

ABSTRACT
Human cytomegalovirus (HCMV), a betaherpesvirus, persists indefinitely in the human host through poorly understood mechanisms. The UL136 gene is carried within a genetic locus important to HCMV latency termed the UL133/8 locus, which also carries UL133, UL135, and UL138. Previously, we demonstrated that UL136 is expressed as five protein isoforms ranging from 33-kDa to 19-kDa, arising from alternative transcription and, likely, translation initiation mechanisms. We previously showed that the UL136 isoforms are largely dispensable for virus infection in fibroblasts, a model for productive virus replication. In our current work, UL136 has emerged as a complex regulator of HCMV infection in multiple contexts of infection relevant to HCMV persistence: in an endothelial cell (EC) model of chronic infection, in a CD34+ hematopoietic progenitor cell (HPC) model of latency, and in an in vivo NOD-scid IL2Rγcnull humanized (huNSG) mouse model for latency. The 33- and 26-kDa isoforms promote replication, while the 23- and 19-kDa isoforms suppress replication in ECs, in CD34+ HPCs, and in huNSG mice. The role of the 25-kDa isoform is context dependent and influences the activity of the other isoforms. These isoforms localize throughout the secretory pathway, and loss of the 33- and 26-kDa UL136 isoforms results in virus maturation defects in ECs. This work reveals an intriguing functional interplay between protein isoforms that impacts virus replication, latency, and dissemination, contributing to the overall role of the UL133/8 locus in HCMV infection.
IMPORTANCE
The persistence of DNA viruses, and particularly of herpesviruses, remains an enigma because we have not completely defined the viral and host factors important to persistence. Human cytomegalovirus, a herpesvirus, persists in the absence of disease in immunocompetent individuals but poses a serious disease threat to transplant patients and the developing fetus. There is no vaccine, and current therapies do not target latent reservoirs. In an effort to define the viral factors important to persistence, we have studied viral genes with no known viral replication function in contexts important to HCMV persistence. Using models relevant to viral persistence, we demonstrate opposing roles of protein isoforms encoded by the UL136 gene in regulating latent and replicative states of infection. Our findings reveal an intriguing interplay between UL136 protein isoforms and define UL136 as an important regulator of HCMV persistence.
INTRODUCTION
Viral persistence, the ability of a virus to persist indefinitely within an immunocompetent host, is a poorly understood phenomenon. The host immune response and viral modulation of the immune response to infection play a critical role in viral persistence. In addition to modulation of the immune response, persistent DNA viruses engage in complex virus-host interactions to optimize the host cell environment for persistence, impacting cellular functions that include survival and differentiation. DNA virus persistence is best defined in the case of herpesviruses, which persist by way of a lifelong latent infection marked by sporadic reactivation events. It is widely appreciated that herpesviruses reactivate in responsive to host or cellular cues, including stress, steroid treatment, or immune deficiency (reviewed in reference 1). However, due to their complexity, the mechanisms by which herpesviruses regulate the establishment of latency or ensure successful reactivation from latency are incompletely defined.
Human cytomegalovirus (HCMV) is a ubiquitous herpesvirus with a worldwide seroprevalence ranging from 40% to 99%, depending on geographical location and socioeconomic factors. In the immunocompetent host, primary infection and reactivation are typically asymptomatic even through periodic reactivation and subsequent virus-shedding episodes (2). However, in the immunocompromised host, opportunistic infection or reactivation can cause significant morbidity and mortality, particularly in stem cell and solid-organ transplant patients (2, 3). In addition, infection of the immune naive fetus in utero is the leading cause of infectious birth defects in the United States, affecting 1 in 150 children each year (4). The effects of lifelong infection with HCMV are beginning to emerge and include atherosclerosis, immune dysfunction, and frailty (5–7). Recent studies in humans and mice indicate that HCMV persistence may actually benefit immune responses to vaccination early in life, while the advantage is lost in the aged (8). A greater understanding of the viral and cellular mechanisms governing latency and persistence is required to appreciate the benefits and costs of viral persistence and to manage or eliminate CMV disease.
HCMV is markedly restricted in its host tropism; however, HMCV infects a variety of cell types within the host. The type of host cell infected largely dictates the pattern of infection, which can be discussed in three general categories: (i) productive infection accompanied by high levels of virus shedding; (ii) chronic/smoldering infection where low levels of virus may be shed over long periods of time; and (iii) latent infections where the genome is maintained in the absence of progeny virus production (3, 9). Endothelial cells (ECs) are thought to be an important site of chronic or smoldering, low-level virus replication and shedding (10). The vascular endothelium is a common target for HCMV infection and serves as a mechanism of virus dissemination to various organs and tissues (11). Infection with HCMV increases inflammation at the endothelium via increased immune cell recruitment, which in turn increases vascular permeability, promotes bidirectional spread of the virus, and may contribute to atherosclerosis and other vascular disease (6, 9, 12).
Reservoirs for the latent virus have not been comprehensively defined, but CD34+ hematopoietic progenitor cells (HPCs) and cells of the myeloid lineage (e.g., CD14+ monocytes) are accepted reservoirs that are commonly studied in vitro (reviewed in reference 1). CMV genomes are harbored in CD14+ monocytes, and differentiation of monocytes into macrophages or dendritic cells can trigger reactivation (13). Further, HCMV genomes have been detected as far back in the hierarchy of hematopoietic differentiation as CD34+ HPCs (14), suggesting that progenitor cells may be a primary reservoir for the virus. Infection of HPCs contributes substantially to virus-related pathogenesis associated with stem cell or solid-organ transplantation (2, 3). Little is known about the cell type-specific requirements for HCMV infection in ECs and HPCs; however, these cell types represent critically important cellular reservoirs for HCMV persistence, latency, and disease in the host.
Viral genes important to HCMV persistence have been identified within a 13- to 15-kb region of the viral genome termed ULb′ that spans genes UL133 to UL154 (15). The ULb′ region is maintained in clinical isolates and low-passage-number strains but is selectively lost upon serial passage of the virus through fibroblasts. Therefore, the ~20 open reading frames (ORFs) that comprise the ULb′ region are hypothesized to support viral persistence through immune evasion and viral dissemination as well as through the establishment and maintenance of latency in cells other than fibroblasts. A 3.6-kb locus spanning UL133 to UL138 (here referred to as the UL133/8 locus) is important for efficient replication in ECs (16, 17) and for the establishment of, or reactivation from latency in vitro in CD34+ HPCs (16, 18–20). UL133 and UL138 are suppressive of virus replication in CD34+ HPCs and aid the establishment or maintenance of latency (16, 18), whereas UL135 overcomes UL138-mediated suppression of replication for reactivation (19). UL135 and UL136 are required for replication in ECs, such that loss of either gene results in defects in virus maturation (17, 21). While the mechanisms by which the UL133/8 genes function have not been fully defined, UL135 and UL138 interact with host factors important to cytoskeleton organization and signaling in the cell (22–25).
UL136 is expressed as five protein isoforms resulting from both alternative transcription and translation initiation (20). In fibroblasts, the pUL136 isoforms are expressed with early to late viral kinetics and have distinct localization patterns during HCMV infection (20). While a virus lacking only the soluble 23- and 19-kDa (23-/19-kDa) isoforms has a replication advantage in both fibroblasts and CD34+ HPCs, a virus null for expression of all UL136 isoforms does not have the same replication advantage in fibroblasts or CD34+ HPCs (20). This finding suggests that interplay between the UL136 isoforms may contribute to the outcome of infection where other pUL136 isoforms promote replication in the absence of the 23-/19-kDa isoforms. However, replication-promoting phenotypes are not apparent in fibroblasts.
To determine how specific isoforms influence the outcome of infection, we explored the interplay between the UL136 isoforms in three models of persistence: a model using ECs, a model using CD34+ HPCs, and an in vivo model using NOD-scid IL2Rγcnull humanized (huNSG) mice. Using a series of recombinant viruses that lack individual isoforms, express individual isoforms, or express combinations of isoforms, we investigated the role and interplay of the UL136 isoforms in multiple infection contexts of infection. Our analyses indicate that two large membrane-associated isoforms (the 33- and 26-kDa isoforms) promote replication, while the two soluble isoforms (the 23-/19-kDa isoforms) suppress replication in ECs, CD34+ HPCs, and huNSG mice. In contrast, the 25-kDa pUL136 isoform exhibits cell type-specific phenotypes, suggesting that it may modify the interplay between replication-promoting and -suppressing pUL136 isoforms in a context-dependent manner. This work reveals an intriguing and complex interplay between pUL136 isoforms that contributes importantly to mediating outcomes of infection. Further, our findings illustrate the complexity and importance of distinct protein isoforms in understanding the overall function of a gene.
RESULTS
UL136 is expressed as five protein isoforms with distinct subcellular localizations in endothelial cells.
