4Δ gal80ΔLYS2::GAL1-HIS3 GAL2-ADE met2::GAL7-LACZ | 26 |
| AJY2161 | MATa bud23Δ::KanMX6 his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 | 22 |
| AJY3711 | MATa KanMX6-PGAL1-3×HA-Dhr1 his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | 18 |
| AJY3243 | MATa KanMX6-PGAL1-3×HA-UTP14 his3Δ1 leu2Δ0 ura3Δ0 | This study |
| AJY3245 | MATa KanMX6-PGAL1-3×HA-UTP14 bud23Δ::Natr his3Δ1 leu2Δ0 ura3Δ0 | This study |
| AJY3243 | MATa KanMX6-PGAL1-3×HA-UTP14 his3Δ1 leu2Δ0 ura3Δ0 | This study |
| L40 | MATa his3Δ200 trp1-901 112 ade2 LYS2::(LexAop)4-His3 URA3 (lexAop)8-LacZ Gal4 | 27 |
TABLE 2.
Plasmids used in this work
| Plasmid | Description | Source or reference |
|---|---|---|
| pACT2 | GAL4AD-HA LEU2 2μ | Clontech |
| pAJ1918 | utp14A758G URA3 CEN ARS | 23 |
| pAJ1919 | UTP14 URA3 CEN ARS | 23 |
| pAJ2311 | DHR1-13myc LEU2 CEN ARS | 24 |
| pAJ2312 | pET21a-DHR1-6×His | 18 |
| pAJ2321 | GAL4AD-HA-UTP14 LEU2 2μ | This study |
| pAJ2330 | GAL4BD-c-myc-dhr11–838 TRP1 2μ | This study |
| pAJ2331 | GAL4BD-c-myc-dhr1319–838 TRP1 2μ | This study |
| pAJ2332 | GAL4BD-c-myc-dhr1319–1267 TRP1 2μ | This study |
| pAJ2334 | GAL4AD-HA-utp141–706 LEU2 2μ | This study |
| pAJ2335 | GAL4AD-HA-utp14707–899 LEU2 2μ | This study |
| pAJ2341 | GAL4AD-HA-utp141–813 LEU2 2μ | This study |
| pAJ2342 | GAL4AD-HA-utp141–654 LEU2 2μ | This study |
| pAJ2343 | GAL4AD-HA-utp141–564 LEU2 2μ | This study |
| pAJ2344 | GAL4AD-HA-utp141–265 LEU2 2μ | This study |
| pAJ2345 | GAL4AD-HA-utp14266–899 LEU2 2μ | This study |
| pAJ2346 | GAL4AD-HA-utp14565–899 LEU2 2μ | This study |
| pAJ2347 | GAL4AD-HA-utp14655–899 LEU2 2μ | This study |
| pAJ2394 | LexABD-c-myc-DHR1 TRP1 2μ | This study |
| pAJ2593 | DHR1 URA3 CEN ARS | 24 |
| pAJ2795 | GAL4AD-HA-dhr1467–1267 LEU2 2μ | 24 |
| pAJ2796 | GAL4AD-HA-dhr11–142 LEU2 2μ | 24 |
| pAJ2797 | GAL4AD-HA-dhr11–467 LEU2 2μ | 24 |
| pAJ2922 | GAL4BD-c-myc-DHR1 | 24 |
| pAJ3081 | dhr1K420A-13myc LEU2 CEN ARS | 18 |
| pAJ3132 | utp14I755T URA3 CEN ARS | This study |
| pAJ3136 | utp14E757G URA3 CEN ARS | This study |
| pAJ3145 | utp14V754G URA3 CEN ARS | This study |
| pAJ3145 | utp14A760P URA3 CEN ARS | This study |
| pAJ3263 | GAL4AD-HA-utp14D753A/V754A/I755A/E757A/F759A LEU2 2μ | This study |
| pAJ3264 | utp14V754G/I755T/E757G/A758G/A760P URA3 CEN ARS | This study |
| pAJ3265 | GAL4AD-HA-utp14Δ719–780 LEU2 2μ | This study |
| pAJ3266 | GAL4AD-HA-utp14V754G/I755T/E757G/A758G/A760P LEU2 2μ | This study |
| pAJ3267 | utp14Δ719–780 URA3 CEN ARS | This study |
| pAJ3276 | utp14D753A/V754A/I755A/E757A/F759A URA3 CEN ARS | This study |
| pAJ3307 | pET16a-6×His-UTP14 | This study |
| pAJ3308 | UTP14-GFP URA3 CEN ARS | This study |
| pAJ3309 | utp14V754G/I755T/E757G/A758G/A760P-GFP URA3 CEN ARS | This study |
| pAJ3310 | utp14D753A/V754A/I755A/E757A/F759A-GFP URA3 CEN ARS | This study |
| pAJ3313 | utp14Δ719–780M-GFP URA3 CEN ARS | This study |
| pAJ3314 | pET16a-6×His-utp14Δ719-780 | This study |
| pAJ3315 | pET16a-6×His-utp14D753A/V754A/I755A/E757A/F759A | This study |
| pAJ3331 | 6×His-UTP14 URA3 CEN ARS | This study |
| pAJ3421 | utp141–813 URA3 CEN ARS | This study |
| pAJ3422 | utp141–706 URA3 CEN ARS | This study |
| pAJ3423 | utp141–654 URA3 CEN ARS | This study |
| pAJ3424 | utp141–564 URA3 CEN ARS | This study |
| pAJ3425 | utp141–265 URA3 CEN ARS | This study |
| pAJ3426 | utp14266–899 URA3 CEN ARS | This study |
| pAJ3427 | utp14565–899 URA3 CEN ARS | This study |
| pAJ3428 | utp14655–899 URA3 CEN ARS | This study |
| pAJ3429 | utp14707–899 URA3 CEN ARS | This study |
| pAJ3455 | dhr1319–1267 URA3 CEN ARS | This study |
| pAJ3456 | dhr1467–1267 URA3 CEN ARS | This study |
| pAJ3457 | dhr11–838 URA3 CEN ARS | This study |
| pAJ3458 | dhr11–142 URA3 CEN ARS | This study |
| pAJ3459 | dhr11–467 URA3 CEN ARS | This study |
| pAJ3460 | GAL4BD-c-myc-dhr1584–1267 TRP1 2μ | This study |
| pAJ3461 | GAL4BD-c-myc-dhr1839–1267 TRP1 2μ | This study |
| pAJ3470 | dhr1319–838 URA3 CEN ARS | This study |
| pGBKT7 | GAL4BD-c-myc TRP1 2μ | Clontech |
TABLE 3.
Oligonucleotides used in this work
| Oligonucleotide | Target(s) | Sequence |
|---|---|---|
| AJO603 | 35S, 27SA2, 23S, 21S | TGTTACCTCTGGGCCCCGATTG |
| AJO130 | 20S | TCTTGCCCAGTAAAAGCTCTCATGC |
| AJO962 | U2 | GCGACCAAAGTAAAAGTCAAGAACGACTCCACAAGTGCGAGGGTCGCGAC |
| AJO2194 | U3 | CTCATCAACCAAGTTGGATTCAGTGGCTC |
| AJO944 | Utp14 | CATAAACTGGACAGTGTTTTCTTCAGACTTTTTATG |
| AJO1399 | Utp14 | ATCACCCATCTCACTCAACTTATCC |
Yeast two-hybrid assays.
Wild-type (WT) and mutant DHR1 and UTP14 were cloned into either pACT2, pGBKT7, or pAJ2394 two-hybrid vectors. These plasmids were transformed into PJ69-4α (26) or L40 (27) and selected on SD medium lacking leucine and tryptophan (SD Leu− Trp− medium). The transformants were then patched on SD Leu− Trp− His− with or without 3-amino-1,2,4-triazole (3AT) to test for activation of UASGAL-HIS3 or (LexAop)4-HIS3 reporter genes.
Screen to identify additional mutants of UTP14 as suppressors of the bud23Δ mutant.
UTP14 was amplified by PCR using oligonucleotides AJO944 and AJO1399 with Taq DNA polymerase. The PCR product was cotransformed with HpaI and AatII-digested pAJ1919 into the bud23Δ mutant strain (AJY2161). The transformants were selected on SD Ura− medium at 30°C. Plasmids were isolated from fast-growing clones and sequenced (24).
Northern blot analysis.
