

ABSTRACT
Geminiviruses are important plant pathogens characterized by circular, single-stranded DNA (ssDNA) genomes. However, in the nuclei of infected cells, viral double-stranded DNA (dsDNA) associates with host histones to form a minichromosome. In phloem-limited geminiviruses, the characterization of viral minichromosomes is hindered by the low concentration of recovered complexes due to the small number of infected cells. Nevertheless, geminiviruses are both inducers and targets of the host posttranscriptional gene silencing (PTGS) and transcriptional gene silencing (TGS) machinery. We have previously characterized a “recovery” phenomenon observed in pepper plants infected with pepper golden mosaic virus (PepGMV) that is associated with a reduction of viral DNA and RNA levels, the presence of virus-related siRNAs, and an increase in the levels of viral DNA methylation. Initial micrococcal nuclease-based assays pinpointed the presence of different viral chromatin complexes in symptomatic and recovered tissues. Using the pepper-PepGMV system, we developed a methodology to obtain a viral minichromosome-enriched fraction that does not disturb the basic chromatin structural integrity, as evaluated by the detection of core histones. Using this procedure, we have further characterized two populations of viral minichromosomes in PepGMV-infected plants. After further purification using sucrose gradient sedimentation, we also observed that minichromosomes isolated from symptomatic tissue showed a relaxed conformation (based on their sedimentation rate), are associated with a chromatin activation marker (H3K4me3), and present a low level of DNA methylation. The minichromosome population obtained from recovered tissue, on the other hand, sedimented as a compact structure, is associated with a chromatin-repressive marker (H3K9me2), and presents a high level of DNA methylation.
IMPORTANCE Viral minichromosomes have been reported in several animal and plant models. However, in the case of geminiviruses, there has been some recent discussion about the importance of this structure and the significance of the epigenetic modifications that it can undergo during the infective cycle. Major problems in this type of studies are the low concentration of these complexes in an infected plant and the asynchronicity of infected cells along the process; therefore, the complexes isolated in a given moment usually represent a mixture of cells at different infection stages. The recovery process observed in PepGMV-infected plants and the isolation procedure described here provide two distinct populations of minichromosomes that will allow a more precise characterization of the modifications of viral DNA and its host proteins associated along the infective cycle. This structure could be also an interesting model to study several processes involving plant chromatin.
INTRODUCTION
Geminiviruses are important plant pathogens consisting of circular, single-stranded DNA (ssDNA) genomes ranging from 2.7 to 5.2 kb packed into 1 or 2 icosahedral twine-shaped particles with a size of 22 by 38 nm (1). Geminiviruses are classified into seven genera (Begomovirus, Mastrevirus, Curtovirus, Becurtovirus, Eragrovirus, Topocuvirus and Turncurtovirus) on the basis of their genome organization, host range, and insect vector (1–4). After transmission by the insect vector, the ssDNA genome is released from the capsid and converted to double-stranded DNA (dsDNA) through complementary strand replication (CSR) assisted by host enzymes (5). Viral DNA is further processed into a covalently closed circular conformation (cccDNA), which is wrapped around nucleosomes to form a minichromosome that is the template for replication and transcription and to interact with other cellular proteins (6–8). In addition, it might associate with viral movement proteins and histone H3 to move from cell to cell (6–8).
RNA silencing is a ubiquitous eukaryotic gene regulation mechanism. In plants, it can act posttranscriptionally (posttranscriptional gene silencing [PTGS]) by cleavage or translation arrest of RNA targets in a sequence-specific manner or at the transcriptional level (transcriptional gene silencing [TGS]) by cytosine methylation of DNA and/or posttranslational modifications of histones (i.e., methylation, acetylation, phosphorylation, ubiquitination) located at the DNA targets (9–11). Geminiviruses are both inducers and targets of the host PTGS and TGS machinery, mediated by the formation of small interfering RNAs (siRNAs) homologous to the viral mRNA and DNA. This interplay of PTGS and TGS has been understood as part of the plant defense pathways, although the precise mechanisms are not yet well characterized (12–14). Correspondingly, geminiviruses have developed potent suppressors of gene silencing (15–17). TGS directed against the viral minichromosome may lead to epigenetic modifications in an attempt to stop or reduce virus replication (18). In a few documented examples, this leads to a process called host plant recovery (19, 20). The natural outcome of this plant-virus interaction (host recovery) is the emergence of new asymptomatic leaves after a typical symptomatic infection. Host recovery is generally accompanied with reduced virus titers and a sequence-specific resistance to a secondary infection, and it has been linked with the induction of an antiviral RNA-silencing process (21, 22).
The hypothesis that the epigenetic arms of the silencing mechanism are also an important defense pathway against geminiviruses is based on several lines of evidence. (i) The efficiency of virus replication in protoplasts is reduced when viral DNA is methylated in vitro before transfection (23, 24). (ii) Arabidopsis mutant lines for key genes of the transcriptional gene silencing pathway (i.e., cytosine methyltransferases, methyl cycle enzymes, Dicer-like and dsRNA binding proteins) are hypersensitive to virus infection and show altered methylation levels (25). (iii) Geminiviral DNA has been shown to be associated with nucleosomes containing both repressive (H3K9me) and active (H3Ac) histone modifications (26). (iv) Transgenes containing geminivirus promoters are silenced and their cytosines methylated when plants are superinfected with the cognate virus. An increased level of DNA methylation on episomal viral progeny has been also observed (18, 27). (v) A suppressor of gene silencing from a geminivirus changed the global methylation profile of infected plants (28). (vi) Host recovery was accompanied by the presence of virus-related siRNAs and elevated cytosine methylation of geminivirus DNA (25).
Geminivirus DNA exists in multiple forms in infected cells: circular ssDNA, which cannot be methylated, is the molecule found in viral particles; circular dsDNA replicative forms (RF), which are the templates for replication and transcription and may or may not be methylated; linear dsDNA forms of heterogeneous length, some of which may be products of recombination-dependent replication and also may or may not be methylated; and several heterogeneous linear ssDNA molecules detected in infected plants with a still-undefined role in the virus cycle (29). Production of ssDNA and dsDNA RF is asymmetric during the geminivirus infective cycle. Using a quantitative PCR (qPCR) approach, it has been reported that the dsDNA/ssDNA ratio is about 1:100 in Nicotiana benthamiana and tomato plants infected with tomato yellow leaf curl Sardinia virus (TYLCSV) (30). Therefore, studies on virus minichromosome structure and genome modifications should use methodologies that consider and distinguish the different molecular structures present in the infected cell. In addition, in phloem-limited viruses (such as pepper golden mosaic virus [PepGMV] and tomato yellow leaf curl virus [TYLCV]), the actual number of infected cells is quite low; thus, the recovery of minichromosome complexes in sufficient quantities to be analyzed is a complicated task (31).
Our group has reported that the recovery phenomenon observed in pepper plants infected with PepGMV is associated with a reduction of viral DNA and RNA levels, the presence of virus-related siRNAs, and an increase in the levels of viral DNA methylation (19, 20). Based on those results and additional evidence, it was suggested that viral minichromosomes in symptomatic and recovered tissues could present differences in structure organization. Therefore, we proceeded to analyze the minichromosome populations in both types of tissue. Our initial analysis, based on micrococcal nuclease sensitivity, supported that hypothesis. We then designed a simple methodology to obtain a viral minichromosome-enriched fraction. The recovered minichromosomes were associated with nucleosome core histones, indicating that the procedure does not disturb the basic chromatin structure. Interestingly, using sucrose gradient sedimentation, we also observed that minichromosomes isolated from recovered tissue migrated as a high-density, compact structure (associated with the histone H3K9me2-repressive marker) whereas the complex isolated from symptomatic tissue migrated as a low-density structure (associated with the histone H3K4me3 activation marker). Finally, a high level of DNA methylation was associated with the compact minichromosome structure obtained from recovered tissue. The importance of geminivirus minichromosome isolation and future applications are discussed below.
MATERIALS AND METHODS
Plant inoculation and collecting of tissue.
Pepper (Capsicum annuum L. var. Sonora Anaheim) seeds were germinated in a bioclimatic chamber at 26 to 28°C with a 16/8-h (light/dark) photoperiod. The fourth leaf on plants at the four-leaf stage (around 30 days postgermination) were inoculated with PepGMV (pepper golden mosaic virus) infectious dimeric clones or with pBluescript for mock inoculations by biolistic bombardment (20). Samples of symptomatic tissue were harvested 10 days postinoculation (dpi), whereas recovered tissue was harvested at 24 dpi. Samples were immediately frozen in liquid nitrogen and stored at −80°C for subsequent analysis.
Micrococcal nuclease sensitivity assay.