We previously mapped unique transcripts and translation initiation sites (TIS) which support the expression of the five pUL136 isoforms in MRC-5 fibroblasts (16, 20). A schematic representing the pUL136 isoforms is shown in Fig. 1A. Similarly to the expression observed in fibroblasts, we detected each of the pUL136 isoforms in human microvascular lung endothelial cells by immunoblotting (HMVECs; Fig. 1B). To define the localization of the pUL136 isoforms within ECs, we infected HMVECs with viruses expressing a single pUL136 isoform where all TIS have been disrupted except for those specific to each isoform (20). Cells were stained at 5 days postinfection (dpi) using a monoclonal antibody specific to the myc epitope tag fused to the C terminus of each pUL136 isoform and GM130 to mark the Golgi apparatus (Fig. 1C), which is rearranged to form the juxtanuclear viral assembly compartment (VAC) (26), a late-stage, infection-induced organelle unique to HCMV infection that is considered important for virus maturation (26, 27). Note that VAC formation and morphology characteristics, and thus Golgi morphologies, differ in ECs and among the UL136 mutants. Because the pUL136 isoforms are expressed at low levels and are unstable (20), representative images with an optimal signal-to-noise ratio were selected. Expressed together in the UL136myc virus infection, the pUL136 isoforms were distributed throughout the cytoplasm as well as in the Golgi apparatus and VAC. Expressed as single isoforms in the context of infection in ECs, the 33-, 26-, and 25-kDa isoforms exhibited Golgi association as well as punctate staining in the cytoplasm, indicating that the isoforms traffic in vesicles and may function outside the Golgi apparatus. The soluble 23-/19-kDa isoforms were Golgi localized but were also diffusely distributed throughout the cell. Together, these data indicate that the pUL136 isoforms may function in distinct subcellular compartments during HCMV infection. The expression and function of the pUL136 isoforms are of particular interest in ECs, as UL136 is uniquely required for efficient VAC formation and late-stage virus maturation events in ECs (21).
FIG 1 .
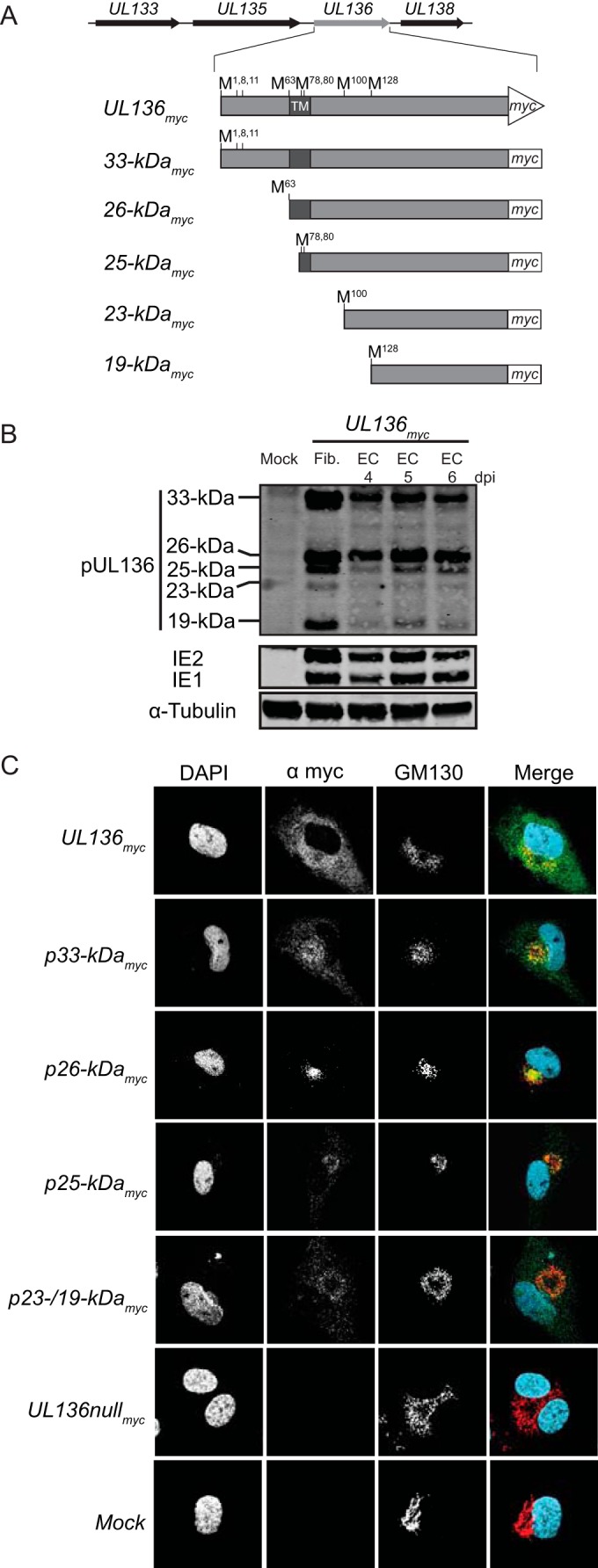
Expression and distinct localization of pUL136 in endothelial cells. (A) Schematic of the protein isoforms of UL136. (B) Expression of the pUL136 isoforms in ECs. HMVECs were either mock infected or infected with the UL136myc virus at an MOI of 2 and harvested at 4 to 6 dpi. As a positive control, MRC-5 fibroblasts were either mock infected or infected with the UL136myc virus at an MOI of 2 and harvested at 3 dpi. Lysates were immunoblotted with an antibody specific to the myc epitope tag. A monoclonal antibody to α-tubulin was used as a loading control. Fib., fibroblasts. EC, endothelial cells. (C) Distinct localization of the pUL136 isoforms in ECs. HMVECs were either mock infected or infected at an MOI of 3 with indicated viruses. At 5 days postinfection, the UL136 proteins were localized by indirect immunofluorescence using an antibody specific to the myc epitope tag or a Golgi apparatus marker, GM130. DAPI staining marks the nuclei. DAPI, blue. Myc, green. GM130, red.
The 23-/19-kDa isoforms suppress while the 33-/26-kDa isoforms promote virus replication in endothelial cells.
We previously reported that disruption of the 23-/19-kDa isoforms results in enhanced virus replication in fibroblasts, while a virus null for expression of all pUL136 isoforms replicated with wild-type (WT) kinetics (20). These data suggest that while the 23-/19-kDa isoforms may suppress replication, other pUL136 isoforms must promote replication. However, we were unable to discern these functions in fibroblasts, as viruses lacking other individual isoforms also replicated with WT kinetics (20), indicating either that multiple pUL136 isoforms promote replication or that the functions of these isoforms may be context specific. Because of the requirement for UL136 for replication in ECs (21), we examined the production of infectious virus in ECs infected with UL136 recombinant viruses lacking a single isoform using multistep viral replication curves (multiplicity of infection [MOI], 0.05) to determine the impact of each pUL136 isoform(s) in infected ECs. The multistep replication curves are shown in Fig. 2A, and the 12-dpi time point is graphed for each infection in Fig. 2B to more simply represent the differences in total yield between viruses. The UL136myc parental virus has been previously shown to replicate with WT (untagged) kinetics in both fibroblasts and ECs (20, 21), indicating that the myc epitope tag does not affect virus replication. UL136nullmyc virus replicated with a substantial defect compared to the UL136myc parental virus, as has been previously shown in ECs for a UL136 ORF substitution virus (21). Similarly to infection in fibroblasts, the UL136Δ23-/19-kDamyc virus replicated with a 20-fold advantage, indicating that these isoforms also suppress replication in ECs. Interestingly, the UL136Δ33-kDamyc and UL136Δ26-kDamyc viruses replicated with 40- and 20-fold defects, respectively, indicating a role for these isoforms in promotion of replication in ECs. The UL136Δ25-kDamyc virus replicated with kinetics similar to those of the UL136myc parental virus, raising the possibility that this isoform is dispensable for replication. These data indicate the existence of dual functions carried within the UL136 gene to both promote and suppress virus replication.
FIG 2 .
