

ABSTRACT
The newly emerging Middle East respiratory syndrome coronavirus (MERS-CoV) causes a severe respiratory infection with a high mortality rate (~35%). MERS-CoV has been a global threat due to continuous outbreaks in the Arabian peninsula and international spread by infected travelers since 2012. From May to July 2015, a large outbreak initiated by an infected traveler from the Arabian peninsula swept South Korea and resulted in 186 confirmed cases with 38 deaths (case fatality rate, 20.4%). Here, we show the rapid emergence and spread of a mutant MERS-CoV with reduced affinity to the human CD26 receptor during the South Korean outbreak. We isolated 13 new viral genomes from 14 infected patients treated at a hospital and found that 12 of these genomes possess a point mutation in the receptor-binding domain (RBD) of viral spike (S) protein. Specifically, 11 of these genomes have an I529T mutation in RBD, and 1 has a D510G mutation. Strikingly, both mutations result in reduced affinity of RBD to human CD26 compared to wild-type RBD, as measured by surface plasmon resonance analysis and cellular binding assay. Additionally, pseudotyped virus bearing an I529T mutation in S protein showed reduced entry into host cells compared to virus with wild-type S protein. These unexpected findings suggest that MERS-CoV adaptation during human-to-human spread may be driven by host immunological pressure such as neutralizing antibodies, resulting in reduced affinity to host receptor, and thereby impairs viral fitness and virulence, rather than positive selection for a better affinity to CD26.
IMPORTANCE
Recently, a large outbreak initiated by an MERS-CoV-infected traveler from the Middle East swept South Korea and resulted in 186 confirmed cases with 38 deaths. This is the largest outbreak outside the Middle East, and it raised strong concerns about the possible emergence of MERS-CoV mutations. Here, we isolated 13 new viral genomes and found that 12 of them possess a point mutation in the receptor-binding domain of viral spike protein, resulting in reduced affinity to the human cognate receptor, CD26, compared to the wild-type virus. These unexpected findings suggest that MERS-CoV adaptation in humans may be driven by host immunological pressure.
INTRODUCTION
Middle East respiratory syndrome coronavirus (MERS-CoV), a newly emerging zoonotic pathogen first identified in the Kingdom of Saudi Arabia in 2012, causes an acute and severe respiratory illness with a high mortality rate in humans (1). As of 20 September 2015, 1,569 laboratory-confirmed human infections have been reported to the World Health Organization (WHO), including 554 deaths (case fatality rate, 35.3%) (2). Although the majority of the reported cases are associated with sporadic outbreaks in the countries of the Middle East (3), more than 200 cases occurred outside the Middle East region and are primarily linked to recent travel to the Middle East (2). These cases include an unexpected large outbreak (186 confirmed cases with 38 deaths) in South Korea from May to July 2015 (4). Similar to other large outbreaks in Saudi Arabia (3, 5), the South Korean MERS outbreak was mainly associated with health care settings and was accelerated by interhospital spread (4).
Although early genomic analysis of MERS-CoV revealed that the respiratory pathogen is closely related to a bat coronavirus belonging to the genus Betacoronavirus (6), accumulating evidence support dromedary camels as a reservoir host and the primary source of human infection (7–9). A viral MERS-CoV spike (S) protein has been suggested to be a critical viral factor for host tropism via its interaction with a host receptor, CD26 (10, 11), but the evolutionary pathway of MERS-CoV for human adaptation remains unclear. The efficacy of direct human-to-human spread in the community seems to be quite low, as the rate of human transmission among household contacts of MERS patients has been approximately 5% based on serological analysis (12). However, ongoing sporadic outbreaks highlight the importance of early nosocomial superspreading events in secondary human infections before active case detection and implementation of interventions (13). There were also typical superspreading events fueling the unexpected large outbreak in South Korea. The index case (patient A) returning from the Middle East generated 38 cases of secondary infections; two patients (patient D and E) of the second wave of infection or generation were linked to at least 81 and 23 cases of the third generation, respectively (Fig. 1) (4). The rapid and wide spread of MERS-CoV during the South Korean outbreak raised strong concerns about the possible generation of mutations with enhanced sequential human infection. In this study, we analyzed MERS-CoV genomes isolated from 13 patients admitted to Chungnam National University Hospital, one of the designated “national safe hospitals” during the South Korean outbreak and compared them to recently reported genomes from two South Korean patients (14, 15) and others deposited in the GenBank database.
FIG 1 .
