

Abstract
Notch activation in aortic endothelial cells (ECs) takes place at embryonic stages during cardiac valve formation and induces endothelial-to-mesenchymal transition (EndMT). Using aortic ECs, we show here that active Notch expression promotes EndMT, resulting in downregulation of vascular endothelial cadherin (VE-cadherin) and upregulation of mesenchymal genes such as those for fibronectin and Snail1/2. In these cells, transforming growth factor β1 exacerbates Notch effects by increasing Snail1 and fibronectin activation. When Notch-downstream pathways were analyzed, we detected an increase in glycogen synthase kinase 3β (GSK-3β) phosphorylation and inactivation that facilitates Snail1 nuclear retention and protein stabilization. However, the total activity of Akt was downregulated. The discrepancy between Akt activity and GSK-3β phosphorylation is explained by a Notch-induced switch in the Akt isoforms, whereby Akt1, the predominant isoform expressed in ECs, is decreased and Akt2 transcription is upregulated. Mechanistically, Akt2 induction requires the stimulation of the β-catenin/TCF4 transcriptional complex, which activates the Akt2 promoter. Active, phosphorylated Akt2 translocates to the nucleus in Notch-expressing cells, resulting in GSK-3β inactivation in this compartment. Akt2, but not Akt1, colocalizes in the nucleus with lamin B in the nuclear envelope. In addition to promoting GSK-3β inactivation, Notch downregulates Forkhead box O1 (FoxO1), another Akt2 nuclear substrate. Moreover, Notch protects ECs from oxidative stress-induced apoptosis through an Akt2- and Snail1-dependent mechanism.
INTRODUCTION
Endothelial-to-mesenchymal transition (EndMT) is a cellular conversion that generates mesenchymal cells from endothelial cells. During embryonic development, EndMT takes place at embryonic day 9.5 (E9.5), when endocardial cells that overlie the atrioventricular (AV) canal and outflow tract regions delaminate from the endocardial sheet and invade the cardiac jelly, to form the endocardial cushions that establish the AV valves (1). EndMT is essential for cardiac valve development and heart septation and requires transforming growth factor β (TGF-β) (2). Generation of mesenchymal cells is a crucial step for the differentiation of endothelial cells into several lineages, including fibroblasts, myofibroblasts, pericytes, osteoblasts, chondrocytes, and adipocytes (3). Pathological EndMT has also been associated with angiogenic sprouting, arteriosclerosis, cardiac fibrotic disorders, and tumor progression (4–6). In tumors, EndMT contributes to generate cancer-associated fibroblasts that alter microenvironments by secreting oncogenic signals, such TGF-β, to induce the epithelial-to-mesenchymal transition (EMT) (7).
Notch signaling has been implicated in EndMT during development of the heart valves, arterial-venous differentiation, and remodeling of the primitive vascular plexus; accordingly, mutations of the Notch pathway are associated with congenital defects of the cardiovascular system (8, 9). Notch genes encode transmembrane receptors with a large extracellular domain that interacts with different membrane-bound ligands of the Delta and Serrate/Jagged families and a Notch intracellular domain (NICD) (9). Notch signaling requires ligand binding, proteolytic processing of the receptor, nuclear translocation of NICD, and a Notch interaction with RBPJ/CBF1/Su(H) to form a complex that activates the expression of target genes such as those for Myc, p21, and the HES family members (Hes1 and Hes2) (10). Notch also interacts functionally with the Wnt/β-catenin pathway, a signaling cascade that is also essential for cardiogenesis (11). β-Catenin interacts with NICD and signals synergistically by forming a ternary complex with RBPJ (RBPJ/NICD/β-catenin) (12–14). Therefore, γ-secretase inhibitors preventing NICD generation also reduce the expression of Wnt-dependent genes such as Axin2 (15). In contrast, inactive Notch negatively regulates active β-catenin accumulation by associating with unphosphorylated β-catenin at the cell membrane in colon cancer cells (16).
Snail family members have been associated with cells undergoing metastatic as well as developmental EMT (17, 18). An important target of Snail1 repression is the E-cadherin (CDH1) gene, the primary cadherin responsible for homotypic adhesion between members of an epithelial sheet (19, 20). Snail1 has additional cellular functions that are independent of EMT, since it also confers resistance to cell death (21–23). Snail1 is a highly unstable protein, very sensitive to proteasome inhibitors. Several E3 ubiquitin ligases target the Snail1 protein (18, 24), such as the E3 ubiquitin ligase β-TrCP1/FBXW1, which requires prior phosphorylation of Snail1 by glycogen synthase kinase 3β (GSK-3β) (25). In addition to phosphorylating the sequence required for β-TrCP1 binding, GSK-3β also phosphorylates other residues in Snail1, thus favoring its nuclear export and indirectly controlling its accessibility to β-TrCP1 and other cytosolic ubiquitin ligases. Therefore, the presence of GSK-3β in the nucleus is particularly relevant for regulating Snail1 expression; accordingly, nuclear export of this kinase is associated with Snail1 stability (26).
Functionally, GSK-3β is controlled by kinases such as Akt, which phosphorylates it at serine 9 to inhibit its activity (27), and by those of the p90 ribosomal S6 kinase (RSK) family (28). The Akt family controls many cellular processes, such as proliferation, growth, metabolism differentiation, migration, angiogenesis, survival, and tumor progression, and has also been implicated in EMT (29, 30). Akt isoforms (Akt1/protein kinase Bα [PKBα], Akt2/PKBβ, and Akt3/PKBγ) are highly conserved, are activated by extracellular signals, such as insulin, insulin-like growth factor 1 (IGF-1), or transforming growth factor β (TGF-β) via phosphatidylinositol 3-kinase, and are negatively regulated by the tumor suppressor PTEN phosphatase (31). To be fully activated, Akt needs to be phosphorylated at Thr-308 by PDK1 and at Ser-473 by mTORC2, after which it can translocate to the nucleus to act on its many substrates, which are involved in cell proliferation, survival, intermediary metabolism, angiogenesis, cell growth, and apoptosis (32, 33). Akt controls the activity of the proapoptotic FoxO proteins (FoxO-1, -3, -4, and -6). Akt-mediated phosphorylation of FoxO proteins induces their translocation from the nucleus to the cytoplasm, where they are degraded; this leads to the downregulation of FoxO target genes, such as those for the Fas ligand, Bim, and Bcl-6 (34, 35). Akt1 and Akt2 are present in all tissues, whereas Akt3 is mostly expressed in the brain (36).
Notch has been shown to promote EndMT during cardiac valve formation via Snail1 induction and vascular endothelial cadherin (VE-cadherin) downregulation (37). We have investigated the role of protein kinases in controlling Snail1 stability during this process. Unexpectedly, we found that Notch induces a general decrease in Akt global levels during EndMT that does not correlate with the detected upregulation of GSK-3β phosphorylated at Ser9. Upon Notch expression, Akt2 is specifically upregulated by β-catenin/TCF4 transcriptional activation and is translocated to the nucleus, where it localizes to the nuclear envelope. We propose that this specific localization provides Akt2 with full access to nuclear substrates, such as FoxO1 or GSK-3β. This would also explain why Akt2, but not Akt1, is related to a more mesenchymal phenotype and resistance to apoptosis in Notch-expressing cells.
MATERIALS AND METHODS
Cell culture, reagents, and antibodies.
Pig aortic endothelial (PAE) cells stably expressing Notch and control cells with an empty vector were previously described (37) and were a generous gift from J. L. de la Pompa (Centro Nacional de Investigación Cardiovascular, Madrid, Spain). The HEK 293T cell line was obtained from our institute's cell bank. Murine embryonic fibroblasts (MEFs), wild type (WT) and knockout (KO) for Akt1 or Akt2, were kindly provided by M. J. Birnbaum (University of Pennsylvania) (38). Cells were maintained in Dulbecco modified Eagle medium (Gibco-BRL) with 10% heat-inactivated fetal bovine serum (Gibco-BRL) at 37°C in 5% CO2. Where indicated, cells were treated with 5 ng of TGF-β1 (PeproTec)/ml, 200 μM H2O2 (Merck), and 20 μg of cycloheximide (CHX; Sigma)/ml. The inhibitors used were MK-2206 (10 μM; Deltaclon) and CCT128930 (10 μM; SelleckChem) for Akt, SL0101 (10 μM; Calbiochem) for RSK, UO126 (10 μM; Cell Signaling) for extracellular signal-regulated kinase 1/2 (Erk-1/2), and iCRT14 (25 μM; Tocris) for β-catenin/TCF4 transcriptional activity. The antibodies used in the present study included mouse monoclonal antibodies against Snail1 (39), Myc (clone 9E10; a gift from Gabriel Gil, Institut Hospital del Mar d'Investigacions Mèdiques [IMIM], Barcelona, Spain), Erk-1/2 (Zymed), and GSK-3β (BD Bioscience); rabbit polyclonal antibodies against pan-Akt, Akt1, Akt2, pAkt (S473), pAkt (T308), FoxO1, pFoxO1 (S256), cleaved caspase-3, pGSK-3β (S9), and pErk-1/2 (T202/Y204) (all from Cell Signaling), β-TrCP1 (Santa Cruz Biotechnology), fibronectin (Dako), N-cadherin, RSK1, pRSK1 (T359/S363) (AbCam), cyclin D1 (Thermo Scientific), tubulin, α-smooth muscle actin (α-Sma), and Flag tag (Sigma); and goat polyclonal antibodies against lamin B (C-20), Snail2 (Slug) (G-18), VE-cadherin (Santa Cruz Biotechnology), and rat antihemagglutinin (anti-HA) tag (Roche).
Transfection, cell lysis, immunoprecipitation, and Western blotting.
