

Abstract
The successful colonization of the majority of the population by human cytomegalovirus is a direct result of the virus's ability to establish and, more specifically, reactivate from latency. The underlying cellular factors involved in viral reactivation remain unknown. Here, we show that the host complex facilitates chromatin transcription (FACT) binds to the major immediate early promoter (MIEP) and that inhibition of this complex reduces MIEP transactivation, thus inhibiting viral reactivation.
TEXT
Human cytomegalovirus (HCMV) infection is lifelong and in healthy individuals remains asymptomatic, residing latently in CD34+ hematopoietic progenitor cells (HPCs) (1–4). Reactivation from latency is an intricate event, dictated by both host and viral components, and causes a variety of symptoms in immunocompromised individuals (reviewed in reference 5). Here, we show that HCMV utilizes the facilitates chromatin transcription (FACT) complex to aid in transactivation of the major immediate early promoter (MIEP) following quiescence. FACT, a heterodimer of suppressor of Ty16 (SPT16) and structure-specific recognition protein 1 (SSRP1) (6), is highly enriched in HPCs (7). This complex repositions histones, rendering nucleosome-containing DNA accessible to RNA polymerase II. We report that inhibition of FACT suppresses MIEP transcription.
The MIEP drives expression of the IE genes UL122 and UL123, viral transactivators that facilitate lytic replication. This promoter is repressed during HPC latency, thus restricting viral replication (8, 9). Using our Kasumi-3 (K3) HPC latency model system, we reported that phorbol ester (e.g., tetradecanoyl phorbol acetate [TPA]) treatment following latent infection stimulates MIEP-driven transcription and the production of infectious virus (10). Also, tumor necrosis factor alpha (TNF-α) or interleukin-1β (IL-1β) treatment of latently infected K3 cells (K3s) (10) or primary HPCs induced MIEP transcription (8, 11). These inflammatory cytokines, as well as TPA, activate NF-κB (12, 13). As the MIEP contains four NF-κB binding sites (14–16), we hypothesized that one could block MIEP activation following TPA or TNF-α treatment of quiescently infected K3s by treatment with quinacrine, which indirectly targets a subset of NF-κB-responsive promoters and activates p53 through direct inhibition of FACT (17). Indeed, quinacrine treatment inhibits TPA- or TNF-α-induced activation of the MIEP in quiescent K3s (Fig. 1), suggesting that quinacrine treatment inhibits viral MIEP transactivation.
FIG 1.
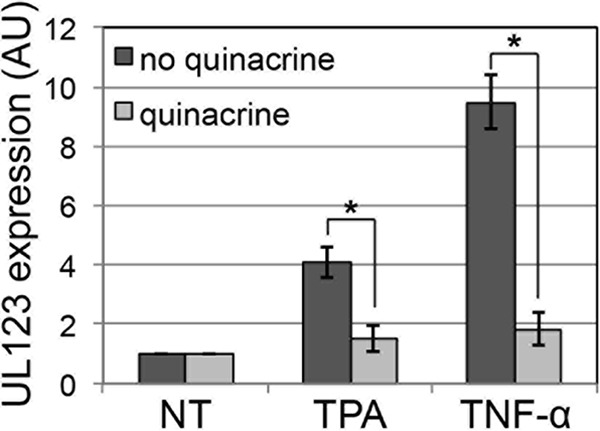
Quinacrine represses MIEP transactivation following stimulation of latently infected K3s. K3s were infected with TB40/E-mCherry (multiplicity of infection, 1.0) under quiescent conditions for 10 days and then treated with quinacrine (20 μM) or not treated for 2 h prior to the addition of stimulant, where indicated (e.g., 20 μM TPA or 20 ng/ml TNF-α) for an additional 2 days. Total RNA was collected, and the expression of UL123 relative to cellular glyceraldehyde-3-phosphate dehydrogenase was assessed. All samples were analyzed in triplicate, and results are presented as fold change relative to the nontreated (NT) infected cells. *, P < 0.05. AU, arbitrary units.