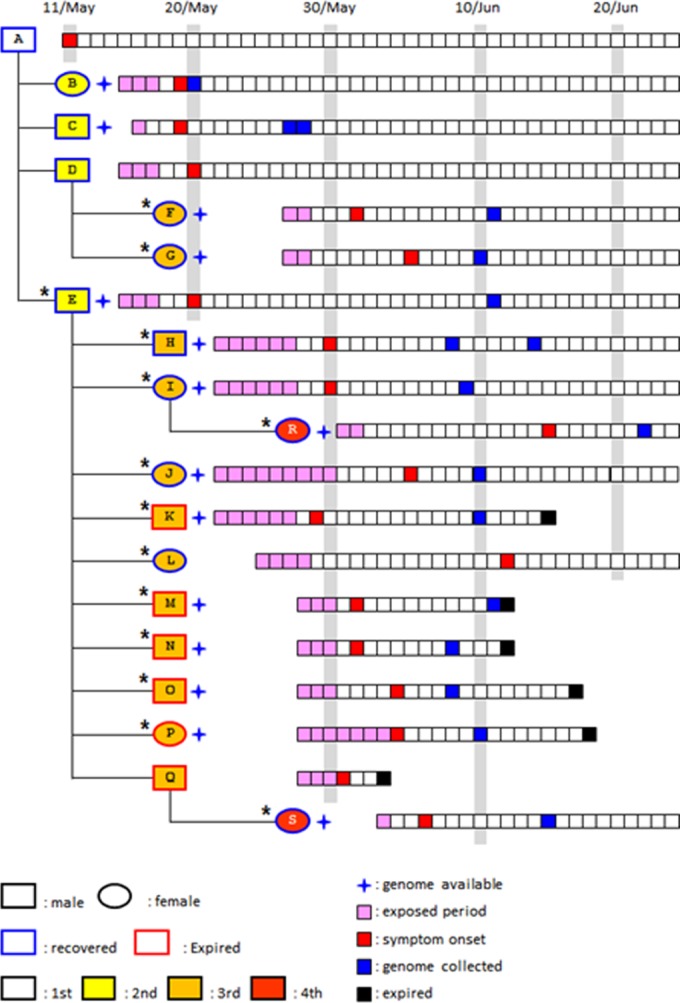
Transmission tree and timeline of potential virus exposure, onset of symptoms, and genome collection. The transmission chain of infection and the timeline of potential viral exposure, symptom onset, and the date of genome collection from patients who were epidemiologically linked to the 14 patients (indicated by an asterisk) analyzed in this study are schematically presented. The wave of infection or generation (first to fourth) is indicated.
RESULTS
Patients and sample history.
The transmission chain of MERS-CoV and the timeline of potential viral exposure, symptom onset, and date of genome collection from the patients admitted to Chungnam National University Hospital are presented in Fig. 1. This patient pool includes a secondary infection case (patient E) who had close contact with the index case (patient A) in hospital P. Eleven cases (patients F, G, H, I, J, K, L, M, N, O, and P) of the third wave of infection or generation and two cases (patients R and S) of the fourth generation are also included in this study. Two (patients F and G) of 11 tertiary cases visited hospital S, where at least 81 infections occurred, infected by patient D, while the remaining 9 tertiary cases (patients H to P) were patients or care givers at hospitals D and G where 23 cases were generated by patient E. Clinical characteristics of the patients, including incubation period, fever duration, and the presence of pneumonia, are summarized in Table S1 in the supplemental material. Among the 14 cases, 9 patients completely recovered and 5 patients expired. Fatal cases include an 82-year-old female and four patients who had comorbid diseases such as chronic obstructive pulmonary disease or liver cirrhosis (Table S1).
Characterization of genomic mutations.
We isolated and sequenced the whole viral genomes from respiratory secretions from the 13 patients (Fig. 1). Genome sequences from South Korean patients B and C that were recently reported by the Republic of Korea Centers for Disease Control and Prevention (Korean CDC) (GenBank accession number KT029139) (14) and the Chinese Center for Disease Control and Prevention (Chinese CDC) (GenBank accession numbers KT006149 and KT036372) (15), respectively, were also analyzed together with the new viral genomes. When we compared the viral genomes isolated from the 13 South Korean patients with all the MERS-CoV genomes available in GenBank as of 20 September 2015 (99 genomes with more than 90% coverage), 26 mutations were found only in the South Korean isolates (Fig. 2; see Table S2 in the supplemental material). Fourteen of these mutations are nonsynonymous mutations that result in changes of amino acids in ORF1a, S, and ORF4b. Six mutations were observed in ORF1a, seven mutations were observed in the S protein, and there was one deletion mutation in ORF4b that causes a frameshift and fusion of ORF4b and ORF5. Four mutations (D977G and G6896S in ORF1a and I529T and V718I in S) were detected in more than one isolate. We observed no change at position R1020 of the S protein, which is the only codon predicted to be under positive selection in the viral genome (16). It is worth noting that the D510G and I529T mutations in the receptor-binding domain (RBD) of the S protein (17) result in a change in amino acid properties: acidic aspartate (D) to hydrophobic glycine (G) and hydrophobic isoleucine (I) to polar threonine (T), respectively. Moreover, the mutations in RBD, especially I529T, emerged in patients of the second generation (patient E) and spread to the third and fourth generations of patients within a month (Fig. 1).
FIG 2 .

Mutations in MERS-CoV observed in the South Korean patients. (Top) Map of the MERS-CoV genome, plotted with synonymous mutations (blue triangles) and nonsynonymous mutations (red triangles) detected in the isolates from South Korean patients, is presented (see Table S2 in the supplemental material). Mutations in the spike (S) gene are mapped separately in the known domains. PLpro/ADRP, papain-like protease/ADP-ribose 1′ -phosphatase; Prim, primase; RdRp, RNA-dependent RNA polymerase; Hel, helicase; ExoN, exonuclease N; NendoU, Nidoviral endoribonuclease specific for U; 2'OMT, S-adenosylmethionine-dependent ribose 2'-O-methyltransferase; E, envelope; N, nucleocapsid; M, matrix; SP, signal peptide; FP, fusion peptide; HR1, heptad repeat1; TM, transmembrane. (Bottom) List of nonsynonymous mutations observed in the genome isolates from each patient are summarized in the table. The group or wave of infection (second to fourth) is indicated. The patient identification (ID) for patients who expired are indicated in red. The accession numbers are GenBank accession numbers. The presence (+) or absence (−) of the mutation in the indicated open reading frames (i.e., ORF1a, S, and ORF4b) is indicated.