For Snail1-HA degradation assays, PAE cells were seeded in 60-mm plates for 24 h and transfected with 200 ng of pCDNA3-Snail1-HA (19), using Lipofectamine Plus reagent (Invitrogen) for 6 h according to the manufacturer's instructions for PAE cells, whereas HEK 293T cells were transfected with polyethylenimine (PEI; Polysciences, Inc.). Cells were treated with CHX for the indicated times after 24 h of transfection and then harvested. Total extracts obtained using total lysis buffer (2% sodium dodecyl sulfate [SDS], Tris-HCl [pH 7.5], glycerol 10%) were boiled and then centrifuged at 13,000 rpm for 15 min. Alternatively, Snail1-F-Luc (Snail1-firefly luciferase fusion protein) and R-Luc (Renilla luciferase) were expressed with a pLEX-Snail-F-Luc and pMSCV-R-Luc vectors (a kind gift from Y. Kang, Princeton University) and used to measure Snail1 stability as previously described (40). Other transfections were carried out using Lipofectamine and plasmid pcDNA4-NICD-Myc (a kind gift from A. Bigas, IMIM), pcDNA3-β-TrCP1-Flag, pcDNA3-Fbxl14-Myc, or pcDNA3-Fbxl5-Myc were previously described (41, 42). For immunoprecipitation experiments of endogenous Akt1/2, total cell extracts were obtained with lysis buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 0.5% Triton X-100, and protease inhibitors) and precleared with protein A-magnetic beads (Millipore) for 1 h at 4°C. Clarified supernatants were incubated with rabbit anti-Akt1 or anti-Akt2 antibodies (1:50) overnight. Immunocomplexes were recovered on protein A-magnetic beads for 1 to 2 h at 4°C and analyzed in Western blotting with anti-pAkt (Ser473 or Thr308) antibodies.
To obtain cell extracts with differentiated cytoplasmic and nucleoplasmic fractions, cells were scrapped in ice-cold buffer A (10 mM HEPES-KOH [pH 7.8], 1.5 mM MgCl2, 10 mM KCl, and protease inhibitors) and kept on ice for 10 min (42). A 1/30 volume of the lysate of 10% Triton X-100 was added, and the tubes were vortex mixed for 20 s. Centrifugation of the sample for 1 min at 11,000 rpm separated out the cytoplasmic extract. The remaining pellet was lysed with 2% SDS buffer for the nuclear fraction. Isolation of the nuclear envelope and associated proteins was performed with a Minute nuclear envelope protein extraction kit (Invent Biotechnologies, Inc.). Western blotting was performed as previously reported (41), quantified with ImageJ software, and normalized with respect to tubulin. Statistical analyses were carried out using SPSS 18.0; P values were calculated using a two-tailed unpaired Student t test.
Cell infection.
Lentiviral expression in different cell lines was used to stably knock down Akt1 and Akt2 using shRNAs cloned in pLKO.1-puro vector (Mission; Sigma) (42). For viral infection, HEK 293T cells were seeded in p100 plates with 10 ml of medium at high confluence and transfected by mixing 1.5 ml of 150 mM NaCl and 78 μl of PEI with a total of 20 μg of DNA (50% was the shRNA or an equally distributed quantity of different shRNAs targeting the same gene, 10% was the pCMV-VSV-G vector, 30% was the pMDLg/pRRE vector, and 10% was the pRSV rev vector). The medium was changed 24 h after transfection, and 5.5 ml fresh medium was added. The supernatant was used for cell transduction at 24 h and 48 h after the medium change by filtering it through 45-μm-pore size membrane filters (Millipore) and then adding 0.8 μg of Polybrene/ml to the viral supernatant, which was used to replace the medium of the target cells. After the second round of infection, the target cell medium was changed, and puromycin (2.5 μg/ml) was added, where indicated, for 72 h to select for infected cells.
Immunofluorescence.
Cells were grown on sterile coverslips for 24 h and fixed with PBS–4% paraformaldehyde for 15 min at room temperature. After washing with PBS, the cells were permeabilized with PBS–0.5% (vol/vol) Triton X-100 for 5 min and blocked with PBS–3% bovine serum albumin (BSA) for 1 h at room temperature. Coverslips were incubated with pGSK-3β (S9) at 1:50 in blocking buffer (PBS–3% BSA) and rinsed with PBS. To detect Akt1 and Akt2 in the nuclear compartment without cytoplasmic interference, the cells were pretreated with CSK buffer as previously described (43) and analyzed with antibodies against Akt1, Akt2, and lamin B (1:50). Briefly, coverslips were incubated on ice for 5 min with CSK buffer (20 mM HEPES [pH 8], 100 mM KCl, 3 mM MgCl2, 300 mM sucrose) containing 0.5% Triton X-100 and rinsed once with CSK buffer without Triton X-100. Cells were then fixed with 4% paraformaldehyde at room temperature and rinsed with PBS. Coverslips were incubated with secondary antibodies (Alexa Fluor 555-conjugated goat anti-rabbit IgGs for Akt1, Akt2, and pS9-GSK-3β and Alexa Fluor 647-conjugated rabbit anti-goat IgGs for lamin B), stained with DAPI (4′,6′-diamidino-2-phenylindole; Sigma) for 2 min, rinsed, and then mounted with Fluoromount G (Southern Biotech). Fluorescence was visualized using the inverted fluorescence microscope DM IRBE (Leica, Wetzlar, Germany) and captured with a TCS-NT argon/krypton confocal laser microscope (Leica). ImageJ software was used for the relative quantification of the amount of each Akt isoform in the nuclear compartment. At least 10 different cells from three independent confocal images were analyzed; the perinuclear compartment was delineated by the lamin B signal.
RNA interference.
For stable Akt1 or Akt2 gene silencing, the Mission shRNA plasmids (Sigma) were used to produce lentiviral particles. After transduction, stable cell lines expressing the shRNA were isolated by puromycin (2.5 μg/ml) selection. For Akt2 or Snail1 depletion, cells were transfected using Lipofectamine RNAiMAX (Invitrogen) with specific synthetic small interfering RNAs (siRNAs) for Akt2 or Snail1 (Dharmacon) or a siRNA control for 72 h.
Luciferase reporter assays.
Cells were transfected with a TOP-Flash plasmid, a synthetic promoter sensitive to the activity of the β-catenin/TCF4 complex that contains three copies of the TCF4 binding site upstream of a firefly luciferase reporter gene. A mutant form of this promoter (FOP plasmid) was used as a control. Activity of the product of the Renilla luciferase gene under the control of a constitutive thymidine kinase promoter (Promega) was used to normalize transfection efficiency. Assays were always performed in triplicate. The pGL3-Akt1 promoter (−4293 to +1888) was provided by P.-J. Lu (National Cheng Kung University, School of Medicine, Tainan, Taiwan) (44) and the pGL3-Akt2 promoter (−2898 to +220) was a kind gift from J. Q. Cheng (Moffitt Cancer Center, Tampa, FL) (45).
RNA analyses.
RNA was extracted and retrotranscribed as described previously (41, 42). Analyses were carried out by quantitative PCR (qPCR) using a LightCycler 480 real-time PCR system (Roche), with 100 ng per condition and always in triplicates. The primers (Sigma) used were as follows: Akt1, forward, 5′-CTCAAGAACGACCCCAAGCA-3′, and reverse, 5′-CCGTGAACTCCTGGTCGAAA-3′; Akt2, forward, 5′-ATTCACCGCCCAATCCATCA-3′, and reverse, 5′-CGAGTAGGAGAACTGGGGGA-3′; Snail1, forward, 5′-CGAGTAGGAGAACTGGGGGA-3′, and reverse, 5′-CCAGGAGAGAGTCCCAGATG-3′; FoxO1, forward, 5′-GCAAATCGAGTTACGGAGGC-3′, and reverse, 5′-AATGTCATTATGGGGAGGAGAGT-3′; Fibronectin (FN1), forward, 5′-AGCAAGCCTGAGCCTGAAGAC-3′, and reverse, 5′-GCGATTTGCAATGGTACAGCT-3′; and Pumilio, forward, 5′-CGGTCGTCCTGAGGATAAA-3′, and reverse, 5′-CGTACGTGAGGCGTGAGTAA-3′.
Annexin V and MTT assays.
The annexin V-allophycocyanin (APC)-conjugated (ImmunoTools) assay was used as an early apoptotic indicator by detecting phosphatidylserine in the plasma membrane. Cells were treated with hydrogen peroxide (200 μM) for 16 h and then trypsinized, and 500,000 cells were resuspended in 70 μl of binding buffer (2.7 mM CaCl2 in PBS) with 5 μl of annexin V that was incubated at room temperature for 15 min in the dark. Prior to flow cytometry analysis, 10 μl of a 10-μg/ml concentration of propidium iodide was added to each sample to detect late apoptotic or necrotic cells. Samples were analyzed with the LSR II flow cytometer (Becton Dickinson) at the CRG/Universitat Pompeu Fabra (UPF) FACS Unit. The percentages of cells in each quadrant were analyzed using FACSDiva software (Becton Dickinson). MTT assays were performed by adding 0.5 mg of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma)/ml for 3 h at 37°C to determine the percentage of viable cells. After solubilization of the cells in dimethyl sulfoxide-isopropanol (1:4), the absorbance of insoluble formazan (purple) at 590 nm was determined. The absorbance at 590 nm was proportional to the number of viable cells.
RESULTS
Notch promotes EndMT in aortic endothelial cells.