Fresh tissue (5 g) was ground into a fine powder with a mortar and pestle using liquid nitrogen. The powder was resuspended in 20 ml buffer consisting of 10 mM Tris-HCl [pH 8], 10 mM NaCl, 3 mM MgCl2, 0.15 mM spermine, 0.5 mM spermidine, and 0.5% Triton X-100. The homogenate was first filtered through three layers of cheesecloth and the resulting filtrate through three layers of Miracloth (catalog number 475855; Calbiochem). The filtrate was centrifuged for 10 min at 1,000 × g (Sorvall super T21, SL50T rotor). The pellet (nucleus-rich fraction) was washed with 5 ml micrococcal nuclease (Mnase) digestion buffer (10 mM Tris-HCl [pH 7.4], 15 mM NaCl, 60 mM KCl, 0.15 mM spermine, and 0.5 mM spermidine). The nuclei were resuspended in 1 ml Mnase digestion buffer containing 10 mM CaCl2. The nuclei (100 μl) were transferred to several microcentrifuge tubes containing different concentrations of Mnase (0.5, 1, 2.5, 5, and 10 units) (EN0181; Thermo Scientific) and 1 μl RNase (10 mg/ml). Samples were incubated at 37°C for 10 min. To stop the reaction, 80 μl of Mnase digestion buffer and 20 μl of Mnase stop buffer (100 mM EDTA and 10 mM EGTA) were added to each sample. To eliminate proteins, the samples were treated with 3 μl of proteinase K (25 mg/ml) and 10 μl of 20% SDS. Samples were then incubated at 37°C overnight. DNA was extracted from each sample by standard phenol-chloroform extraction and ethanol precipitation. DNA was separated by agarose gel (1.5%) electrophoresis (60 V/3 h). Nucleic acids were then transferred to a nylon membrane (10× SSC, where 1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). The blots were hybridized against a biotin-labeled (K0651; Thermo Scientific) DNA probe (full-length PepGMV A). Detection was performed using a biotin chromogenic detection kit (K0661; Thermo Scientific). Both labeling and detection were performed according to the manufacturer's protocol. The image was analyzed using ImageJ software (http://imagej.nih.gov/ij/).
Isolation of PepGMV minichromosome.
Fresh tissue (20 g) was ground into a fine powder with a mortar and pestle using liquid nitrogen. The powder was immediately transferred into an ice-cold 500-ml beaker containing 200 ml buffer A (10 mM Tris-HCl, pH 9, 80 mM KCl, 1 mM spermidine, 1 mM spermine, 0.5% Triton X-100, 500 mM sucrose, and 15 mM β-mercaptoethanol) supplemented with one Sigmafast protease inhibitor cocktail tablet (catalog number S8830). The homogenate was first filtered through two layers of cheesecloth and the resulting filtrate through two layers of Miracloth (475855; Calbiochem). The filtrate was centrifuged for 15 min at 2,000 × g (Sorvall super T21, SL250T rotor). The nucleus-rich pellet was resuspended in 1 ml buffer B (10 mM Tris-HCl [pH 8], 1% Triton X-100) and centrifuged again for 5 min at 2,000 × g (Sorvall super T21, SL50T rotor). The pellet was resuspended in 1 ml extraction buffer (10 mM Tris-HCl [pH 8], 0.05% Sarkosyl, 1% Triton X-100) and incubated on ice for 15 min. The suspension was centrifuged for 15 min at 2,000 × g. The supernatant that contained the viral minichromosome was recovered (32).
ChIP.
The chromatin immunoprecipitation (ChIP) protocol was based on the method described by Saleh et al. (33). Symptomatic and recovered tissues (1 g) were used for isolation of PepGMV minichromosome (500 μl). Protein A beads (16-157; Millipore) were used for preclearing (2 h at 4°C). Immunoprecipitation was carried out overnight at 4°C using anti-histone H2B (07-371; Millipore) and anti-histone H3 (06-755; Millipore) antibodies. DNA was extracted using phenol-chloroform, followed by ethanol precipitation. Purified DNA was quantified by real-time PCR as described below. For the sucrose gradient fraction analysis, the fractions were first concentrated 10-fold using Centricon tubes (nominal molecular weight limit [NMWL], 10,000; Amicon Ultra-4) to obtain a final volume of 50 μl. Then, 10 μl was used for each antibody assay. Protein A beads (16-157; Millipore) were used for preclearing (2 h at 4°C). Immunoprecipitation was carried out overnight at 4°C using anti-histone H3K9me2 (17-648; Millipore) and anti-histone H3K4me3 (17-614; Millipore) antibodies. DNA was extracted using phenol-chloroform, followed by ethanol precipitation. Purified DNA was quantified by real-time PCR.
Sucrose gradient centrifugation.
The supernatant containing the viral minichromosome (1 ml) was separated using a discontinuous sucrose gradient (10 mM Tris-HCl [pH 8], 1× protease inhibitor cocktail, and 15%, 25%, or 35% [wt/vol] sucrose). Samples were centrifuged at 250,000 × g and 4°C for 1 h (SW55Ti rotor; Sorvall). Six fractions were collected. Fractions were washed two times with 5 ml Tris buffer (10 mM Tris-HCl [pH 8]) and concentrated 10-fold using Centricon tubes (NMWL, 10,000; Amicon Ultra-4) at 4,000 × g and 4°C for 15 min (8947 rotor and 6563E adapter; IEC Multi Thermo Electron Corporation). Aliquots of 20 μl were treated with 2% SDS, 25 mM EDTA, and 3 μl proteinase K (20 mg/ml) and incubated at 37°C overnight. DNA was extracted for each fraction by standard phenol-chloroform extraction and ethanol precipitation.
Quantification of viral DNA by real-time PCR.
PCR was performed on a StepOne System (Applied Biosystems) using SYBR green JumpStart Taq ReadyMix (S4438; Sigma). For 20-μl reaction volumes, 2 μl of DNA solution was used along with 1 μl of each 10 μM primer. Amplification was done as follows: the denaturation was at 95°C for 2 min, the amplification and quantification program (95°C for 15 s, 57°C for 1 min, with a single fluorescence measurement) was repeated 40 times, and the melting curve program was 60 to 95°C with a heating rate of 0.1°C per s and a continuous fluorescence measurement. The primers used were PepGMVRepq5′ (5′-CAAAGCTGGTGATCCGAAAAACG-3′) and PepGMVRepq3′ (5′-GTTAAACGAGGATAATGGATAAGG-3′) for an expected PCR product of 120 bp. To create standard curves for the absolute quantification of viral DNA, serial dilutions containing PepGMV dsDNA in the range of 103 to 107 molecules per μl were prepared (monomeric PepGMV A cloned in pBluescript). Standard curves were obtained by linear regression analysis of the quantification cycle (Cq) value of each of the three technical replicates over the log10 of the amount of DNA. qPCR efficiency (E) was calculated as follows: E = e(ln10/−s) − 1, where a slope (s) of −3.570 represents an efficiency of 96.5%. Quantification of viral DNA was obtained by extrapolation of Cq data with the corresponding standard curve.
PCR assay of Alien transposon.
For the detection of Alien transposable elements, a PCR assay was carried out under the following conditions. Primers used were CaAlienFw, TTAGACGTG AATGCCCCGAG, and CaAlienRv, GGTAGAGTGTATTGGCTAACTTTG. The amplification program was an initial denaturing treatment at 95°C for 5 min and 30 cycles of the following steps: 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s.
Quantification of viral ssDNA and dsDNA.
Quantification of ssDNA and dsDNA molecules was performed with a procedure similar to the one described previously for TYLCV and TYLCSV (30). The monomer PepGMV A standard was used to prepare standard curves for the absolute quantification. Viral strand-specific primers for T4 DNA polymerase DNA synthesis containing a 5′ virus-unrelated TAG sequence were VS-TAG (5′-AGTTTAAGAACCCTTCCCGCCATGACGCTGTTGGTGTGGTTCT TG-3′) for viral strand (VS) determination and CS-TAG (5′-AGTTTAAGAACCCTTCCCGCCCCATCGTGTAGGCAAGCGTTTCTG-3′) for viral complementary strand (CS) determination. Strand-specific qPCR primers were CS (5′-CATGACGCTGTTGGTGTGGTTCTTG-3′) and VS (5′-CCCATCGTGTAGGCAA GCGTTTCTG-3′). Strand-specific primers were combined with the TAG oligonucleotide (5′-AGTTTAAGAACCCTTCCCGC-3′) for qPCR assays as described previously (30). qPCR efficiency (E) was calculated as follows: E = e(ln10/−s) − 1, where a slope (s) of −3.113 represents an efficiency of 109.5% to CS and s of −3.302 represents an efficiency of 100.8% to VS. Quantification of viral DNA was obtained by extrapolation of Cq data with the corresponding standard curve.