Structural prediction of S RBD mutations.
We thus assessed whether nonsynonymous mutations in the RBD of the S protein affect interactions with the cognate human receptor, CD26, by structural analysis. Both mutations (D510G and I529T) are located on the interfacial region of the S protein (Fig. 3A). We found that both mutations introduced energetically unfavorable interactions to the S-protein/CD26 interface. Specifically, D510 of the S protein is located on the interfacial loop and interacts directly with CD26 residues (Fig. 3B; see Table S3 in the supplemental material). The D510G mutation disrupts the ionic interaction with R317 of CD26. Moreover, I529 is located on the backside of the S-protein/CD26 interface (Fig. 3C; Table S4). The I529T mutation introduces a polar side chain to a hydrophobic residue cluster (L506, L554, and A556) and therefore potentially affects the energetic stability of the interfacial beta sheets of RBD.
FIG 3 .

Structural prediction of D510G and I529T mutations in the RBD of S protein and their binding affinity to CD26. (A to C) Locations of D510 and I529 on the structure of the MERS-CoV RBD (red) and CD26 (blue). Amino acid residues that potentially interact with D510 or I529 are shown in gray. (D) Results of a surface plasmon resonance assay characterizing the specific binding between CD26 and wild-type (wt) and mutant MERS-CoV RBDs.
Effects of RBD mutation on CD26 binding and viral entry.
In order to quantify the binding affinity of the mutant RBDs (D510G and I529T) to CD26, we performed surface plasmon resonance (SPR) experiments with purified RBD proteins (Fig. 3D). In our studies, wild-type RBD binds to CD26 with a dissociation constant (Kd) of 64.5 nM (Kon, 1.45 × 104 M−1 s−1; Koff, 9.36 × 10−4 s−1), which is slightly lower but comparable to the 16.7 nM measured in previous studies (17). The difference might be due to having fixed CD26 on the chip for the SPR assay, whereas RBD was fixed in the previous experiment (17). The I529T mutant RBD binds to CD26 with a Kd of 293 nM (Kon, 4.03 × 103 M−1 s−1; Koff, 1.18 × 10−3 s−1), which is almost 4.5 times lower than wild-type RBD. In the case of D510G RBD, it binds to CD26 with a Kd of 316 nM (Kon, 5.02 × 103 M−1 s−1; Koff, 1.59 × 10−3 s−1), which is almost 4.9 times lower than wild-type RBD and as expected by structural prediction.
To further confirm the reduced affinity, purified RBD mutants were analyzed for CD26 binding in 293T cells stably expressing CD26 (see Fig. S1 in the supplemental material) by flow cytometry assay. As shown in Fig. 4A, mutated RBDs bind to 293T cells overexpressing CD26 with different efficacies. The D510G substitution increased the 50% effective concentration (EC50) by twofold (EC50 = 0.24 µg/ml), whereas the EC50 of the I529T RBD mutant was almost 20 times higher (EC50 = 2.37 µg/ml) than that of wild-type RBD (EC50 = 0.12 µg/ml). In addition, the maximum binding of the two mutant RBDs was saturated at approximately 80% of wild-type RBD. Finally, we investigated the biological relevance of the I529T mutation in S protein by using a pseudotyped lentivirus bearing the mutant S protein (Fig. 4B). The I529T mutation in S protein significantly lowered (~25%) the efficiency of viral entry into 293T cells overexpressing CD26 compared to that of virus bearing wild-type S protein. The basal level of viral entry in the control cells (293T-vector) was not changed, and the infectivity of vesicular stomatitis virus G protein (VSV-G)-pseudotyped virus was not significantly affected by the expression of CD26 in the host cells, suggesting that the reduction in viral entry by the I529T mutation is specifically dependent on its interaction with CD26.
FIG 4 .