To study the role of Notch in normal aortic endothelial cells, we used pig aortic endothelial (PAE) cells stably expressing moderate or high levels of activated Notch intracellular domain (NICD1 and NICD2 cells, respectively) or the empty vector (control [CTL]) and analyzed the expression of genes related to EndMT (Fig. 1A). Active Notch expression resulted in a partial (in the case of NICD1) or a complete (NICD2) downregulation of VE-cadherin and an upregulation of the mesenchymal proteins N-cadherin, fibronectin, α-Sma, Snail1, and Snail2 (Fig. 1A). Concomitantly, cells expressing Notch displayed a more fibroblastic phenotype than cells transfected with the control plasmid (Fig. 1B). We compared these results to those obtained with a well-established inducer of the mesenchymal phenotype, TGF-β1 (46, 47). In PAE cells, TGF-β1 was unable to induce Snail1 or fibronectin or to downregulate VE-cadherin (Fig. 1C). Interestingly, although TGF-β1 did not upregulate the Snail1 protein, it did increase Snail1 mRNA in control PAE cells (compare Fig. 1C and D to Fig. 1E). In contrast to the effects of TGF-β1, Notch activation barely affected Snail1 mRNA (with a 1.1- and 2-fold increases in NICD1 and NICD2 cells, respectively, compared to CTL cells) (Fig. 1E), although it significantly induced Snail1 protein, especially in NICD2 cells (4-fold) (Fig. 1D). Therefore, the increased expression of Snail1 in Notch-expressing cells was not mainly a consequence of an upregulated Snail1 transcription, suggesting instead a posttranscriptional regulation. Probably as a result of these transcriptional (TGF-β1) and posttranscriptional (Notch) effects, both factors cooperatively upregulated the Snail1 protein (Fig. 1C and D). The cooperation between Notch and TGF-β1 was also detected on fibronectin RNA (Fig. 1E) and protein (Fig. 1C and D). These experiments suggest that Notch and TGF-β1 jointly trigger Snail1 and fibronectin expression and EndMT.
FIG 1.

Notch and TGF-β1 cooperate in EndMT and in the stimulation of Snail1 and fibronectin expression. (A) The indicated proteins were analyzed in PAE total extracts from control (CTL) or NICD-transfected (NICD1 and NICD2) cells by Western blotting. Tubulin was used as a loading control. (B) Representative micrographs of PAE-CTL and PAE-NICD2 cells. (C and D) VE-cadherin, Snail1, and fibronectin levels were determined by Western blotting in total extracts of the indicated cells treated with TGF-β1 (5 ng/ml) for 24 h. A representative result is shown in panel C, and the averages ± the standard deviations (SD; n = 3) of the quantification of Snail1 and fibronectin are shown in panel D. Densitometric values were normalized with respect to tubulin. (E) Snail1 and fibronectin (FN1) RNA in the indicated cells treated with TGF-β1 for 24 h was analyzed by RT-qPCR. Averages ± the SD (n = 4) are shown. In panels D and E, Snail1 values obtained in nontreated control cells or fibronectin values in nontreated NICD1 cells were used as a reference. (F) PAE cells were transfected with pcDNA3-Snail1-HA and, after 24 h, the cells were treated with 20 μg of CHX/ml for the indicated times and analyzed by Western blotting. (G) Snail1-HA degradation was quantified with ImageJ software, normalized with respect to tubulin, and represented with respect to the value at 0 h. The averages ± the SD (n = 4) are shown. (*, P < 0.05; **, P < 0.01).
Snail1 stability is increased by Notch and requires β-TrCP1/GSK-3β inactivation.
Since the Snail1 protein is highly unstable (see the introduction), we examined whether Notch increased Snail1 by upregulating the Snail1 protein stability. We measured the half-life of exogenous Snail1-HA transiently transfected in PAE-CTL or PAE-NICD1 and NICD2 cells after adding CHX. Indeed, the relative half-life of Snail1-HA increased by >2-fold in NICD1 or NICD2 cells compared to the control cells (Fig. 1F and G). These results suggest that Notch posttranscriptionally affects Snail1 by enhancing its protein stability.
Notch stabilization of Snail1 protein was also detected in HEK 293T cells (Fig. 2A). We analyzed the capability of different Snail1 E3 ubiquitin ligases to promote Snail1 degradation in NICD-expressing cells. Transfection of the E3 ligases Fbxl14 or Fbxl5 induced Snail1 downregulation in NICD2 cells; however, ectopic β-TrCP1 was not effective (Fig. 2B), indicating that the action of β-TrCP1 ligase was impaired after Notch expression.
FIG 2.

Notch impairs Snail1 degradation by β-TrCP1. (A) Snail1-HA stability was checked in HEK 293T cells transfected with NICD-myc or with an empty vector (CTL). (B) Three known, tagged Snail1 ubiquitin ligases, namely, β-TrCP1-flag, Fbxl5-myc, and Fbxl14-HA, were also cotransfected. After 24 h, cells were treated with CHX for the indicated times, and cell extracts were analyzed by Western blotting.
β-TrCP1 protein levels were not affected by NICD expression (Fig. 3A). β-TrCP1-dependent degradation of Snail1 requires its previous phosphorylation by GSK-3β. The levels of this protein kinase were not decreased by Notch, either in total extracts (Fig. 3A) or in the cytoplasm or nucleus (Fig. 3C); however, phosphorylation of GSK-3β at S9, indicative of inactivation, was highly increased in NICD1 and NICD2 cells (Fig. 3A and B). These results suggest that the stabilization of the Snail1 protein in Notch-expressing PAE cells is a consequence of decreased GSK-3β activity.
FIG 3.

Notch promotes a switch in Akt isoform expression, inactivates GSK-3β, and accumulates Snail1 in the nucleus. (A and C) The indicated proteins were analyzed by Western blotting in total (A) or cytosolic (lanes CE) and nuclear (lanes NE) extracts (C). Tubulin and lamin B were used as a control for cytosolic and nuclear compartments, respectively. (B) pS9-GSK-3β levels were quantified by densitometric analysis and normalized respect to total GSK-3β; the averages ± the SD (n = 4) are shown (*, P < 0.05). (D) Active levels of Akt1 and Akt2 were determined after immunoprecipitation (IP) of both isoforms, which were blotted against the respective phosphorylated proteins. Irr Ab, irrelevant control antibody; IgG, immunoglobulin band. (E) pS9-GSK-3β levels were analyzed by immunofluorescence in PAE-CTL and PAE-NICD2 cells (red); DAPI was used to identify nuclei. (F) PAE cells stably transfected with NICD-myc (NICD2) or empty vector (CTL) were treated with TGF-β1 for 24 h, and the indicated proteins were analyzed by Western blotting.
Akt1 and Akt2 are inversely modulated by Notch.
Since GSK-3β is inactivated in NICD cells, we also determined the levels of Akt, the kinase responsible for GSK-3β S9 phosphorylation. Unexpectedly, both total Akt and the active form of this kinase (as determined by the presence of the S473 phosphorylation) were decreased upon Notch activation (Fig. 3A). However, independently analyzing the Akt1 and Akt2 isoforms showed an inverse modulation for these, with Notch downregulating Akt1 but increasing Akt2 (Fig. 3A); the third isoform, Akt3, was not expressed in these cells. The fact that total Akt and Akt1, but not Akt2, was downregulated by Notch suggests that Akt1 is the most abundant isoform in these cells and also that Akt2 is likely to be the isoform causing GSK-3β phosphorylation upon Notch expression, since it is the only one induced under these conditions. We also determined the levels of active Akt1 and Akt2 individually by immunoprecipitating each isoform and analyzing the phosphorylation at S473 and T308, which correlate with activity. The levels of pS473-Akt2 and pT308-Akt2 were increased by Notch expression, whereas the levels of pS473-Akt1 were downregulated (Fig. 3D). These results indicate that Akt2 is the active isoform upon expression of NICD.
Snail1 destabilization by GSK-3β requires the presence of this protein kinase in the nucleus (25). Therefore, we determined if GSK-3β phosphorylation and inactivation was promoted by nuclear Akt1/2. As expected, Snail1 was detected in the nucleus (Fig. 3C). Akt1 and Akt2, although mostly cytosolic, were also present in the nuclear fraction (Fig. 3C). Expression of NICD promoted an Akt1 downregulation in the nucleus, as well as an Akt2 increase in both the nuclear and the cytoplasmic compartments, that paralleled the increase in GSK-3β phosphorylation (Fig. 3C). Immunofluorescence staining also showed a remarkable increase in pS9-GSK-3β in the nucleus (Fig. 3E). These results suggest a clear relationship between Notch activation, nuclear Akt2 accumulation, and phosphorylation and inactivation of GSK-3β in this compartment, as well as Snail1 protein stabilization. Accordingly, TGF-β1, which enhances the Notch effect on Snail1 (and fibronectin) expression but does not rise Snail1 stability (see above), did not alter Akt1 or Akt2 levels and did not increase GSK-3β phosphorylation (Fig. 3F).
Apart from Akt, the p90 ribosomal S6 kinase RSK can also phosphorylate and inactivate GSK-3β (28). RSK activity is dependent on its prior phosphorylation by Erk (28). Interestingly, phosphorylation of RSK and Erk-1/2 is increased in NICD1 and -2 cells (Fig. 3A). To evaluate the relative contributions of different kinases in Snail1 induction upon Notch expression, we used a general Akt inhibitor MK-2206 (48) and compared the results to those obtained with the Erk and RSK inhibitors U0126 (49) and SL0101 (50), respectively. The three compounds decreased Snail1 protein levels in NICD2 cells (Fig. 4A and B). As expected, MK-2206 completely downregulated Akt phosphorylation, whereas U0126 efficiently inhibited pErk-1/2 and pRSK; cyclin D1 downregulation was used to verify SL0101 activity on RSK (51). MK-2206 exhibited the strongest effect on GSK-3β phosphorylation, although the other two inhibitors also significantly affected it (Fig. 4A and C). Overall, these experiments suggest that although both the Akt and Erk/RSK pathways additively phosphorylate and inactivate GSK-3β, Akt—and most likely, Akt2—exerted the strongest effect.
FIG 4.