Methylation density comparisons.
Aliquots of the sedimented minichromosome extract containing 100 ng of viral DNA were treated according the manufacturer's instructions (EZ DNA Methylation-Gold kit D5005). Bisulfite-treated DNA was used as the template for PCR using primers and conditions previously described (19). Viral complementary strand (CS)-specific primers were designed to direct the amplification of a 580-bp fragment that includes the first 60 bp of the Rep coding region, the 337 bp of the PepGMV A intergenic region (IR), and the initial 183 bp of the CP coding region. As a bisulfite conversion control, 100 ng of plasmid containing a PepGMV A monomeric clone was used for test samples. PCR products were cloned with the CloneJET PCR Cloning kit (catalog number K1231), and several clones were sequenced. Data were analyzed using Kismeth software (34) (http://katahdin.mssm.edu/kismeth/revpage.pl).
RESULTS
Micrococcal analysis of viral minichromosomes from symptomatic and recovered tissues.
Previous results have suggested that viral minichromosomes from symptomatic and recovered tissues could present differences in structure organization (19, 20). Therefore, we decided to compare the minichromosome populations from both types of tissue using a micrococcal nuclease sensitivity assay. Nuclei from symptomatic, recovered, and mock tissues were isolated and treated with several concentrations of micrococcal nuclease. Digestion products were analyzed by agarose gel electrophoresis and Southern blot hybridization using PepGMV A as a probe. Figure 1 shows the results of the analysis. Figure 1A shows the typical DNA band ladder showing monomeric (ca. 150 bp) and multimeric bands. In the GelRed-stained gel, most of the DNA observed is related to the plant genome. The hybridization signal observed in Fig. 1B is related to viral DNA. To be able to compare the nuclease effects in the two cases, the hybridization image was analyzed using ImageJ software, and results are shown in Fig. 1C. Using the presence and relative concentration of the bands corresponding to the mono-, di-, and trimeric DNA bands as a reference, the minichromosome from symptomatic tissue (S) presents a higher sensitivity to nuclease degradation than the one observed from the minichromosome extracted from recovered tissue. Even at the lowest nuclease concentration (0.5 U), the monomeric form shows already the highest relative concentration. In the treatment with the highest nuclease concentration (10 U), most of the viral DNA (80%) is found in the monomeric fraction. In general, a good correlation between the nuclease concentration and the relative concentration of mononucleosome concentration is observed for the S tissue (Fig. 1C, left side). On the other hand, the viral minichromosome from the recovered tissue seems to be more resistant to nuclease digestion. For example, it is possible to detect mono- and dimeric bands only at the nuclease concentration of 2.5 U. More striking, the dimeric fraction is still the major form detected at the highest nuclease treatment (10 U).
FIG 1.

Micrococcal nuclease sensitivity assay of minichromosomes obtained from samples of symptomatic and recovered plant tissue. Nuclei from mock-inoculated plant tissue (M) and symptomatic (S) and recovered (R) tissue from a PepGMV-infected pepper plant were treated with increasing concentrations of micrococcal nuclease for 10 min at 37°C. After treatment, the resulting DNA fragments were purified and analyzed by 1.5% agarose gel electrophoresis and Southern blot hybridization using PepGMV A DNA as a probe. (A) Migration of the DNA fragments on a GelRed-stained agarose gel. A typical ladder of nucleosome multimers is observed in all samples. (B) Hybridization results of the PepGMV DNA fragments generated after micrococcal nuclease digestion. (C) Relative concentrations (%) of the monomeric (ca. 150 bp), dimeric (300 to 350 bp), and trimeric (ca. 500 bp) fractions observed in panel B after nuclease digestion.
These results support the hypothesis that, indeed, PepGMV DNA is organized as a minichromosome. In addition, they also show that the minichromosome from symptomatic tissue is more sensitive to nuclease digestion, suggesting a more relaxed, accessible structure than the one obtained from recovered tissue.
Solubilization of a nuclear extract enriched for viral nucleoprotein complexes.
One of the constraints to study viral minichromosome is the low concentration. This is especially noticeable in phloem-limited viruses such as our model, PepGMV. In addition, the problem of contamination with plant chromatin fragments with varied structure, size, and composition is probably the most difficult burden.
Starting with a nucleus-rich subcellular fraction, we have developed a procedure to obtain a nuclear extract enriched with nucleoprotein complexes associated with viral dsDNA. This extract has a minimal contamination of plant chromatin or viral ssDNA; therefore, it offers to facilitate the study of viral processes such as replication and transcription as well as viral genomic structure. After testing several salts (NaCl, KCl, MgCl2), detergents (individually or in mixtures, such as SDS, Tween 20, Sarkosyl, Triton X-100), and different pH buffers (from pH 5 to pH 9), the optimal protocol to recover most of the dsDNA associated with histones was an incubation of a nucleus-rich fraction with a mixture of Triton X-100 (1%) and Sarkosyl (0.05%) at pH 8. Use of detergents such as SDS and Sarkosyl (concentrations of >0.05%) or high NaCl concentrations usually resulted in the extraction of free viral DNA, i.e., without associated proteins (histones). Treatment with other detergents such as Tween 20 or Triton X-100 failed to extract viral DNA from the nucleus-rich fraction. In addition, it has been reported that when buffers with pH <7 are used, chromatin tends to condense, whereas a buffer with pH >9 favors histone dissociation from chromatin (35).
A typical starting material for this method is a nucleus-rich extract obtained after grinding 20 g of tissue from approximately 100 infected plants. A sequential treatment with 1% Triton X-100 (mainly to reduce chloroplast contamination) and 0.05% Sarkosyl at pH 8.0 released, from the nucleus-rich fraction, viral nucleoprotein complexes that were associated mostly with viral dsDNA and almost no plant chromatin. Standard DNA extraction (chloroform and ethanol precipitation) of the nuclear extract yielded 10 μg of viral DNA. Viral DNA (dsDNA and ssDNA) was observed and identified by agarose (1%) gel electrophoresis and hybridization (using PepGMV A DNA as a probe) (Fig. 2A).
FIG 2.
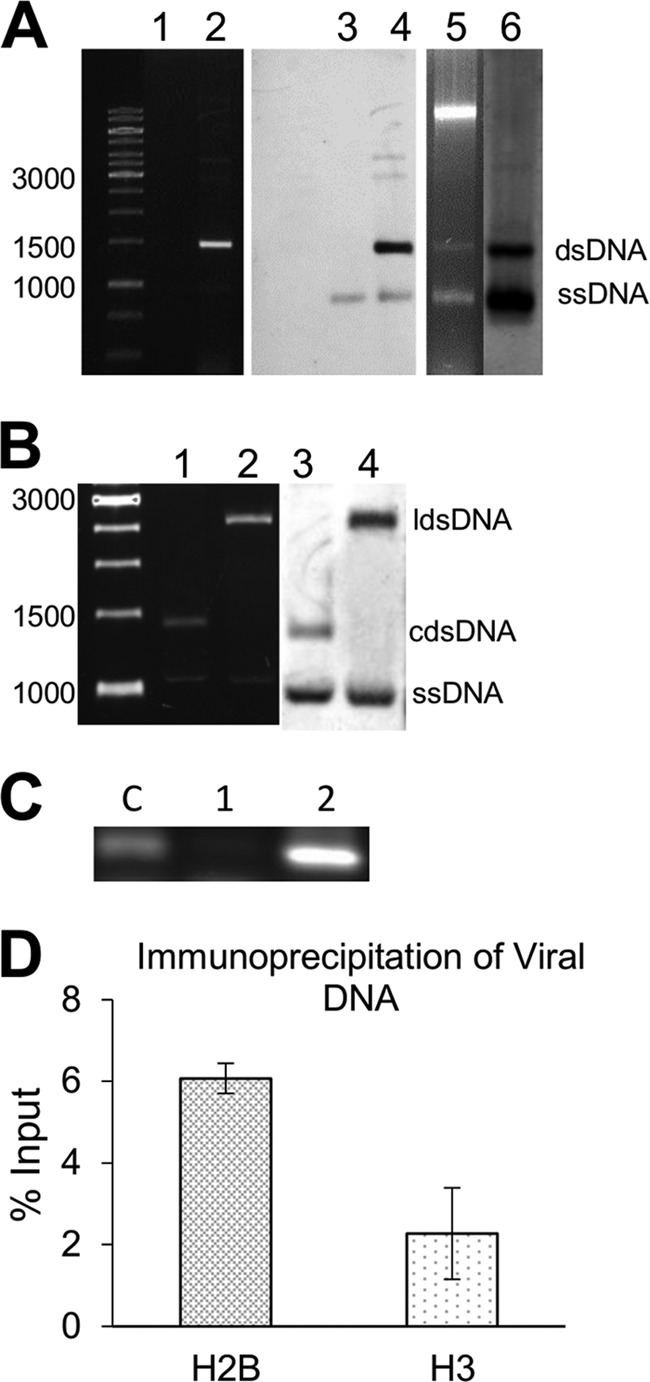
Extracts from Sarkosyl-treated nuclei are enriched for viral dsDNA. (A) GelRed-stained 1% agarose gel containing total DNA solubilized after detergent treatment of a nuclei preparation. Lane 1, DNA obtained from nuclei treated with Triton X-100; lane 2, DNA obtained after treatment with Triton X-100 and Sarkosyl. Nucleic acids in lanes 1 and 2 were transferred to a nylon membrane and analyzed by Southern blotting hybridization using a PepGMV A probe (lanes 3 and 4). As a comparison, a total nucleic acid extract from a symptomatic tissue is shown in lane 5 as well as its corresponding hybridization image (lane 6). DNA bands corresponding to viral dsDNA and ssDNA are labeled. (B) To verify the identity of viral DNA molecules, samples as in lanes 2 and 4 in panel A were digested with EcoRI and HindIII restriction enzymes and analyzed by agarose electrophoresis and Southern blotting. Lane 1, undigested control; lane 2, EcoRI-digested extract; lanes 3 and 4, hybridization results of lanes 1 and 2. DNA forms are labeled as linear double-stranded (lds), circular double-stranded (cds), and single-stranded (ss) DNA. Numbers on the left of panels A and B represent molecular size markers in base pairs. (C) To verify the levels of “contaminant” host genomic DNA, a PCR assay designed to detect a mobile element reported in pepper (Alien transposon) was carried out. Lane c, pepper DNA as a positive control; lane 1, DNA from a Triton X-100 plus Sarkosyl supernatant; lane 2, DNA from the Triton X-100 plus Sarkosyl pellet (sediment). (D) To confirm the nature of the nucleoprotein complexes extracted by Sarkosyl, a ChIP-qPCR analysis was carried out using antibodies against histones H2B and H3 and PepGMV-specific primers. Data are presented as percentages of the input.