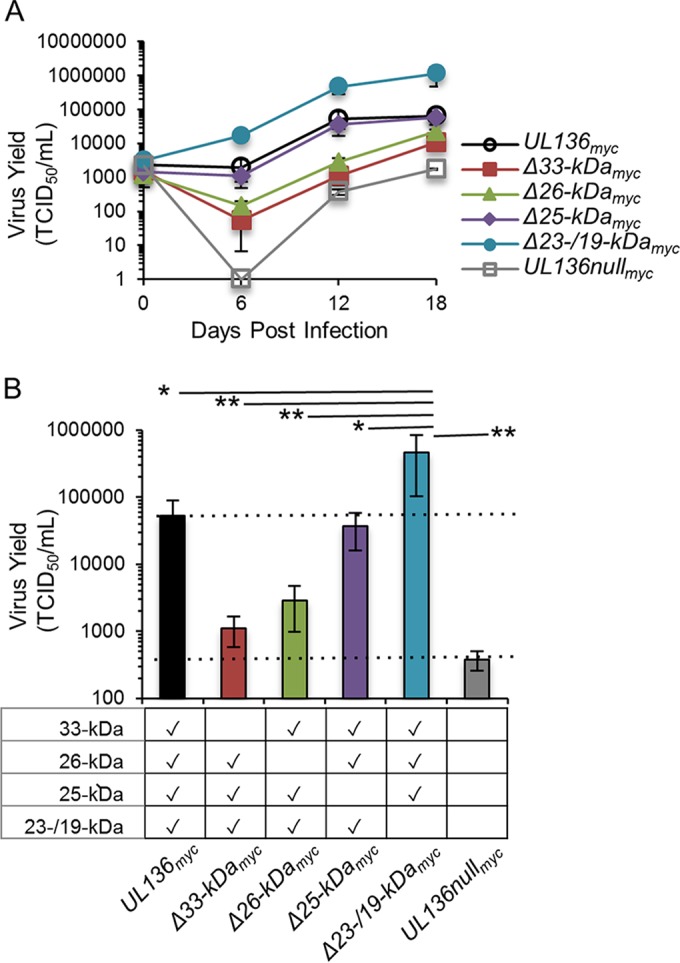
The 33- and 26-kDa isoforms promote replication in endothelial cells, while the 23-/19-kDa isoforms suppress replication. HMVECs were infected with UL136myc variants as indicated at an MOI of 0.05. Virus yields in cell lysates were measured over a time course by TCID50. Data points represent the averages of the results from at least three experiments. Error bars represent standard deviations. (A) Replication curve of UL136 mutants over time. (B) Replication of UL136 mutants at 12 dpi (data adapted from the experiment whose results are shown in panel A). Statistical significance was determined using one-way analysis of variance, followed by Tukey’s posttest. *, P < 0.05; **, P < 0.01.
The pUL136 33-kDa isoform is required for efficient viral assembly compartment formation.
Genes in the UL133/8 locus, specifically, UL135 and UL136, are required for efficient virus maturation in ECs, including the rearrangement of cytoplasmic membranes to form the juxtanuclear VAC (17, 21). To determine whether a specific pUL136 isoform(s) is required for formation of the VAC in ECs, we examined VAC formation in ECs infected with UL136 recombinant viruses lacking a single isoform at 5 dpi by indirect immunofluorescence (Fig. 3). The VAC was labeled using antibodies specific to pp28 and the Golgi compartment (GM130), as viral and cellular markers of the VAC, respectively (27). Cells were scored for the presence or absence of VAC architecture based on both Golgi and pp28 morphology and localization, as has been previously described (21). Cells with a rearranged Golgi compartment that was organized, concentric, and contained pp28 were considered positive for VAC formation, regardless of the size of the VAC. In contrast, cells with dispersed localization of pp28 or Golgi compartments were scored negative for VAC formation. WT-infected ECs formed a VAC in 75% to 100% of cells, among 3 independent experiments, consistent with our previous observations (21). We never observed dispersed membranes in WT infection. As previously reported for a virus lacking the entire UL136 ORF, the UL136nullmyc virus infection resulted in VAC formation in 45% of the infected cells (Fig. 3) (21). Fifty-five percent of the cells infected with UL136nullmyc virus exhibited diffuse pp28 and Golgi staining as if the membrane architecture had been shattered. ECs infected with viruses lacking the 26-, 25-, or 23-/19-kDa isoforms formed the VAC as efficiently as ECs infected with the UL136myc parental virus (Fig. 3) and the WT (untagged) virus (data not shown), indicating that these isoforms are not required for the efficient establishment of the HCMV-induced VAC. However, the VAC was disrupted in 60% of ECs infected with UL136Δ33-kDamyc virus, suggesting a unique requirement for the 33-kDa isoform in virus-induced membrane reorganization or in maintaining membrane integrity that supports VAC formation in ECs.
FIG 3 .
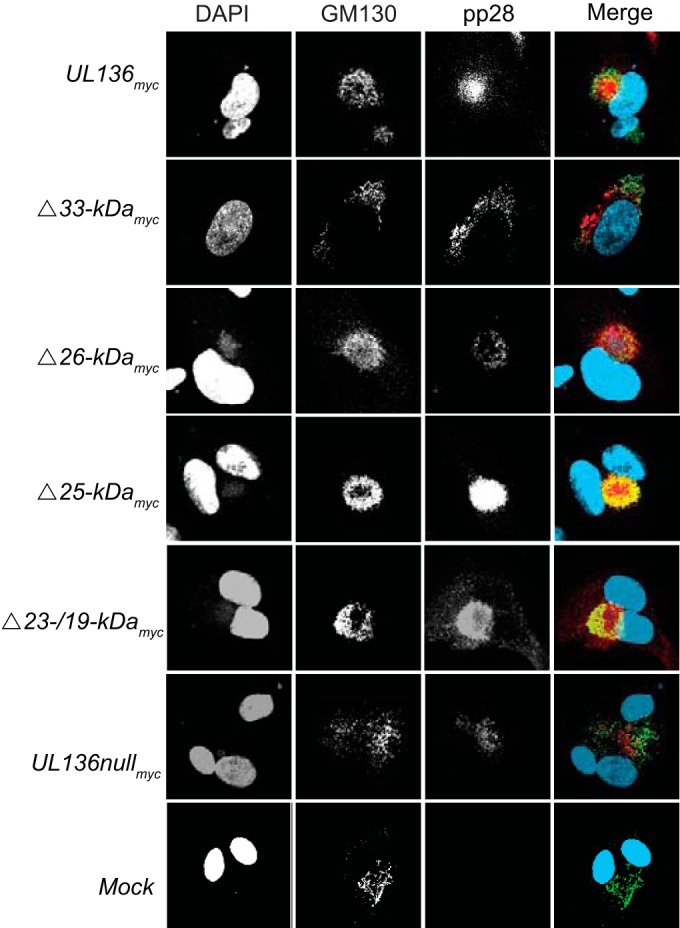
The 33-kDa isoform of pUL136 is required for efficient viral assembly compartment formation in endothelial cells. HMVECs were either mock infected or infected at an MOI of 3 with the indicated viruses. At 5 days postinfection, the HCMV pp28 protein was localized by indirect immunofluorescence using a protein-specific antibody. GM130 marks the Golgi apparatus. DAPI staining marks the nuclei. DAPI, blue. GM130, green. pp28, red.
The pUL136 isoforms contribute to virus maturation and organization of intracellular membranes in endothelial cells.
The defect in VAC formation observed in infections lacking the UL136 ORF is accompanied by defects in progeny virion maturation in ECs (21). Without UL136 in ECs, capsids accumulate in the cytoplasm but do not appear to acquire tegument or envelopes (21). To assess the requirement of each individual isoform for virion maturation, we analyzed ECs infected with TB40/E-WT or UL136 recombinant viruses lacking a single isoform at 5 dpi using transmission electron microscopy (Fig. 4). Quantification of the virus particles present in each infection is shown in Fig. 4G. UL136Δ33-kDamyc virus-infected cells exhibited a defect in virion maturation, as the vast majority of particles lacked envelopes and tegument relative to the WT-infected cells (Fig. 4B compared to A, arrowheads), resulting in ~90% of virus progeny in the cytoplasm being nonenveloped capsids compared to 4.5% in the WT (Fig. 4G). This phenotype resembled that of infection where the entire UL136 ORF disrupted (21) or where all isoforms were disrupted via discrete mutations within the ORF (Fig. 4F and G). Further, the defect in tegumentation and envelopment is consistent with the failure of the UL136Δ33-kDamyc virus to form a VAC (Fig. 3). The UL136Δ26-kDamyc virus-infected cells also exhibited substantial defects in tegumentation and envelopment (Fig. 4C compared to A, arrowheads), with 35% of virus progeny in the cytoplasm being nonenveloped capsids (Fig. 4G). Interestingly, the defect associated with UL136Δ26-kDamyc virus infection occurred even though these cells formed WT-like VACs, suggesting that the failure to reorganize membranes in the VAC does not alone account for the defects in virion maturation and indicating a unique role for the 26-kDa isoform in tegumentation and envelopment that does not impact the formation of the VAC. In contrast, the envelopment of maturing virions in the cytoplasm of UL136Δ25-kDamyc virus- and UL136Δ23-/19-kDamyc virus-infected ECs was similar to that seen with WT infection (Fig. 4D and E compared to A, arrowheads), with less than 10% of progeny particles lacking envelopes (Fig. 4G). UL136Δ23-/19-kDamyc virus-infected ECs produced a large number of virions with prototypical morphology (Fig. 4E compared to A), consistent with the higher virus yields in UL136Δ23-/19-kDamyc virus infection (Fig. 2).
FIG 4 .

The pUL136 isoforms differentially impact the maturation of virus progeny formed in infected ECs. HMVECs were infected at an MOI of 4 with TB40/E-WT (A), UL136Δ33-kDamyc (B), UL136Δ26-kDamyc (C), UL136Δ25-kDamyc (D), UL136Δ23-/19-kDamyc (E), or UL136nullmyc (F) virus. At 5 days postinfection, cells were fixed, embedded, and sectioned for transmission electron microscopy (TEM). Representative micrographs are shown to illustrate the accumulation of virus particles in the cytoplasm. The inset shows magnified virions. Two distinct types of virus particles are shown: virions (arrowheads) and dense bodies (arrows). Scale bar, 500 nm. (F) Total particles (200 to 300) were counted in 15 to 20 cells. The percentages of aberrantly enveloped virions, noninfectious enveloped particles (NIEPs), nonenveloped capsids, and normal virions in each infection are shown.