In order to ensure that quinacrine's impact on MIEP-driven transcription was not due to pleotropic effects beyond targeting FACT, we utilized three different curaxins, compounds that bind to and tether FACT to DNA (17). We first asked if curaxins could inhibit lytic replication by pretreating permissive fibroblasts, which express FACT (Fig. 2A), with three different curaxin compounds or vehicle before infecting the cells with TB40/E-mCherry-UL99eGFP. This bacterial artificial chromosome (BAC)-derived clinical isolate has the simian virus 40 (SV40) promoter driving mCherry expression (18) and enhanced green fluorescent protein (eGFP) fused to the C terminus of UL99 encoding the true late protein, pp28. Surprisingly, curaxin treatment impacted MIEP transcription within 6 h postinfection (hpi) (Fig. 2B), suggesting that at least in fibroblasts, FACT transcriptional regulation may be important. Additionally, all three curaxin compounds significantly decreased lytic infection, as demonstrated by the reduction of mCherry-positive cells. Additionally, eGFP expression in the mCherry-positive cells is reduced in the curaxin-treated infected cells (Fig. 2C), as is UL123 mRNA expression at 96 hpi under both high- and low-serum conditions (Fig. 2D), suggesting that these compounds impact lytic replication prior to viral late gene transcription. Importantly, at the concentrations used, none of the curaxins impacted cell viability (Fig. 2E).
FIG 2.

Robust HCMV lytic replication in fibroblasts is decreased with curaxin treatment. (A) Total human foreskin fibroblast lysates were collected and assessed by immunoblotting with an antibody specific for SPT16 (Santa Cruz Biotechnology). Cellular tubulin is shown as a control (Sigma-Aldrich). (B) Human foreskin fibroblasts were treated with 1 μM CBLC000, 10 nM CBLC100, 100 nM CBLC137, or vehicle (dimethyl sulfoxide [DMSO]) for 2 h prior to infection with TB40/E-mCherry-UL99eGFP (multiplicity of infection, 0.1). Total RNA was collected at 6 hpi, and UL123 transcripts were measured by reverse transcriptase quantitative PCR using protocols previously described (10). UL123 expression is shown relative to cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All samples were analyzed in triplicate. (C) Human foreskin fibroblasts were infected as described for panel B and monitored for mCherry expression as a measure of lytic infection as well as pUL99eGFP expression, which denotes successful DNA replication and late-stage protein translation. Representative images were collected at 96 h postinfection. (D) Human foreskin fibroblasts were grown to confluence and then cultured in either 10% or 0.5% serum-containing medium. Cells were then pretreated with the compounds and infected as described for panel B. Total RNA was analyzed at 96 hpi for UL123 expression relative to cellular glyceraldehyde-3-phosphate dehydrogenase. Samples were analyzed in triplicate. (E) Equal numbers of human foreskin fibroblasts were cultured in the respective serum-containing medium for 24 h prior to the addition of drug compounds. Cell viability was quantified by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay at 48 h posttreatment according to the manufacturer's protocol (Promega; CellTiter 96 Aqueous One Solution cell proliferation assay). AU, arbitrary units; FBS, fetal bovine serum.
We next assessed the ability of curaxins to block MIEP-driven transcription following stimulation of quiescent/latent infections. We quiescently infected K3s with TB40/E-mCherry and then added curaxins or vehicle prior to stimulation with or without TNF-α. Quiescently infected cultures pretreated with vehicle and then TNF-α demonstrated activation of UL123 transcription compared to parallel cultures lacking additional stimuli (Fig. 3A). However, curaxin pretreatment was sufficient to suppress MIEP transcription in the presence of TNF-α, as UL123 mRNA expression was not significantly upregulated upon cytokine addition. Although K3s are a suitable in vitro latency model system with which to interrogate HCMV, it is nonetheless critical to confirm salient findings, when possible, in ex vivo systems that utilize primary HPCs. We latently infected cord blood-derived ex vivo-cultured CD34+ HPCs with TB40/E-mCherry. We treated these cells with either CBLC100 or vehicle upon culturing them in reactivation medium (19). As expected, UL123 expression was suppressed in latently infected primary CD34+ HPCs, regardless of treatment (Fig. 3B), and yet was increased in the vehicle-treated infected cells cultured in reactivation medium. However, this increase was blocked in the presence of CBLC100 (Fig. 3B). Importantly, none of the curaxins impacted HPC viability (Fig. 3C). Together, these data indicate that the curaxin compounds successfully suppress HCMV lytic replication in fibroblasts and MIEP-dependent transcription following stimulation of in vitro quiescently infected K3s or ex vivo-latently infected primary HPCs following reactivation.
FIG 3.