To confirm the identity of viral dsDNA and ssDNA bands, DNA from the Sarkosyl supernatant was digested with EcoRI and HindIII restriction enzymes. These enzymes recognize unique sites in PepGMV A and B components, respectively (20). After digestion of circular dsDNA, viral DNA would migrate as monomeric, linear dsDNA molecules (ca. 2,600 bp), whereas ssDNA molecules would remain undigested. Figure 2B shows the typical migration of those molecular entities under our standard gel electrophoresis conditions. Considering that around 80% of the pepper genome is related to transposable elements (36), we tried unsuccessfully to detect, in the solubilized fraction, DNA from a transposable element using PCR assays (37) (Fig. 2C). This suggested that contamination of that fraction with genomic DNA was minimal.
To verify if viral DNA was being released as nucleoprotein complexes (i.e., minichromosome structures), a chromatin immunoprecipitation assay coupled with a quantitative PCR amplification (ChIP-qPCR) was carried out. For the analysis, we used antibodies against H3 and H2B histones and specific primers that direct the amplification of a fragment of PepGMV A DNA. Both histones are part of the typical octameric core of nucleosomes (38). The immunoprecipitation assays were carried out with minichromosome extracts obtained from symptomatic tissue. The results of the qPCR analysis of the immunoprecipitation complexes are shown in Fig. 2D. The fact that both antibodies (against H3 and H2A) were able to pull down viral DNA suggests that viral DNA is indeed associated with histones and that solubilization from the nuclei did not disturb the basic minichromosomal structure. The variation on the quantification of viral DNA is probably due to the different specificities and concentrations of these commercial antibodies. Similar results were also obtained with H4 antibody (data not shown).
Comparison of viral minichromosome populations from symptomatic and recovered stages.
Recent studies with a system based on an L2-deficient mutant of beet curly top virus (BCTV) and Arabidopsis thaliana suggested that host recovery is the consequence of an interplay of plant gene silencing-related proteins (i.e., AGO4, DCL3, DRB3) and geminivirus silencing suppressors (C2, L2) (26). In our model, we have also shown differential methylation levels of viral DNA in S and R tissues that might lead to differences in minichromosome structure as suggested by the data shown in Fig. 1. We hypothesized that viral DNA with a higher level of methylation could be associated with minichromosomal structures arranged in a condensed structure, typical of silent, nonexpressing chromatin. On the other hand, viral DNA present in symptomatic tissues and showing a lower level of methylation could be associated with a relaxed chromatin structure to facilitate transcription and replication processes. To verify these differences, we decided to carry out an additional characterization of both minichromosome populations. This analysis was performed by taking advantage of the fact that different structures of viral minichromosomes can be differentiated by a discontinuous sucrose gradient (39, 40), because the sedimentation rate is defined by the density of the minichromosome (weight/volume ratio). The sedimentation rate of a viral minichromosome would be higher if it were organized in a compact structure. On the other hand, a more relaxed structure should present a lower sedimentation rate (41).
Extracts from symptomatic and recovered tissues were centrifuged side by side as described in Materials and Methods. Six fractions were collected from each sucrose gradient for viral DNA quantification by qPCR. In extracts from symptomatic tissue, 50 to 70% of the viral dsDNA was present in fractions 1 and 2 (Fig. 3B) (low-density fraction; 15% sucrose). In contrast, in extracts from recovered tissue, 55 to 75% of dsDNA was found in the bottom, in fraction 6 (high density, 35% sucrose) (Fig. 3C). This suggested that most of the viral dsDNA released from the nuclei obtained from the symptomatic tissue is present in a relaxed minichromosome structure; on the other hand, viral dsDNA from recovered tissue showed a higher sedimentation rate, suggesting a condensed minichromosomal structure.
FIG 3.
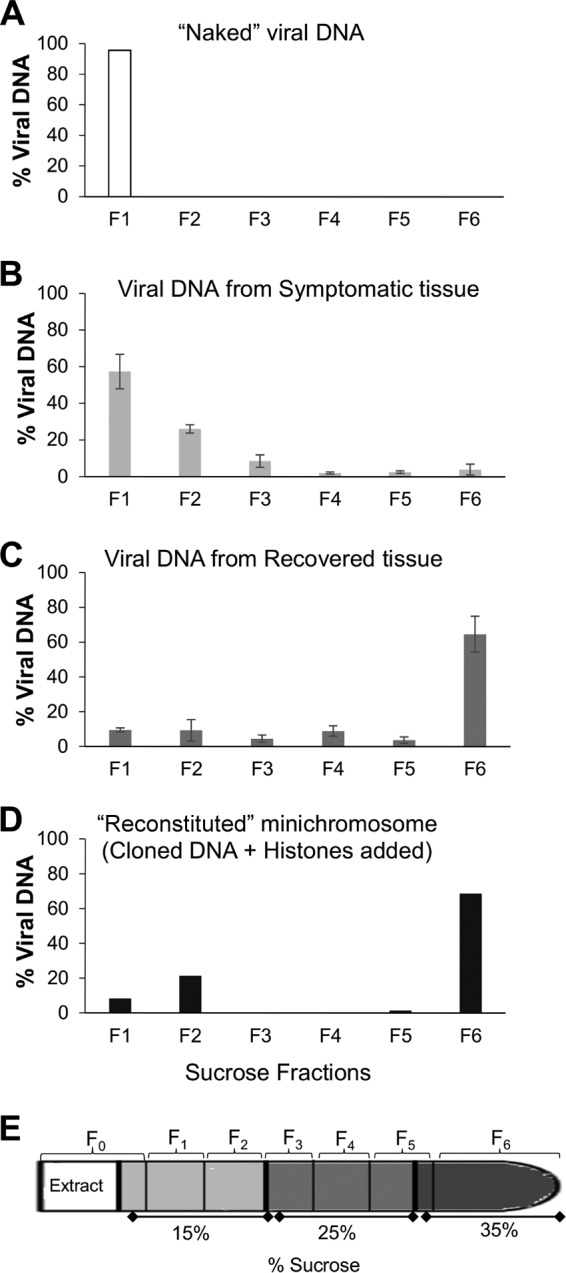
Identification of two viral minichromosomal populations in PepGMV-infected tissue. Quantification by qPCR of viral DNA molecules present in each fraction of discontinuous sucrose gradient (15%, 25%, and 35% sucrose). (A) “Naked” viral DNA (dimer). (B) Viral DNA from symptomatic tissue extract. (C) Viral DNA from recovered tissue extract. (D) “Reconstituted” minichromosome (monomer plus histones added). Fraction 1 (F1) corresponding to the top (15% sucrose) and fraction 6 (F6) corresponding to the bottom (35% sucrose) of the tube. (E) Sucrose gradient scheme; the fractions collected are indicated. Error bars represent the averages for three independent replicates (B and C). In each case (A to D), the scale in the y axis represents the relative concentration of viral DNA in the fraction of the respective gradient.