Total RNA was prepared from yeast cells grown at 30°C to a density of ∼1 × 107 cells/ml using the hot acidic phenol method (28). For RNA isolation from sucrose gradient fractions, 1/10 volume of 20% SDS, 1/10 volume of 3 M sodium acetate (pH 5.5), and 2.375 volumes of 100% ethanol were added to each fraction. Samples were stored at −20°C for 24 h before centrifugation at 14,000 rpm at 4°C for 30 min. The RNA pellet was washed once with 70% ethanol and resuspended in H2O. Northern blotting was performed as described previously (28) using oligonucleotide probes listed in Table 3. The hybridization signals were detected by phosphorimaging on a Typhoon FLA9500.
Sucrose density gradient sedimentation.
Cells were grown at 30°C to a density at of ∼1 × 107 cells/ml. Cycloheximide was added to a final concentration of 100 μg/ml, and the cultures were incubated at 30°C shaker for 10 min. The cells were then poured onto ice and collected by centrifugation. Sucrose density gradient ultracentrifugation was done as described previously (24). To precipitate proteins from fractions, trichloroacetic acid (TCA) was added to a 10% final concentration and samples were stored at −20°C for 24 h. Precipitated proteins were collected by centrifugation, resuspended in 1× Laemmli buffer, and heated at 99°C for 3 min prior to separation on 8% SDS-PAGE gels. Proteins were transferred to a nitrocellulose membrane and subjected to Western blot analysis.
Immunoprecipitation.
Cells were grown in 300 ml of Ura− Leu− galactose medium to an optical density at 600 nm (OD600) of 0.1, followed by the addition of 2% glucose and continued growth for 6 h. Cells were harvested and extracts prepared in IP buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1.5 mM MgCl2, 0.15% NP-40, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml of leupeptin, 1 μg/ml of pepstatin A) by vortexing with glass beads and clarified by centrifugation at 14,000 × g at 4°C. Placental RNase inhibitor (New England BioLabs) was added, and samples were incubated with protein G magnetic beads (New England BioLabs) prebound with anti-green fluorescent protein (anti-GFP) antibody for 2 h at 4°C. The beads were washed with IP buffer, and proteins and RNA were extracted with equal volumes of LETS (10 mM Tris-HCl [pH 7.4], 100 mM LiCl, 10 mM EDTA, and 0.2% SDS) and acid-phenol-chloroform. RNA was precipitated from the aqueous phase with ethanol, and protein was precipitated from the organic phase with acetone.
Western blot analysis.
Primary antibodies used in this study were polyclonal rabbit anti-GFP antibody (1:5,000; Rout Lab), anti-c-myc monoclonal antibody (9e10, 1:10,000; Biolegend), polyclonal anti-Mpp10 antibody (1:10,000), polyclonal guinea pig anti-Imp4 antibody (1:3,000; Baserga Lab), and monoclonal mouse antihemagglutinin (anti-HA) antibody (1:5,000). Secondary antibodies used were polyclonal goat anti-mouse, goat anti-rabbit, and goat anti-guinea pig horseradish peroxidase (HRP)-coupled antibodies (1:30,000), and immunodetection was performed with ECL solution (Thermo Scientific).
Protein expression and purification.
Dhr1 and Dhr1D516A/E517A were expressed and purified as described before (18). His6-Utp14, His6-Utp14multi-Ala, and His6-Utp14Δ719-780 were expressed from pAJ3307, pAJ3315, and pAJ3314, respectively, overnight at 15°C in BL21 Star (DE3) (Life Technologies) cells supplemented with a vector expressing the tRNA genes argU, ileY, and leuW. Cells were washed once and resuspended with extraction buffer (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 10% [vol/vol] glycerol, 5 mM β-mercaptoethanol [BME], 7 U/ml of RNase A, and 10 U/ml of RNase I). The extensive RNase treatment ensured the removal of tightly bound RNA. A French press was used to lyse cells, and cell extracts were clarified for 10 min at 10,000 × g followed by 30 min at 50,000 × g. The supernatant was loaded on nickel-nitrilotriacetic acid (Ni-NTA) resin (Invitrogen) and washed once with extraction buffer without RNase. The resin was then resuspended with 3 column volumes (CV) of extraction buffer and incubated for 15 min. The resin was washed extensively with extraction buffer without RNase, and protein was eluted with extraction buffer in which NaCl was replaced with 250 mM imidazole. Fractions containing Utp14 were pooled, supplemented with 1 mM dithiothreitol (DTT), and applied to a CM Hitrap column (GE Healthcare Life Sciences). The column was washed with buffer A (30 mM Tris [pH 8.0], 5% [vol/vol] glycerol, 5 mM sodium acetate, and 1 mM DTT). Protein was eluted with a 21-CV gradient from 0% to 60% buffer B (buffer A plus 1 M NaCl). Utp14-containing fractions were pooled, dialyzed (30 mM Tris [pH 8.0], 10% [vol/vol] glycerol, 5 mM sodium acetate, 150 mM NaCl, and 1 mM DTT), and concentrated to ∼5 μM. Aliquots were flash-frozen and stored at −80°C. The yield for WT and mutant Utp14 was approximately 1 mg/liter.
Preparation of Dhr1-Utp14 complex.
All reactions were performed at room temperature (RT). A 2 μM concentration of Dhr1 in the presence or absence of 2 μM Utp14 (WT or mutant) was preincubated for 5 min, and then bis(sulfosuccinimidyl) suberate (BS3) was added to a concentration of 2 mM. The reaction mixtures were incubated for 30 min, and then the reaction was stopped by addition of 200 mM Tris (pH 8.0). The reaction mixture was analyzed by SDS-PAGE or mass spectrometry.
Mass spectrometric analysis.
Protein samples were subjected to analysis by surface-enhanced laser desorption and ionization–time of flight (SELDI-TOF) mass spectrometry to examine the distribution of masses of protein components. Samples were spotted in three successive 2-μl aliquots onto individual spots of a ProteinChip H50 array (Bio-Rad) and allowed to partially air dry for 1 h or overnight between applications. The chip was then washed three times with 5 μl of distilled water and with 5% MeOH and air dried prior to the addition of 3 μl of 10-mg/ml sinapinic acid (Bio-Rad) in 60% acetonitrile–0.1% formic acid. Controls omitting the air drying for multiple applications of the sample as well as omitting the formic acid with matrix crystallization indicated no difference in the distribution of multimers (data not shown). Samples were analyzed using a Bio-Rad ProteinChip System 4000 Enterprise mass spectrometer calibrated against Bio-Rad's protein standards. The acquisition mode was positive with a source at 25 kV. The matrix attenuation was set up as 500 Da. The sampling rate was 800 MHz. Each spot was divided into 10 partitions, with 210 shots/partition, and the data were collected at a laser energy of 3,300 nJ with a mass range between 10 and 200 kDa.
RNA substrates.
U3 snoRNA was transcribed as previously described (29, 30), purified by gel electrophoresis, and refolded. ETS2 (5′-GGA UUU GGU GG-3′) was purchased from IDT. The 5′ end of ETS2 was phosphorylated with [γ-32P]ATP (PerkinElmer; 150 μCi/μl) by using T4 polynucleotide kinase (New England BioLabs). Radiolabeled ETS2 was purified by gel electrophoresis, precipitated by ethanol, resuspended in water, and stored at −20°C. Poly(A) was purchased from Sigma.
ATPase assay.
ATPase assays were performed as described before (18), except that some reaction mixtures included 0.5 μM Utp14 and final reaction concentrations were 1 mM ATP, 1 mM MgCl2, 32 mM Tris (pH 8), 60 mM NaCl, 4% glycerol, 2 mM sodium acetate, and 2.4 mM DTT.
Unwinding reactions.
All unwinding reactions were performed at RT. For pre-steady-state reactions, to form the U3-ETS2 duplex, bottom-strand U3 snoRNA was incubated for 20 min at 45°C, then refolded on ice for 10 min, and subsequently annealed with 32P-ETS2 at RT for 10 min. The reaction mixture was further incubated for 10 min with buffer supplemented with MgCl2 at RT. The preformed U3-ETS2 duplex was incubated with Dhr1 (WT or mutant) in the presence or absence of Utp14 (WT or mutant) for 5 min, and reactions were initiated by rapid addition of a 1 mM concentration of a mixture of ATP and MgCl2. The final concentrations were 1 mM ATP, 1.5 mM MgCl2, 1.5 nM bottom-strand RNA, ≤0.3 nM 32P-ETS2, and 50 nM Dhr1 in the presence or absence of 200 nM Utp14, 20 to 35 mM Tris (pH 8.0), 50 mM NaCl, 7% (vol/vol) glycerol, 2.7 mM DTT, 0.8 U/μl of RNasin, and 0.2 mg/ml of bovine serum albumin (BSA). Reaction mixtures were sampled, and reactions were quenched by addition of one-half volume of stop buffer (150 mM Tris [pH 8.0], 0.3% [wt/vol] SDS, and 150 mM EDTA). The amounts of duplex and single-stranded RNAs for both of the above helicase reactions were resolved by electrophoretic mobility shift assays (EMSAs).