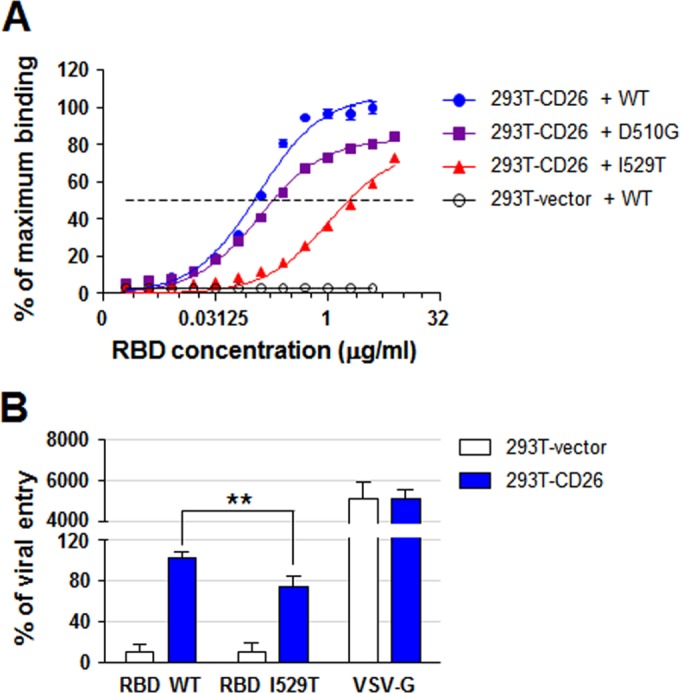
Effects of RBD mutations on cellular binding and viral entry. (A) Analysis of 50% effective concentration (EC50) of RBD mutant proteins binding to 293T cells expressing CD26. Each purified RBD protein tagged with His was serially diluted 2-fold (dilutions starting from 4 or 8 µg/ml) and then incubated with 2 × 105 cells. The binding of the RBD proteins was measured by flow cytometry analysis after staining with anti-His antibody. The dashed line indicates 50% of maximum binding of wild-type RBD to 293T-CD26 cells. (B) The efficiency of viral entry was analyzed on the basis of luciferase activity in cells infected with pseudotyped viruses bearing the wild-type (WT) or I529T mutant S protein. The efficiency of viral entry was measured by setting the luciferase activity in 293T-CD26 cells incubated with pseudotyped virus bearing the wild-type S protein at 100%. VSV-G-pseudotyped virus was used as a pseudovirus control. This assay was performed in triplicate. 293T-vector cells are the control cells. Values that are significantly different (P < 0.01) by Student’s t test are indicated by a bar and two asterisks.
DISCUSSION
Considering the genetic stability of the S protein in MERS-CoV genomes reported thus far (16), it is quite interesting to note the rapid emergence and subsequent conservation of the RBD I529T mutation in consecutive human infections. Since it is generally thought that interspecies transmission of coronaviruses is primarily mediated by mutations in the S protein with enhanced affinity toward human receptors (18), this unexpected emergence and wide spread of an RBD mutation with reduced affinity to human CD26 during human-to-human spread in the South Korean MERS outbreak raises several critical questions. First, the mutation rate of MERS-CoV may be different during sequential human spread. Based on the results of our genomic analysis, however, genomes from each generation have only one to three nucleotide changes at most (see Table S2 in the supplemental material). In addition, when we isolated and sequenced two viral genomes in a patient (patient H) (Fig. 1) separated by a 6-day interval, we failed to observe any detectable changes (data not shown). It has also been reported that only a few or no differences were found in nucleotide sequences between the genomes from samples from a single patient taken 1 day to 2 weeks later (19, 20). Second, the mutations in the RBD of S protein might be generated to better suit polymorphisms of CD26 specific to the South Korean population. Indeed, it has been shown that the difference in interface residues among the host receptors of different mammalian species is critical to triggering interspecies transmission of coronavirus (18). However, our preliminary analysis of CD26 sequences encoding the interface domain in the South Korean patients failed to show any substantial difference (data not shown). Third, the mutations observed in the South Korean outbreak could affect the severity of disease caused by MERS. Since the number of patients whose viral genomes are currently available is limited, direct demonstration of a correlation is not yet possible; we could not distinguish a specific correlation using the epidemiological and clinical information of the 15 cases listed in Table S1 in the supplemental material. Functional analyses of the mutations (Fig. 2) in viral pathogenesis or infectivity need to be performed. Considering that the case fatality rate (19.9%) of the South Korean outbreak is lower than the overall mortality rate of all MERS cases (~35%), the spread of the RBD I529T mutation with reduced affinity to human CD26 receptor, as well as earlier diagnosis and quarantine during the outbreak (21), is potentially associated with milder consequences, although the differences in health care settings among the nations where MERS is endemic need to be taken into account to make a direct comparison. In addition to mutations in the S protein, a deletion of three nucleotides (ΔTTA) in ORF4b (expired patient K), leading to a fusion of ORF4b and ORF5 (Fig. 1; Table S2), is notable, since these proteins are known to function as antagonists of type 1 interferon signaling (22). Finally, is the reduced affinity to host receptor CD26 by RBD mutations linked to transmissibility of MERS-CoV, and how are these viral mutations selected and maintained during human-to-human transmission? Currently, we are not able to calculate and compare the reproduction rate of MERS-CoV infections during the South Korean outbreak due to the limited availability of information on the patients infected with the mutant viruses. However, the available s gene sequences isolated from Korean patients revealed that 72% (18/25) of the patients, including three superspreaders (patients 1, 14, and 16), were infected with MERS-CoV carrying the I529T mutated S protein (Fig. 5) (20, 23). Remarkably, wild-type RBD sequences were found in several second- and