

Abstract
Adult respiratory distress syndrome (ARDS) is an acute lung injury of high mortality rate, and the molecular mechanisms underlying it are poorly understood. Acid aspiration–induced lung injury is one of the most common causes of ARDS, characterized by an increase in lung permeability, enhanced polymorphonuclear neutrophil (PMN) sequestration, and respiratory failure. Here, we investigated the role of platelet-activating factor (PAF) and the PAF receptor (PAFR) gene in a murine model of acid aspiration–induced lung injury. Overexpression of the PAFR gene in transgenic mice enhanced lung injury, pulmonary edema, and deterioration of gas exchange caused by HCl aspiration. Conversely, mice carrying a targeted disruption of the PAFR gene experienced significantly less acid-induced injury, edema, and respiratory failure. Nevertheless, the efficiency of PMN sequestration in response to acid aspiration was unaffected by differences in PAFR expression level. The current observations suggest that PAF is involved in the pathogenesis of acute lung injury caused by acid aspiration. Thus, inhibition of this pathway might provide a novel therapeutic approach to acute lung injury, for which no specific pharmaceutical agents are currently available.
Introduction
Adult respiratory distress syndrome (ARDS) is an acute lung injury with a mortality rate of 40–70% despite intensive care using currently available drugs. Acid aspiration–induced injury is one of the most common causes of ARDS. Potential mechanisms causing acid aspiration–associated lung injury include HCl-induced damage to the alveolar-capillary membrane, and polymorphonuclear neutrophil (PMN) adhesion, activation, and sequestration, leading to pulmonary edema and deterioration of gas exchange (1–3).
Platelet-activating factor (PAF) is a proinflammatory phospholipid mediator that has various biologic effects, including cell adhesion, endothelial cell activation, and the production of cytokines and eicosanoids via activation of G protein–coupled PAF receptor (PAFR) (4–13). Considering its potent biologic activity, PAF is potentially involved in the development of inflammatory disorders, including acute lung injury. To examine the pathophysiological role of PAF in the pathogenesis of diseases, previous studies have depended on various types of PAFR antagonists, although there exist significant problems regarding the specificity of these agents. For example, some PAFR antagonists have inhibitory effects on histamine via interaction with its G protein–coupled receptor (14, 15). To elucidate the pathophysiological role of PAF in vivo without using potentially nonspecific PAFR antagonists, we have recently developed genetically engineered mice (16, 17).
The purpose of this report was to investigate the exact role of PAF in a murine model of acid aspiration–induced lung injury. To address this question, we used 2 different mutant mice established in our laboratory: transgenic mice overexpressing the PAFR gene (PAFR-Tg) (16) and controls (PAFR-Con), and PAFR gene–disrupted mice (PAFR-KO) (17) and controls (PAFR-WT). We observed that overexpression of the PAFR gene exaggerated the acute lung injury induced by acid aspiration, whereas disruption of the PAFR gene significantly attenuated the injury, indicating that the PAFR, and presumably PAF, may mediate important features of acid-induced lung injury.
Methods
Mice.
PAFR-Tg mice were established as described previously (16). Male PAFR-Tg mice were mated to BDF1 females. The genetic background was C57BL/6 × DBA/2 hybrid. Northern blot analysis demonstrated the expression of the PAFR transgene in trachea, lung, heart, skeletal muscle, eye, skin, and aorta (16).
PAFR-KO mice were established by gene targeting (17). Mice heterozygous for the PAFR mutant allele with the genetic background of the C57BL/6J × 129/Ola hybrid were mated. Offspring were genotyped at 4 weeks of age. For genotyping, genomic DNAs were isolated from biopsied tail and subjected to 35 cycles of PCR amplification (10 seconds at 98°C; 2 seconds at 65°C; 30 seconds at 74°C). The primers were as follows: forward 5′-TATGGCTGACCTGCTCTTCCTGAT-3′ and reverse 5′-TATTGGGCACTAGGTTGGTGGAGT-3′ for detecting the intact PAFR allele; and forward 5′-GCCTGCTTGCCGAATATCATGGTGGAAAAT-3′ and reverse 5′-GCGATGCGCTGCGAATCGGGAGCGGCGATA-3′ for detecting the disrupted PAFR allele. The former set of primers amplified a 287-bp DNA fragment, and the latter PCR product consisted of 194 bp.