RESULTS
Utp14 and Dhr1 interact by yeast 2-hybrid analysis.
To examine the interaction between Dhr1 and Utp14, we performed two-hybrid studies and detected a robust interaction between these proteins (Fig. 1A). To narrow down the regions of Dhr1 needed for this interaction, we first made a series of truncations. Dhr1 can be divided into functional domains based on sequence homology and by extension of structural homology to Prp43 (31, 32) (Fig. 1A), which we used to construct a model (see Fig. S1 in the supplemental material). Previous studies showed that the N-terminal domain (amino acids [aa] 1 to 356) contains a binding site for Bud23 (24). This domain is followed by the helicase core (aa 375 to 839) composed of tandem RecA-like domains, designated RecA1 and RecA2. The core is followed by a winged-helix domain (aa 840 to 905), a ratchet domain (aa 906 to 1056), an OB fold (aa 1057 to 1192), and a small C-terminal extension (aa 1193 to 1267). Deletion of the N-terminal domain of Dhr1 had no effect on its interaction with Utp14 (Fig. 1A). However, deletion of an additional 148 aa, extending into the RecA1 domain, or larger deletions completely eliminated the two-hybrid interaction (Fig. 1A). Likewise, deletion of the winged-helix domain through the C terminus completely eliminated two-hybrid interaction. Western blotting confirmed that all truncations were expressed as well as the wild-type protein (see Fig. S2A in the supplemental material), demonstrating that loss of two-hybrid interaction was not due to poor expression of the mutant proteins. Thus, all but the N-terminal domain of Dhr1 (aa 1 to 318) is needed for two-hybrid interaction with Utp14. The winged-helix domain, ratchet domain, and OB fold are expected to create a base to orient the tandem RecA-like domains. Therefore, a relatively compact region of Utp14 could interact with disparate domains of Dhr1. We also tested these truncations of Dhr1 for their ability to complement loss of Dhr1 (Fig. 1A; see also Fig. S3A). Dhr1 lacking the N terminus weakly complemented loss of Dhr1. Further deletions into the RecA-like domains or deletion of the remaining C-terminal region completely inactivated the protein.
FIG 1.

Mapping the Dhr1-Utp14 interaction by yeast two-hybrid system. (A) Mapping the Dhr1 binding site on Utp14. The middle image is a cartoon of the domain organization of Dhr1. Strain PJ69-4α was transformed with Gal4AD-Utp14 and a second plasmid encoding Gal4BD fusions of either full-length Dhr1, or truncated proteins containing aa 1 to 813, aa 319 to 838, and aa 319 to 1267. The truncated Dhr1 mutant proteins containing aa 1 to 467, aa 1 to 142, and aa 467 to 1267 were fused with Gal4AD and were coexpressed with Gal4BD-Utp14 in PJ69-4α. The transformants were patched on SD medium lacking either leucine and tryptophan (L−W−) or lacking leucine, tryptophan, and histidine (L−W−H−) and incubated for 3 days at 30°C. (B) Mapping the Utp14 binding site on Dhr1. The middle image is a cartoon of the amino acid conservation of Utp14. Multiple-sequence alignment was done with Geneious software. Black, 100% identity; dark gray, 80 to 100% similarity; light gray, 60 to 80% similarity; white, <60% similarity. Strain PJ69-4α was transformed with Gal4BD-Dhr1 and a second plasmid encoding Gal4AD fusions of either full-length Utp14 or truncated Utp14. The transformants were patched on SD L−W−, SD L−W−H−, and SD L−W−H− media with different concentrations of 3AT and incubated for 3 days at 30°C.
In contrast to Dhr1, Utp14 has no structural homologs and thus no identifiable sequence motifs suggesting functions for Utp14. However, there are regions of strong conservation in the N terminus (∼aa 180 to 520) and in the C terminus (∼aa 750 to 895) (Fig. 1B). To narrow down the region of Utp14 that interacts with Dhr1, we made serial N- and C-terminal truncations (Fig. 1B). We tested these in two-hybrid assays with full-length Dhr1 and titrated with 3-aminotriazole (3AT) to better differentiate relative strengths of interaction (26). Utp141-813 and Utp141-706 (deletions of 86 and 193 aa, respectively, from the C terminus) showed increasingly severe reductions in two-hybrid interaction with Dhr1, and larger deletions showed no detectable interaction (Fig. 1B). Utp14266-899 (deletion of 265 aa from the N terminus) had no discernible effect on Dhr1 interaction, whereas Utp14565-899 and Utp14655-899 (deletion of 564 and 654 aa, respectively, from the N terminus) gave increasingly severe losses of interaction. These results show that aa 565 to 813 of Utp14 are necessary for efficient two-hybrid interaction with Dhr1.
We also tested the Utp14 truncations for function. We observed a correlation between the ability of Utp14 variants to interact with Dhr1 and to complement loss of Utp14 (Fig. 1B; see also Fig. S3B in the supplemental material). Only the smallest deletions, expressing aa 1 to 813 or 265 to 899, which retained Dhr1 interaction were able to complement to a modest degree. All other tested truncations were lethal.
Mutations in Utp14 that suppress bud23Δ map to a highly conserved peptide in the C terminus of Utp14.
We found previously that an alanine-to-glycine change at position 758 in Utp14 suppressed the growth and rRNA processing defects of bud23Δ (23). To better define the functional domain of Utp14 important for bud23Δ suppression, we screened for additional suppressing mutations. We randomly mutagenized the entire open reading frame of UTP14 by PCR and recombined the mutant product into a UTP14 expression vector in bud23Δ cells. We assumed that additional suppressing mutations in UTP14 could be identified in the presence of genomic WT UTP14 because UTP14A758G is a dominant suppressor. Therefore, fast-growing colonies were isolated and UTP14 was sequenced to identify suppressing mutations. In cases where multiple mutations were identified, we subcloned these to identify the specific mutation conferring suppression. We identified four additional unique mutations that suppressed bud23Δ: V754G, I755T, E757G, and A760P (Fig. 2A). All single point mutations suppressed bud23Δ to similar extents (Fig. 2B), and all fully complemented loss of Utp14 (Fig. 2C). The clustering of mutations identified a short highly conserved peptide as a functionally important element of Utp14.
FIG 2.

Suppressing mutations map to a highly conserved motif in Utp14. (A) Cartoon showing conservation of amino acid sequence across Utp14 (top) as described in the legend to Fig. 1. Multiple-sequence alignment of the region of Utp14 containing mutations that suppress bud23Δ (bottom) was performed with T-coffee. S cerev, Saccharomyces cerevisiae; A thali, Arabidopsis thaliana; D melan, Drosophila melanogaster; X tropi, Xenopus tropicalis; D rerio, Danio rerio; G gallus, Gallus gallus; H sapiens, Homo sapiens. Positions of single amino acid substitutions that suppress bud23Δ are indicated by triangles. The amino acid changes of Utp14multi-Sup and Utp14multi-Ala are indicated. The consensus sequence of G-patch proteins is shown below the alignment. Perfect matches with invariant residues are shaded in black, and similar residues are shaded in gray. (B) WT UTP14 or the indicated mutants were expressed in bud23Δ PGAL1-UTP14 strain AJY3245, and 10-fold serial dilutions of cells were spotted onto glucose-containing medium and grown for 2 days at 30°C. (C) WT UTP14 or the indicated mutants were expressed in PGAL1-UTP14 strain AJY3243, and 10-fold serial dilutions of cells were spotted onto glucose-containing medium and grown for 2 days at 30°C. (D) Combining suppressing mutations, multiple alanine substitutions, or deletion of the region of Utp14 identified by suppressing mutations causes an increasing defect in Dhr1 interaction scored by two-hybrid assay.
Combining suppressing mutations or deletion of the region containing the mutations impairs Utp14 function.