To support these conclusions, two control samples of the sucrose gradient sedimentation analysis were included in parallel tubes. The first one consisted of an infective dimeric clone of PepGMV A as a “naked,” protein-free DNA sample. After sedimentation, fractions were collected and analyzed as mentioned above. Free DNA showed a low sedimentation rate under these conditions and was found primarily in the interphase formed between the loading sample and the top segment of gradient fraction 1 (low density) (Fig. 3A). The second control consisted of an artificially reconstituted minichromosome obtained with cloned viral DNA (monomer copy plus plasmid) and commercial calf histones (H9250; Sigma) (H2A, H2B, H3, H4, and H1). Nucleosome assembly was carried out by mixing DNA and histones in a 0.6:1 ratio followed by a dialysis in an NaCl gradient (2 M to 0.1 M) as described previously (42). To verify the structure of the reconstituted minichromosome, the resulting complex was digested with micrococcal nuclease and analyzed by agarose gel electrophoresis (data not shown). This artificial minichromosome was then sedimented as mentioned above. Viral DNA quantification showed that, in this case, most viral DNA was recovered in the high-density fraction 6 (Fig. 3D).
Viral DNA in F1S and F6R samples is primarily in dsDNA form.
To corroborate the dsDNA nature of the viral DNA present in F1S and F6R samples, we implemented a procedure to quantify viral ssDNA and dsDNA molecules using a qPCR analysis following the protocol described for TYLCV (30). This procedure is based on the independent labeling of both strands of viral DNA (sense and complementary orientations) and its quantification by real-time qPCR. According to Rodríguez-Negrete et al. (30), in the case of TYLCV, more than 99% of the complementary (−) strand is present as part of the dsDNA (RF intermediate), whereas the virion sense (+) strand is present mainly in two forms: (i) associated with RF in equimolar concentration with the complementary strand and (ii) as circular, ssDNA free form (and probably most of it encapsidated). Another set of virion sense, ssDNAs could be the molecules identified in two-dimensional (2D) gels for several geminiviruses and proposed to be part of the replicative process (29).
To verify the efficiency of the procedure, we first analyzed the concentrations of ssDNA and dsDNA present in a total DNA extract from apical, newly emerged leaves of a PepGMV-infected plant expressing typical severe symptoms. Figure 4A shows that almost 90% of the viral DNA isolated from an infected plant is in the ssDNA form. This result is similar to the one reported for TYLCV in Nicotiana benthamiana and tomato, where around 99% of the TYLCV DNA was in the ssDNA form. Therefore, we proceeded to analyze the DNA present in the F1S and F6R samples. As seen in Fig. 4B and C, most of the DNA present in both fractions is in dsDNA form, supporting the notion that the viral DNA released from the nucleus-rich fraction with the Sarkosyl treatment is found mainly as a dsDNA nucleoprotein complex. This was also confirmed by analyzing the viral DNA obtained from F1S and F6R using agarose (1%) gel electrophoresis and digestion with restriction enzymes.
FIG 4.

Quantification of viral ssDNA and dsDNA in F1S and F6R shows that dsDNA is preferentially released from the nuclei. Quantification of ssDNA and dsDNA from the total DNA extract from apical PepGMV-infected tissue, similar to the one shown in Fig. 2A, lanes 5 and 6 (A) and DNA extract from sucrose gradient fractions F1S and F6R as shown in Fig. 2B and C (B and C). Data are expressed as percentages of quantified total molecules.
Bisulfite sequencing confirmed differences in methylation levels between minichromosome populations from S and R samples.
Nuclease sensitivity assay digest and sedimentation data (Fig. 1 and 3) suggest that the minichromosome structure obtained from S tissue presents an open or relaxed conformation; on the other hand, the minichromosomes obtained from R tissue showed a “condensed” conformation. It has been reported that a condensed chromatin correlates with high levels of DNA methylation (43). To verify if there was a difference in DNA methylation levels in the two minichromosome populations (S and R), we conducted a high-resolution methylation analysis of PepGMV A intergenic region/CP using a bisulfite sequencing approach. The comparison of contrasting samples, fraction 1 of S samples (F1S) and fraction 6 of R samples (F6R), is shown in Fig. 5.
FIG 5.

High-resolution methylation analysis of F1S and F6R samples by bisulfite sequencing. Bisulfite-treated DNA from minichromosome-enriched fractions (F1S and F6R) was used for the analysis. (A) Dot graphics represent all cytosines present in the 580-bp fragment corresponding to the 337 nucleotides (nt) of PepGMV A intergenic region (IR) and the first 183 nt of the CP gene. Colors represent different cytosine contexts (CG, red; CHG, green; CHH, blue). Filled circles indicate methylation, and each line represents the sequence of an individual clone (12 clones per condition). (B) The percentage of methylation in each cytosine in both stages was calculated. Each bar represents the position of a cytosine in the IR/CP fragment. Map and main motifs (Loop, stem-loop structure; TATA, TATA boxes for Rep and CP promoters; CLE, conserved late element in CP promoter) are included at the bottom. (C) Distribution of methylation in symmetric (CG/CHG) and asymmetric (CHH) cytosines in the analyzed sequences.
The distribution of the methylated cytosines in the analyzed fragment from both minichromosome populations is shown in Fig. 5A and B. Figure 5A shows all cytosines using a color code to distinguish cytosine types (CG, CHG, and CHH). Full symbols represent a methylated C. Figure 5B aligns the cytosine distribution along the corresponding viral genomic region and includes the percentage of methylated Cs in the analyzed samples. It was noteworthy that, contrasting with previously published results (19), the methylation patterns obtained here were rather homogeneous. In the previous work, the DNA samples used for methylation analysis were total DNA extracts from S and R tissues with any further purification procedure (as the one performed in this case), representing perhaps a mixture of all minichromosome structures presented in that tissue. The overall percentage of methylated cytosines in F1S was low (6.4%), whereas the percentage of methylated cytosines in F6R was quite high (89.3%) (Fig. 5C). As generally observed in plant systems, all types of cytosines were methylated at similar rates: 4.72, 6.54, and 6.99% for CG, CHG, and CHH, respectively, for the F1S sample; and 91.21, 91.23, and 87.88% for CG, CHG, and CHH for the F6R sample. The results shown in Fig. 5 strengthen the correlation between the structure of the minichromosomes, their methylation levels, and the recovery phenomenon (14, 25).
Modified histones are present in F1S and F6R samples in different ratios.
Chromatin structures associated with active (relaxed) or repressed (condensed) stages of transcription/replication activities are also correlated with specific histone modifications (44, 45). Therefore, we proceeded to analyze the presence of two histone modifications associated with active or repressed chromatin in F1S and F6R samples. The assay consisted of an immunoprecipitation protocol with antibodies that recognize two histone posttranslational modifications: α-H3K4me3 (active) and α-H3K9me2 (repressed). Then, viral DNA pulled down in the immunoprecipitation complex was quantified by qPCR. The results are shown in Fig. 6. Figure 6A shows that in S tissue more viral DNA is associated with H3K4me3 than with H3K9me2. This result correlates with a relaxed, active chromatin. On the other hand, Fig. 6B shows that in sample F6R more viral DNA is associated with the histone modification related to a condensed chromatin structure H3K9me2. These antibodies were used to verify their specificity (Fig. 6C) and establish the immunoprecipitation protocol (33). The concentration of viral DNA obtained from S tissue is usually several orders of magnitude higher than the one recovered from R tissue (19, 20). Since the Ab concentration is the same in both cases, the difference in the scales of Fig. 6A and B might just reflect the concentration disparity.
FIG 6.