The animals were maintained on a light-dark cycle with light from 0900 to 2100 hours at 25°C. Mice were fed with a standard laboratory diet and water ad libitum. Mutant mice and their littermate controls (13–24 weeks old) were used in the current study.
Protocols.
One minute before aspiration challenge, 2 deep inhalations (3 times tidal volume) were delivered to standardize volume history, and measurements were made as baseline. Then, anesthetized and mechanically ventilated mice received 2 mL/kg HCl (pH = 1.5) intratracheally, followed by a bolus of air (30 mL/kg). In the saline-treated group, animals received saline instead of HCl in the same manner and served as controls. In all groups, measurements were made at 30-minute intervals for 2 hours. In some animals, the observation period was extended up to 5 hours. To assess the development of lung injury physiologically, lung elastance (EL; a reciprocal of lung compliance) was measured as described previously (18–20). Briefly, we measured the tracheal pressure (Ptr), flow, and volume (V). EL and lung resistance (RL) were calculated by adjusting the equation of motion: Ptr = EL•V + RL(dV/dt) + K, where K was a constant. Changes in EL reflect lung parenchymal alterations and stiffening of the lungs (21).
Assessment of respiratory failure.
At the end of the experiment, blood samples for gas analysis were obtained from the left ventricle. The measurements of PaO2, PaCO2, and pH were then made to assess the extent of respiratory failure (blood gas analyzer, Compact 3; AVL Medical Systems, Schaffhousen, Switzerland).
Assessment of pulmonary edema.
At the end of the experiment, the lung wet/dry weight ratios (W/D) were calculated to assess pulmonary edema. After the trapped blood was drained from the excised lungs, the lung wet weight was measured. Then, we heated the lungs at 90°C to constant weight in a gravity convection oven for 72 hours and weighed the residuum as the lung dry weight.
Bronchoalveolar lavage fluid.
At the end of the experiment, bronchoalveolar lavage (BAL) was performed (1 mL of PBS × 5) in each group. After BAL fluid (BALF) was centrifuged at 450 g for 10 minutes, the total and differential cell counts of the BALF were determined from the cell fraction. The supernatant was stored at –70°C until the protein was determined. The concentration of protein was measured by Lowry’s method using BSA as a standard.
Histology.
At the end of the experiment, the lungs were removed intact and frozen with liquid nitrogen (22). A constant transpulmonary pressure of 3 cm H2O was maintained during freezing by delivering constant flow into the trachea. Frozen lungs were fixed in Carnoy’s solution (60% ethyl alcohol, 30% chloroform, and 10% acetic acid) at –70°C for 18 hours. Progressively increasing concentrations of ethanol at –20°C were then substituted for the Carnoy’s solution until 100% ethanol was reached. The tissue was maintained at –20°C for 4 hours, warmed to 4°C for 12 hours, and then allowed to reach and remain at room temperature for 2 hours. After fixation, the tissue blocks obtained from midsagittal slices of the lungs were embedded in paraffin. Blocks were cut 4-μm thick using a microtome. Slides were stained with hematoxylin-eosin.
Data analysis.
Comparisons of data among each experimental group were carried out with ANOVA (Scheffe’s test). Data are expressed as mean ± SEM. P values less than 0.05 were taken as significant.
Results
Figure 1 demonstrates the time course of changes in lung elasticity in HCl- and saline-treated groups. After HCl administration, the survival periods of PAFR-Tg and control mice were 2 hours and 3 hours, respectively, whereas PAFR-KO mice were all alive throughout the experiment and their survival was significantly prolonged (P < 0.02 vs. the other groups; χ2 analysis). In terms of changes in EL and RL, HCl-treated PAFR-Tg mice demonstrated a significantly greater response than the other groups, whereas the HCl-induced responses in PAFR-KO mice were significantly reduced compared with their WT controls (Figure 2 and Table 1).