Individually, the mutations in Utp14 fully complemented loss of Utp14 and therefore are expected to only slightly perturb a protein-protein or protein-RNA interaction to suppress bud23Δ. To make more severely disruptive mutants, we combined multiple suppressing mutations (Utp14multi-sup), changed multiple residues within this motif to alanine (Utp14multi-Ala), or deleted this region entirely (Utp14Δ719-780). We tested whether these additional mutants had more severe phenotypes than the single point mutations. Indeed, these mutants displayed a gradation of function in the order WT, Utp14multi-sup, Utp14multi-Ala, and Utp14Δ719-780, which was lethal (Fig. 2C). None of the three mutants suppressed bud23Δ (Fig. 2B). These more disruptive utp14 mutants allowed us to separate functions of Utp14, which could not be done with a complete-loss-of-function mutant.
Impaired function of Utp14 correlates with loss of Dhr1 interaction.
Because the mutations that suppressed bud23Δ mapped within the region that we identified as important for interaction with Dhr1, we tested if they affected interaction using yeast two-hybrid studies. Although the individual point mutants and combined Utp14multi-sup mutant had no obvious effect on Dhr1 interaction (Fig. 2D and data not shown), Utp14multi-Ala and Utp14Δ719-780 displayed increasing losses of interaction (Fig. 2D) that correlated with their reduced abilities to complement loss of Utp14 (Fig. 2C). These results suggest that the motif identified by mutations that suppress bud23Δ contributes to interaction with Dhr1.
rRNA processing in utp14 mutants.
Cleavage at site A2 produces 20S and 27SA2 pre-rRNAs (Fig. 3A) and is the primary event that separates the RNAs of the pre-40S subunit from the pre-60S subunit. We previously showed that deletion of BUD23, loss of UTP14, or a catalytically inactive dhr1 mutant results in loss of the 27SA2 pre-rRNA intermediate (18, 22, 23), indicating either a failure in A2 cleavage or a delay in A2 cleavage such that cleavage at A3 precedes cleavage at A2. We compared pre-rRNA processing in WT and utp14 mutant cells. As we have shown previously, WT cells displayed a strong signal for 27SA2 pre-rRNA, which was absent from Utp14-depleted cells (Fig. 3B, lane 1). Utp14multi-Ala, Utp14multi-sup, and Utp14Δ719-708 cells contained decreasing amounts of 27SA2 RNA, indicating an increasingly severe defect in cleavage at A2 (Fig. 3B, lanes 3 to 5). The mutants also showed an increase in 35S and a modest increase in 23S RNA (Fig. 3C, lanes 3 to 5), which results from cleavage at A3 without earlier cleavages at A0, A1, or A2. We also detected low levels of 21S, which increased with the severity of the growth defect of the mutant (Fig. 3B, lanes 3 to 5). 21S results from correct processing at A0 and A1 to generate the mature 5′ end of 18S but is cleaved at A3 rather than A2 on the 3′ end. Despite the absence of cleavage at A2, 20S levels were not significantly altered. This is consistent with the notion that A2 cleavage occurs after A3 cleavage in these mutants. A similar phenotype of loss of 27A2 but not 20S pre-rRNA was seen for bud23Δ cells (22). These results suggest that the Utp14multi-Ala, Utp14multi-sup, and Utp14Δ719-708 mutants are hypomorphic alleles of UTP14 with defects intermediate between WT and complete loss of Utp14 function. The loss of 27SA2 in these utp14 mutants was not due to loss of Utp14 protein, as these proteins were readily detected by Western blotting (Fig. 4). Rather, loss of 27SA2 must be due to a specific defect in the function of the mutant Utp14 proteins.
FIG 3.

Utp14 mutants show reduced 27SA2 pre-rRNA. (A) Cartoon for rRNA processing. (B and C) Northern blots: PGAL1-Utp14 (AJY3243) containing empty vector (pAJ100) (lane 1), wild-type UTP14 (pAJ1919) (lane 2), utp14multi-Sup(pAJ3264) (lane 3), utp14multi-Ala(pAJ3276) (lane 4), and utp14Δ719-780 (pAJ3276) (lane 5) were grown in SD Ura− galactose medium and then shifted to SD Ura− glucose medium for 6 h at 30°C. All cultures were harvested at an OD600 of ∼0.3. Total RNA was extracted using hot phenol, separated on a 1% agarose-formaldehyde denaturing gel, transferred to a membrane, and probed with probe 1 (AJO603, site A2-A3) (B) and probe 2 (AJO130, site D-A2) (C). U2 RNA (probed with AJO962) was used as a loading control.
FIG 4.
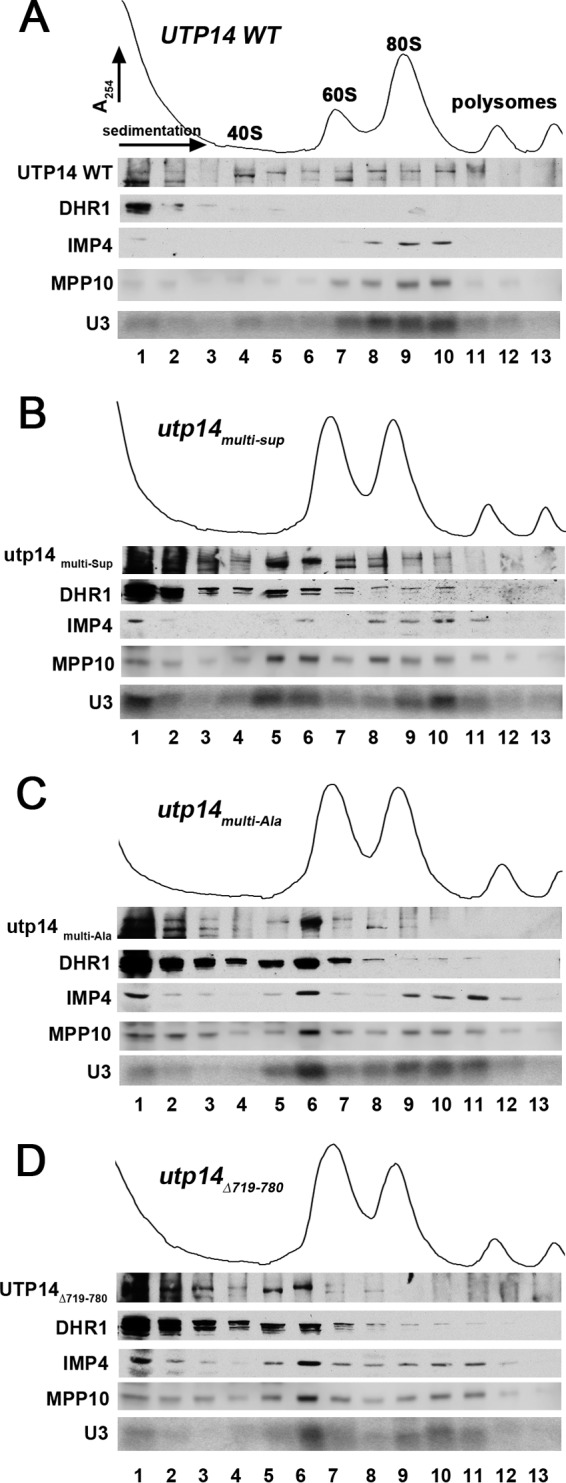
Dhr1 sedimentation in utp14 mutants. Whole-cell extracts were prepared from a PGAL1-Utp14 strain (AJY3243) containing plasmids expressing wild-type UTP14 (pAJ3308) (A), utp14multi-Sup (pAJ3309) (B), utp14multi-Ala (pAJ3313) (C), and utp14Δ719-780 (pAJ3310) (D) grown in glucose medium for 6 h. Extracts were subjected to sucrose density gradient ultracentrifugation. Proteins were precipitated from fractions and subjected to Western blotting, and RNA was extracted and subjected to Northern blotting. Utp14, Dhr1, Imp4, and Mpp10 were detected using anti-GFP (Utp14), anti-myc (Dhr1), anti-Imp4, and anti-Mpp10 antibodies, respectively. U3 was probed with AJO2194.
Impaired function of Utp14 phenocopies a Dhr1 catalytic mutant.