Akt and Erk/RSK collaborate in GSK-3β inactivation. (A to C) Total extracts of CTL or NICD2 were analyzed by Western blotting. Where indicated, cells were treated with MK-2206 (MK), UO126 (UO), or SL0101 (SL) (all at 10 μM) for 24 h. (B and C) Densitometric quantification of Snail1 normalized to tubulin (B) and pS9-GSK-3β normalized to total GSK-3β (C). Expression was related to maximal expression in NICD2 cells treated with vehicle (−) (averages ± the SD; n = 3); (*, P < 0.05).
Akt2 upregulation by Notch is dependent on β-catenin transcriptional activity.
We next analyzed the mechanism controlling Akt expression in endothelial cells. Akt2 induction and Akt1 downregulation by NICD were verified by mRNA analyses (Fig. 5A and B). The activity of a 6-kbp fragment of Akt1 promoter was downregulated by Notch (Fig. 5C), indicating that the Notch effect on this gene was transcriptional. In contrast, Notch transfection increased the activity of a fragment of Akt2 promoter corresponding to −2898 to +220 with respect the transcription start site (Fig. 5D). These experiments suggest that Notch inversely modulates the transcription of Akt1 and Akt2 genes.
FIG 5.

Notch inversely modulates Akt1 and Akt2 RNA and promoter activities. (A and B) Akt1 and Akt2 mRNA were analyzed by RT-qPCR in CTL and Notch-expressing cells. The averages ± the SD (n = 4) are shown. (C and D) PAE-CTL cells were transiently transfected with NICD and Akt1 or Akt2 promoters for 24 h. Firefly luciferase was measured and normalized with respect to thymidine kinase (TK)-Renilla. (**, P < 0.01; *, P < 0.05).
Because active Akt2 is the predominant isoform in Notch expressing cells, we further investigated the mechanism by which its expression is increased. It has been previously described that Akt2 transcription is activated by the β-catenin/TCF4 transcriptional complex (52). By quantifying this complex activity using the TOP-FLASH reporter plasmid, we found that NICD expression stimulated the β-catenin transcriptional activity by 2.5-fold (Fig. 6A). This increase was abolished if cells were supplemented with iCRT14 inhibitor (53), which blocks β-catenin/TCF4 interaction to prevent β-catenin transcriptional activity (Fig. 6A). This inhibitor precluded the NICD-induced upregulation in Akt2 mRNA (Fig. 6B) and decreased other Notch effects. Specifically, it diminished total and nuclear Snail1 protein (Fig. 6C and D) and decreased the presence of nuclear Akt2 and nuclear phosphorylation of GSK-3β (Fig. 6D), without affecting Akt1 protein levels or nuclear accumulation (Fig. 6D and E). Downregulation of the Akt2 protein caused by iCRT14 was also accompanied by a decrease in pS473-Akt2, indicative of lower activity of this kinase (Fig. 6E). iCRT14 did not affect active Akt1 levels (Fig. 6E). The iCRT14 inhibitor did affect Snail1 stability, since it decreased the expression of ectopic Snail1-HA in HEK 293T cells transfected with NICD (Fig. 6F). Similar to PAE cells, this cell line responded to Notch ectopic expression with Akt2 upregulation and GSK-3β inactivation (Fig. 6G). Overall, these data suggest that β-catenin/TCF4 activity is essential for Akt2 transcriptional upregulation by Notch and for Snail1 stabilization.
FIG 6.

Notch upregulates Akt2 and GSK-3β phosphorylation by stimulating β-catenin/TCF4 transcriptional activity. (A) Firefly luciferase activity of TOP-FLASH was measured in PAE-CTL and PAE-NICD2 cells after 48 h of plasmid transfection; where indicated, iCRT14 (25 μM) was added to the cell medium for the last 24 h. (B to D) mRNA levels of Akt2 (n = 3) (B), total Snail1 protein from densitometric quantification of independent blots (n = 4) (C), or the indicated nuclear proteins (D) were analyzed as above at 24 h (B) or 72 h (C and D) after iCRT14 addition. (E) Active levels of Akt1 and Akt2 were determined as described for Fig. 3D after treatment with iCRT14 for 72 h. (F) HEK 293T cells were transiently transfected with pcDNA3-Snail1-HA and pcDNA3-NICD-myc; after 24 h, the cells were treated with iCRT14 for an additional 24 h. Exogenous Snail1-HA or NICD-myc were analyzed by Western blotting. (G) HEK 293T cells were transfected with NICD-myc or an empty vector as control (vector), and cell extracts were analyzed by Western blotting with the indicated antibodies. The averages ± the SD respect to CTL untreated cells are shown in panels A to C (**, P < 0.01; *, P < 0.05).
Notch activation of Akt2 blocks GSK-3β activity.
To specifically confirm that Akt2 was involved in Notch-dependent Snail1 stabilization, we downregulated Akt2 by infecting a shRNA that selectively affected Akt2 but not Akt1 (Fig. 7A and B). Akt2 depletion did not modify pS473-Akt global levels, suggesting that Akt1 is the most abundant isoform also in NICD2 cells, but it did decrease pS9-GSK-3β and Snail1 levels (Fig. 7B). We verified that Akt2 downregulation decreased Snail1 stability in NICD2 cells by expressing a Snail1-firefly luciferase fusion protein in cells treated with CHX; the results were normalized with a Renilla luciferase cotransfected as an internal control (54). Indeed, Snail1 degradation was accelerated in NICD2 cells depleted of Akt2 compared to NICD2 control cells (Fig. 7C). We also investigated the role of Akt2 in the Notch/TGF-β1 cooperation. Besides inhibiting Snail1 upregulation, Akt2 attenuation decreased fibronectin induction (Fig. 7D, compare lanes 4 and 8; quantification in Fig. 7E). Similar results were obtained by transfecting an Akt2 siRNA that promoted a higher downregulation in Akt2, which also prevented Snail1 and fibronectin upregulation by Notch and TGF-β1 (Fig. 7F).
FIG 7.

Akt2 is required for the Notch induction of Snail1 protein stability. (A) After selecting the most appropriate shRNA for each isoform, Akt1 and Akt2 were downregulated transducing the corresponding lentiviral shRNA and selected with puromycin. Akt1 and Akt2 were determined by Western blotting; tubulin was used as a loading control. (B to E) CTL and NICD2 cells were infected with shRNA corresponding to Akt2 or a scrambled control, and the indicated proteins were analyzed by Western blotting. (C) The indicated cells were transfected with a fusion protein of Snail1 and firefly luciferase. The luciferase activity was measured and normalized against Renilla luciferase at different time points after CHX supplementation. Data are represented with respect to the value at time zero; the averages ± the SD (n = 3) are shown. In panel D, cells stably infected with shCtl and shAkt2 were treated with 5 ng of TGF-β/ml for 24 h. (E) The levels of protein shown in panel D were quantified as indicated above (n = 3). **, P < 0.01; *, P < 0.05; au, arbitrary units. (F) The experiment was carried out as for panel D, but using transfection with siRNA corresponding to Akt2 or a scrambled control.
Finally, these results were confirmed in a different cellular model, using MEFs. TGF-β1 enhanced the levels of Snail1 with a concomitant increase in pS9-GSK-3β (Fig. 8). Although both Akt1 and Akt2 depletion affected Snail1 levels in control cells, only Akt2 was relevant in TGF-β1-treated cells, since MEFs depleted of this isoform (but not of Akt1) showed an impaired upregulation of Snail1 protein and GSK-3β phosphorylation in response to this cytokine (Fig. 8). Therefore, Akt2 but not Akt1 is involved in GSK-3β inactivation and Snail1 stabilization.
FIG 8.
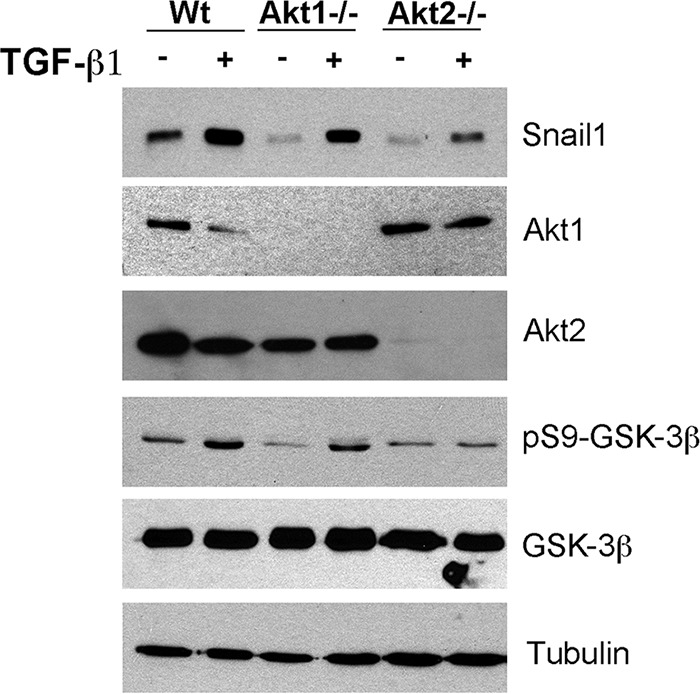
Akt2 deletion prevents GSK-3β inactivation and Snail1 induction by TGF-β1 in MEFs. MEFs, either wild-type (WT) or knocked out for Akt1 (Akt1–/–) or for Akt2 (Akt2–/–), were treated for 1 h with TGF-β1. The indicated proteins were analyzed by Western blotting.
Notch effects on oxidative stress-induced apoptosis are Akt2 dependent.