Figure 1.

The time course of response in lung elastance (EL) in HCl- and saline-treated groups (n = 3 for each group). After HCl aspiration, all animals died at 2–2.5 hours in PAFR-Tg and at 3–3.5 hours in controls, whereas all animals survived in the PAFR-KO group.
Figure 2.

Roles of the PAFR gene in acid-induced lung injury. Changes in EL 2 hours after aspiration (n = 8–10). Responses after the administration of HCl (filled bars) or saline (open bars) are shown. +P < 0.05, *P < 0.001 vs. saline-treated groups. #P < 0.001 vs. HCl-treated controls.
Table 1.
RL, PaCO2, and pH levels 2 hours after aspiration in the experimental groups

HCl aspiration induced deterioration of gas exchange, which was not observed in the saline-treated groups. HCl-induced hypoxemia was most prominent in PAFR-Tg mice, whereas it was significantly attenuated in PAFR-KO mice (Figure 3). After HCl treatment, increases in PaCO2 and decreases in pH were observed in PAFR-Tg mice, although there were no differences in PaCO2 or pH levels between PAFR-KO mice and saline-treated groups (Table 1).
Figure 3.

Roles of the PAFR gene in acid-induced hypoxemia. PaO2 level 2 hours after aspiration (n = 8–10). Responses after the administration of HCl (filled bars) or saline (open bars) are shown. *P < 0.001 vs. saline-treated groups. #P < 0.001 vs. HCl-treated controls.
To assess the severity of lung edema, lung W/D was measured in each group (Figure 4). In control groups, HCl challenge significantly increased W/D compared with all saline-treated groups. The W/D in HCl-treated PAFR-Tg was significantly greater than that in any other group, whereas the W/D in HCl-treated PAFR-KO was markedly decreased and fell to virtually the same level as for the saline-treated groups.
Figure 4.

Roles of the PAFR gene in acid-induced lung edema. The lung W/D 2 hours after aspiration (n = 4–5). Responses after the administration of HCl (filled bars) or saline (open bars) are shown. *P < 0.05 vs. saline-treated groups. #P < 0.01 vs. HCl-treated controls.
HCl administration increased the amount of protein and the number of PMNs in BALF, indicating HCl-induced protein leakage and PMN infiltration. The amount of BALF protein was significantly greater in HCl-treated PAFR-Tg than that in its control, and HCl-induced protein leakage was significantly attenuated in PAFR-KO mice (Figure 5). Total cell counts and differential cell fractions are shown in Table 2. Of note, HCl aspiration elicited no significant differences in PMN infiltration between PAFR-Tg and its control or between PAFR-KO and PAFR-WT (Figure 6).
Figure 5.

Roles of the PAFR gene in acid-induced protein leakage. Total protein amounts of BALF 2 hours after aspiration (n = 4–5). Responses after the administration of HCl (filled bars) or saline (open bars) are shown. +P < 0.01, *P < 0.001 vs. saline-treated groups. #P < 0.001 vs. HCl-treated controls.
Table 2.
Total cell counts and cell fractions in BALF 2 hours after aspiration in the experimental groups

Figure 6.

Roles of the PAFR gene in acid-induced neutrophil infiltration. PMN counts of BALF 2 hours after aspiration (n = 4–5). Responses after the administration of HCl (filled bars) or saline (open bars) are shown. *P < 0.001 vs. saline-treated groups.
Figure 7 illustrates the lung histology after HCl administration. The HCl-induced lesions included alveolar thickening, distortion, protein leakage, and cellular infiltration and were most prominent in the PAFR-Tg mice. In contrast, the alveolar architecture was well preserved, and the histological changes were minimal in the PAFR-KO mice.
Figure 7.