We reasoned that Utp14 mutants with reduced interaction with Dhr1 should inhibit recruitment of Dhr1 to the preribosome. To explore this idea, we examined the sedimentation of Dhr1 in sucrose gradients in the presence of WT or mutant Utp14. WT and mutant forms of Utp14 were expressed in cells in which genomic Utp14 was under the control of the glucose-repressible GAL1 promoter. In the presence of WT Utp14, Dhr1 sedimented primarily as free protein at the top of the gradient (Fig. 4A, fractions 1 and 2). However, in the presence of each of the three Utp14 mutants, we observed a shift in the sedimentation of Dhr1 from the top of the gradient to ∼55S (Fig. 4B to D, fractions 5 and 6). The effect was greater for Utp14multi-Ala than for Utp14multi-sup, correlating with their severity of impact on function. Clearly, mutant Utp14 did not prevent recruitment of Dhr1 to the preribosome. Rather, the data suggest that mutant Utp14 caused the accumulation of wild-type Dhr1 in the preribosome. Evidently, the function of Utp14 is more than merely binding to and recruiting Dhr1. The accumulation of Dhr1 was somewhat less pronounced for Utp14Δ719-780 despite its more severe growth defect. We suspect that this mutant is partially defective in recruitment of Dhr1 in addition to failing to activate Dhr1 (see below).
We previously showed that mutant Dhr1K420A, which is defective for ATP binding and hydrolysis, arrests SSU maturation by trapping a U3-bound ∼55S particle that also contains the U3-associated proteins Mpp10 and Imp4 (18). Therefore, we monitored the sedimentation of U3, Mpp10, and Imp4 in the presence of the Utp14 mutants. Indeed, these trans-acting factors also shifted in distribution, with a significant fraction shifting from the 90S region (Fig. 4A to D, fractions 9 and 10) to cosediment with Dhr1 at ∼55S (Fig. 4A to D, fractions 5 and 6). To confirm that the cosedimentation of Dhr1 with mutant Utp14 and U3 (Fig. 4) reflected association with a pre-40S particle, we immunoprecipitated WT Utp14 or Utp14multi-Ala and probed for the presence of Dhr1, Imp4, Mpp10, U3, and pre-rRNAs. We observed an accumulation of Dhr1, Imp4, and Mpp10 in the Utp14multi-Ala sample compared to WT Utp14 (Fig. 5A, compare lanes 3 and 4), demonstrating that these proteins accumulate in a common complex. We also observed a striking accumulation of 20S pre-rRNA as well as U3 in the Utp14multi-Ala sample compared to the WT (Fig. 5B, compare lanes 3 and 4). However, we did not detect accumulation of 21S (Fig. 5C). The lack of 27SA2 product (Fig. 3B, lane 4) and accumulation of 20S pre-rRNA in the Utp14 particle could be explained if A2 cleavage is delayed relative to A3 cleavage in the Utp14multi-Ala mutant, so that 21S pre-rRNA is processed into 20S in the stalled particle, as we suggested for the catalytic dhr1 mutant dhr1K420A (18). Alternatively, a failure to recycle U3 from the stalled mutant Utp14 particle would also lead to reduction in U3-dependent cleavages at A0, A1, and A2. The relative contributions to pre-rRNA processing of delayed cleavage at A2 and the consequences of failing to recycle U3 remain to be determined. We conclude that the altered sedimentation of Dhr1, U3, Imp4, Mpp10, and Utp14 is due to accumulation of these factors on a stalled pre-40S particle. The similarity in phenotype between the utp14 mutants and the catalytic dhr1K420A mutant led us to posit that Utp14 activates Dhr1 helicase activity.
FIG 5.

Utp14multi-Ala coimmunoprecipitates a pre-40S particle containing U3 and Dhr1. (A) Utp14 WT and the multi-alanine mutant were immunoprecipitated (IP) from extracts prepared from strains as described in the legend to Fig. 4A. Immunoprecipitated proteins were analyzed by SDS-PAGE and Western blotting for Utp14, Dhr1, Imp4, and Mpp10. The asterisk indicates a probable proteolytic product of Imp4. (B and C) RNAs were extracted from the immunoprecipitated samples and analyzed by Northern blotting with probe 1 to detect pre-rRNAs containing the D-A2 fragment (B) and probe 2 to detect pre-rRNAs containing the A2-A3 fragment (C), as described in the legend to Fig. 3.
Utp14 stimulates the unwinding activity but not the ATPase activity of Dhr1.
To assess whether Utp14 binds to and stimulates unwinding activity of Dhr1, we expressed and purified both proteins. Dhr1 was expressed with a C-terminal His6 tag in Escherichia coli and the recombinant protein was purified as described previously (18). Utp14, Utp14multi-Ala, and Utp14Δ719-780 were expressed with an N-terminal His6 tag in E. coli and purified (see Materials and Methods). In yeast, Dhr1-His6 fully complemented a dhr1 null mutant (18) and His6-Utp14 fully complemented the utp14 null mutant (see Fig. S4 in the supplemental material), demonstrating that the tags did not interfere with the function of either protein in vivo.
To test if Utp14 stimulates unwinding activity of Dhr1, we used established assays with a substrate that mimics one of the three genetically verified U3-pre-rRNA duplexes: the U3-ETS2 duplex (18). The U3-ETS2 duplex comprises full-length U3 snoRNA bound to nt 281 to 291 of the 5′ ETS of the pre-rRNA (Fig. 6A) and is required for subsequent U3-pre-rRNA interactions in vivo (16). This duplex forms spontaneously and is stable in vitro (33). The U3-ETS2 duplex unwinding reactions were performed under pre-steady-state conditions with an excess of enzyme over the duplex substrate and low substrate concentrations to minimize duplex reformation. To enhance detection of Utp14-dependent stimulation of Dhr1 unwinding activity, the enzyme concentration was lowered to the point where unwinding activity by Dhr1 alone was barely detectable (Fig. 6B, lane 4). Addition of Utp14 to a reaction mixture containing Dhr1 resulted in a noteworthy increase in unwinding activity that was dependent on ATP-Mg2+ (Fig. 6B, compare lanes 4, 5, and 6). No activity was observed with Utp14 alone in the presence of ATP-Mg2+ (Fig. 6B, lane 8), indicating that this protein has no intrinsic helicase activity. Stimulation of Dhr1-dependent unwinding by Utp14 was also seen when both proteins were at 500 nM, although the fold stimulation was reduced (data not shown). Compared with WT Utp14, Utp14multi-Ala and the Utp14Δ719-780 showed reduced activity (Fig. 6B, compare lane 6 with lanes 9 and 10). To quantitate these activities, we performed time course assays and fit the data to determine the unwinding (kunw) rate constant as described in reference 34. Addition of WT Utp14 to the Dhr1 reactions stimulated kunw 98-fold, whereas the mutants showed less stimulation. Addition of Utp14multi-Ala and of Utp14Δ719-780 to the Dhr1 reactions stimulated the kunw 41-fold and 18-fold, respectively, in accordance with the reduced function of these mutants in vivo (Fig. 2 and 3).
FIG 6.

Utp14 activates Dhr1 unwinding activity in vitro. (A) Cartoon of U3-ETS2 substrate used for unwinding assays. Full-length U3 snoRNA was used for the reactions in panels B and C. (B) Representative unwinding reactions stopped after 20 min. EMSAs separated the 32P-labeled ETS2 free (unwound) from its duplex form. The RT reaction mixture contained 50 nM Dhr1 (WT or mutant), 200 nM Utp14 (WT or mutant), 1 mM ATP, and ≤0.3 nM U3-ETS2 duplex with other reagents described in Materials and Methods. (C) Fraction unwound was plotted as a function of time after addition of ATP and either Utp14, Dhr1, or Dhr1 in the presence of either Utp14, Utp14Multi-Ala, or Utp14Δ719-780 or after addition of Dhr1 and Utp14 in the absence of ATP. (D) Fraction unwound was plotted as a function of time after addition of ATP and Dhr1D516A/E517A in the absence or presence of Utp14.
Previously, we established that Dhr1 shares mechanistic similarities with DEAD box helicases in that unwinding requires ATP binding but not hydrolysis, whereas product recycling requires ATP hydrolysis (18). To explore whether Utp14 stimulates Dhr1-dependent unwinding, we tested the activity of the Dhr1D516A/E517A mutant, which eliminates the catalytic and metal binding carboxylates and thus probes the unwinding and not the recycling step (18). Utp14 stimulated the kunw of Dhr1D516A/E517A 26-fold (Fig. 6D), which is less than the 96-fold stimulation observed for WT Dhr1 (Fig. 6C). Thus, Utp14 accelerates the Dhr1-dependent unwinding step; however, because Utp14 stimulated WT Dhr1 to a greater extent than Dhr1D516A/E517A, Utp14 must also contribute to other steps, such as substrate recruitment and/or product recycling.