To evaluate the physiological significance of Notch-mediated Akt2 induction, we used endothelial cells that are sensitive to oxidative stress, since they undergo apoptosis after H2O2 exposure (55, 56). At 16 h after incubation of PAE cells with 200 μM H2O2, a remarkable percentage (36%) of the cells were apoptotic, as determined by staining with the specific marker annexin V and propidium iodide (Fig. 9A). In contrast, Notch expression in NICD2 cells protected from H2O2-induced cell death, since only 7% of the cells were apoptotic under these treatment conditions. A similar protection was observed when the levels of processed caspase-3 were determined: at 24 h after H2O2 addition, the levels of this protease were significantly increased in control PAE cells but not in NICD-expressing cells (Fig. 9B and C). H2O2 did not significantly modify the levels of either total or active Akt1 or Akt2 (Fig. 9C and D). The apoptotic regulator and Akt-substrate FoxO1 was downregulated in NICD2 cells (Fig. 9C, F, and G). This protein undergoes nuclear export and degradation upon phosphorylation (34, 35). Accordingly, FoxO1 S256 phosphorylation was higher in NICD2 cells compared to CTL cells, whereas the mRNA levels were similar (Fig. 9E, F, and H). The higher FoxO1 phosphorylation observed in NICD2 cells further indicated that nuclear Akt activity was stimulated in these cells.
FIG 9.

Notch expression in PAE cells protects from apoptosis. (A) FACS analysis by annexin V-APC and propidium iodide (PI) staining at 16 h after adding 200 μM hydrogen peroxide (H2O2) to CTL and NICD2 cells. PI−/annexin V+ cells (Q4) were considered early apoptotic. Representative results from one of the three experiments (which all gave similar results) are shown. (B and C) Total protein extracts of indicated cells, treated where indicated with H2O2 for 24 h, were analyzed by Western blotting. A representative experiment is shown (C), as well as the quantification of cleaved caspase-3 (B), using the averages ± the SD (n = 4). (D) Active levels of Akt1 and Akt2 were analyzed as described for Fig. 3D. (E and F) Changes in FoxO1 were determined in control (Ctl) and NICD2-expressing cells by RT-qPCR (E) or by Western blotting (F). Phosphorylated FoxO1 (pS256-FoxO1) was also determined (F). (G and H) The quantification of three different Western blot analyses. *, P < 0.05 compared to CTL.
To evaluate the effects of Akt1, Akt2, and Snail1 in H2O2-induced apoptosis, we used PAE cell lines stably infected with shRNAs (for Akt1 and Akt2), or transfected with siRNAs (for Snail1), to block the expression of these proteins. Akt shRNAs are specific and did not modify the levels of the not-targeted isoform (Fig. 7A). In control PAE cells, downregulation of Akt1 or Akt2 but not of Snail1 resulted in a higher activation of caspase-3 upon H2O2 treatment, suggesting that both isoforms limit apoptosis in endothelial cells in basal conditions (Fig. 10A and B, lanes 2 and 6). However, only downregulation of Akt2 (but not of Akt1) prevented the Notch-dependent protection against apoptosis. As shown above, NICD2 cells did not activate caspase-3 after H2O2 treatment (Fig. 10A and B, compare lanes 2 and 4); downregulation of Akt2 but not of Akt1 restored caspase-3 activation after this insult in Notch cells (Fig. 10A and B, compare lanes 4 and 8), confirming our previous observations. Snail1 was not induced in NICD2 cells in which Akt2 had been downregulated (Fig. 10B, lanes 3 and 7); in contrast, it was upregulated by Notch in Akt1-depleted cells (Fig. 10A, lanes 3 and 7).
FIG 10.

Akt2-induced Snail1 stability is essential for apoptosis prevention by Notch in endothelial cells. (A, B, D, and E) Akt1 and Akt2 were downregulated using specific shRNAs targeting each kinase and Snail1 with a specific siRNA in both CTL and NICD2 PAE cells. Cells were treated with H2O2 for 24 h, and the indicated proteins were analyzed by Western blotting. Tubulin was used as a loading control. (C) Control or NICD2 PAE cells were treated with H2O2 for 24 h, when indicated, and Akt2 was inhibited by treatment with CCT128930 (CCT; 10 μM). (E) After 32 h of H2O2 treatment, the cell viability was quantified as indicated in Materials and Methods and is represented with respect to the value in nontreated cells. Averages ± the SD (n = 3) are shown. (*, P < 0.05).
We also determined the levels of FoxO1 that, as shown before (Fig. 9), was downregulated in NICD2 cells. Akt1 attenuation increased basal FoxO1 but only prevented its downregulation by Notch to a low extent (Fig. 10A). In contrast, Akt2 downmodulation did not affect basal FoxO1 levels but blocked the decrease observed in NICD2 cells (Fig. 10B). These results suggest that Akt1 controls the basal levels of FoxO1, whereas Akt2 acts on this protein upon Notch activation. These results were confirmed using CCT128930, an ATP competitive and selective inhibitor of Akt2 (57). This compound also prevented NICD2 from affecting caspase-3 activation and blocked GSK-3β inactivation in these cells (Fig. 10C).
Snail1 depletion altered the levels of active caspase-3 in a manner similar to Akt2 downmodulation (Fig. 10D). In Snail1-depleted cells, NICD was unable to counteract H2O2 and to preclude caspase-3 activation (Fig. 10D, compare lane 8 to lane 4). Finally, to confirm that caspase-3 activation results in cell death, we determined cell viability after H2O2 treatment by MTT incorporation (Fig. 10E). Cell viability was notably reduced by H2O2 in CTL cells, with only 55% of cells viable after this insult. However, NICD2 cells had a much higher resistance, and their viability decreased only by 13% after H2O2 treatment (Fig. 10E). Akt2 depletion only slightly decreased control cell viability (from 55 to 52%) but significantly affected NICD2 cell viability (from 87 to 50%) after H2O2 exposure (Fig. 10E). Based on these results, we conclude that Akt2 and Snail1 mediate the protection of Notch against H2O2-induced apoptosis.
Notch promotes nuclear localization of Akt2 and accumulation at the nuclear membrane.
Our results suggest that the effects of Notch on endothelial cell apoptosis and Snail1 stability are mediated by Akt2 but not by Akt1. However, the mechanism underlying this difference in action of the two Akt isoforms is unknown. Although the increased Akt2/Akt1 ratio might explain this effect, a considerable amount of Akt1 is still present in endothelial cells upon Notch expression (Fig. 3A). We hypothesized that the presence of one the two isoforms in the nucleus should be particularly relevant for GSK-3β phosphorylation, since this protein acts on Snail1 in the nucleus, similar to Akt's actions on FoxO1. To explore this, we determined the localization of the endogenous Akt isoforms in control and NICD2 cells.
Using an immunostaining procedure to enhance the detection of nuclear proteins (43), we determined that nuclear Akt1 was expressed to a lower degree in Notch-expressing cells compared to control cells (Fig. 11A and B), in line with previous results. In contrast, nuclear Akt2 expression was enhanced upon Notch expression (Fig. 11C and D). Interestingly, nuclear Akt2 staining was concentrated in the nuclear lamina, as shown by lamin B costaining (Fig. 11C). Confocal quantification analyses of Akt2 and lamin B in nuclear sections corroborated that Akt2 accumulated in the nuclear membrane upon NICD expression (Fig. 11D, right panel). A complete quantification of perinuclear and nucleoplasmic Akt2 determined in different sections along the cell nucleus revealed that the nuclear membrane-associated Akt2 was strongly enhanced upon NICD expression (Fig. 11E), in contrast to Akt1, which was not significantly present in the nuclear lamina in either control or NICD2 cells (Fig. 11B and E). Finally, we isolated the nuclear envelope proteins by biochemical subfractionation. The nuclear lamina fraction was not contaminated with cytoplasmic and nucleoplasmic proteins, since it did not contain tubulin or Sin3A proteins, but was enriched with lamin B (Fig. 11F). Importantly, Akt2, but not Akt1, as well as a significant part of pS9-GSK-3β, was detected in the nuclear lamina in NICD2 cells (Fig. 11F). We did not identify Snail1 in the nuclear membrane fraction, suggesting that it is a nucleoplasmic substrate of GSK-3β (Fig. 11F).
FIG 11.

Akt2 but not Akt1 is localized in the nuclear envelope in endothelial cells stimulated by Notch expression. (A to D) Nuclear immunofluorescence in PAE-CTL and PAE-NICD2 cells previously treated with CSK buffer to eliminate cytosolic proteins. Akt1, Akt2, lamin B, and DAPI staining was analyzed using a confocal microscope. Representative graphs of the colocalization of Akt1 (A and B) or Akt2 (C and D) with lamin B in both PAE-CTL and PAE-NICD2 cells are presented. (B and D) Each graph shows a line profile indicating areas of colocalization of Akt1 (B) or Akt2 (D) and lamin B in both PAE-CTL (left) and PAE-NICD2 (right) cells. Each line profile represents the intensity of the signal versus the pixel distance. The maximal lamin B staining is represented by a dashed line indicated by “NE” and corresponds to the nuclear envelope. (E) Quantification of the different immunofluorescence of Akt1 and Akt2 in the perinuclear compartment was performed as described in Materials and Methods. (F) Nuclear envelope proteins (NEnv) were isolated and analyzed by Western blotting. Total extracts (TE) were used as an input. Lamin B was used as a nuclear membrane marker; Sin3A and tubulin, present in the nucleoplasm and cytosol, respectively, were used to verify the absence of contamination in the nuclear lamina fraction.
To determine whether Akt2 activation is required for nuclear envelope localization, NICD2 cells were treated with the Akt inhibitor MK-2206. This inhibitor completely blocks phosphorylation of Akt at S473 (Fig. 4A), which results in an impaired Akt2 localization in the nuclear membrane, as determined by immunofluorescence (Fig. 12). In fact, Akt2 was not observed in the nucleus, suggesting that only active phosphorylated Akt2 is present in this compartment (Fig. 12). As a control, we determined that the Erk-1/2 inhibitor UO126 did not affect Akt2 localization to the nuclear membrane (Fig. 12). Interestingly, CCT128930 did not inhibit the presence of Akt2 in the nuclear membrane (Fig. 12), although it blocked the biological effects of Akt2 on apoptosis (Fig. 10C). In contrast to MK-2206, this Akt inhibitor does not prevent phosphorylation of Akt at S473 (57), which suggests that Akt2 phosphorylation regulates interaction with the nuclear membrane. Altogether, these results indicate that phosphorylated Akt2 is localized in the nuclear membrane of NICD2 cells, which might facilitate the interaction and phosphorylation of nuclear substrates, such as FoxO1 and GSK-3β, resulting in Snail1 stabilization and thereby preventing apoptosis.