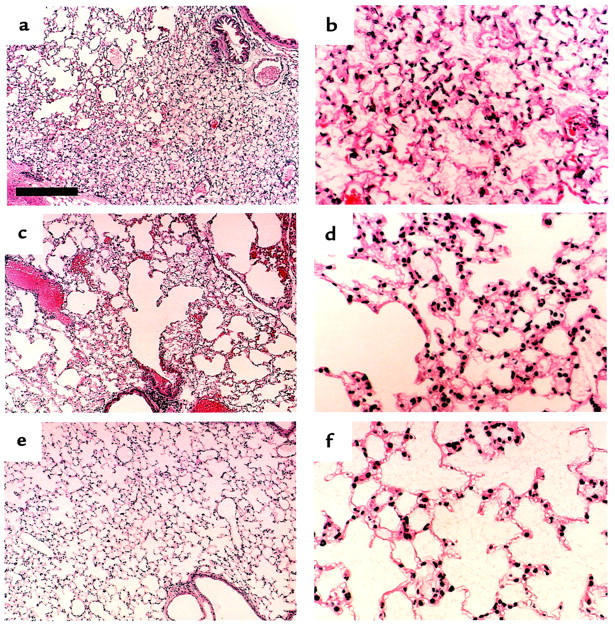
Photomicrograph of lung tissues from PAFR-Tg (a and b), PAFR-WT (c and d), and PAFR-KO (e and f) mice 2 hours after HCl administration. Hematoxylin-eosin stain. Scale bar in a represents 200 μm in a, c, and e and 50 μm in b, d, and f.
Discussion
The results of the current study suggest that the PAFR and its physiological ligand, PAF, are crucially involved in the pathogenesis of acid aspiration–induced lung injury. Overexpression of the PAFR gene enhanced pulmonary edema and deterioration of gas exchange caused by HCl aspiration. Disruption of the PAFR gene significantly attenuated the acid-induced edema and respiratory failure. However, PMN sequestration was not regulated by the PAFR gene. These observations indicate that PAF and the PAFR gene may mediate important features of acid-induced lung injury.
Aspiration of gastric contents is reported to be associated with a 26–36% incidence of ARDS (23, 24). Acute lung injury induced by massive acid aspiration is characterized by increased permeability in the lung, decreased lung compliance, enhanced PMN sequestration, and respiratory failure. Aspirated HCl may evoke direct damage to the alveolar-capillary membrane and promote PMN adhesion, activation, and sequestration. It has been reported that thromboxane (TX) synthesis and generation of oxygen radicals associated with PMN activation are potential mechanisms to induce pulmonary edema and respiratory failure (1, 2). It has been demonstrated that inhibition of either TX or H2O2 attenuates acid aspiration–induced increase in protein in BALF in rats (2, 25). After acid aspiration, there is generation of eicosanoids, including TXA2, from activated tissues (26, 27). It is speculated that activation and adhesion of PMNs might be the initial action, leading to generation of radical oxygen species and increases in permeability. The current findings suggest that PAF has a main role in PMN activation, but not in PMN sequestration. Potentially, leukotriene B4, a potent neutrophil chemoattractant, has a dominant role in PMN sequestration (28). The current observation that neither overexpression nor disruption of the PAFR gene affected PMN sequestration may be explained by the possibility that acid-induced release of chemotactic mediators, including leukotriene B4, would not be affected by relative expression of the PAFR gene.
Thromboxanes and leukotrienes are potent mediators that are generated from arachidonic acid by cyclooxygenase and 5-lipoxygenase, respectively. A key enzyme for the production of these inflammatory mediators, including eicosanoids and PAF, is phospholipase A2. Especially, cytosolic PLA2 (cPLA2) is of importance, as it is activated by submicromolar concentration of Ca2+ and by phosphorylation via mitogen-activated protein kinases (29–33). There exists a possibility that cPLA2 per se is related to the formation of PAF (34), as the cPLA2 hydrolysis of alkyl-phospholipids produces lyso-PAF, which is further converted to PAF by the action of acetyltransferase. Potential interaction between PAF and eicosanoids may further upregulate the development of acute lung injury (20, 35).