DEAH helicases are often activated by G-patch proteins that stimulate both unwinding and ATPase activity. To examine whether Utp14 shares these properties, we first analyzed sequence conservation within the Utp14 family. G-patch proteins are defined by the consensus sequence hhX3GaX2GXGhGX4G, where a is an aromatic residue, h is hydrophobic, and X can be any amino acid (20). While many positions allow some divergence from this sequence, the first glycine followed by an aromatic residue is invariant. Utp14 proteins contain a highly glycine-rich conserved motif, hPGWGXWXGXG. Even though the dipeptide GW is invariant, the larger motif does not fit the consensus of a G-patch protein. Dhr1 has RNA-stimulated ATPase activity (18). Consequently, we tested whether Utp14 could stimulate the ATPase activity of Dhr1 like other G-patch proteins that activate the ATPase activities of helicases. The ATPase activity of Dhr1 was indistinguishable in the presence and absence of Utp14, regardless of the presence or absence of poly(A) (Fig. 7). Furthermore, Utp14 alone showed no intrinsic ATPase activity, in contrast to its annotation as an ATPase (2). These results indicate that Utp14 stimulates Dhr1 unwinding activity without increasing its RNA-dependent ATPase activity. Thus, we conclude that Utp14 is not a G-patch protein or represents a divergent class of G-patch proteins.
FIG 7.
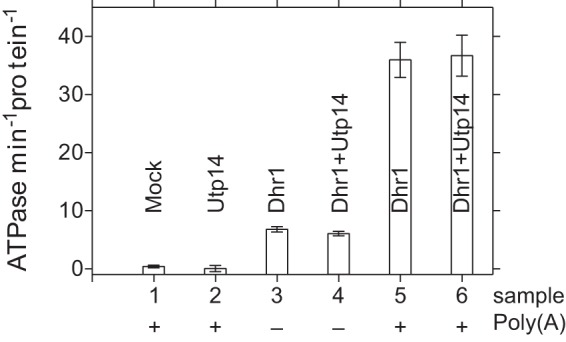
Utp14 does not stimulate the ATPase activity of Dhr1. Initial velocities of Pi released after addition of 1 mM ATP at RT in the presence or absence of the poly(A) added to either Dhr1, Dhr1 with Utp14, Utp14, or a control (mock) with other reagents described in Materials and Methods are shown. Each protein had a final concentration of 0.5 μM. “Protein” on the y-axis label refers to either Dhr1, Utp14, or the complex of the two. Similar results were obtained at 50 nM Dhr1 in the presence or absence of 200 nM Utp14 (see Fig. S5 in the supplemental material) or at a lower poly(A) concentration (data not shown).
Dhr1 and Utp14 form a complex in vitro.
To determine the stoichiometry of the Dhr1-Utp14 complex, we performed cross-linking assays with BS3, a reagent that reacts with surface lysine residues, and used mass spectrometry to probe its stoichiometry (see Materials and Methods). Cross-linking slightly altered migration of the individual proteins on SDS-PAGE (Fig. 8A, compare lanes 8 to 11 with lanes 1 and 4). When both proteins were present, a slower-migrating band was observed, correlating with loss of the individual protein bands (Fig. 8A, compare lanes 5 to 7 with lanes 1 to 4), consistent with a Dhr1-Utp14 complex. Interestingly, neither Utp14multi-Ala nor Utp14Δ719-780 produced as much of the higher-order cross-linked species with Dhr1 as was observed with WT Utp14 and Dhr1 (Fig. 8A, compare lane 5 with lanes 6 and 7). Mass spectrometry verified that individual proteins modified by BS3 showed increased mass: 166 kDa for Dhr1 and 122 kDa for Utp14. A heavier species was observed at 287 kDa only in the presence of both proteins, which was consistent with a 1:1 stoichiometry of Dhr1 and Utp14 (Fig. 8B, compare red trace with black and blue traces). The low intensity of the 287-kDa species likely arose from the inefficiency of ionizing such a large complex. Together, these results provide evidence that Dhr1 and Upt14 form a 1:1 complex and that both Utp14 mutants weaken this interaction in accord with the two-hybrid data (Fig. 1 and 2). While a 1:1 complex was observed, the technical limitation of SELDI-TOF mass spectrometry cannot rule out the presence of higher-order complexes as well.
FIG 8.
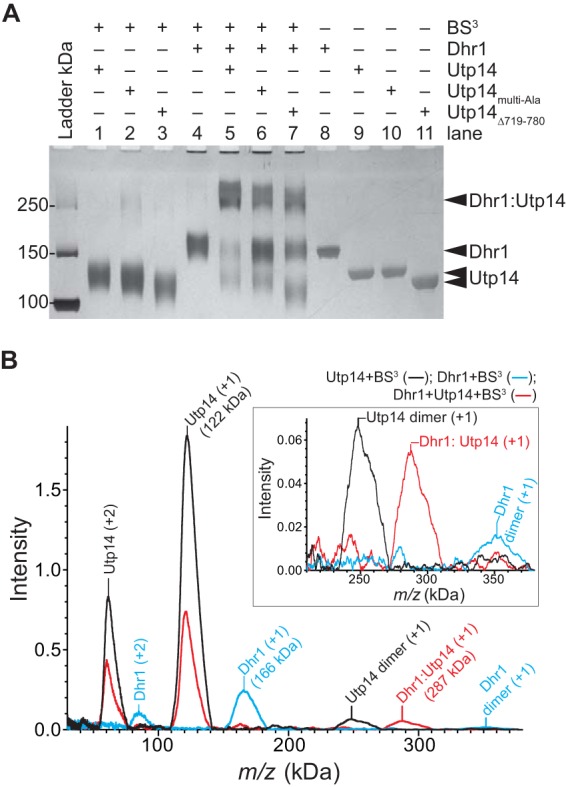
Dhr1 and Utp14 form a 1:1 complex in vitro. (A) Representative cross-linking reactions stopped after 30 min. SDS-PAGE separated the cross-linked protein complex from individual protein. The RT reaction mixture contained a 2 μM concentration of each protein with other reagents described in Materials and Methods. (B) Mass spectrometry analysis of the same samples as in lane 1 (Utp14), lane 4 (Dhr1), and lane 5 (both proteins) in panel A. Masses of the individual proteins, Dhr1 (166 kDa) and Utp14 (122 kDa), have increased by more than 10% due to modification of the large numbers of lysine residues in each protein: 123 and 99, respectively. Upon addition of both proteins, a new mass appears, 287 kDa, consistent with formation of a 1:1 complex. The inset shows an enlargement of the intensity of the masses centered on 300 kDa.
Utp14 and Bud23 together are required for efficient association of Dhr1 with the preribosome.
A major question is how is Dhr1 recruited to and activated at the appropriate time during SSU biogenesis. To address this question, we investigated whether Dhr1 recruits Utp14 to the preribosome or vice versa. Because WT Dhr1 does not stably associate with preribosomes, we used the Dhr1K420A mutant that does stably interact with an ∼55S preribosomal particle (18) (Fig. 9A, fraction 6). We previously showed that Bud23 binds to the N-terminal domain of Dhr1 and that mutations in Dhr1 partially suppress the growth defect of bud23Δ cells (24). Despite the physical and functional interaction between Bud23 and Dhr1, loss of Bud23 has no observed effect on the sedimentation of Dhr1K420A (24) (Fig. 9B). This suggests that Bud23 alone is not necessary for recruitment of Dhr1 to the preribosome, contrary to a recent report (21). Given that Utp14 also interacts with Dhr1, we asked if Utp14 is required for association of Dhr1 with the preribosome. Again, we did not observe any obvious change in the sedimentation of Dhr1K420A in the absence of Utp14 (Fig. 9C). However, when we eliminated both Utp14 and Bud23 by depleting Utp14 from Bud23-deficient cells, we saw a significant loss of Dhr1K420A from the ∼55S region of the gradient (Fig. 9D, fractions 5 and 6) and corresponding accumulation of free protein at the top of the gradient (Fig. 9D, fractions 1 and 2). These findings suggest that efficient association of Dhr1 with the preribosome requires both Bud23 and Utp14.
FIG 9.