third-generation patients, and the D510G mutation was found in two patients of the third and fourth generations (Fig. 5). The original virus carried by the index patient may have carried the I529T mutation, and it may have converted to wild-type or mutant viruses bearing the D510G mutation in the S protein during human-to-human spread. In order to elucidate the evolutionary pathway of the s gene mutations during sequential human infection, more-detailed analysis needs to be conducted using genomic sequences isolated from more South Korean patients. Nevertheless, it was shown that the South Korean outbreak followed a progression similar to those of previously described hospital clusters involving coronaviruses, with early superspreading events generating a disproportionately large number of secondary and tertiary infections, and the transmission potential diminishing greatly in subsequent generations (13). It is thought that human adaptation of animal coronavirus might be achieved through sequential mutations that enhance the affinity of S protein to the human receptor as demonstrated in severe acute respiratory syndrome coronavirus (SARS-CoV) evolution (24). However, the results of our current study showing the rapid and wide spread of an RBD mutant with reduced affinity to human receptor generates evidence that do not support this hypothesis. Interestingly, previous reports showed that a neutralizing antibody against MERS-CoV may contribute not only to MERS-CoV clearance but also to viral evolution (25). Treatment of a neutralizing antibody to in vitro infection media generated several escape mutations in the RBD of MERS-CoV; the majority of the escape mutations had a negative impact on CD26 receptor binding and viral fitness (25) in line with what we observed in our current study. It was shown that RBD mutations (L506F, T512A, Y540C, and R542G) generated by neutralizing antibody pressure decreases RBD binding affinity to CD26, resulting in a profound loss of neutralizing antibody binding activity (25). Based on these results, it was proposed that the immunodominance of human neutralizing antibody response to RBD-CD26 interface can restrict MERS-CoV evolution by driving the virus down an escape pathway that predominantly results in a significant cost in viral fitness. Considering that I529T and D510G mutations emerged and were conserved in the majority of viral genomes isolated from South Korean patients (Fig. 5), these natural variations with reduced affinity to CD26 may also have been produced in response to neutralizing antibodies exerting an in vivo selection pressure. Decreased virulence of the neutralization escape mutants also coincided with reduced affinity of the mutant SARS S protein for its ACE2 receptor (26). Further analysis on the interaction of neutralizing antibodies generated in infected patients and the mutant viruses needs to be conducted to confirm this hypothesis.
FIG 5.
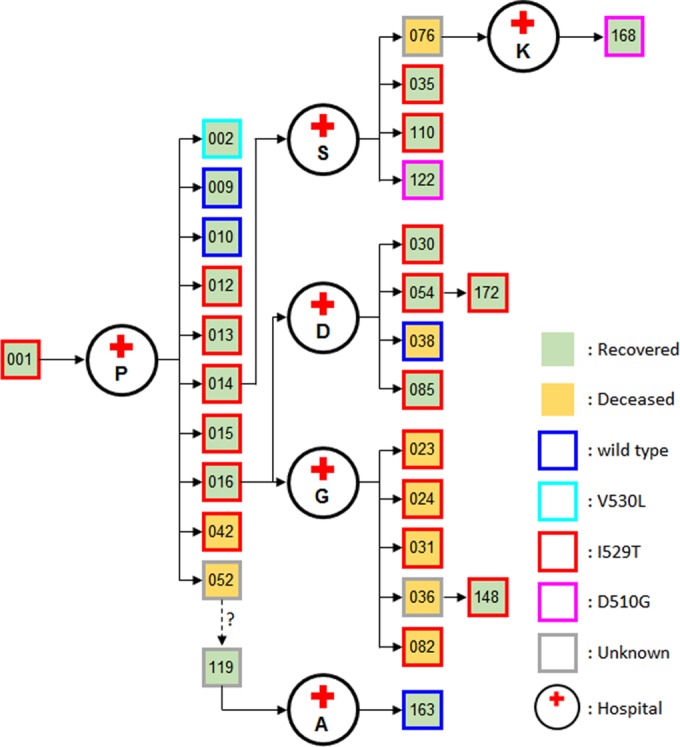
Spread of MERS-CoV bearing the I529T mutation in the S protein during the South Korean outbreak. The transmission of infection and the identified mutation in the S protein of MERS-CoV isolated from the patients are presented. The patients are indicated by their epidemiological number annotated during the South Korean outbreak.
MATERIALS AND METHODS
Ethics statement.
MERS-CoV genome sequences were obtained from the patients’ respiratory samples after ethical approval granted by the institutional review boards of Chungnam National University Hospital and Seoul National University Hospital. The clinical samples collected for viral diagnosis before consent of the patients were used for viral sequencing analysis after the ethical approval. These procedures were approved by the institutional review boards. All the patients who recovered provided their written, informed consent to participation. In the cases of patients who died, we obtained exemption of patients’ consent for the analysis of viral sequences from the institutional review boards of Chungnam National University Hospital and Seoul National University Hospital.
Viral genome analysis.
Nucleic acid extracts from PCR-confirmed MERS patients were processed for reverse transcription and PCR amplification using a modified version of methods described previously (16). Briefly, RNAs were extracted from sputum or tracheal aspirate samples using TRIzol LS reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. The MERS-CoV RNA genome was converted to DNA and amplified by PCR in 30 overlapping amplicons using the primer sets listed in Table S5 in the supplemental material. All amplicons were gel purified and sequenced directly using an ABI-3730xl DNA analyzer (Applied Biosystems), and the contigs were assembled using SeqMan version 7.0 (DNAStar Lasergene, USA). The 13 new genomes were aligned with the 101 published MERS-CoV genomes using MUSCLE (27) and MEGA6 software (28).
Structural analysis of D510G and I529T mutations.