After HCl treatment, the survival rate (100%) and levels of W/D, PaCO2, and pH in PAFR-KO mice were the same as for saline-treated controls. However, we observed that acid aspiration–induced alterations in EL, PaO2, BALF protein, and PMNs were not completely eliminated in PAFR-KO mice, although they were markedly attenuated. These observations suggest that factors other than PAF also play a role and contribute to physiological alteration, whereas PAF is closely related to the development of acute lung injury. It has been postulated that adhesion molecules and cytokines are also involved in this mechanism (22, 36). Considering that there are yet no drugs to manage pulmonary edema and improve survival rates, these mediators are possible targets to develop agents. The present study indicates that the specific intervention of the PAFR could make an important contribution to management of acid-induced lung injury, whereas the previous studies have also shown that antagonism of the PAFR might be effective in acute lung injury (37, 38). Moreover, it has been recently demonstrated that PAF acetylhydrolase, which abolishes the PAF activity, would be a potential therapeutic of severe acute inflammation (39). These observations suggest that the intervention of the biologic activity of PAF is a useful therapy for acute lung injury.
It has been reported that oxidized phospholipids, lyso-phosphatidylcholine, or bacterial endotoxin can activate the PAFR (40–43). Acid aspiration could potentially induce the formation of these compounds, which might be involved in the injured lung, edema, and respiratory failure. Therefore, there might be a possibility that PAF as well as the PAF-like ligands could be related to the responses elicited by acid aspiration, although this study clearly shows a crucial role of the PAFR in acid-induced injury.
In summary, disruption of the PAFR gene significantly attenuated acid aspiration–induced lung damage and respiratory failure, whereas overexpression of the PAFR gene exaggerated these phenomena. The current observations suggest that PAF might be involved in the pathogenesis of acute lung injury caused by acid aspiration. Taken together, specific antagonism of the PAFR might provide a novel and potential therapeutic approach to acute lung injury, for which no drugs are currently available.
Acknowledgments
The writers thank Y. Hara, Y. Matsumoto, H. Sakamoto, Y. Suzuki, C. Suzuki, C. Jin, M. Hata, and M. Ito (The University of Tokyo) for their technical assistance. We are also grateful to F. Takaku (Jichi Medical School) and N. Ito (The University of Tokyo) for valuable suggestions. This work was supported in part by grants-in-aid from the Ministry of Education, Science, Sports and Culture of Japan and by grants from the Sankyo Foundation of Life Science and the Organization for Pharmaceutical Safety and Research.
References
- 1.Eijking EP, Gommers D, So KL, Vergeer M, Lachmann B. Surfactant treatment of respiratory failure induced by hydrochloric acid aspiration in rats. Anesthesiology. 1993;78:1145–1151. doi: 10.1097/00000542-199306000-00019. [DOI] [PubMed] [Google Scholar]
- 2.Goldman G, et al. Reactive oxygen species and elastase mediate lung permeability after acid aspiration. J Appl Physiol. 1992;73:571–575. doi: 10.1152/jappl.1992.73.2.571. [DOI] [PubMed] [Google Scholar]
- 3.Pittet JF, Mackersie RC, Martin TR, Matthay MA. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med. 1997;155:1187–1205. doi: 10.1164/ajrccm.155.4.9105054. [DOI] [PubMed] [Google Scholar]
- 4.Prescott SM, Zimmerman GA, McIntyre TM. Platelet-activating factor. J Biol Chem. 1990;265:17381–17384. [PubMed] [Google Scholar]
- 5.Chao W, Olson MS. Platelet-activating factor: receptors and signal transduction. Biochem J. 1993;292:617–629. doi: 10.1042/bj2920617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izumi T, Shimizu T. Platelet-activating factor receptor: gene expression and signal transduction. Biochim Biophys Acta. 1995;1259:317–333. doi: 10.1016/0005-2760(95)00171-9. [DOI] [PubMed] [Google Scholar]