Interestingly, there was an increase in the number of noninfectious enveloped particles (NIEPs), morphologically normal virus particles that lack a viral genome, in the cytoplasm of ECs infected with viruses lacking the 26-, 25-, and 23-/19-kDa isoforms. Further, many of the nonenveloped capsids of ECs infected with a virus lacking the 33-kDa isoform also lacked genomes, suggesting that there may also be defects in viral genome packaging in the nucleus; however, more work is needed to understand how the UL136 isoforms may impact capsid assembly in the nucleus and maturation events in the cytoplasm.
We previously reported that dense bodies (DBs), vesicles containing tegument proteins produced during infection, are on average 2.5-fold larger in UL136-null virus infection than in WT infection (21). Interestingly, we observed enlarged DBs in ECs infected with either UL136Δ33-kDamyc or UL136Δ26-kDamyc virus, while ECs infected with UL136Δ25-kDamyc or UL136Δ23-/19-kDamyc virus produced DBs that were similar in size to those seen in WT infection (Fig. 4, arrows). The DBs in UL136Δ33-kDamyc virus and UL136Δ26-kDamyc virus also appeared to have defective envelopes or lacked envelopes compared to WT DBs. The alterations in DB formation and virion maturation observed with UL136 isoform-mutant virus infection indicate roles for the pUL136 isoforms in commandeering host trafficking machinery, which may directly or indirectly affect virus maturation.
The interplay of the pUL136 isoforms alters replication in endothelial cells.
Protein isoforms often function to affect the activity of one another (28–31). The opposing effects of pUL136 isoforms in HCMV infection suggest that the pUL136 isoforms may antagonize or synergize with each other. To investigate the interplay between the pUL136 isoforms and their impact on infection, we compared the kinetics and yields of virus replication in EC infection by UL136 mutant viruses lacking individual isoforms and combinations of isoforms by analysis of the 50% tissue culture infective dose (TCID50) in fibroblasts. For simplicity, only the 12-dpi time point for each replication curve is shown in Fig. 5.
FIG 5 .

Interplay of the pUL136 isoforms governs replication in endothelial cells. HMVECs were infected with UL136myc variants as indicated at an MOI of 0.05. Virus yields in cell lysates were determined at 12 days postinfection by TCID50. (A) The 33- and 26-kDa isoforms promote replication in a cooperative manner. (B) The 25-kDa isoform enhances the effects of the combined 33-/26-kDa isoforms. (C) The 23-/19-kDa isoforms consistently suppress replication in the context of other pUL136 isoforms. Data points represent the averages of the results from at least three experiments. Error bars represent standard deviations.
To determine if the 33- and 26-kDa isoforms function cooperatively to promote replication, we analyzed a recombinant virus in which these isoforms were disrupted in combination. This virus replicated with a defect equivalent to but not greater than that seen with the Δ33- or Δ26-kDa mutant viruses (Fig. 5A). However, a virus expressing only the 33- and 26-kDa isoforms replicated to nearly WT levels, suggesting that the 33- and 26-kDa isoforms synergize to promote virus replication. While disruption of the 25-kDa isoform alone had no effect on virus replication in ECs (Fig. 2A and B and 5B), the 25-kDa isoform enhanced the combined functions of the 33-/26-kDa replication-promoting isoforms (Fig. 5B) (up to ~45-fold) compared to the results seen with the virus expressing only the 33-/26-kDa isoforms. These data suggest that the 25-kDa isoform may enhance replication by stimulating the effects of the 33-/26-kDa isoforms in the absence of the 23-/19-kDa isoforms. The 23-/19-kDa isoforms are suppressive of virus replication in the context of multiple combinations of pUL136 isoforms (Fig. 2A and B and 5C). We examined the effect of the 25-kDa isoforms on replication-promoting isoforms using viruses that lack the 33- or 26-kDa isoforms with or without the 23-/19-kDa isoforms. While the changes did not reach statistical significance, viruses expressing the 33- and 25-kDa or 26- and 25-kDa isoforms replicated with an ~4- or ~2-fold advantage compared to viruses also expressing the 23-/19-kDa isoforms, respectively. Taking these results together with the finding that a virus expressing the 33-, 26-, and 25-kDa isoforms replicated with an ~20-fold increase over the UL136myc parental virus expressing the 23-/19-kDa isoforms suggests that the 33- and 26-kDa isoforms, together with the 25-kDa isoform, promote virus replication and are antagonized by the 23-/19-kDa isoforms. These findings demonstrate complex interplay between the pUL136 isoforms.
The pUL136 isoforms differentially impact viral latency and reactivation in CD34+ hematopoietic progenitor cells.
The 23-/19-kDa isoforms are required for the efficient establishment of latency in an in vitro model in CD34+ HPCs (Fig. 6) (20). Because the abilities of the UL136Δ23-/19-kDamyc and the UL136nullmyc viruses to replicate in CD34+ HPCs are not equivalent, we hypothesized that other pUL136 isoforms must contribute to the replication advantage observed in UL136Δ23-/19-kDamyc virus infection, as is the case in ECs. To discern the role of the other pUL136 isoforms in latency, CD34+ HPCs were infected with the UL136myc parental virus, the UL136nullmyc virus, and viruses lacking the individual pUL136 isoforms. Populations of infected green fluorescent protein-positive (GFP+) CD34+ HPCs were purified by fluorescence-activated cell sorting (FACS) at 24 hpi and seeded into long-term bone marrow culture (LTBMC) over stromal cell support as previously described (32). After the HPCs were cultured for 10 days in LTBMC, we seeded cells or an equivalent cell lysate onto monolayers of permissive fibroblasts by limiting dilution in cytokine-rich media to promote myeloid differentiation. The cell lysate distinguishes virus formed during the LTBMC period prior to reactivation stimuli (prereactivation) from virus formed as a result of reactivation (postreactivation). After cocultures with fibroblasts were incubated for 14 days, we used the fraction of GFP+ wells at each dilution to determine the frequency of infectious centers (Fig. 6). The UL136Δ23-/19-kDamyc virus failed to establish latency and instead replicated with increased efficiency under conditions of both prereactivation and mock reactivation relative to the UL136myc parental virus, consistent with our previous observations (20). Consistent with the requirement of the 33- and 26-kDa isoforms for replication and maturation in ECs, these isoforms were required for reactivation in CD34+ HPCs; few infectious centers were produced prior to or following reactivation of both UL136Δ33-kDamyc virus and UL136Δ26-kDamyc virus. In contrast to its phenotype in ECs, UL136Δ25-kDamyc virus had an enhanced ability to reactivate in CD34+ HPCs. This phenotype of augmented reactivation may be the result of a less tightly controlled or “leaky” latent infection. Together, these data indicate that the replication-influencing functions of some pUL136 isoforms (the 33-, 26-, and 23-/19-kDa isoforms) are similar across multiple cell types relevant to persistence, whereas the 25-kDa isoform has context-dependent roles.
FIG 6 .
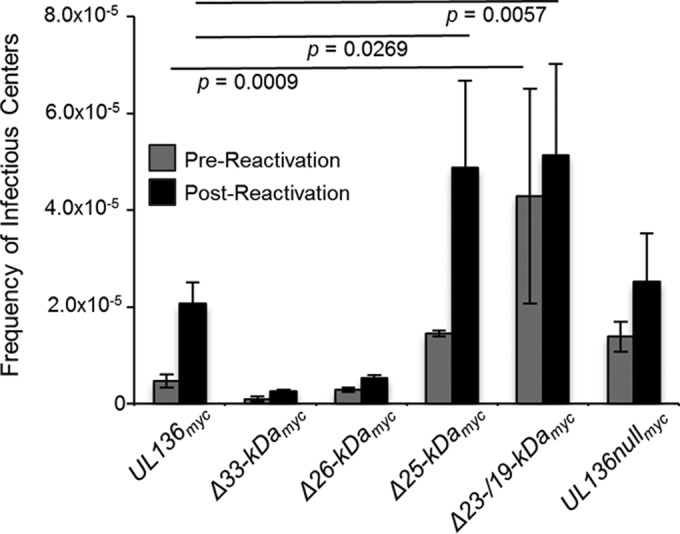
The pUL136 isoforms differentially influence the establishment of latency and virus reactivation in CD34+ HPCs. CD34+ HPCs were infected at an MOI of 2 with the indicated virus and sorted by FACS to isolate pure, infected populations. HPCs were maintained in LTBMC over stromal cell support for 10 days. Subsequently, HPCs were cocultured with an MRC-5 fibroblast cell monolayer (mock reactivation). In parallel, lysates from an equal number of HPCs were plated with an MRC-5 fibroblast cell monolayer (prereactivation). At 14 days later, 96-well dishes were scored for GFP+ wells, and the frequency of infectious centers was determined using ELDA software. Statistical significance was determined using two-way analysis of variance followed by Dunnett’s posttest for comparisons to UL136myc virus results. P values are shown.
The pUL136 isoforms differentially impact reactivation and dissemination in vivo.