The complex structure of MERS-CoV spike RBD bound to CD26 (PDB identification [ID] or accession number 4KR0) were used to analyze the effects of D510G and I529T mutations (17). Mutations were highlighted using PyMOL (http://www.pymol.org). FoldX (http://foldx.crg.es) was used to calculate the changes in the energetic stability of residues (ΔΔG) in wild-type and mutant RBD structures (29). Interfacial residues in RBD were identified by the change in solvent-accessible surface area (SASA) greater than 1 Å2 upon the formation of complexes (30). The SASA of each residue was calculated by using NACCESS (31). The contacts of particular residues were identified using CSU software (http://ligin.weizmann.ac.il/lpccsu/) (32).
Protein expression and purification.
The human CD26 and MERS-CoV RBD (wild-type and mutant) proteins used for surface plasmon resonance experiments were prepared using the Bac-to-Bac baculovirus expression system (Invitrogen, Life Technologies). CD26 (residues 39 to 766) and MERS-CoV RBD (spike residues 367 to 606, wild type), which were kindly provided by George F. Gao, were cloned into pFastBac1 vector (17), which contains the gp67 signal peptide at the N terminus for protein secretion and the hexahistidine tag at the C terminus for further protein purifications. Two MERS-CoV RBD mutants (D510G and I529T) were generated by using the QuikChange kit (Stratagene). CD26 and RBD constructs were expressed in Spodoptera frugiperda (Sf9) insect cells in a secreted form and collected after culture at 27°C for 3 days through centrifugation to remove Sf9 cells. Media were concentrated and adjusted to a pH of 8.0 before purification. All the proteins were purified using 5 ml HisTrap HP column (GE Healthcare), with washing buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 20 mM imidazole) and elution buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 300 mM imidazole). Finally, each protein was loaded onto Superdex 200 columns (GE Healthcare) equilibrated with 20 mM Tris-HCl (pH 8.0) and 150 mM NaCl to check homogeneity.
Surface plasmon resonance assay.
The SPR experiments were performed at 25°C by using a ProteOnXPR36 machine with GLC83F10NO1 chip (Bio-Rad). PBST (phosphate-buffered saline containing Tween 20) buffer (10 mM PO4 [pH 7.4], 2.4 mM KCl, 138 mM NaCl, 0.05% [vol/vol] Tween 20) was used throughout the SPR experiments. The CD26 protein was immobilized on the chip with approximately 590 response units. Each RBD protein (wild-type protein and D510G and I529T mutant proteins) with gradient concentrations (0, 80, 160, 320, and 640 nM) was prepared and passed over the chip surface. At the end of each cycle, the chip surface was regenerated with 10 mM glycine-HCl (pH 2.5). A 1:1 Langmuir binding model was used to analyze the binding kinetics of each RBD (wild type, D510G, and I529T) to CD26 protein.
Stable 293T cells expressing human CD26 and RBD binding assay.
Full-length human CD26 protein with a C-terminal hemagglutinin (HA) tag was cloned into the pEFIRES-P vector to express both CD26 and the puromycin resistance gene via an internal ribosome entry site sequence (33). Overexpression of CD26 in 293T cells was confirmed by flow cytometry analysis using anti-human CD26 antibody (BioLegend) after transfection with the plasmid constructs and selection with puromycin (see Fig. S1 in the supplemental material). RBD-CD26 binding was further examined by flow cytometry analysis. Purified wild-type or mutant RBD proteins were serially diluted 2-fold and incubated with 2 × 105 293T cells overexpressing CD26 in 200 µl of Dulbecco modified Eagle medium (DMEM) and incubated at 37°C for 1 h. Cells were washed three times with phosphate-buffered saline (PBS) and stained with rabbit anti-His antibody (Santa Cruz) and anti-rabbit IgG conjugated to Alexa Fluor 488 (Life Technologies) for flow cytometric analysis.
Pseudotyped virus and viral entry.
Pseudotyped virus was generated from 293FT cells (Invitrogen) by cotransfection of human immunodeficiency virus backbone plasmids expressing firefly luciferase as described previously (34). We used the packaging plasmids (pLP1, pLP2, and pLP/VSV-G; all from Invitrogen) and pLVX-Luc-IRES-ZsGreen1 (Luc stands for luciferase, and IRES stands for internal ribosomal entry site) (Clontech). For S-protein pseudotyping, full-length cDNA of the s gene (Sino Biological Inc.) was cloned into pcDNA3 and used for transfection instead of pLP/VSV-G. A plasmid carrying the gene encoding the I529T mutation in S protein was generated by using the QuikChange kit (Stratagene) based on the wild-type construct, and the point mutation was confirmed by sequencing. Viral supernatants were harvested 48 h after transfection and normalized by p24 enzyme-linked immunosorbent assay (ELISA) kit (Clontech) before infecting 293T cells for viral entry assay. The infected 293T cells were lysed 48 h after infection, and the efficiency of viral entry was measured by comparing the luciferase activity between pseudotyped viruses bearing the wild-type or mutant S protein (34). VSV-G-pseudotyped lentivirus was used as the positive control. The relative luciferase activity in cell lysates was measured using a luciferase assay kit (Promega) and Infinite 200 PRO microplate reader (Tecan).