- 7.Honda Z-I, et al. Cloning by functional expression of platelet-activating factor receptor from guinea-pig lung. Nature. 1991;349:342–346. doi: 10.1038/349342a0. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura M, et al. Molecular cloning and expression of platelet-activating factor receptor from human leukocytes. J Biol Chem. 1991;266:20400–20405. [PubMed] [Google Scholar]
- 9.Ye RD, Prossnitz ER, Zou AH, Cochrane CG. Characterization of a human cDNA that encodes a functional receptor for platelet activating factor. Biochem Biophys Res Commun. 1991;180:105–111. doi: 10.1016/s0006-291x(05)81261-6. [DOI] [PubMed] [Google Scholar]
- 10.Kunz D, Gerard NP, Gerard C. The human leukocyte platelet-activating factor receptor. cDNA cloning, cell surface expression, and construction of a novel epitope-bearing analog. J Biol Chem. 1992;267:9101–9106. [PubMed] [Google Scholar]
- 11.Sugimoto T, et al. Molecular cloning and characterization of the platelet-activating factor receptor gene expressed in the human heart. Biochem Biophys Res Commun. 1992;189:617–624. doi: 10.1016/0006-291x(92)92245-s. [DOI] [PubMed] [Google Scholar]
- 12.Bito H, Honda Z-I, Nakamura M, Shimizu T. Cloning, expression and tissue distribution of rat platelet-activating-factor-receptor cDNA. Eur J Biochem. 1994;221:211–218. doi: 10.1111/j.1432-1033.1994.tb18731.x. [DOI] [PubMed] [Google Scholar]
- 13.Ishii S, et al. A murine platelet-activating factor receptor gene: cloning, chromosomal localization and up-regulation of expression by lipopolysaccharide in peritoneal resident macrophages. Biochem J. 1996;314:671–678. doi: 10.1042/bj3140671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merlos M, et al. Rupatadine, a new potent, orally active dual antagonist of histamine and platelet-activating factor (PAF) J Pharmacol Exp Ther. 1997;280:114–121. [PubMed] [Google Scholar]
- 15.Carceller E, et al. [(3-Pyridylalkyl) piperidylidene]benzocycloheptapyridine derivatives as dual antagonists of PAF and histamine. J Med Chem. 1994;37:2697–2703. doi: 10.1021/jm00043a009. [DOI] [PubMed] [Google Scholar]
- 16.Ishii S, et al. Bronchial hyperreactivity, increased endotoxin lethality and melanocytic tumorigenesis in transgenic mice overexpressing platelet-activating factor receptor. EMBO J. 1997;16:133–142. doi: 10.1093/emboj/16.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii S, et al. Impaired anaphylactic responses but intact sensitivity to endotoxin in mice lacking a platelet-activating factor receptor. J Exp Med. 1998;187:1779–1788. doi: 10.1084/jem.187.11.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagase T, Aoki T, Oka T, Fukuchi Y, Ouchi Y. ET-1–induced bronchoconstriction is mediated via ETB receptor in mice. J Appl Physiol. 1997;83:46–51. doi: 10.1152/jappl.1997.83.1.46. [DOI] [PubMed] [Google Scholar]
- 19.Nagase T, et al. Airway hyperresponsiveness to methacholine in mutant mice deficient in endothelin-1. Am J Respir Crit Care Med. 1998;157:560–564. doi: 10.1164/ajrccm.157.2.9706009. [DOI] [PubMed] [Google Scholar]
- 20.Nagase T, et al. Airway responsiveness in transgenic mice overexpressing platelet-activating factor receptor: roles of thromboxanes and leukotrienes. Am J Respir Crit Care Med. 1997;156:1621–1627. doi: 10.1164/ajrccm.156.5.9703016. [DOI] [PubMed] [Google Scholar]
- 21.Nagase T, Matsui H, Aoki T, Ouchi Y, Fukuchi Y. Lung tissue behavior in the mouse during constriction induced by methacholine and endothelin-1. J Appl Physiol. 1996;81:2373–2378. doi: 10.1152/jappl.1996.81.6.2373. [DOI] [PubMed] [Google Scholar]
- 22.Nagase T, et al. Intercellular adhesion molecule-1 mediates acid aspiration-induced lung injury. Am J Respir Crit Care Med. 1996;154:504–510. doi: 10.1164/ajrccm.154.2.8756829. [DOI] [PubMed] [Google Scholar]