We previously demonstrated that the UL133/8 locus modulates virus reactivation and dissemination in vivo utilizing the huNSG mouse model of HCMV latency, reactivation, and dissemination of infected cells in vivo (16, 33). Viruses lacking the UL133/8 locus replicate or disseminate to a greater extent than the WT, as demonstrated by a 2- to 3-fold increase in the number of viral genomes in the spleen (16). Given the complexity of the interplay between the pUL136 isoforms in ECs and CD34+ HPCs, we reasoned that the huNSG mouse model would further discern the differential roles of the pUL136 isoforms in vivo (Fig. 7). huNSG mice were sublethally irradiated and engrafted with human CD34+ HPCs. After CD34+ engraftment, mice were injected with human fibroblasts infected with UL136myc or the UL136 recombinant viruses indicated. Mice injected with uninfected fibroblasts served as a negative control. At 4 weeks postinfection, 5 mice from each group of 10 were treated with G-CSF and AMD-3100 to induce stem cell mobilization and virus reactivation. At 1 week postmobilization, viral genome loads were assessed in the spleen and liver to evaluate amplification of viral genomes and dissemination of infected cells.
FIG 7 .
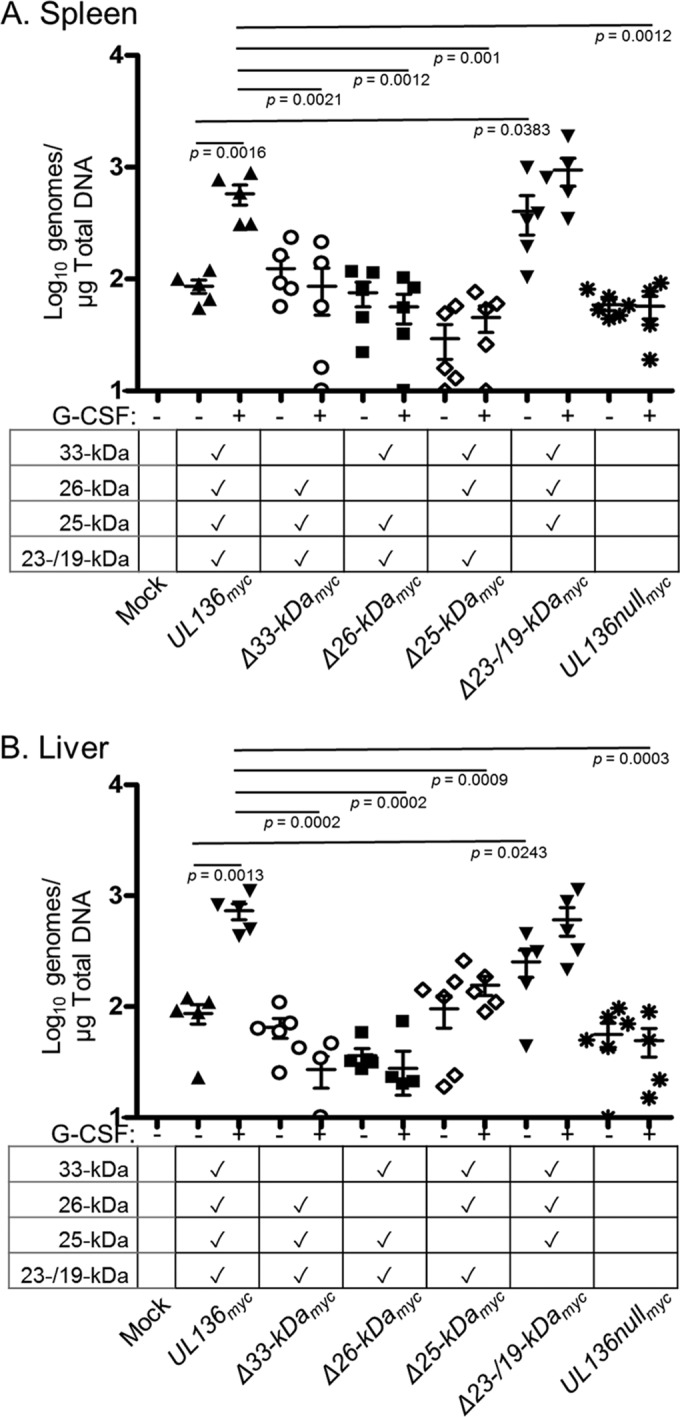
The pUL136 isoforms modulate virus reactivation and dissemination in vivo. Sublethally irradiated NOD-scid IL2Rγcnull mice were engrafted with CD34+ HPCs and subsequently injected with human fibroblasts previously infected with UL136myc or UL136 recombinant viruses as indicated. Mice injected with uninfected fibroblasts served as a negative control (n = 5). At 1 week postinfection, half of the mice were treated with G-CSF and AMD-3100 and the other half left untreated (n = 5/group). At 1 week posttreatment, mice were euthanized and tissues harvested. Total genomic DNA was isolated from spleen (A) and liver (B) tissues, and HCMV genomes were quantified using quantitative PCR with primers and probe specific for the UL141 gene. Statistical significance was determined using two-way analysis of variance, followed by Bonferroni’s posttest. P values are shown.
HCMV genomes were detected in the spleen and liver of all infected huNSG mice, both nonmobilized and mobilized (Fig. 7). Numbers of splenic and liver viral genomes increased upon mobilization in parental UL136myc virus-infected mice, consistent with reactivation from latency and dissemination of infected cells. Interestingly, lower levels of viral genomes in the spleen and liver were detected in UL136nullmyc virus-infected mice than in UL136myc virus-infected mice following G-CSF/AMD-3100 treatment and the presence of G-CSF/AMD-3100 did not increase the numbers of genomes relative to those seen with untreated mice. These data suggest that UL136 is not required for the maintenance of viral genomes but is required for efficient reactivation or dissemination of infected cells in vivo and are in contrast to our findings in CD34+ HPCs infected in vitro (Fig. 6).
Consistent with our in vitro data in ECs and CD34+ HPCs, viral loads in unmobilized or mobilized mice infected with UL136Δ23-/19-kDamyc virus were greater than those in mice infected with the UL136myc parental virus (Fig. 6). In fact, viral loads were higher in both spleen and liver, even in the absence of reactivation and mobilization stimuli, consistent with the suppressive role of these soluble isoforms (Fig. 2, 5, and 6) (20). Substantially fewer genomes were detected in mobilized mice infected with Δ33- or Δ26-kDa viruses than in those infected with the UL136myc parental virus, consistent with their role in promoting replication and reactivation in multiple cell types (Fig. 2, 5, and 6). Mobilization also failed to increase viral loads in the spleen or liver of mice infected with UL136Δ25-kDa virus relative to the UL136myc parental virus. This finding stands in striking contrast to the phenotype of augmented reactivation in CD34+ HPCs infected in vitro (Fig. 6), as well as in contrast to the WT-like phenotype observed in ECs (Fig. 2), further indicating context-dependent functions of the 25-kDa isoform. Importantly, the levels of genomes measured in the spleen and liver of unmobilized mice infected with UL136Δ33-kDamyc virus, UL136Δ26-kDamyc virus, and UL136Δ25-kDamyc virus were not significantly different from those measured in the unmobilized UL136myc mice (spleen, P = 0.14, P = 0.31, and P = 0.06; liver, P = 0.17, P = 0.06, and P = 0.41), suggesting that there was no defect in maintenance of the genomes. Together, these results point to important and distinct roles for the UL136 isoforms in balancing states of latency and reactivation in vivo. These results further reveal functions for the 25-kDa isoforms unique to infection in a host organism. The context-dependent functions of the pUL136 isoforms are summarized in Fig. 8A.
FIG 8 .
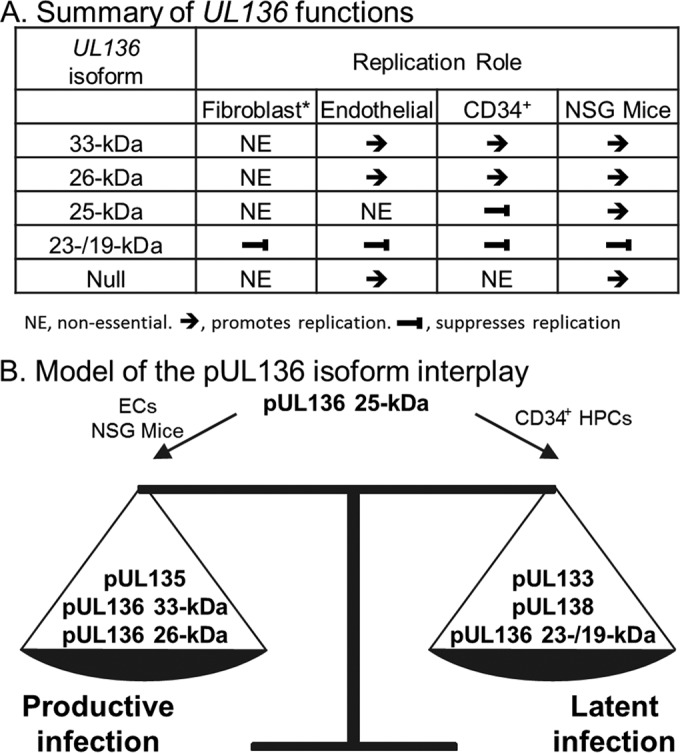
Proposed model hypothesizing the role of pUL136 in modulating the outcome of infection (productive/latent). (A) Table summarizing the functions of the pUL136 isoforms in multiple cell type-specific infection states. (B) Model of pUL136 isoform interplay. Together, the 33- and 26-kDa isoforms, along with pUL135, cooperatively promote replication, while the 23-/19-kDa isoforms suppress replication in conjunction with pUL133 and pUL138. The 25-kDa isoform functions in a cell type-dependent manner, likely governing the balance of both the 33- and 26-kDa and the 23-/19-kDa isoforms. How the multiple replication-promoting and -suppressing proteins of the UL133/8 locus work together is not yet known. *, adapted from reference 20.