SUPPLEMENTAL MATERIAL
Stable 293T cells expressing CD26. Expression of CD26 in 293T cell line was confirmed by flow cytometry after staining with anti-CD26 antibody. Download
Clinical characteristics of MERS patients included in this study.
List of mutations observed only in MERS-CoV isolated from the Korean patients.
CD26 residues contacted with D510 of S protein.
Potential residues that change the energetic stability upon I529T mutation in S protein.
List of primer sets for the amplification and sequencing of MERS-CoV genome.
ACKNOWLEDGMENTS
This work was supported by funds from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare and Family Affairs (HI15C3034 and HI15C3227).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Funding Statement
This work was supported by funds from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs (HI15C3034 and HI15C3227). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Kim Y, Cheon S, Min C-K, Sohn KM, Kang Y, Cha Y-J, Kang J-I, Han SK, Ha N-Y, Kim G, Aigerim A, Shin HM, Choi M-S, Kim S, Cho H-S, Kim Y-S, Cho N-H. 2016. Spread of mutant Middle East respiratory syndrome coronavirus with reduced affinity to human CD26 during the South Korean outbreak. mBio 7(2):e00019-16. doi:10.1128/mBio.00019-16.
REFERENCES
- 1.Chan JF, Lau SK, To KK, Cheng VC, Woo PC, Yuen KY. 2015. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev 28:465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reference deleted.
- 3.Oboho IK, Tomczyk SM, Al-Asmari AM, Banjar AA, Al-Mugti H, Aloraini MS, Alkhaldi KZ, Almohammadi EL, Alraddadi BM, Gerber SI, Swerdlow DL, Watson JT, Madani TA. 2015. 2014 MERS-CoV outbreak in Jeddah—a link to health care facilities. N Engl J Med 372:846–854. doi: 10.1056/NEJMoa1408636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korean Society of Infectious Diseases, Korean Society for Healthcare-associated Infection Control and Prevention . 2015. An unexpected outbreak of Middle East respiratory syndrome coronavirus infection in the Republic of Korea, 2015. Infect Chemother 47:120-122. doi: 10.3947/ic.2015.47.2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Assiri A, McGeer A, Perl TM, Price CS, Al Rabeeah AA, Cummings DA, Alabdullatif ZN, Assad M, Almulhim A, Makhdoom H, Madani H, Alhakeem R, Al-Tawfiq JA, Cotten M, Watson SJ, Kellam P, Zumla AI, Memish ZA, KSA MERS-CoV Investigation Team . 2013. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 369:407–416. doi: 10.1056/NEJMoa1306742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3:00473-12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Memish ZA, Cotten M, Meyer B, Watson SJ, Alsahafi AJ, Al Rabeeah AA, Corman VM, Sieberg A, Makhdoom HQ, Assiri A, Al Masri M, Aldabbagh S, Bosch BJ, Beer M, Müller MA, Kellam P, Drosten C. 2014. Human infection with MERS coronavirus after exposure to infected camels, Saudi Arabia, 2013. Emerg Infect Dis 20:1012–1015. doi: 10.3201/eid2006.140402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haagmans BL, Al Dhahiry SH, Reusken CB, Raj VS, Galiano M, Myers R, Godeke GJ, Jonges M, Farag E, Diab A, Ghobashy H, Alhajri F, Al-Thani M, Al-Marri SA, Al Romaihi HE, Al Khal A, Bermingham A, Osterhaus AD, AlHajri MM, Koopmans MP. 2014. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 14:140–145. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azhar EI, El-Kafrawy SA, Farraj SA, Hassan AM, Al-Saeed MS, Hashem AM, Madani TA. 2014. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 370:2499–2505. doi: 10.1056/NEJMoa1401505. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Qi J, Yuan Y, Xuan Y, Han P, Wan Y, Ji W, Li Y, Wu Y, Wang J, Iwamoto A, Woo PC, Yuen KY, Yan J, Lu G, Gao GF. 2014. Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 16:328–337. doi: 10.1016/j.chom.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Doremalen N, Miazgowicz KL, Milne-Price S, Bushmaker T, Robertson S, Scott D, Kinne J, McLellan JS, Zhu J, Munster VJ. 2014. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 88:9220–9232. doi: 10.1128/JVI.00676-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drosten C, Meyer B, Müller MA, Corman VM, Al-Masri M, Hossain R, Madani H, Sieberg A, Bosch BJ, Lattwein E, Alhakeem RF, Assiri AM, Hajomar W, Albarrak AM, Al-Tawfiq JA, Zumla AI, Memish ZA. 2014. Transmission of MERS-coronavirus in household contacts. N Engl J Med 371:828–835. doi: 10.1056/NEJMoa1405858. [DOI] [PubMed] [Google Scholar]