FIG 12.

Nuclear Akt2 staining induced by Notch is blocked by inhibition of Akt phosphorylation. Akt2 and lamin B were analyzed by immunofluorescence as done in Fig. 11 in control PAE cells and Notch cells (NICD2) that had been treated with MK-2206 (MK), UO126 (UO), or CCT128930 (CCT) (all at 10 μM) for 24 h. Merge images indicate colocalization.
DISCUSSION
We demonstrate here that the expression of activated Notch1 in endothelial cells causes β-catenin activation, Akt2 upregulation, GSK-3β inactivation, and Snail1 stabilization. Furthermore, Notch expression results in phenotypic changes characteristic of EndMT and in resistance to oxidative stress-induced apoptosis. These responses require Akt2 and Snail1 but not Akt1. We also describe that Notch produces a switch in the expression of the Akt isoforms and that Akt2, but not Akt1, is associated with the nuclear membrane when present in the nucleus.
Notch activation in endothelial cells resulted in morphological changes associated with a conversion to a mesenchymal phenotype, with VE-cadherin levels downregulated and fibronectin and α-Sma levels increased. In PAE cells, Notch activates Snail1 by a mechanism that mainly involves protein stabilization, although a small increase in mRNA was also detected at high concentrations of NICD (Fig. 1E). We also observed this effect of Notch on Snail1 stability in other cell types, such as HEK 293T. Previous studies in cells undergoing hypoxia indicated that the Notch intracellular domain is recruited to the Snail1 promoter (although this mechanism does not seem to be relevant in our cells) and that Snail1 is stabilized by Notch through the upregulation of LOX (58). However, in most cellular systems, LOX expression is induced by Snail1 but does not contribute to its stabilization. As an alternative, we propose that Notch1 inactivates GSK-3β by blocking the β-TrCP1-mediated degradation of Snail1.
In epithelial cells, TGF-β1 induces an EMT characterized by a rapid Snail1 induction and a later fibronectin upregulation (47). In our model of endothelial cells (that is, in PAE cells), TGF-β1 enhanced Snail1 transcription but was not sufficient to upregulate the Snail1 protein. In line with this, it has been previously suggested that TGF-β1 and -β2 are not essential for Notch-mediated EMT in microvascular endothelial cells (59). Our results also suggest that TGF-β1 acts cooperatively with Notch in Snail1 induction, since the transcriptional effect of the former complements the protein stabilization effect of the latter. This cooperation between Notch and TGF-β1 does not occur on Snail2 (Slug) that is activated only by Notch (60) (Fig. 3F). During cardiac cushion cellularization, both Snail1 and -2 are required for EndMT, but Snail2 is first expressed at E9.5, whereas Snail1 is expressed at E10.5; Snail1 expression at this time point requires the parallel activation of Notch and TGF-β1 (60). It is possible that, in this process, Snail2 is the main factor involved in VE-cadherin repression, whereas Snail1 is required for the induction of mesenchymal markers. Accordingly, in our PAE cells, Snail1 levels correlated better with the activation of fibronectin than with the downregulation of VE-cadherin (Fig. 1C). It should be noted that Snail1 has a direct positive effect on activating fibronectin and other mesenchymal genes in EMT models (61, 62).
In PAE cells, GSK-3β inactivation is associated with its phosphorylation at Ser9, a modification carried out by Akt and RSK (27, 28). Although both Akt2 and RSK are activated by NICD, our studies with pharmacological inhibitors indicated that Akt2 is more relevant for pS9-GSK-3β (Fig. 4). Other experiments downregulating Akt2 (Fig. 7) also confirmed the impact of this isoform on GSK-3β activity in NICD cells, although we do not discard that RSK also cooperates in inactivating this kinase.
Notch induces the transcriptional upregulation of Akt2. Akt2 is a target gene of β-catenin/TCF4 complex that binds to this promoter (52). In endothelial cells, Akt2 activation downstream of Notch signaling was mainly dependent on β-catenin/TCF4 activity, as blocking β-catenin/TCF4 activity with the iCRT14 inhibitor prevented both Akt2 gene expression and Snail1 stabilization. Active β-catenin is the molecular link between Notch and Wnt signals (63). Simultaneous activation of Notch and β-catenin signaling is observed during endothelial cell differentiation after forming a complex with RBP-J (14). In fact, the two pathways have a very high degree of interplay, and it has been proposed that both comprise a unique signaling module (64).
An increase in Akt2 levels is associated with EndMT. TGF-β1 did not boost the levels of this kinase, neither in PAE nor in MEFs (Fig. 3 and 8, respectively). In both cell types, downmodulation of Akt2 influenced the expression of Snail1 more than did that of Akt1. In fact, Akt1 downregulation did not decrease Snail1 expression in NICD2 cells (Fig. 10A, lanes 3 and 7) but reduced it in MEFs (Fig. 8). It is possible that Akt1 participates in Snail1 transcription in these fibroblastic cells (30).
It has also been reported that expression of the two Akt isoforms is coordinated in other cellular systems. Akt1 downregulation induces EMT and Akt2 upregulation in mammary epithelial cells by a mechanism that requires Erk activation (29). In our cell system, Akt2 upregulation by Notch does not seem to be due to an Akt1 decrease, since Akt1 downregulation did not stimulate Akt2 in either PAE cells or MEF cells (Fig. 7 and 8). Inversely, Akt2 depletion did not alter Akt1 levels. Our results also show that Notch induced the Akt1 promoter downregulation. This was not due to increased β-catenin/TCF4 transcriptional activity, since the addition of iCRT14 did not restore Akt1 levels in NICD2 cells, although it decreased Akt2. Alternatively, it has been reported that Notch increases Runx3 (65), a potent repressor of Akt1 (66). Further studies are required to demonstrate whether this mechanism works in PAE cells.
The Notch-induced Akt1-to-Akt2 switch is also relevant for its antiapoptotic role. In particular, our results demonstrate that Notch protects endothelial cells from oxidative stress-induced cell death. Although the Notch effect might be isoform dependent (67), the majority of published data suggest a role for most Notch family members in apoptosis protection. For instance, constitutively active Notch4 inhibits endothelial cell apoptosis induced by lipopolysaccharide by a dual mechanism that requires JNK activation and Bcl-2 upregulation (68). Similarly, impaired Notch4 activity is associated with endothelial cell apoptosis (69). The molecular basis of Notch apoptosis protection is not fully understood and may involve multiple players. Our results suggest that Notch1 protects endothelial cells from H2O2-induced apoptosis by upregulating Akt2 and Snail1, since depletion of these two components abrogated the Notch effect, as determined by analyzing caspase-3 activation and cell viability (Fig. 10). Notch1 expression also caused a decrease in FoxO1, and this downregulation showed a higher sensitivity to the downregulation of Akt2 than Akt1 (Fig. 10B). Therefore, Notch protection mainly requires Akt2-dependent Snail1 activation and FoxO1 downmodulation. It should be noted that the antiapoptotic role of Snail1 has been demonstrated in different systems through the inhibition of several proapoptotic genes, either directly or indirectly through the activation of other repressors (70).
Little information has been available concerning the contribution of the different Akt isoforms in apoptosis protection. We now show that this effect is mainly dependent on Akt2. This conclusion is supported by other results indicating that Akt2 inhibits apoptosis during myogenic differentiation or UV irradiation (71, 72). Moreover, depletion of Akt2 but not of Akt1 induces tumor regression (73). However, it is likely that both Akt1 and Akt2 have alternative roles in inhibiting apoptosis in different conditions; in fact, depletion of Akt1 in control PAE cells stimulated cleaved caspase-3 (Fig. 10A). Therefore, Akt1 might be relevant in endothelial not-stimulated cells and, following Notch induction, this role might be transferred to Akt2, which is then the main isoform controlling apoptosis under these conditions.
Another point of discussion is the specific nuclear localization of Akt2 when preventing apoptosis. Transgenic mice expressing Akt targeted to the nuclei of cardiac cells show apoptosis resistance in cardiomyocytes (74). Hydrogen peroxide mimics oxidative stress-mediated vascular damage that occurs during atherosclerosis and hypertension. For example, arteries exposed to high pressure generate substantial increases of vascular H2O2 (75). It has been recently shown that aortas from Akt2-deficient mice display high levels of apoptotic cell death that lead to increased aortic aneurysms (76). This Akt2-dependent protection is caused by an elevated expression of matrix metalloproteinase 9 (MMP-9) and reduced expression of tissue inhibitor of metalloproteinase 1 (TIMP-1), a consequence of the decreased binding of FoxO1 to the MMP-9 and TIMP-1 promoters (76). In our cell model, it is likely that Snail1 also plays a role in the induction of some of these genes, such as that for MMP-9, which is induced during Snail1-controlled EMT (77).