DISCUSSION
Reevaluation of HCMV coding capacity has recently revealed a much greater potential than previously appreciated (15, 34, 35). A single gene encoding several protein isoforms is one mechanism to expand coding capacity. While protein isoforms may have independent functions, they often impact the function or activity of each other. The antagonistic or synergistic roles of protein isoforms are poorly understood in biology but represent an important aspect of post-translational control. Viruses provide a model that is highly amenable to genetic manipulation to investigate the function of protein isoforms within the context of infection. In the present report, we demonstrate that the UL136 isoforms have unique, antagonistic, and synergistic functions in the contexts of important HCMV infection or viral latency and persistence. The interplay between the pUL136 isoforms impacts not only virus replication in ECs and latency in vitro but also viral latency, reactivation, and dissemination in an in vivo model. Understanding how the interplay of protein isoforms encoded by a single gene can functionally balance multiple states of HCMV infection is important for fully understanding the mechanisms of latency and persistence.
Our in vitro and in vivo data from the present study suggest that the 23-/19-kDa isoforms function to suppress while the 33- and 26-kDa isoforms function to promote replication in CD34+ HPCs, huNSG mice, and ECs. The functions of the pUL136 isoforms are summarized in Fig. 8A. Because the pUL136 isoforms are dispensable for virus replication in fibroblasts (20), a model for productive HCMV infection where the entire ULb′ region of the genome is also dispensable, we are struck by the consistency in the phenotypes associated with these replication-suppressing or replication-promoting isoforms across multiple, distinct in vitro and in vivo contexts of infection relevant to persistence. The conservation of phenotypes among the 33- and 26-kDa mutant viruses might indicate that the role of these proteins in virus maturation is also important in other contexts of infection. The UL136 isoforms may represent an evolved network where the 33-, 26, and 23-/19-kDa isoforms have a highly conserved function in promoting or suppressing replication.
The role of the 25-kDa isoform is unique in that the phenotypes associated with the 25-kDa mutant virus were highly dependent on context. Although the 25-kDa isoform was largely dispensable for replication in ECs (Fig. 2), it augmented the replication-promoting effects of the 33-/26-kDa isoforms in ECs (Fig. 5). However, the 25-kDa isoform was required for the virus to maintain latency in CD34+ HPCs (Fig. 6). In contrast, the 25-kDa isoform was required for reactivation in huNSG mice, as its absence resulted in fewer genomes detected in the spleen and liver post mobilization (Fig. 7). One possible explanation for failure of the numbers of viral genomes to increase in the liver and spleen of infected huNSG mice treated with G-CSF and AMD-3100 is that infected cells reactivated but did not mobilize to tissues. Analysis of viral genomes in other tissues such as the blood and bone marrow will be important to differentiate these possibilities. It is equally possible that the 25-kDa isoform is critical for reactivation in vivo but not in the context of infection in vitro due to factors specific to infection in an organism. Given the distinct phenotypes and context-dependent roles of the 25-kDa isoform, it may function to shift the balance toward replicative or latent states depending on context-specific cues. Whatever the reason for these differing phenotypes, this property of the 25-kDa isoform likely allows the conserved network of replication-promoting (33- and 26-kDa) and replication-suppressing (23-/19-kDa) isoforms to be dynamic and to respond to changes in the host state. These findings underscore the importance of investigating HCMV protein function and infection in contexts of infection other than fibroblasts.
Understanding the basis of the differences between in vitro and in vivo latency models will be important for defining mechanisms that allow the shift between productive and latent states. Taken together, our results suggest a model whereby the 25-kDa isoform governs the balance between replication-promoting 33-/26-kDa isoforms and replication-suppressing 23-/19-kDa isoforms in a context-specific manner (Fig. 8B). We have previously shown that other proteins encoded within the UL133/8 locus also function in this balance. UL133 and UL138 both function to suppress virus replication for latency (16, 18), while UL135 functions to overcome this suppression for virus reactivation and replication (19). We are interested in exploring how the pUL136 isoforms specifically contribute to or antagonize the function of the gene products encoded within the UL133/8 locus in future studies. To our knowledge, this work defines the first HCMV gene that encodes multiple antagonistic protein isoforms to govern states of HCMV replication, latency, and dissemination.
It is intriguing to speculate on the mechanisms governing the interplay and functions of the pUL136 isoforms. The distinct functions of the pUL136 isoforms are intriguing, considering that the isoforms share dramatic similarity in their primary protein sequences. This shared primary protein sequence could allow the pUL136 isoforms to target each other or the same host cell proteins in a dominant-negative manner. The distinct localization patterns of the pUL136 isoforms within the secretory pathway may allow the isoforms to function in distinct subcellular compartments (Fig. 1), as has been previously suggested for other viral protein isoforms (31, 36–38). Further, host cell protein relocalization via trafficking to distinct pUL136 isoform-specific subcellular compartments could allow the isoforms to sequester or control the activity of host cell proteins during HCMV infection. More work is needed to understand how the pUL136 isoforms interact with the host cell during HCMV infection and the significance of these interactions to the outcome of infection.
We previously reported that UL136 is required for the formation of the VAC and virion envelopment and maturation (21). Our current studies revealed that only the 33-kDa isoform of UL136 was required for the efficient formation of the VAC whereas both the 33- and 26-kDa isoforms were required for virion tegumentation, envelopment, and maturation (Fig. 3 and 4). It is possible that the 33-kDa isoform of pUL136 functions synergistically with pUL135 to redirect membrane organization in order to form the VAC and, in turn, to promote virus maturation and envelopment. As the 26-kDa isoform was not required for the formation of the VAC (Fig. 3), the defects in virion maturation in viruses lacking the 26-kDa isoform most likely stem from a function distinct from that of the 33-kDa isoform. Interestingly, UL136 is not required for VAC formation or maturation and virion envelopment in fibroblasts (21), further revealing the cell type-specific roles of the pUL136 isoforms. Viral determinants outside the UL133/8 locus that control VAC compartment formation have been previously described in fibroblasts (39); however, their functions in ECs have not been explored. Defining the cellular interactions specific to the role of the 33-kDa isoform in mediating VAC formation and membrane trafficking and reorganization will be important for our understanding of virus control of membrane trafficking and virus maturation in ECs. It is possible that the observed defects in maturation and membrane organization are symptomatic of global alterations of trafficking and membrane organization/architecture and that defining these mechanisms could, in turn, shed light on host pathways that are critical to HCMV infection in multiple infection states in vitro and in vivo.
The protein isoforms encoded by the UL136 gene impact virus maturation in ECs, latency and reactivation in CD34+ HPCs, and virus replication or dissemination in vivo in huNSG mice. These phenotypes suggest an intriguing link between membrane trafficking and latency. The regulation of membrane organization and trafficking is critical to intracellular and intercellular signaling, stress responses, and biosynthesis. The ability of HCMV to interface with these host pathways is a means by which the virus can sense and respond to changes in host homeostasis or perhaps influence the regulation of signaling and stress. The role of the multiple pUL136 isoforms encoded from a single gene in regulating other proteins encoded within the UL133/8 locus and in balancing states of HCMV infection is a rich area of investigation which will produce novel insights into the mechanisms governing viral latency and persistence. UL133/8 gene functions have coevolved with the host through millennia to result in an intricate molecular switch to control HCMV infection and persistence in multiple cell types within the host.
MATERIALS AND METHODS
Cells.
Human primary embryonic lung fibroblasts (MRC-5; purchased from ATCC, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-alanyl-glutamine, 0.1 mM nonessential amino acids, 100 U/ml penicillin, and 100 µg/ml streptomycin. Primary human lung microvascular lung endothelial cells (HMVEC-L; purchased from Lonza, Walkersville, MD) were cultured with microvascular endothelial cell growth BulletKit medium (EGM-2; Lonza). Human cord blood and bone marrow were obtained from donors at the University of Arizona Medical Center and processed using an Institutional Review Board-approved protocol or were isolated from fetal liver tissue obtained from Advanced Bioscience Resources as previously described (16, 32). All specimens were deidentified and provided as completely anonymous samples. Briefly, fetal liver tissue was preprocessed via manual disruption and digestion with DNase, collagenase, and hyaluronidase. All CD34+ HPCs were isolated using a CD34 MicroBead kit (magnetically activated cell sorting [MACS]; Miltenyi Biotec, San Diego, CA). Pure populations of CD34+ HPCs were cultured in MyeloCult H5100 (Stem Cell Technologies) and maintained in long-term coculture with M2-10B4 and s.l./s.l. murine stromal cell lines (kind gift from Stem Cell Technologies on behalf of D. Hogge, Terry Fox Laboratory, University of British Columbia, Vancouver, BC, Canada) (16, 32). All cells were maintained at 37°C with 5% CO2.