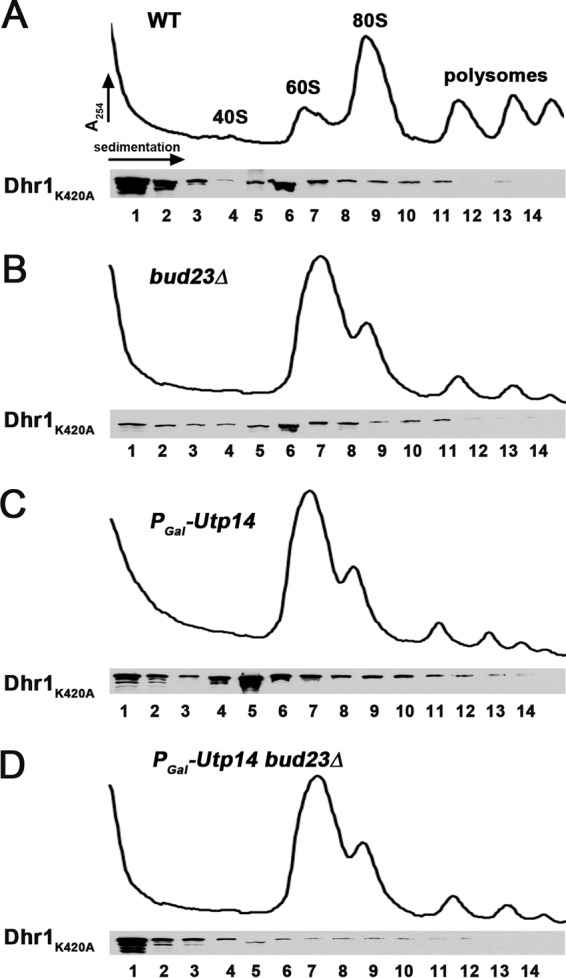
Utp14 and Bud23 together are necessary for efficient Dhr1 recruitment to the preribosome. Plasmid pAJ3081 (dhr1K420A-13myc) was transformed into wild-type (BY4741) (A), bud23Δ (AJY2161) (B), PGAL1-Utp14 (AJY3243) (C), and PGAL1-Utp14 bud23Δ (AJY3245) (D) strains. Whole-cell extracts from strains grown in Leu− galactose medium that were shifted to Leu− glucose medium for 6 h were subjected to sucrose density gradient ultracentrifugation. Proteins were precipitated from fractions and subjected to Western blotting. Dhr1K420A was detected with anti-myc antibody.
DISCUSSION
Our results identify Utp14 as an essential factor required for activating the DEAH/RHA helicase Dhr1, which removes U3 from the preribosome to allow folding the CPK and to promote A2 cleavage. Both Utp14 and the RNA methyltransferase Bud23 are required for stable association of Dhr1 with the preribosome. Thus, Utp14 has two functions: together with Bud23 it recruits Dhr1 to the preribosome, and it activates Dhr1 in the context of the preribosome.
We uncovered the connection between Utp14 and Dhr1 because mutations in either protein suppress the growth and biogenesis defects of a bud23Δ mutant (23, 24). In earlier work, we showed that the utp14A758G mutation partially suppressed the growth defect of bud23Δ. In this study, we found that additional mutations that suppress bud23Δ map to a short segment necessary for stimulation of Dhr1 unwinding activity, and our results suggest that this is through direct physical interaction with Dhr1.
Mutations in BUD23, DHR1, or UTP14 primarily impact cleavage at A2. Interestingly, whereas the single point mutation utp14A758G suppresses the growth and A2 cleavage defects of bud23Δ cells, simultaneous mutation of multiple residues in Utp14 eliminated suppression and resulted in an A2 cleavage defect. Apparently, subtle perturbation of the function of this peptide suppresses bud23Δ, whereas more severe perturbation leads to a loss of function. What, then, is the function of Bud23 and how do mutations in Utp14 suppress bud23Δ? Since mutations in Utp14 reduce the activity of Dhr1 and suppress bud23Δ, one possibility is that Bud23 slows or limits the activity of Dhr1. Bud23 and Utp14 are both needed for stable association of Dhr1 with the preribosome. Perhaps in the absence of Bud23, ATP hydrolysis by Dhr1 leads to unproductive events and premature release of Dhr1. Thus, mutations that slow its activity could allow more time for productive engagement with its substrate. This thinking is analogous to what has been proposed for Prp5, a DEAD box helicase required for spliceosome assembly. Mutations in Prp5 that reduce its intrinsic ATPase activity increased the fidelity of splicing a suboptimal intron (35). Alternatively, Bud23 may stabilize an RNA structure in the preribosome, consistent with the fact that Bud23 protein, but not its methyltransferase activity, is important for supporting ribosome assembly (22). Dhr1 action on a destabilized pre-40S may lead to misfolding or misassembly that is recognized by surveillance systems, triggering discard of the subunit. Reducing the rate of Dhr1 unwinding could allow sufficient time for productive RNA-RNA or RNA-protein rearrangements in the absence of Bud23.
How does Utp14 stimulate Dhr1 unwinding activity?
Various DEAH/RHA RNA helicases depend on G-patch proteins for their recognition of substrates and for their activation (20). Here, we have shown that Utp14 binds to and activates Dhr1, but it neither possesses the canonical G-patch sequence nor stimulates ATPase activity upon activation of unwinding activity, two hallmarks of known G-patch proteins that activate RNA helicases. In contrast, we found that Utp14 stimulated the helicase activity of Dhr1 without stimulating its ATPase activity. Such stimulation of unwinding without activation of ATPase activity is not without precedent. The Ski2-like RNA helicase Brr2 is required for pre-mRNA splicing, and its unwinding activity can be stimulated by Prp8, which also reduces its ATPase activity (36). In this case, Prp8 increases the coupling between ATP hydrolysis and productive unwinding by Brr2 (37) by a mechanism that is unclear. Utp14 also increases the coupling between ATP hydrolysis and productive unwinding by Dhr1, because the presence of Utp14 stimulates unwinding activity by Dhr1 without affecting rates of ATPase hydrolysis.
A possible molecular basis of such stimulation is provided by the DNA helicase PcrA, whose activator, RepD, stimulates the unwinding of PcrA without affecting ATPase activity (38, 39). RepD exploits a common feature of the reaction cycle of helicases with tandem RecA-like domains—these domains cycle between an open, inactive conformation and a closed, active conformation (40). In the active form, the two RecA-like domains come together to form the NTP and RNA duplex binding cleft. Cross-linking and fluorescence resonance energy transfer (FRET) studies suggest that RepD stimulates the helicase activity of PcrA by locking the tandem RecA-like domains in an active, closed conformation (39), and this is accomplished without stimulated ATPase activity (38). Perhaps the Utp14 contact to Dhr1 similarly stabilizes a closed conformation of the RecA-like domains of Dhr1 to couple unwinding to ATPase activity.
How is the activity of Dhr1 regulated to release U3 at the appropriate time in ribosome assembly?
Base pairing between U3 and the pre-rRNA orchestrates early RNA folding and cleavage events. However, these early events, leading to the cleavage at A2, occur very rapidly; in actively growing yeast, cleavage at A2 occurs within 50 s of initiation of transcription (4). Thus, the binding and release of U3 are highly dynamic and must be driven by factors such as Imp3, which promotes its annealing with pre-rRNA (33), and Dhr1, which promotes its dissociation (18). Utp14 was initially characterized as a component of the SSU processome (8) and likely associates with the preribosome before Bud23 (23). Bud23 methylates G1575 in the 3′ major head domain of the subunit (21, 22) and presumably binds to its RNA target site only after it is properly folded but before A2 cleavage, as bud23Δ mutants are defective for cleavage at this site (22). Both Utp14 and Bud23 remain associated with the subunit after A2 cleavage (23) and thus mark the transition from the 90S particle to the pre-40S particle. As both Bud23 and Utp14 are needed for stable association of Dhr1 with the preribosome, we suggest that these two proteins together couple Dhr1 activity to the status of rRNA transcription and folding within the preribosome. Thus, Utp14 may help position Dhr1 in the preribosome, but U3 release would not be triggered until the proper folding of the Bud23 binding site within the 3′ major head domain.
While cleavages at A0, A1, and A2 are all U3 dependent, these three cleavage sites appear to separate into two functionally distinct groups. Production of the mature 5′ end of 18S by A0 and A1 cleavage requires U3 docking, whereas efficient A2 cleavage is expected to require undocking of U3. Thus, U3 docking produces the 5′ end of 18S, whereas U3 release signals SSU biogenesis by liberating the SSU 20S precursor from the pre-rRNA. In this model, U3 docking and undocking provide distinct structural switches to mark steps during SSU biogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Rout for anti-GFP and S. Baserga for anti-Mpp10 and anti-Imp4. In addition, we thank Xinli Yang of the Midwest Proteome Center at Rosalind Franklin University of Medicine and Science for performing the SELDI-TOF measurements and providing detailed experimental information.
We have no conflicts of interest to declare.
Funding Statement
The Midwest Proteome Center is supported by the following grants: NIH grant T35DK074390 and HRSA grant C76 HF03610-01-00.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00773-15.