The specific mechanism that links Akt2 but not Akt1 with EndMT is not known. The balance between the Akt isoforms might be relevant for driving a differential phosphorylation of specific substrates (78, 79); thus, it might promote the regulation of specific Akt2 nuclear targets (80). Akt2 is located in both the cytoplasm and the nucleus; nuclear localization requires a previous activation by phosphorylation, suggesting that these modifications release Akt2 from an interaction with a cytosolic chaperone that precludes its traffic to the nucleus. This specific localization of Akt2 at the nuclear membrane in Notch-expressing cells is not observed for Akt1 in control PAE cells. Although Akt1 and Akt2 are highly homologous, they mostly differ in the linker region between the pleckstrin homology and catalytic domains (81). It is possible that an adaptor protein specific for Akt2 is expressed or exposed in the nuclear envelope upon Notch expression and is responsible for the specific binding. Alternatively, this nuclear scaffold protein might present similar affinity for both proteins but, since it is only active in NICD cells, it would preferably bind the Akt isoform with the highest expression in these cells—thus, Akt2. In any case, this specific Akt2 subnuclear localization might facilitate its action on FoxO1 and GSK-3β, promoting the inactivation of this kinase and the subsequent stabilization of Snail1. Since Snail1 also interacts with and increases Akt2 activity (43), it is possible that it creates a stimulatory loop to further enhance Akt2 activity on nuclear targets that regulate apoptosis. In any case, our results explain how Snail1 is controlled by Notch to regulate EndMT and apoptosis.
ACKNOWLEDGMENTS
We thank J. L. de la Pompa, A, Bigas, M. J. Birnbaum, J. Q. Cheng, P.-J. Lu, G. Gil, and Y. Kang for reagents. The technical support of Xavier Sanjuan (Advanced Light Microscopy Unit, CRG) and Oscar Fornas (CRG/UPF FACS Unit) was greatly appreciated. We also thank J. Baulida for advice.
We declare that we have no conflict of interest.
REFERENCES
- 1.Lim J, Thiery JP. 2012. Epithelial-mesenchymal transitions: insights from development. Development 139:3471–3486. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- 2.Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, Klewer SE. 2002. Temporal and distinct TGFβ ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol 248:170–181. doi: 10.1006/dbio.2002.0731. [DOI] [PubMed] [Google Scholar]
- 3.Medici D, Kalluri R. 2012. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol 22:379–384. doi: 10.1016/j.semcancer.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu W, Liu Z, An S, Zhao J, Xiao L, Gou Y, Lin Y, Wang J. 2014. The endothelial-mesenchymal transition (EndMT) and tissue regeneration. Curr Stem Cell Res Ther 9:196–204. doi: 10.2174/1574888X09666140213154144. [DOI] [PubMed] [Google Scholar]
- 5.Potenta S, Zeisberg E, Kalluri R. 2008. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer 99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. 2007. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 7.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. 2007. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67:10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 8.High FA, Epstein JA. 2008. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet 9:49–61. doi: 10.1038/nrg2279. [DOI] [PubMed] [Google Scholar]
- 9.MacGrogan D, Luna-Zurita L, de la Pompa JL. 2011. Notch signaling in cardiac valve development and disease. Birth Defects Res A Clin Mol Teratol 91:449–459. doi: 10.1002/bdra.20815. [DOI] [PubMed] [Google Scholar]
- 10.Kopan R, Ilagan MX. 2009. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzahor E. 2007. Wnt/beta-catenin signaling and cardiogenesis: timing does matter. Dev Cell 13:10–13. doi: 10.1016/j.devcel.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu T, Kagawa T, Inoue T, Nonaka A, Takada S, Aburatani H, Taga T. 2008. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jκ complex to promote proliferation and suppress differentiation of neural precursor cells. Mol Cell Biol 28:7427–7441. doi: 10.1128/MCB.01962-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gopalakrishnan N, Saravanakumar M, Madankumar P, Thiyagu M, Devaraj H. 2014. Colocalization of beta-catenin with Notch intracellular domain in colon cancer: a possible role of Notch1 signaling in activation of cyclin D1-mediated cell proliferation. Mol Cell Biochem 396:281–293. doi: 10.1007/s11010-014-2163-7. [DOI] [PubMed] [Google Scholar]
- 14.Yamamizu K, Matsunaga T, Uosaki H, Fukushima H, Katayama S, Hiraoka-Kanie M, Mitani K, Yamashita JK. 2010. Convergence of Notch and beta-catenin signaling induces arterial fate in vascular progenitors. J Cell Biol 189:325–338. doi: 10.1083/jcb.200904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arcaroli JJ, Quackenbush KS, Purkey A, Powell RW, Pitts TM, Bagby S, Tan AC, Cross B, McPhillips K, Song EK, Tai WM, Winn RA, Bikkavilli K, Vanscoyk M, Eckhardt SG, Messersmith WA. 2013. Tumours with elevated levels of the Notch and Wnt pathways exhibit efficacy to PF-03084014, a gamma-secretase inhibitor, in a preclinical colorectal explant model. Br J Cancer 109:667–675. doi: 10.1038/bjc.2013.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon C, Cheng P, King IN, Andersen P, Shenje L, Nigam V, Srivastava D. 2011. Notch posttranslationally regulates beta-catenin protein in stem and progenitor cells. Nat Cell Biol 13:1244–1251. doi: 10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia de Herreros A, Baulida J. 2012. Cooperation, amplification, and feed-back in epithelial-mesenchymal transition. Biochim Biophys Acta 1825:223–228. doi: 10.1016/j.bbcan.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Diaz VM, Vinas-Castells R, Garcia de Herreros A. 2014. Regulation of the protein stability of EMT transcription factors. Cell Adh Migr 8:418–428. doi: 10.4161/19336918.2014.969998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. 2000. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 20.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. 2000. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature cell biology 2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 21.Valdes F, Alvarez AM, Locascio A, Vega S, Herrera B, Fernandez M, Benito M, Nieto MA, Fabregat I. 2002. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol Cancer Res 1:68–78. [PubMed] [Google Scholar]
- 22.Escriva M, Peiro S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentis B, Murray SA, Franci C, Gridley T, Virtanen I, Garcia de Herreros A. 2008. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol 28:1528–1540. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajita M, McClinic KN, Wade PA. 2004. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 24:7559–7566. doi: 10.1128/MCB.24.17.7559-7566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diaz VM, de Herreros AG. 2015. F-box proteins: Keeping the epithelial-to-mesenchymal transition (EMT) in check. Semin Cancer Biol pii:S1044-579X(15)00100-5. doi: 10.1016/j.semcancer.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. 2004. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 26.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. 2006. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol 8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 27.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. 2005. Role that phosphorylation of GSK3 plays in insulin and Wnt signaling defined by knockin analysis. EMBO J 24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anjum R, Blenis J. 2008. The RSK family of kinases: emerging roles in cellular signaling. Nat Rev Mol Cell Biol 9:747–758. doi: 10.1038/nrn2493. [DOI] [PubMed] [Google Scholar]
- 29.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. 2005. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol 171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, van Roy F, Lee-Kwon W, Donowitz M, Tsichlis PN, Larue L. 2003. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res 63:2172–2178. [PubMed] [Google Scholar]
- 31.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. 1998. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 32.Ananthanarayanan B, Fosbrink M, Rahdar M, Zhang J. 2007. Live-cell molecular analysis of Akt activation reveals roles for activation loop phosphorylation. J Biol Chem 282:36634–36641. doi: 10.1074/jbc.M706227200. [DOI] [PubMed] [Google Scholar]
- 33.Testa JR, Bellacosa A. 2001. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A 98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tzivion G, Dobson M, Ramakrishnan G. 2011. FoxO transcription factors: regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 1813:1938–1945. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Tang N, Hadden TJ, Rishi AK. 2011. Akt, FoxO, and regulation of apoptosis. Biochim Biophys Acta 1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 36.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. 2005. Role for Akt3/protein kinase Bγ in attainment of normal brain size. Mol Cell Biol 25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL. 2004. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. 2006. Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J Biol Chem 281:36443–36453. doi: 10.1074/jbc.M600788200. [DOI] [PubMed] [Google Scholar]
- 39.Franci C, Takkunen M, Dave N, Alameda F, Gomez S, Rodriguez R, Escriva M, Montserrat-Sentis B, Baro T, Garrido M, Bonilla F, Virtanen I, Garcia de Herreros A. 2006. Expression of Snail protein in tumor-stroma interface. Oncogene 25:5134–5144. [DOI] [PubMed] [Google Scholar]