Viruses.
The HCMV TB40/E bacterial artificial chromosome (BAC) was previously engineered to express green fluorescent protein (GFP) as a visual marker of infection. To engineer the recombinant viruses used in the present study, an intermediate ΔUL136<GalK> BAC was created as previously described (16, 20). The construction of ΔUL136<GalK>, ΔUL136myc, UL136Δ33-kDamyc, UL136Δ26-kDamyc, UL136Δ25-kDamyc, UL136Δ23-/19-kDamyc, UL136p33-kDamyc, UL136p26-kDamyc, UL136p25-kDamyc, UL136p23-/19-kDamyc, and UL136nullmyc viruses has been previously described (16, 20).
All recombinant viruses were created as previously described using a two-step, positive/negative selection approach that leaves no trace of recombination (18). To engineer UL136Δ26-/23-/19-kDamyc and UL136Δ33-/26-kDamyc viruses, a previously described shuttle vector was created using viral sequences from the UL136myc BAC (20). Briefly, a region of TB40/E BAC was PCR amplified from UL135 (bp 854) to UL138 (bp 139), A-tailed with Klenow exo- fragment (NEB), and TA cloned into pGEM-T Easy (Promega). Subsequently, serial site-directed Phusion mutagenesis (NEB) was used to mutate specific methionine codons in UL136 to either stop or alanine codons. Methionine codons 1, 8, and 11 were substituted with stop codons, while methionine codons 63, 78, 80, 100, and 128 were substituted with alanine codons in the recombinant BACs. Desired mutations were confirmed by sequencing the mutagenized pGEM-T plasmids, and then a PCR product of the UL135 (bp 854)-UL138 (bp 139) region of the plasmid was created and subsequently recombined into the intermediate ΔUL136<GalK> BAC. BAC integrity was examined by enzyme digest analysis and sequencing of the entire UL133/8 viral genomic region. All oligonucleotide primers used to engineer recombinant viruses have been previously described (20).
BAC genomes were maintained in SW102 Escherichia coli. Virus stocks were produced by transfecting the BAC genomes (15 to 20 µg) and 2 µg of a plasmid carrying UL82 (pp71) into 5 × 106 MRC-5 fibroblasts and incubating until 100% cytopathic effects (CPE) were observed. Virus stocks were purified and stored as previously described (18), and the 50% tissue culture infectious dose (TCID50) was used to determine virus titers on MRC-5 fibroblasts.
Immunoblotting.
Immunoblotting was performed as previously described (20).
Indirect immunofluorescence.
Localization of both cellular and viral proteins via immunofluorescence was performed as described previously (16, 18). Briefly, HMVEC-Ls were seeded onto coverslips (2 × 104 cells/well in 24-well plates) and allowed to settle overnight. The next day, cells were infected with HCMV at an MOI of 2-3 for 5 to 6 days. Cells were fixed in 2% to 4% paraformaldehyde–phosphate-buffered saline (PBS) and stained with antibodies as previously described (20, 21). To visualize cell structures, the Golgi apparatus was stained using GM130 and nuclei were marked using 1 µg/ml DAPI (4′,6-diamidino-2-phenylindole). Cells were visualized with a Zeiss 510 Meta confocal microscope (Carl Zeiss Microimaging, Inc.).
Quantification of infectious virus.
Quantification of infectious virus produced by ECs over time was determined by infecting HMVEC-Ls at an MOI of 0.05 and subsequently collecting cells and medium over an 18-day infection time course. Virus titers were determined by TCID50 on MRC-5 fibroblasts as previously described (18).
Transmission electron microscopy.
HMVEC-Ls were either mock infected or infected at an MOI of 4, harvested at 5 days postinfection, and processed as described previously (17). Briefly, cells were fixed in 2.5% glutaraldehyde buffered in 0.1 M piperazine–N,N′-bis(2-ethanesulfonic acid) (PIPES) for 20 min and then pelleted. Cell pellets were mock fixed with osmium tetroxide buffered in 0.1 M PIPES and subsequently dehydrated in a graded ethanol series. Resin-infiltrated cell pellets were cut into 100-nm sections and floated onto copper grids. Sections were imaged using a Phillips CM-12 transmission electron microscope. Cells were embedded and sectioned at Arizona Research Laboratories, Arizona Health Sciences Center Core Facility, University of Arizona.
Assays of infectious centers for latency.
Quantitation of latency and reactivation in vitro was performed with CD34+ HPCs obtained from cord blood and/or bone marrow as described previously (16, 26). Briefly, CD34+ HPCs were infected at an MOI of 2 for ~20 h prior to FACS (FACSAria; BD Biosciences Immunocytometry Systems, San Jose, CA), in which a (>97%) pure population of infected (GFP+) CD34+ cells was obtained by utilizing a phycoerythrin-conjugated CD34 (PE-CD34)-specific antibody (BD Biosciences). Infected CD34+ cells were cocultured for 10 days in transwells above irradiated M2-10B4 and s.l./s.l. stromal cells (137Cs gamma cell-40 irradiator type B; Atomic Energy of Canada Limited, Ottawa, Canada) (3,000 rads). An extreme limiting dilution assay was used to quantify the frequency of the production of infectious centers as previously described (16, 26). Briefly, infected CD34+ cells were split into two groups: pre- and post-reactivation. Prereactivation cells were lysed, and the lysate was used to infect MRC-5 fibroblasts in 96-well plates to quantify infectious virus present during the 10-day long-term culture. Post-reactivation cells were cocultured with MRC-5 fibroblasts in 96-well plates to promote differentiation of the cells and reactivation of HCMV. At 14 days postinfection of MRC-5 fibroblasts, the frequency of infectious centers was calculated on the basis of the number of GFP+ wells by the use of extreme limiting dilution analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/) (40).
Engraftment and infection of humanized mice.
All animal studies were carried out in strict accordance with the recommendations of the American Association for Accreditation of Laboratory Animal Care (AAALAC). The protocol was approved by the Institutional Animal Care and Use Committee (number IS00001049) at Oregon Health and Science University. NOD-scid IL2Rγcnull humanized (huNSG) mice were maintained at a pathogen-free facility at Oregon Health and Science University in accordance with procedures approved by the Institutional Animal Care and Use Committee (IACUC). Newborn (0-to-3-day-old) mice were sublethally irradiated with 50 cGy by 137Cs gamma irradiation and subsequently engrafted via intrahepatic injection with 1 × 105 human CD34+ HPCs obtained from fetal liver as described above. CD34+ HPCs were allowed to engraft for a minimum of 8 weeks, and the efficiency of engraftment was determined using FACS treatment as previously described (33). Briefly, the efficiency of engraftment is calculated as the percentage of human CD45+ cells present in the total circulating lymphocytes in the blood. Mice were screened at 4-week intervals, and groups were normalized for human cell engraftment prior to infection. At 12 to 14 weeks after human CD34+ HPC engraftment, mice were treated with 25 ng of lipopolysaccharide (LPS)/mouse and infected after 6 h via intraperitoneal (IP) injection using normal human dermal fibroblasts previously infected with HCMV UL136myc or UL136 recombinant viruses as designated at approximately 1.6 × 105 PFU per mouse. A control group of engrafted mice were mock infected using uninfected fibroblasts. At 4 weeks postinfection, the infected mice were split into two groups and half of the mice were treated with 100 µl of granulocyte colony-stimulating factor (G-CSF) (Amgen) (300 mg/ml) using a subcutaneous micro-osmotic pump (1007D; Alzet) and 125 µg AMD3100 {1,1′-[1,4-phenylene bis(methylene)]bis-1,4,8,11-tetraazacyclotetradecane octahydrochloride or plerixafor} administered IP to mobilize HPCs. The remaining half of the mice served as a direct comparison for analysis of the effects of virus reactivation and dissemination that followed HPC mobilization. At 1 week postmobilization, the mice were sacrificed, lymphoid organs harvested, and samples for PCR frozen in RNAlater and stored at −80°C for subsequent analysis.
Quantitative PCR for viral genomes.
Total DNA was extracted from approximately 1-mm2 sections of mouse spleen or liver using a DNAzol kit (Life Technologies). HCMV genomes were analyzed using quantitative PCR (TaqMan) performed on 1 µg of total DNA and TaqMan FastAdvance PCR master mix (Applied Biosystems, Foster City, CA), according to the manufacturer’s instructions. Primers and a probe recognizing HCMV UL141 were used to quantify HCMV genomes (probe, 5′-CGAGGGAGAGCAAGTT; forward primer, 5′-GATGTGGGCCGAGAATTATGA; reverse primer, 5′-ATGGGCCAGGAGTGTGTCA). The probe contained a 5′ 6-carboxyfluorescein (FAM) reporter molecule and a 3′ quencher molecule (Applied Biosystems). The reaction was initiated using TaqMan Fast Advanced master mix (Applied Biosystems) activated at 95°C for 10 min followed by 40 cycles (15 s at 95°C and 1 min at 60°C) using a StepOnePlus TaqMan PCR machine. Results were analyzed using ABI StepOne software.