- 13.Chowell G, Abdirizak F, Lee S, Lee J, Jung E, Nishiura H, Viboud C. 2015. Transmission characteristics of MERS and SARS in the healthcare setting: a comparative study. BMC Med 13:210. doi: 10.1186/s12916-015-0450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YJ, Cho YJ, Kim DW, Yang JS, Kim H, Park S, Han YW, Yun MR, Lee HS, Kim AR, Heo DR, Kim JA, Kim SJ, Jung HD, Kim N, Yoon SH, Nam JG, Kang HJ, Cheong HM, Lee JS, Chun J, Kim SS. 2015. Complete genome sequence of Middle East respiratory syndrome coronavirus KOR/KNIH/002_05_2015, isolated in South Korea. Genome Announc 3(4):00787-15. doi: 10.1128/genomeA.00787-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu R, Wang Y, Wang W, Nie K, Zhao Y, Su J, Deng Y, Zhou W, Li Y, Wang H, Wang W, Ke C, Ma X, Wu G, Tan W. 2015. Complete genome sequence of Middle East respiratory syndrome coronavirus (MERS-CoV) from the first imported MERS-CoV case in China. Genome Announc 3(4):00818-15. doi: 10.1128/genomeA.00818-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cotten M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, Ong SH, Al Rabeeah AA, Alhakeem RF, Assiri A, Al-Tawfiq JA, Albarrak A, Barry M, Shibl A, Alrabiah FA, Hajjar S, Balkhy HH, Flemban H, Rambaut A, Kellam P, Memish ZA. 2014. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio 5:01062-13. doi: 10.1128/mBio.01062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, Zhang Y, Zhang W, Yuan Y, Bao J, Zhang B, Shi Y, Yan J, Gao GF. 2013. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500:227–231. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu G, Wang Q, Gao GF. 2015. Bat-to-human: spike features determining “host jump” of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol 23:468–478. doi: 10.1016/j.tim.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Q, Cao Y, Su J, Wu X, Wan C, Ke C, Zhao W, Zhang B. 2015. Genomic sequencing and analysis of the first imported Middle East respiratory syndrome coronavirus (MERS CoV) in China. Sci China Life Sci 58:818–820. doi: 10.1007/s11427-015-4903-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seong MW, Kim SY, Corman VM, Kim TS, Cho SI, Kim MJ, Lee SJ, Lee JS, Seo SH, Ahn JS, Yu BS, Park N, Oh MD, Park WB, Lee JY, Kim G, Joh JS, Jeong I, Kim EC, Drosten C, Park SS. 2016. Microevolution of outbreak-associated Middle East respiratory syndrome coronavirus, South Korea, 2015. Emerg Infect Dis 22:151700. doi: 10.3201/eid2202.151700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korea Centers for Disease Control and Prevention 2015. Middle East respiratory syndrome coronavirus outbreak in the Republic of Korea, 2015. Osong Public Health Res Perspect 6:269-278. doi: 10.1016/j.phrp.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Zhang L, Geng H, Deng Y, Huang B, Guo Y, Zhao Z, Tan W. 2013. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim DW, Kim YJ, Park SH, Yun MR, Yang JS, Kang HJ, Han YW, Lee HS, Kim HM, Kim H, Kim AR, Heo DR, Kim SJ, Jeon JH, Park D, Kim JA, Cheong HM, Nam JG, Kim K, Kim SS. 2016. Variations in spike glycoprotein gene of MERS-CoV, South Korea, 2015. Emerg Infect Dis 22:100–104. doi: 10.3201/eid2201.151055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Wong SK, Li F, Kuhn JH, Huang IC, Choe H, Farzan M. 2006. Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. J Virol 80:4211–4219. doi: 10.1128/JVI.80.9.4211-4219.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang XC, Agnihothram SS, Jiao Y, Stanhope J, Graham RL, Peterson EC, Avnir Y, Tallarico AS, Sheehan J, Zhu Q, Baric RS, Marasco WA. 2014. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc Natl Acad Sci U S A 111:E2018–E2026. doi: 10.1073/pnas.1402074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rockx B, Donaldson E, Frieman M, Sheahan T, Corti D, Lanzavecchia A, Baric RS. 2010. Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. J Infect Dis 201:946–955. doi: 10.1086/651022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. 2005. The FoldX web server: an online force field. Nucleic Acids Res 33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi YS, Han SK, Kim J, Yang JS, Jeon J, Ryu SH, Kim S. 2010. ConPlex: a server for the evolutionary conservation analysis of protein complex structures. Nucleic Acids Res 38:W450–W456. doi: 10.1093/nar/gkq328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubbard S, Thornton J. 1993. NACCESS. Department of Biochemistry and Molecular Biology, University College, London; London, United Kingdom. [Google Scholar]
- 32.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics 15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 33.Hobbs S, Jitrapakdee S, Wallace JC. 1998. Development of a bicistronic vector driven by the human polypeptide chain elongation factor 1alpha promoter for creation of stable mammalian cell lines that express very high levels of recombinant proteins. Biochem Biophys Res Commun 252:368-372. [DOI] [PubMed] [Google Scholar]
- 34.Wang N, Shi X, Jiang L, Zhang S, Wang D, Tong P, Guo D, Fu L, Cui Y, Liu X, Arledge KC, Chen YH, Zhang L, Wang X. 2013. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res 23:986–993. doi: 10.1038/cr.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stable 293T cells expressing CD26. Expression of CD26 in 293T cell line was confirmed by flow cytometry after staining with anti-CD26 antibody. Download
Clinical characteristics of MERS patients included in this study.
List of mutations observed only in MERS-CoV isolated from the Korean patients.
CD26 residues contacted with D510 of S protein.
Potential residues that change the energetic stability upon I529T mutation in S protein.
List of primer sets for the amplification and sequencing of MERS-CoV genome.