- 40.Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY, Kang Y. 2014. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell 26:358–373. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vinas-Castells R, Beltran M, Valls G, Gomez I, Garcia JM, Montserrat-Sentis B, Baulida J, Bonilla F, de Herreros AG, Diaz VM. 2010. The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J Biol Chem 285:3794–3805. doi: 10.1074/jbc.M109.065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinas-Castells R, Frias A, Robles-Lanuza E, Zhang K, Longmore GD, Garcia de Herreros A, Diaz VM. 2014. Nuclear ubiquitination by FBXL5 modulates Snail1 DNA binding and stability. Nucleic Acids Res 42:1079–1094. doi: 10.1093/nar/gkt935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villagrasa P, Diaz VM, Vinas-Castells R, Peiro S, Del Valle-Perez B, Dave N, Rodriguez-Asiain A, Casal JI, Lizcano JM, Dunach M, Garcia de Herreros A. 2012. Akt2 interacts with Snail1 in the E-cadherin promoter. Oncogene 31:4022–4033. doi: 10.1038/onc.2011.562. [DOI] [PubMed] [Google Scholar]
- 44.Park S, Kim D, Kaneko S, Szewczyk KM, Nicosia SV, Yu H, Jove R, Cheng JQ. 2005. Molecular cloning and characterization of the human AKT1 promoter uncovers its up-regulation by the Src/Stat3 pathway. J Biol Chem 280:38932–38941. doi: 10.1074/jbc.M504011200. [DOI] [PubMed] [Google Scholar]
- 45.Kaneko S, Feldman RI, Yu L, Wu Z, Gritsko T, Shelley SA, Nicosia SV, Nobori T, Cheng JQ. 2002. Positive-feedback regulation between Akt2 and MyoD during muscle differentiation: cloning of Akt2 promoter. J Biol Chem 277:23230–23235. doi: 10.1074/jbc.M201733200. [DOI] [PubMed] [Google Scholar]
- 46.Fuxe J, Vincent T, Garcia de Herreros A. 2010. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle 9:2363–2374. doi: 10.4161/cc.9.12.12050. [DOI] [PubMed] [Google Scholar]
- 47.Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, de Herreros AG. 2011. Functional cooperation between Snail1 and Twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem 286:12024–12032. doi: 10.1074/jbc.M110.168625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H. 2010. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 49.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 50.Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. 2005. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res 65:1027–1034. [PubMed] [Google Scholar]
- 51.Eisinger-Mathason TS, Andrade J, Groehler AL, Clark DE, Muratore-Schroeder TL, Pasic L, Smith JA, Shabanowitz J, Hunt DF, Macara IG, Lannigan DA. 2008. Codependent functions of RSK2 and the apoptosis-promoting factor TIA-1 in stress granule assembly and cell survival. Mol Cell 31:722–736. doi: 10.1016/j.molcel.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Huang K, Shi Z, Zou J, Wang Y, Jia Z, Zhang A, Han L, Yue X, Liu N, Jiang T, You Y, Pu P, Kang C. 2011. High beta-catenin/Tcf-4 activity confers glioma progression via direct regulation of AKT2 gene expression. Neuro Oncol 13:600–609. doi: 10.1093/neuonc/nor034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, Nagourney R, Cardozo T, Brown AM, DasGupta R. 2011. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A 108:5954–5963. doi: 10.1073/pnas.1017496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin Y, Shenoy AK, Doernberg S, Chen H, Luo H, Shen H, Lin T, Tarrash M, Cai Q, Hu X, Fiske R, Chen T, Wu L, Mohammed KA, Rottiers V, Lee SS, Lu J. 2015. FBXO11 promotes ubiquitination of the Snail family of transcription factors in cancer progression and epidermal development. Cancer Lett 362:70–82. doi: 10.1016/j.canlet.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuda G, Paterno R, Ceravolo R, Candigliota M, Perrotti N, Perticone F, Faniello MC, Schepis F, Ruocco A, Mele E, Cassano S, Bifulco M, Santillo M, Avvedimento EV. 2002. Protection of human endothelial cells from oxidative stress: role of Ras-ERK1/2 signaling. Circulation 105:968–974. doi: 10.1161/hc0802.104324. [DOI] [PubMed] [Google Scholar]
- 56.Hogg N, Browning J, Howard T, Winterford C, Fitzpatrick D, Gobe G. 1999. Apoptosis in vascular endothelial cells caused by serum deprivation, oxidative stress and transforming growth factor-beta. Endothelium 7:35–49. [DOI] [PubMed] [Google Scholar]
- 57.Yap TA, Walton MI, Hunter LJ, Valenti M, de Haven Brandon A, Eve PD, Ruddle R, Heaton SP, Henley A, Pickard L, Vijayaraghavan G, Caldwell JJ, Thompson NT, Aherne W, Raynaud FI, Eccles SA, Workman P, Collins I, Garrett MD. 2011. Preclinical pharmacology, antitumor activity, and development of pharmacodynamic markers for the novel, potent AKT inhibitor CCT128930. Mol Cancer Ther 10:360–371. doi: 10.1158/1535-7163.MCT-10-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. 2008. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A 105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R, Shahidi R, Dorovini-Zis K, Li L, Beckstead B, Durand RE, Hoodless PA, Karsan A. 2004. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res 94:910–917. doi: 10.1161/01.RES.0000124300.76171.C9. [DOI] [PubMed] [Google Scholar]
- 60.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. 2008. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol 182:315–325. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsu DS, Wang HJ, Tai SK, Chou CH, Hsieh CH, Chiu PH, Chen NJ, Yang MH. 2014. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell 26:534–548. doi: 10.1016/j.ccell.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 62.Stanisavljevic J, Porta-de la Riva M, Batlle R, de Herreros AG, Baulida J. 2011. The p65 subunit of NF-κB and PARP1 assist Snail1 in activating fibronectin transcription. J Cell Sci 124:4161–4171. doi: 10.1242/jcs.078824. [DOI] [PubMed] [Google Scholar]
- 63.Andersen P, Uosaki H, Shenje LT, Kwon C. 2012. Non-canonical Notch signaling: emerging role and mechanism. Trends Cell Biol 22:257–265. doi: 10.1016/j.tcb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Munoz Descalzo S, Martinez Arias A. 2012. The structure of Wntch signaling and the resolution of transition states in development. Semin Cell Dev Biol 23:443–449. doi: 10.1016/j.semcdb.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 65.Fu Y, Chang AC, Fournier M, Chang L, Niessen K, Karsan A. 2011. RUNX3 maintains the mesenchymal phenotype after termination of the Notch signal. J Biol Chem 286:11803–11813. doi: 10.1074/jbc.M111.222331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin FC, Liu YP, Lai CH, Shan YS, Cheng HC, Hsu PI, Lee CH, Lee YC, Wang HY, Wang CH, Cheng JQ, Hsiao M, Lu PJ. 2012. RUNX3-mediated transcriptional inhibition of Akt suppresses tumorigenesis of human gastric cancer cells. Oncogene 31:4302–4316. doi: 10.1038/onc.2011.596. [DOI] [PubMed] [Google Scholar]
- 67.Quillard T, Devalliere J, Chatelais M, Coulon F, Seveno C, Romagnoli M, Barille Nion S, Charreau B. 2009. Notch2 signaling sensitizes endothelial cells to apoptosis by negatively regulating the key protective molecule survivin. PLoS One 4:e8244. doi: 10.1371/journal.pone.0008244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacKenzie F, Duriez P, Wong F, Noseda M, Karsan A. 2004. Notch4 inhibits endothelial apoptosis via RBP-Jκ-dependent and -independent pathways. J Biol Chem 279:11657–11663. doi: 10.1074/jbc.M312102200. [DOI] [PubMed] [Google Scholar]
- 69.Quillard T, Coupel S, Coulon F, Fitau J, Chatelais M, Cuturi MC, Chiffoleau E, Charreau B. 2008. Impaired Notch4 activity elicits endothelial cell activation and apoptosis: implication for transplant arteriosclerosis. Arterioscler Thromb Vasc Biol 28:2258–2265. doi: 10.1161/ATVBAHA.108.174995. [DOI] [PubMed] [Google Scholar]
- 70.Tiwari N, Gheldof A, Tatari M, Christofori G. 2012. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol 22:194–207. doi: 10.1016/j.semcancer.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 71.Kim MA, Kim HJ, Jee HJ, Kim AJ, Bae YS, Bae SS, Yun J. 2009. Akt2, but not Akt1, is required for cell survival by inhibiting activation of JNK and p38 after UV irradiation. Oncogene 28:1241–1247. doi: 10.1038/onc.2008.487. [DOI] [PubMed] [Google Scholar]
- 72.Fujio Y, Mitsuuchi Y, Testa JR, Walsh K. 2001. Activation of Akt2 Inhibits anoikis and apoptosis induced by myogenic differentiation. Cell Death Differ 8:1207–1212. doi: 10.1038/sj.cdd.4400919. [DOI] [PubMed] [Google Scholar]
- 73.Chin YR, Yuan X, Balk SP, Toker A. 2014. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer Discov 4:942–955. doi: 10.1158/2159-8290.CD-13-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shiraishi I, Melendez J, Ahn Y, Skavdahl M, Murphy E, Welch S, Schaefer E, Walsh K, Rosenzweig A, Torella D, Nurzynska D, Kajstura J, Leri A, Anversa P, Sussman MA. 2004. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res 94:884–891. doi: 10.1161/01.RES.0000124394.01180.BE. [DOI] [PubMed] [Google Scholar]
- 75.Csiszar A, Smith KE, Koller A, Kaley G, Edwards JG, Ungvari Z. 2005. Regulation of bone morphogenetic protein-2 expression in endothelial cells: role of nuclear factor-κB activation by tumor necrosis factor-α, H2O2, and high intravascular pressure. Circulation 111:2364–2372. doi: 10.1161/01.CIR.0000164201.40634.1D. [DOI] [PubMed] [Google Scholar]
- 76.Shen YH, Zhang L, Ren P, Nguyen MT, Zou S, Wu D, Wang XL, Coselli JS, LeMaire SA. 2013. AKT2 confers protection against aortic aneurysms and dissections. Circ Res 112:618–632. doi: 10.1161/CIRCRESAHA.112.300735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jorda M, Olmeda D, Vinyals A, Valero E, Cubillo E, Llorens A, Cano A, Fabra A. 2005. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci 118:3371–3385. doi: 10.1242/jcs.02465. [DOI] [PubMed] [Google Scholar]
- 78.Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A, Simons M, Kyriakides TR, Morales-Ruiz M, Sessa WC. 2014. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci U S A 111:12865–12870. doi: 10.1073/pnas.1408472111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Girardi C, James P, Zanin S, Pinna LA, Ruzzene M. 2014. Differential phosphorylation of Akt1 and Akt2 by protein kinase CK2 may account for isoform specific functions. Biochim Biophys Acta 1843:1865–1874. doi: 10.1016/j.bbamcr.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 80.Lee SB, Xuan Nguyen TL, Choi JW, Lee KH, Cho SW, Liu Z, Ye K, Bae SS, Ahn JY. 2008. Nuclear Akt interacts with B23/NPM and protects it from proteolytic cleavage, enhancing cell survival. Proc Natl Acad Sci ja U S A 105:16584–16589. doi: 10.1073/pnas.0807668105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumar CC, Madison V. 2005. AKT crystal structure and AKT-specific inhibitors. Oncogene 24:7493–7501. doi: 10.1038/sj.onc.1209087. [DOI] [PubMed] [Google Scholar]