- 23.Fowler AA, et al. Adult respiratory distress syndrome: risk with common predispositions. Ann Intern Med. 1983;98:593–597. doi: 10.7326/0003-4819-98-5-593. [DOI] [PubMed] [Google Scholar]
- 24.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 25.Goldman G, et al. Synergism between leukotriene B4 and thromboxane A2 in mediating acid-aspiration injury. Surgery. 1992;111:55–61. [PubMed] [Google Scholar]
- 26.Utsunomiya T, et al. Modification of inflammatory response to aspiration with ibuprofen. Am J Physiol. 1982;243:H903–H910. doi: 10.1152/ajpheart.1982.243.6.H903. [DOI] [PubMed] [Google Scholar]
- 27.Nagase T, et al. Intravenous bolus of prednisolone decreases 15-hydroxyeicosatetraenoic acid formation in the rat model of acid aspiration. Crit Care Med. 1991;19:950–954. doi: 10.1097/00003246-199107000-00021. [DOI] [PubMed] [Google Scholar]
- 28.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387:620–624. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 29.Leslie CC. Properties and regulation of cytosolic phospholipase A2. J Biol Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 30.Clark JD, et al. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 31.Sharp JD, et al. Molecular cloning and expression of human Ca2+-sensitive cytosolic phospholipase A2. J Biol Chem. 1991;266:14850–14853. [PubMed] [Google Scholar]
- 32.Lin LL, et al. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 33.Kramer RM, et al. p38 mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. J Biol Chem. 1996;271:27723–27729. doi: 10.1074/jbc.271.44.27723. [DOI] [PubMed] [Google Scholar]
- 34.Uozumi N, et al. Roles of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 35.Stewart AG, Dubbin PN, Harris T, Dusting GJ. Platelet-activating factor may act as a second messenger in the release of icosanoids and superoxide anions from leukocytes and endothelial cells. Proc Natl Acad Sci USA. 1990;87:3215–3219. doi: 10.1073/pnas.87.8.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid aspiration–induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest. 1995;96:107–116. doi: 10.1172/JCI118009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang S-W, Feddersen CO, Henson PM, Voelkel NF. Platelet-activating factor mediates hemodynamic changes and lung injury in endotoxin-treated rats. J Clin Invest. 1987;79:1498–1509. doi: 10.1172/JCI112980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miotla JM, Jeffery PK, Hellewell PG. Platelet-activating factor plays a pivotal role in the induction of experimental lung injury. Am J Respir Cell Mol Biol. 1998;18:197–204. doi: 10.1165/ajrcmb.18.2.2846. [DOI] [PubMed] [Google Scholar]
- 39.Tjoelker LW, et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature. 1995;374:549–553. doi: 10.1038/374549a0. [DOI] [PubMed] [Google Scholar]
- 40.Smiley PL, Stremler KE, Prescott SM, Zimmerman GA, McIntyre TM. Oxidatively fragmented phosphatidylcholines activate human neutrophils through the receptor for platelet-activating factor. J Biol Chem. 1991;266:11104–11110. [PubMed] [Google Scholar]
- 41.Heery JM, et al. Oxidatively modified LDL contains phospholipids with platelet-activating factor-like activity and stimulates the growth of smooth muscle cells. J Clin Invest. 1995;96:2322–2330. doi: 10.1172/JCI118288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogita T, et al. Lysophosphatidylcholine transduces Ca2+ signaling via the platelet-activating factor receptor in macrophages. Am J Physiol. 1997;272:H17–H24. doi: 10.1152/ajpheart.1997.272.1.H17. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura M, et al. Endotoxin transduces Ca2+ signaling via platelet-activating factor receptor. FEBS Lett. 1992;314:125–129. doi: 10.1016/0014-5793(92)80957-i. [DOI] [PubMed] [Google Scholar]