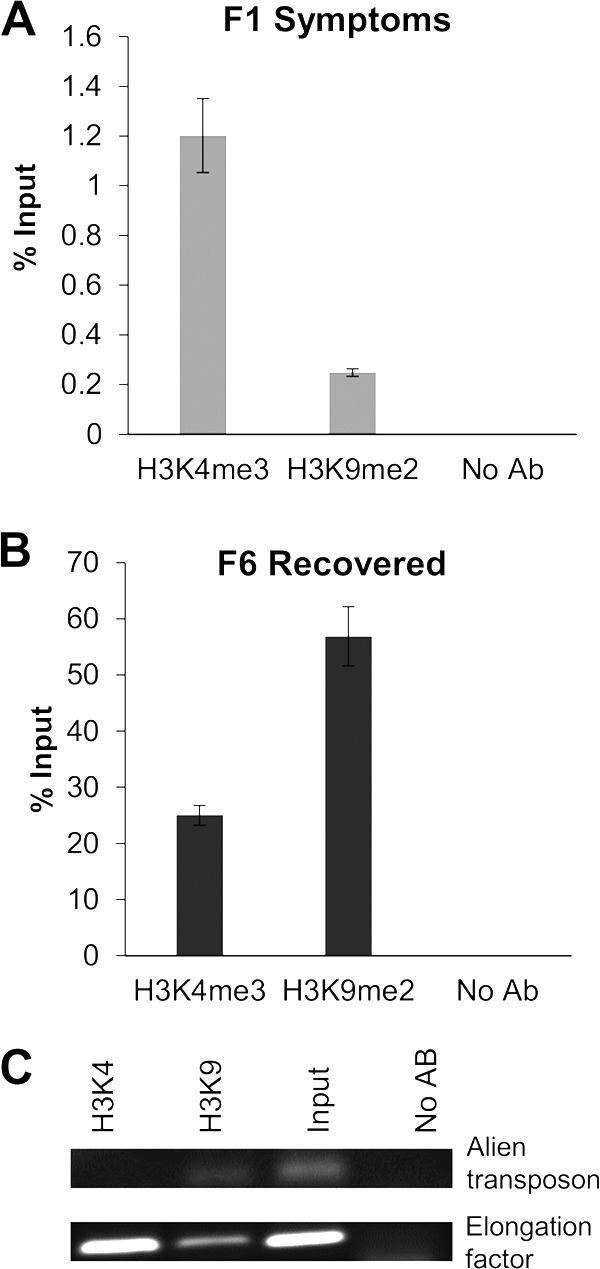
The minichromosome populations from F1S and F6R fractions present an opposite association to active and repressive chromatin histone markers. ChIP-qPCR experiments using antibodies against active (H3K4me3) and repressed (H3K9me2) chromatin markers were carried out for both minichromosome populations from F1S (A) and F6R (B) samples. Input (before immunoprecipitation, 100%) and viral pulled-down DNA were quantified by qPCR (and data are presented as percentages of the input). (C) As a control, the same antibodies (Abs) were used for ChIP assays for genes/elements known to be present in usually condensed/repressed (alien transposon) or relaxed/active (elongation factor) chromatin regions. Total DNA from an equivalent aliquot of the chromatin sample was used as the “input” control. Similarly, an aliquot of the same chromatin sample was included in the ChIP assay, but no Abs were added.
DISCUSSION
Viral DNA exists in several molecular forms in infected cells: circular ssDNA (encapsidated form), heterogeneous-length linear ssDNA (intermediate products of recombination-dependent replication), and circular dsDNA (generally referred to as RF), which is the template for replication and transcription (29). Of these types, the most abundant is ssDNA, which is the product of replication that is encapsidated (30). Geminivirus studies focused on the regulatory mechanisms of transcription and replication processes have been limited by the low concentration of viral nucleoprotein complexes that can be obtained from infected tissue. We implemented here a simple methodology to obtain a solubilized fraction that is enriched for viral dsDNA that still presents an association with histones as verified with coimmunoprecipitation assays with antibodies against several histones (H2B and H3 [Fig. 2] and H4 [not shown]). On the other hand, the majority of the host chromosomal DNA remains in the nuclei after the detergent treatment.
Detergents that solubilize viral complexes have been used to release active nucleoprotein complexes in several systems (46). In our case, since one of our objectives was to study the posttranslational modifications of the histones associated with viral DNA, it was important to ensure that the contamination with host chromatin was minimal. Therefore, an additional sucrose gradient centrifugation step was used. Taking advantage of the recovery phenomenon observed in our pepper-PepGMV model, we compared the minichromosome populations obtained in both symptomatic and recovered tissues (S and R samples). In summary, nuclease sensitivity assays (Fig. 1) and the sedimentation rate suggest a relaxed structure for the minichromosome isolated from S tissue (Fig. 3B); in addition, it presents a low methylation level (Fig. 5) and it is associated with the presence of markers for an active chromatin structure (H3K4me3) (Fig. 6A). On the other hand, the minichromosome population released from R tissue showed a reduced sensitivity in the nuclease assays (Fig. 1), along with a sedimentation rate that corresponds to a compact structure (Fig. 3C). The minichromosome from R tissue also presented a high level of cytosine methylation (Fig. 5), and it was associated primarily to inactive chromatin histone marker (H3K9me2) (Fig. 6B).
The sedimentation analysis, in addition to identifying two main minichromosome populations, also showed some heterogeneity in the minichromosome population, especially the one from S tissue. The fact that free, “naked” DNA, twice the size of the PepGMV genome, remained in fraction 1 (Fig. 3A) whereas viral dsDNA-containing complexes from S samples were distributed in fractions 1, 2, and 3 suggested a heterogeneous population that might reflect a variable number of nucleosomes associated with viral DNA. Such a variability has been observed in other geminiviruses using different methodologies. For example, in the case of abutilon mosaic virus (AbMV), minichromosomes with 5 to 15 nucleosomes were observed by electron microscopy (7). More-recent work with several geminiviruses (mainly AbMV and TYLCV) suggested that most cccDNA was organized in minichromosomes with 12 and 13 nucleosomes based on the topoisomers observed in 1D and 2D gel electrophoresis analysis (47). This type of heterogeneity has been also observed in other viral systems. For example, in the infection cycle of herpes simplex virus, the virus may be in two different states: lysis and latency. It has been found that when virus is in the lytic process, there is a high level of transcriptional activity and replication. This activity has been associated with a structure of a relaxed minichromosome containing only a few nucleosomes, referred as “nucleosome unstable,” whereas in latency, virus transcriptional activity is very low and the chromatin is condensed (48–50). A similar trend is found in hepatitis B virus (HBV) (51). In this case, it has been observed that cccDNA methylation in regulatory regions can regulate the expression of their genes (52–54), but if methylation by RNA silencing (TGS) is induced, it can suppress viral transcription and adversely affect its replication mechanism (55, 56). Similarly, different minichromosome populations were also observed in duck hepatitis B virus infection. Although 21 topoisomers were reported, the two most abundant populations are the ones with 8 to 10 nucleosomes for active minichromosomes and a second group with 18 to 20 nucleosomes in less active or inactive minichromosomes (51, 57).
Overall, the data support a recent suggestion that “form follows function in geminivirus minichromosome architecture” (47). Nevertheless, conformations are dynamic and can be affected by host defense factors, virus self-regulation mechanisms, or both.
Transcriptional gene silencing (TGS) is a widespread mechanism for both defense and self-regulation processes. Cytosine methylation and modification of histones are recognized hallmarks for TGS. Upon analysis of methylated cytosine of both populations of viral minichromosomes, we observed an important difference (82.9%) in methylation levels obtained with S (low) and R (high) samples (Fig. 5). Methylation of viral DNA has been reported before in association with a recovery process in geminivirus-infected plants (25, 26, 58). However, the role and relevance of those methylated sequences in the geminivirus replication have been questioned (12, 59).
There are several points to consider in this matter. First, every experimental model may present individual characteristics that might not be extrapolated to, or repeated with, all geminiviruses. An example of this was recently observed with AbMV, TYLCV, African cassava mosaic virus (ACMV), and Indian cassava mosaic virus (ICMV) (47). Another example is the fact that not all geminivirus infections present a recovery stage; therefore, it is important, when discussing results, to consider the specific experimental model. Second, as also noted recently (47), all geminivirus experimental models consist of asynchronous infections, meaning that when tissue is analyzed it will include infected cells at different stages and all mixed with uninfected cells. In other words, even samples collected from apical tissue include cells that were recently infected as well as cells that were infected several hours/days earlier. This means that some cells might be actively expressing and replicating viral DNA whereas in other cases the replication and/or expression of the viral genome might be already in a later phase, i.e., there may be no replication/expression. This heterogeneity might also affect the interpretation of studies on chromatin structure.
An experimental advantage of our pepper-PepGMV model and the protocol described here is that this combination allowed us to separate two distinct populations of minichromosome complexes. Although our system still presents the “inconveniences” of an asynchronous infection, the presence of two evident infection stages facilitates the separation of two distinct minichromosome populations. The first population (S sample) might include minichromosomes with a low number of nucleosomes or even the “nucleosome-free” status suggested by some authors (12) and mostly associated with no-recovery models. On the other hand, the results obtained with the R samples clearly showed that the viral minichromosome, at least in this model, does present an epigenetic regulation that results in viral DNA methylation and the presence of modified histones associated with inactive chromatin, generating a condensed chromatin structure.