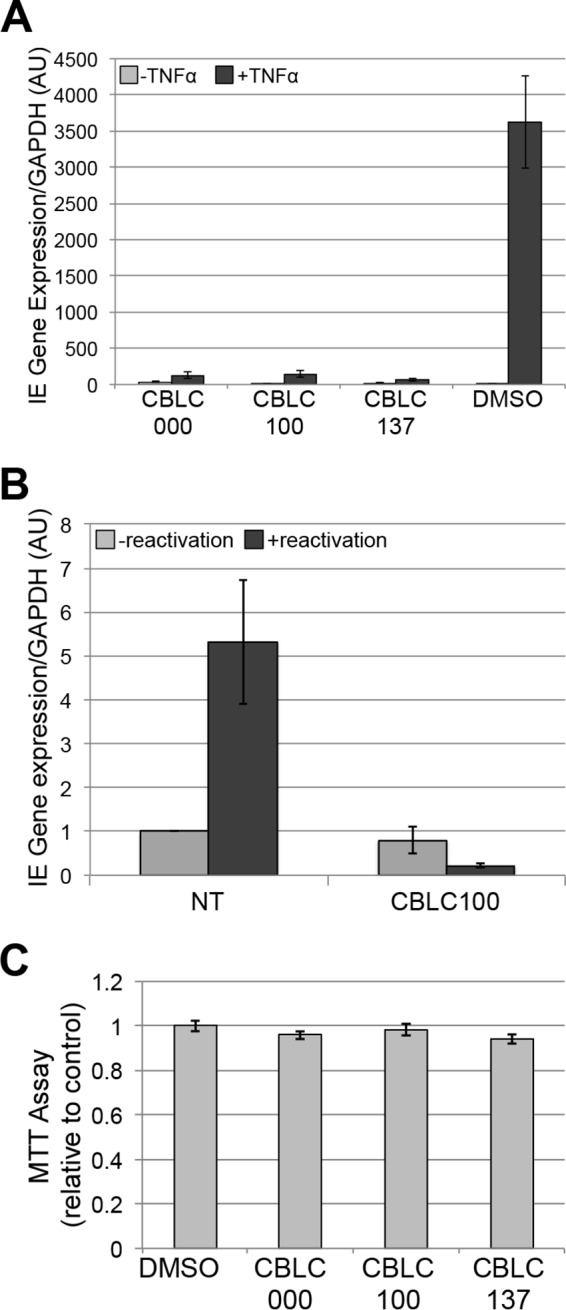
Curaxins inhibit MIEP transactivation in HPCs. (A) K3s were infected with TB40/E-mCherry (multiplicity of infection, 1.0) for 18 days under quiescent conditions. The infections were then treated with 1 μM CBLC000, 10 nM CBLC100, or 100 nM CBLC137, concentrations previously determined to have no effect on cell viability, or vehicle (dimethyl sulfoxide [DMSO]) for 2 h prior to treatment with 20 ng/ml TNF-α or no treatment for an additional 2 days. (B) Immunopurified primary CD34+ HPCs isolated from pooled patient deidentified cord blood collections (19) were infected with TB40/E-mCherry (multiplicity of infection, 2) under latency conditions for 10 days. The infected cells were then treated with 10 nM CBLC100 or vehicle (dimethyl sulfoxide) for 2 h prior to an additional 2 days in medium favoring reactivation or latency conditions. For both panels A and B, cells were harvested and UL123 mRNA was analyzed by reverse transcriptase quantitative PCR. All samples were assessed in triplicate and normalized to the cellular transcript, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). AU, arbitrary units; NT, not treated. (C) An equal number of K3s were cultured for 24 h and then treated with the respective compounds as in panel A for 48 h, after which cell viability was quantified by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay as in Fig. 2E.
We next hypothesized that FACT's presence on the MIEP is a requirement for transcription. To determine if FACT functions directly on the MIEP as opposed to exerting its effects epistatically, we monitored SPT16 levels and found that while its expression is high in K3s, total SPT16 decreases as these cells differentiate (Fig. 4A), confirming previous findings in other cell types (7). We next assessed the presence of FACT on the MIEP by chromatin immunoprecipitation (ChIP). FACT is enriched on the MIEP relative to a nonpromoter region of the virus, UL69, prior to and during stimulation of infected K3s (Fig. 4B). This is not surprising, as FACT binds to DNA in the presence or absence of active transcription (17, 20). Thus, to determine if FACT functions to aid MIEP transactivation, we transduced K3s with lentiviral constructs expressing either a nonspecific or a validated SPT16-specific short hairpin RNA (shRNA). SPT16 knockdown reduced SPT16 expression (Fig. 4C) without affecting cell viability (Fig. 4D). TNF-α treatment of these cells following quiescent infection resulted in significantly reduced UL123 transcripts compared to similarly treated infected K3s transduced with nonspecific shRNA (Fig. 4E). Thus, although FACT is present at the MIEP under both quiescent and transactivated conditions, this complex is critical in facilitating transcription from this promoter upon treatment with the appropriate transactivation stimulus.