ACKNOWLEDGMENTS
We acknowledge Paula Campbell and the Arizona Cancer Center/Arizona Research Laboratories Division of Biotechnology Cytometry Core Facility for expertise and assistance in flow cytometry and Patricia Jansma of the Molecular and Cellular Biology Imaging Facility and William Day of the Arizona Research Laboratories for their expertise in and assistance in fluorescent imaging and transmission electron microscopy, respectively. Special thanks to Terry Fox Laboratory for providing the M2-10B4 and s.l./s.l. cells. We acknowledge Tom Shenk for the gift of antibodies.
This work was supported by Public Health Service grant A1079059 (to F.G.) and AI21640 (to J.A.N.) from the National Institute of Allergy and Infectious Disease. F.G. was a 2008 Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts.
The content of this paper is solely our responsibility and does not necessarily represent the official views of the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, or the Pew Charitable Trusts.
Footnotes
Citation Caviness K, Bughio F, Crawford LB, Streblow DN, Nelson JA, Caposio P, Goodrum F. 2016. Complex interplay of the UL136 isoforms balances cytomegalovirus replication and latency. mBio 7(2):e01986-15. doi:10.1128/mBio.01986-15.
REFERENCES
- 1.Goodrum F, Caviness K, Zagallo P. 2012. Human cytomegalovirus persistence. Cell Microbiol 14:644–655. doi: 10.1111/j.1462-5822.2012.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. doi: 10.1007/978-3-540-77349-8_23. [DOI] [PubMed] [Google Scholar]
- 3.Mocarski ES, Shenk TE, Griffiths PD, Pass RF. 2013. Cytomegaloviruses. In Fields’ virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang GC, Kao WH, Murakami P, Xue Q-L, Chiou RB, Detrick B, McDyer JF, Semba RD, Casolaro V, Walston JD, Fried LP. 2010. Cytomegalovirus infection and the risk of mortality and frailty in older women: a prospective observational cohort study. Am J Epidemiol 171:1144–1152. doi: 10.1093/aje/kwq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Streblow DN, Orloff SL, Nelson JA. 2001. Do pathogens accelerate atherosclerosis? J Nutr 131:2798S–2804S. [DOI] [PubMed] [Google Scholar]
- 7.Sansoni P, Vescovini R, Fagnoni FF, Akbar A, Arens R, Chiu Y-L, Cičin-Šain L, Dechanet-Merville J, Derhovanessian E, Ferrando-Martinez S, Franceschi C, Frasca D, Fulöp T, Furman D, Gkrania-Klotsas E, Goodrum F, Grubeck-Loebenstein B, Hurme M, Kern F, Lilleri D, López-Botet M, Maier AB, Marandu T, Marchant A, Matheï C, Moss P, Muntasell A, Remmerswaal EBM, Riddell NE, Rothe K, Sauce D, Shin E-C, Simanek AM, Smithey MJ, Söderberg-Nauclér C, Solana R, Thomas PG, van Lier R, Pawelec G, Nikolich-Zugich J. 2014. New advances in CMV and immunosenescence. Exp Gerontol 55:54–62. doi: 10.1016/j.exger.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 8.Furman D, Jojic V, Sharma S, Shen-Orr SS, Angel CJ, Onengut-Gumuscu S, Kidd BA, Maecker HT, Concannon P, Dekker CL, Thomas PG, Davis MM. 2015. Cytomegalovirus infection enhances the immune response to influenza. Sci Transl Med 7:281ra43. doi: 10.1126/scitranslmed.aaa2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol 325:63–83. [DOI] [PubMed] [Google Scholar]
- 10.Jarvis MA, Nelson JA. 2002. Human cytomegalovirus persistence and latency in endothelial cells and macrophages. Curr Opin Microbiol 5:403–407. doi: 10.1016/S1369-5274(02)00334-X. [DOI] [PubMed] [Google Scholar]
- 11.Adler B, Sinzger C. 2009. Endothelial cells in human cytomegalovirus infection: one host cell out of many or a crucial target for virus spread? Thromb Haemost 102:1057–1063. doi: 10.1160/TH09-04-0213. [DOI] [PubMed] [Google Scholar]
- 12.Caposio P, Orloff SL, Streblow DN. 2011. The role of cytomegalovirus in angiogenesis. Virus Res 157:204–211. doi: 10.1016/j.virusres.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Söderberg-Nauclér C, Fish KN, Nelson JA. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126. doi: 10.1016/S0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- 14.Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687–695. doi: 10.1182/blood-2003-12-4344. [DOI] [PubMed] [Google Scholar]
- 15.Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7:e1002444. doi: 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bughio F, Elliott DA, Goodrum F. 2013. An endothelial cell-specific requirement for the UL133-UL138 locus of human cytomegalovirus for efficient virus maturation. J Virol 87:3062–3075. doi: 10.1128/JVI.02510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol 83:5615–5629. doi: 10.1128/JVI.01989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD. 2014. Antagonistic determinants controlling replicative and latent States of human cytomegalovirus Infection. J Virol 88:5987–6002. doi: 10.1128/JVI.03506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caviness K, Cicchini L, Rak M, Umashankar M, Goodrum F. 2014. Complex expression of the UL136 gene of human cytomegalovirus results in multiple protein isoforms with unique roles in replication. J Virol 88:14412–14425. doi: 10.1128/JVI.02711-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bughio F, Umashankar M, Wilson J, Goodrum F. 2015. Human cytomegalovirus UL135 and UL136 genes are required for postentry tropism in endothelial cells. J Virol 89:6536–6550. doi: 10.1128/JVI.00284-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanton RJ, Prod’homme V, Purbhoo MA, Moore M, Aicheler RJ, Heinzmann M, Bailer SM, Haas J, Antrobus R, Weekes MP, Lehner PJ, Vojtesek B, Miners KL, Man S, Wilkie GS, Davison AJ, Wang EC, Tomasec P, Wilkinson GW. 2014. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 16:201–214. doi: 10.1016/j.chom.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, Lehner PJ, Gygi SP. 2014. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le VT, Trilling M, Hengel H. 2011. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb’-encoded modulation of TNF-α signaling. J Virol 85:13260–13270. doi: 10.1128/JVI.06005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montag C, Wagner JA, Gruska I, Vetter B, Wiebusch L, Hagemeier C. 2011. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor alpha (TNF-alpha) signaling by upregulating TNF-alpha receptor 1 cell surface expression. J Virol 85:11409–11421. doi: 10.1128/JVI.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol 85:5864–5879. doi: 10.1128/JVI.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y-J, Jan Y-J, Ko B-S, Liang S-M, Liou J-Y. 2015. Involvement of 14-3-3 proteins in regulating tumor progression of hepatocellular carcinoma. Cancers (Basel) 7:1022–1036. doi: 10.3390/cancers7020822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Marsh JL, Holcombe RF, Waterman ML. 2001. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet 28:53–57. doi: 10.1038/88264. [DOI] [PubMed] [Google Scholar]
- 30.Mathers C, Spencer CM, Munger J. 2014. Distinct domains within the human cytomegalovirus U(L)26 protein are important for wildtype viral replication and virion stability. PLoS One 9:e88101. doi: 10.1371/journal.pone.0088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rechsteiner MP, Bernasconi M, Berger C, Nadal D. 2008. Role of latent membrane protein 2 isoforms in Epstein-Barr virus latency. Trends Microbiol 16:520–527. doi: 10.1016/j.tim.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Umashankar M, Goodrum F. 2014. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol Biol 1119:99–112. doi: 10.1007/978-1-62703-788-4_7. [DOI] [PubMed] [Google Scholar]
- 33.Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, Othieno FA, Streblow DN, Garcia JV, Fleming WH, Nelson JA. 2010. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 8:284–291. doi: 10.1016/j.chom.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang S-X, Ma M, Shen B, Qian S-B, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webel R, Hakki M, Prichard MN, Rawlinson WD, Marschall M, Chou S. 2014. Differential properties of cytomegalovirus pUL97 kinase isoforms affect viral replication and maribavir susceptibility. J Virol 88:4776–4785. doi: 10.1128/JVI.00192-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toptan T, Fonseca L, Kwun HJ, Chang Y, Moore PS. 2013. Complex alternative cytoplasmic protein isoforms of the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 generated through noncanonical translation initiation. J Virol 87:2744–2755. doi: 10.1128/JVI.03061-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leelawong M, Lee JI, Smith GA. 2012. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J Virol 86:6303–6314. doi: 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das S, Ortiz DA, Gurczynski SJ, Khan F, Pellett PE. 2014. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J Virol 88:9086–9099. doi: 10.1128/JVI.01141-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]