REFERENCES
- 1.Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65:2334–2359. doi: 10.1007/s00018-008-8027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolford JL Jr, Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195:643–681. doi: 10.1534/genetics.113.153197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tschochner H, Hurt E. 2003. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol 13:255–263. doi: 10.1016/S0962-8924(03)00054-0. [DOI] [PubMed] [Google Scholar]
- 4.Kos M, Tollervey D. 2010. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37:809–820. doi: 10.1016/j.molcel.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sykes MT, Williamson JR. 2009. A complex assembly landscape for the 30S ribosomal subunit. Annu Rev Biophys 38:197–215. doi: 10.1146/annurev.biophys.050708.133615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodson SA. 2011. RNA folding pathways and the self-assembly of ribosomes. Acc Chem Res 44:1312–1319. doi: 10.1021/ar2000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karbstein K. 2011. Inside the 40S ribosome assembly machinery. Curr Opin Chem Biol 15:657–663. doi: 10.1016/j.cbpa.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA, Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, Beyer AL, Hunt DF, Baserga SJ. 2002. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417:967–970. doi: 10.1038/nature00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schafer T, Kuster B, Tschochner H, Tollervey D, Gavin AC, Hurt E. 2002. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell 10:105–115. doi: 10.1016/S1097-2765(02)00579-8. [DOI] [PubMed] [Google Scholar]
- 10.Schäfer T, Strauss D, Petfalski E, Tollervey D, Hurt E. 2003. The path from nucleolar 90S to cytoplasmic 40S pre-ribosomes. EMBO J 22:1370–1380. doi: 10.1093/emboj/cdg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venema J, Tollervey D. 1999. Ribosome synthesis in Saccharomyces cerevisiae. Annu Rev Genet 33:261–311. doi: 10.1146/annurev.genet.33.1.261. [DOI] [PubMed] [Google Scholar]
- 12.Beltrame M, Henry Y, Tollervey D. 1994. Mutational analysis of an essential binding site for the U3 snoRNA in the 5′ external transcribed spacer of yeast pre-rRNA. Nucleic Acids Res 22:5139–5147. doi: 10.1093/nar/22.23.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beltrame M, Tollervey D. 1995. Base pairing between U3 and the pre-ribosomal RNA is required for 18S rRNA synthesis. EMBO J 14:4350–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes JM. 1996. Functional base-pairing interaction between highly conserved elements of U3 small nucleolar RNA and the small ribosomal subunit RNA. J Mol Biol 259:645–654. doi: 10.1006/jmbi.1996.0346. [DOI] [PubMed] [Google Scholar]
- 15.Beltrame M, Tollervey D. 1992. Identification and functional analysis of two U3 binding sites on yeast pre-ribosomal RNA. EMBO J 11:1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dutca LM, Gallagher JE, Baserga SJ. 2011. The initial U3 snoRNA:pre-rRNA base pairing interaction required for pre-18S rRNA folding revealed by in vivo chemical probing. Nucleic Acids Res 39:5164–5180. doi: 10.1093/nar/gkr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma K, Tollervey D. 1999. Base pairing between U3 small nucleolar RNA and the 5′ end of 18S rRNA is required for pre-rRNA processing. Mol Cell Biol 19:6012–6019. doi: 10.1128/MCB.19.9.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sardana R, Liu X, Granneman S, Zhu J, Gill M, Papoulas O, Marcotte EM, Tollervey D, Correll CC, Johnson AW. 2015. The DEAH-box helicase Dhr1 dissociates U3 from the pre-rRNA to promote formation of the central pseudoknot. PLoS Biol 13:e1002083. doi: 10.1371/journal.pbio.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silverman E, Edwalds-Gilbert G, Lin RJ. 2003. DExD/H-box proteins and their partners: helping RNA helicases unwind. Gene 312:1–16. doi: 10.1016/S0378-1119(03)00626-7. [DOI] [PubMed] [Google Scholar]
- 20.Robert-Paganin J, Rety S, Leulliot N. 2015. Regulation of DEAH/RHA helicases by G-patch proteins. Biomed Res Int 2015:931857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Létoquart J, Huvelle E, Wacheul L, Bourgeois G, Zorbas C, Graille M, Heurgue-Hamard V, Lafontaine DL. 2014. Structural and functional studies of Bud23-Trm112 reveal 18S rRNA N7-G1575 methylation occurs on late 40S precursor ribosomes. Proc Natl Acad Sci U S A 111:E5518–E5526. doi: 10.1073/pnas.1413089111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White J, Li Z, Sardana R, Bujnicki JM, Marcotte EM, Johnson AW. 2008. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol Cell Biol 28:3151–3161. doi: 10.1128/MCB.01674-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sardana R, White JP, Johnson AW. 2013. The rRNA methyltransferase Bud23 shows functional interaction with components of the SSU processome and RNase MRP. RNA 19:828–840. doi: 10.1261/rna.037671.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sardana R, Zhu J, Gill M, Johnson AW. 7 April 2014. Physical and functional interaction between the methyltransferase Bud23 and the essential DEAH-box RNA helicase Ecm16. Mol Cell Biol doi: 10.1128/MCB.01656-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sondalle SB, Baserga SJ. 2014. Human diseases of the SSU processome. Biochim Biophys Acta 1842:758–764. doi: 10.1016/j.bbadis.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M. 1996. A three-hybrid system to detect RNA-protein interactions in vivo. Proc Natl Acad Sci U S A 93:8496–8501. doi: 10.1073/pnas.93.16.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Lee I, Moradi E, Hung NJ, Johnson AW, Marcotte EM. 2009. Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol 7:e1000213. doi: 10.1371/journal.pbio.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gérczei T, Correll CC. 2004. Imp3p and Imp4p mediate formation of essential U3-precursor rRNA (pre-rRNA) duplexes, possibly to recruit the small subunit processome to the pre-rRNA. Proc Natl Acad Sci U S A 101:15301–15306. doi: 10.1073/pnas.0406819101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gérczei T, Shah BN, Manzo AJ, Walter NG, Correll CC. 2009. RNA chaperones stimulate formation and yield of the U3 snoRNA-pre-rRNA duplexes needed for eukaryotic ribosome biogenesis. J Mol Biol 390:991–1006. doi: 10.1016/j.jmb.2009.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Y, Andersen GR, Nielsen KH. 2010. Structural basis for the function of DEAH helicases. EMBO Rep 11:180–186. doi: 10.1038/embor.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walbott H, Mouffok S, Capeyrou R, Lebaron S, Humbert O, van Tilbeurgh H, Henry Y, Leulliot N. 2010. Prp43p contains a processive helicase structural architecture with a specific regulatory domain. EMBO J 29:2194–2204. doi: 10.1038/emboj.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah BN, Liu X, Correll CC. 2013. Imp3 unfolds stem structures in pre-rRNA and U3 snoRNA to form a duplex essential for small subunit processing. RNA 19:1372–1383. doi: 10.1261/rna.039511.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Q, Jankowsky E. 2005. ATP- and ADP-dependent modulation of RNA unwinding and strand annealing activities by the DEAD-box protein DED1. Biochemistry 44:13591–13601. doi: 10.1021/bi0508946. [DOI] [PubMed] [Google Scholar]
- 35.Xu YZ, Query CC. 2007. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 28:838–849. doi: 10.1016/j.molcel.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maeder C, Kutach AK, Guthrie C. 2009. ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat Struct Mol Biol 16:42–48. doi: 10.1038/nsmb.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mozaffari-Jovin S, Wandersleben T, Santos KF, Will CL, Luhrmann R, Wahl MC. 2014. Novel regulatory principles of the spliceosomal Brr2 RNA helicase and links to retinal disease in humans. RNA Biol 11:298–312. doi: 10.4161/rna.28353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toseland CP, Martinez-Senac MM, Slatter AF, Webb MR. 2009. The ATPase cycle of PcrA helicase and its coupling to translocation on DNA. J Mol Biol 392:1020–1032. doi: 10.1016/j.jmb.2009.07.071. [DOI] [PubMed] [Google Scholar]
- 39.Arslan S, Khafizov R, Thomas CD, Chemla YR, Ha T. 2015. Protein structure. Engineering of a superhelicase through conformational control. Science 348:344–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozgur S, Buchwald G, Falk S, Chakrabarti S, Prabu JR, Conti E. 2015. The conformational plasticity of eukaryotic RNA-dependent ATPases. FEBS J 282:850–863. doi: 10.1111/febs.13198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.