Further analysis of viral DNA from the solubilized complexes using methodologies such as the 1D/2D gel electrophoresis used for AbMV might generate additional important information. For example, it will be interesting to characterize the ssDNA fraction still present in the solubilized sample (before the separation through sucrose gradients). Are those molecules part of a heterogeneous-size ssDNA predicted in the recombination-dependent replication (RDR) model (12, 29)? Another attractive possibility is a deep mass spectrometry (MS) analysis of both minichromosome populations. We are interested in pursuing that additional characterization.
A major problem is still the low number of infected cells that results in low concentrations of viral complexes. A typical experiment required 20 g of apical tissue, equivalent in our system to around 100 infected pepper plants. We are exploring different approaches to increase this yield to facilitate further analysis. A possibility is to look for other pepper-infecting viruses that are not phloem limited or to use some transgenic plants with viral components (or genes) that have been shown to generate higher concentrations of viral DNA (R. Rivera-Bustamante, data not shown).
With the data presented here, we can conclude that in our pepper-PepGMV model, a high portion of the minichromosome population present in symptomatic tissue tends to be a relaxed structure with low methylation levels and associated with active chromatin markers. This correlates with previous studies (20), in which this tissue showed high accumulation of viral DNA and RNA, suggesting high replication and transcriptional activities. On the contrary, most of the minichromosome population obtained from recovered tissue presented a condensed structure, high levels of methylation, and an association of inactive chromatin markers.
ACKNOWLEDGMENT
E. A. Rodríguez-Negrete is a Conacyt Research Fellow at the Instituto Politécnico Nacional, CIIDIR-Unidad Sinaloa, Departamento de Biotecnología Agrícola, Guasave, Sinaloa, Mexico.
Funding Statement
Edgar Antonio Rodríguez-Negrete acknowledges a Ph.D. fellowship from Conacyt-Mexico.
REFERENCES
- 1.Zhang W, Olson NH, Baker TS, Faulkner L, Agbandje-McKenna M, Boulton MI, Davies JW, McKenna R. 2001. Structure of the Maize streak virus geminate particle. Virology 279:471–477. doi: 10.1006/viro.2000.0739. [DOI] [PubMed] [Google Scholar]
- 2.Adams MJ, King AMQ, Carstens EB. 2013. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch Virol 158:2023–2030. doi: 10.1007/s00705-013-1688-5. [DOI] [PubMed] [Google Scholar]
- 3.Varsani A, Martin DP, Navas-Castillo J, Moriones E, Hernández-Zepeda C, Idris A, Zerbini FM, Brown JK. 2014. Revisiting the classification of curtoviruses based on genome-wide pairwise identity. Arch Virol 159:1873–1882. doi: 10.1007/s00705-014-1982-x. [DOI] [PubMed] [Google Scholar]
- 4.Brown JK, Zerbini FM, Navas-Castillo J, Moriones E, Ramos-Sobrinho R, Silva JCF, Fiallo-Olivé E, Briddon RW, Hernández-Zepeda C, Idris A, Malathi VG, Martin DP, Rivera-Bustamante RF, Ueda S, Varsani A. 2015. Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch Virol 160:1593–1619. doi: 10.1007/s00705-015-2398-y. [DOI] [PubMed] [Google Scholar]
- 5.Kittelmann K, Jeske H. 2008. Disassembly of African cassava mosaic virus. J Gen Virol 89:2029–2036. doi: 10.1099/vir.0.2008/000687-0. [DOI] [PubMed] [Google Scholar]
- 6.Pilartz M, Jeske H. 1992. Abutilon mosaic geminivirus double-stranded DNA is packed into minichromosomes. Virology 189:800–802. doi: 10.1016/0042-6822(92)90610-2. [DOI] [PubMed] [Google Scholar]
- 7.Abouzid AM, Frischmuth T, Jeske H. 1988. A putative replicative form of the abutilon mosaic virus (gemini group) in a chromatin-like structure. Mol Gen Genet 212:252–258. doi: 10.1007/BF00334693. [DOI] [Google Scholar]
- 8.Zhou Y, Rojas MR, Park M-R, Seo Y-S, Lucas WJ, Gilbertson RL. 2011. Histone H3 interacts and colocalizes with the nuclear shuttle protein and the movement protein of a geminivirus. J Virol 85:11821–11832. doi: 10.1128/JVI.00082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parent J-S, Martínez de Alba AE, Vaucheret H. 2012. The origin and effect of small RNA signaling in plants. Front Plant Sci 3:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bologna NG, Voinnet O. 2014. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu Rev Plant Biol 65:473–503. doi: 10.1146/annurev-arplant-050213-035728. [DOI] [PubMed] [Google Scholar]
- 11.Pumplin N, Voinnet O. 2013. RNA silencing suppression by plant pathogens: defence, counter-defence and counter-counter-defence. Nat Rev Microbiol 11:745–760. doi: 10.1038/nrmicro3120. [DOI] [PubMed] [Google Scholar]
- 12.Pooggin MM. 2013. How can plant DNA viruses evade siRNA-directed DNA methylation and silencing? IJMS 14:15233–15259. doi: 10.3390/ijms140815233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Csorba T, Kontra L, Burgyán J. 2015. Viral silencing suppressors: tools forged to fine-tune host-pathogen coexistence. Virology 479-480:85–103. doi: 10.1016/j.virol.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 14.Raja P, Wolf JN, Bisaro DM. 2010. RNA silencing directed against geminiviruses: post-transcriptional and epigenetic components. Biochim Biophys Acta 1799:337–351. doi: 10.1016/j.bbagrm.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Voinnet O, Pinto YM, Baulcombe DC. 1999. Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc Natl Acad Sci USA 96:14147–14152. doi: 10.1073/pnas.96.24.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luna AP, Morilla G, Voinnet O, Bejarano ER. 2012. Functional analysis of gene-silencing suppressors from tomato yellow leaf curl disease viruses. Mol Plant Microbe Interact 25:1294–1306. doi: 10.1094/MPMI-04-12-0094-R. [DOI] [PubMed] [Google Scholar]
- 17.Zrachya A, Glick E, Levy Y, Arazi T, Citovsky V, Gafni Y. 2007. Suppressor of RNA silencing encoded by Tomato yellow leaf curl virus-Israel. Virology 358:159–165. doi: 10.1016/j.virol.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 18.Bian X-Y, Rasheed MS, Seemanpillai MJ, Rezaian MA. 2006. Analysis of silencing escape of tomato leaf curl virus: an evaluation of the role of DNA methylation. Mol Plant Microbe Interact 19:614–624. doi: 10.1094/MPMI-19-0614. [DOI] [PubMed] [Google Scholar]
- 19.Rodríguez-Negrete EA, Carrillo-Tripp J, Rivera-Bustamante RF. 2009. RNA silencing against geminivirus: complementary action of posttranscriptional gene silencing and transcriptional gene silencing in host recovery. J Virol 83:1332–1340. doi: 10.1128/JVI.01474-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carrillo-Tripp J, Lozoya-Gloria E, Rivera-Bustamante RF. 2007. Symptom remission and specific resistance of pepper plants after infection by pepper golden mosaic virus. Phytopathology 97:51–59. doi: 10.1094/PHYTO-97-0051. [DOI] [PubMed] [Google Scholar]
- 21.Chellappan P, Vanitharani R, Pita J, Fauquet CM. 2004. Short interfering RNA accumulation correlates with host recovery in DNA virus-infected hosts, and gene silencing targets specific viral sequences. J Virol 78:7465–7477. doi: 10.1128/JVI.78.14.7465-7477.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghoshal B, Sanfaçon H. 2015. Symptom recovery in virus-infected plants: revisiting the role of RNA silencing mechanisms. Virology 479-480:167–179. [DOI] [PubMed] [Google Scholar]
- 23.Brough CL, Gardiner WE, Inamdar NM, Zhang XY, Ehrlch M, Bisaro DM. 1992. DNA methylation inhibits propagation of tomato golden mosaic-virus DNA in transfected protoplasts. Plant Mol Biol 18:703–712. doi: 10.1007/BF00020012. [DOI] [PubMed] [Google Scholar]
- 24.Ermak G, Paszkowski U, Wohlmuth M, Mittelsten Scheid O, Paszkowski J. 1993. Cytosine methylation inhibits replication of African cassava mosaic virus by two distinct mechanisms. Nucleic Acids Res 21:3445–3450. doi: 10.1093/nar/21.15.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raja P, Sanville BC, Buchmann RC, Bisaro DM. 2008. Viral genome methylation as an epigenetic defense against geminiviruses. J Virol 82:8997–9007. doi: 10.1128/JVI.00719-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raja P, Jackel JN, Li S, Heard IM, Bisaro DM. 2014. Arabidopsis double-stranded RNA binding protein DRB3 participates in methylation-mediated defense against geminiviruses. J Virol 88:2611–2622. doi: 10.1128/JVI.02305-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seemanpillai M, Dry I, Randles J, Rezaian A. 2003. Transcriptional silencing of geminiviral promoter-driven transgenes following homologous virus infection. Mol Plant Microbe Interact 16:429–438. doi: 10.1094/MPMI.2003.16.5.429. [DOI] [PubMed] [Google Scholar]
- 28.Buchmann RC, Asad S, Wolf JN, Mohannath G, Bisaro DM. 2009. Geminivirus AL2 and L2 proteins suppress transcriptional gene silencing and cause genome-wide reductions in cytosine methylation. J Virol 83:5005–5013. doi: 10.1128/JVI.01771-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeske H, Lütgemeier M, Preiss W. 2001. DNA forms indicate rolling circle and recombination-dependent replication of Abutilon mosaic virus. EMBO J 20:6158–6167. doi: 10.1093/emboj/20.21.6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodríguez-Negrete EA, Sánchez-Campos S, Cañizares MC, Navas-Castillo J, Moriones E, Bejarano ER, Grande-Pérez A. 2014. A sensitive method for the quantification of virion-sense and complementary-sense DNA strands of circular single-stranded DNA viruses. Sci Rep 4:6438. doi: 10.1038/srep06438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renteria-Canett I, Xoconostle-Cazares B, Ruiz-Medrano R, Rivera-Bustamante RF. 2011. Geminivirus mixed infection on pepper plants: synergistic interaction between PHYVV and PepGMV. Virol J 8:104. doi: 10.1186/1743-422X-8-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera-Bustamante RF, Semancik J. 1989. Properties of a viroid-replicating complex solubilized from nuclei. J Gen Virol 70:2707. doi: 10.1099/0022-1317-70-10-2707. [DOI] [Google Scholar]
- 33.Saleh A, Alvarez-Venegas R, Avramova Z. 2008. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat Protoc 3:1018–1025. doi: 10.1038/nprot.2008.66. [DOI] [PubMed] [Google Scholar]
- 34.Gruntman E, Qi Y, Slotkin RK, Roeder T, Martienssen RA, Sachidanandam R. 2008. Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics 9:371. doi: 10.1186/1471-2105-9-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo XW, Cole RD. 1989. Chromatin aggregation changes substantially as pH varies within the physiological range. J Biol Chem 264:11653–11657. [PubMed] [Google Scholar]
- 36.Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y, Yang Y, Wu Z, Mao L, Wu H, Ling-Hu C, Zhou H, Lin H, González-Morales S, Trejo-Saavedra DL, Tian H, Tang X, Zhao M, Huang Z, Zhou A, Yao X, Cui J, Li W, Chen Z, Feng Y, Niu Y, Bi S, Yang X, Li W, Cai H, Luo X, Montes-Hernández S, Leyva-González MA, Xiong Z, He X, Bai L, Tan S, Tang X, Liu D, Liu J, Zhang S, Chen M, Zhang L, Zhang L, Zhang Y, Liao W, Zhang Y, Wang M, Lv X, Wen B, Liu H, Luan H, Zhang Y, Yang S, Wang X, Xu J, Li X, Li S, Wang J, Palloix A, Bosland PW, Li Y, Krogh A, Rivera-Bustamante RF, Herrera-Estrella L, Yin Y, Yu J, Hu K, Zhang Z. 2014. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci U S A 111:5135–5140. doi: 10.1073/pnas.1400975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pozueta-Romero J, Klein M, Houlné G, Schantz M-L, Meyer B, Schantz R. 1995. Characterization of a family of genes encoding a fruit-specific wound-stimulated protein of bell pepper (Capsicum annuum): identification of a new family of transposable elements. Plant Mol Biol 28:1011–1025. doi: 10.1007/BF00032663. [DOI] [PubMed] [Google Scholar]
- 38.Richmond TJ, Davey CA. 2003. The structure of DNA in the nucleosome core. Nature 423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 39.Tachiwana H, Kagawa W, Osakabe A, Kawaguchi K, Shiga T, Hayashi-Takanaka Y, Kimura H, Kurumizaka H. 2010. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc Natl Acad Sci U S A 107:10454–10459. doi: 10.1073/pnas.1003064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamakaka RT, Thomas JO. 1990. Chromatin structure of transcriptionally competent and repressed genes. EMBO J 9:3997–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA. 2004. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell 118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 42.Richmond TJ, Searles MA, Simpson RT. 1988. Crystals of a nucleosome core particle containing defined sequence DNA. J Mol Biol 199:161–170. doi: 10.1016/0022-2836(88)90386-5. [DOI] [PubMed] [Google Scholar]
- 43.Kashiwagi K, Nimura K, Ura K, Kaneda Y. 2011. DNA methyltransferase 3b preferentially associates with condensed chromatin. Nucleic Acids Res 39:874–888. doi: 10.1093/nar/gkq870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swygert SG, Peterson CL. 2014. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta 1839:728–736. doi: 10.1016/j.bbagrm.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res 21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gariglio P, Llopis R, Oudet P, Chambon P. 1979. Template of the isolated native simian virus-40 transcriptional complexes is a mini-chromosome. J Mol Biol 131:75–105. doi: 10.1016/0022-2836(79)90302-4. [DOI] [PubMed] [Google Scholar]
- 47.Paprotka T, Deuschle K, Pilartz M, Jeske H. 2015. Form follows function in geminiviral minichromosome architecture. Virus Res 196:44–55. doi: 10.1016/j.virusres.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Toth Z, Maglinte DT, Lee SH, Lee H-R, Wong L-Y, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 6:e1001013. doi: 10.1371/journal.ppat.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nevels M, Nitzsche A, Paulus C. 2011. How to control an infectious bead string: nucleosome-based regulation and targeting of herpesvirus chromatin. Rev Med Virol 21:154–180. doi: 10.1002/rmv.690. [DOI] [PubMed] [Google Scholar]
- 50.Cliffe AR, Knipe DM. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol 82:12030–12038. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi L, Li S, Shen F, Li H, Qian S, Lee DHS, Wu JZ, Yang W. 2012. Characterization of nucleosome positioning in hepadnaviral covalently closed circular DNA minichromosomes. J Virol 86:10059–10069. doi: 10.1128/JVI.00535-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo Y, Li Y, Mu S, Zhang J, Yan Z. 2009. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol 81:1177–1183. doi: 10.1002/jmv.21525. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Mao R, Yan R, Cai D, Zhang Y, Zhu H, Kang Y, Liu H, Wang J, Qin Y, Huang Y, Guo H, Zhang J. 2014. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS One 9:e110442. doi: 10.1371/journal.pone.0110442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vivekanandan P, Thomas D, Torbenson M. 2009. Methylation regulates hepatitis B viral protein expression. J Infect Dis 199:1286–1291. doi: 10.1086/597614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J-W, Lee SH, Park YS, Hwang J-H, Jeong S-H, Kim N, Lee DH. 2011. Replicative activity of hepatitis B virus is negatively associated with methylation of covalently closed circular DNA in advanced hepatitis B virus infection. Intervirology 54:316–325. doi: 10.1159/000321450. [DOI] [PubMed] [Google Scholar]
- 56.Park HK, Min BY, Kim NY, Jang ES, Shin CM, Park YS, Hwang J-H, Jeong S-H, Kim N, Lee DH, Kim J-W. 2013. Short hairpin RNA induces methylation of hepatitis B virus covalently closed circular DNA in human hepatoma cells. Biochem Biophys Res Commun 436:152–155. doi: 10.1016/j.bbrc.2013.04.108. [DOI] [PubMed] [Google Scholar]
- 57.Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. 1995. The covalently closed duplex form of the hepadnavirus genome exists in-situ as a heterogeneous population of viral minichromosomes. J Virol 69:3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hagen C, Rojas MR, Kon T, Gilbertson RL. 2008. Recovery from cucurbit leaf crumple virus (family Geminiviridae, genus Begomovirus) infection is an adaptive antiviral response associated with changes in viral small RNAs. Phytopathology 98:1029–1037. doi: 10.1094/PHYTO-98-9-1029. [DOI] [PubMed] [Google Scholar]
- 59.Paprotka T, Deuschle K, Metzler V, Jeske H. 2011. Conformation-selective methylation of geminivirus DNA. J Virol 85:12001–12012. doi: 10.1128/JVI.05567-11. [DOI] [PMC free article] [PubMed] [Google Scholar]