FIG 4.

FACT is tightly bound to the MIEP in infected K3s, and the reduction of FACT levels limits activation of the MIEP following quiescent infection conditions. (A) K3s were treated with 20 ng/ml TNF-α or not treated (NT) for the indicated times. Total cell lysates were then analyzed for SPT16 expression by immunoblotting. Cellular tubulin is shown as a control. (B) K3s were infected with TB40/E-mCherry (multiplicity of infection, 1.0) for 10 days under quiescent conditions, after which 20 μM TPA or 20 ng/ml TNF-α was added for an additional 2 days to activate MIEP-driven transcription. Cells were then harvested, and the binding of FACT to either the MIEP or nonpromoter region of UL69 was determined by ChIP according to an established protocol (21). NT, not treated. (C) K3s were transduced with either a nonspecific (NS) or an SPT16-specific (KD1) shRNA lentivirus (Sigma-Aldrich Mission shRNA, catalog no. TRCN0000001257). SPT16 levels were then assessed by immunoblotting. Cellular tubulin levels are shown as a control (bottom panel). (D) The nonspecific (NS) and KD1 K3s were subjected to 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay as described for Fig. 2E. (E) These cells were then infected with TB40/E-mCherry (multiplicity of infection, 1.0) under quiescent conditions for 10 days and then treated for an additional 2 days with 20 ng/ml TNF-α or not treated. Next, the cells were harvested, and UL123 transcript levels were determined by reverse transcriptase quantitative PCR. Samples were analyzed in triplicate and normalized to cellular glyceraldehyde-3-phosphate dehydrogenase levels. AU, arbitrary units.
Our findings demonstrate that HCMV evolved to utilize the FACT complex to aid in successfully activating the MIEP. MIEP transactivation following latency is the “switch” that allows the virus to subsequently enter the lytic replicative cycle, and it is evident that these processes are multifaceted and likely quite complicated events. We propose that FACT's presence on the MIEP aids in transactivation of this promoter when conditions favor reactivation (i.e., inflammatory cytokine influx), which is limited by curaxins, thereby suppressing MIEP transcription. Curaxins may represent novel therapeutics that could dampen HCMV reactivation, posing a significant advancement in clinical treatment of HCMV, whose current therapies target lytic replication where disease is primed to occur.
Funding Statement
The content is solely the responsibility of the authors. The funding agency had no role in study design, data collection and interpretation, or the decision to submit the findings for publication.
REFERENCES
- 1.Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77:3099–3102. doi: 10.1099/0022-1317-77-12-3099. [DOI] [PubMed] [Google Scholar]
- 2.Zhuravskaya T, Maciejewski JP, Netski DM, Bruening E, Mackintosh FR, St Jeor S. 1997. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: model of HCMV latency. Blood 90:2482–2491. [PubMed] [Google Scholar]
- 3.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci U S A 99:16255–16260. doi: 10.1073/pnas.252630899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soderberg-Naucler C, Fish KN, Nelson JA. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126. doi: 10.1016/S0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- 5.Mocarski E, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. 1999. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400:284–288. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- 7.Garcia H, Fleyshman D, Kolesnikova K, Safina A, Commane M, Paszkiewicz G, Omelian A, Morrison C, Gurova K. 2011. Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget 2:783–796. doi: 10.18632/oncotarget.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy JC, Fischle W, Verdin E, Sinclair JH. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 21:1112–1120. doi: 10.1093/emboj/21.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81:9109–9120. doi: 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol 86:9854–9865. doi: 10.1128/JVI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91:599–604. doi: 10.1099/vir.0.015602-0. [DOI] [PubMed] [Google Scholar]
- 12.Zaragoza DB, Wilson RR, Mitchell BF, Olson DM. 2006. The interleukin 1beta-induced expression of human prostaglandin F2alpha receptor messenger RNA in human myometrial-derived ULTR cells requires the transcription factor, NFkappaB. Biol Reprod 75:697–704. doi: 10.1095/biolreprod.106.053439. [DOI] [PubMed] [Google Scholar]
- 13.Wajant H, Scheurich P. 2011. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J 278:862–876. doi: 10.1111/j.1742-4658.2011.08015.x. [DOI] [PubMed] [Google Scholar]
- 14.Cherrington JM, Mocarski ES. 1989. Human cytomegalovirus ie1 transactivates the alpha promoter-enhancer via an 18-base-pair repeat element. J Virol 63:1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeMeritt IB, Milford LE, Yurochko AD. 2004. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J Virol 78:4498–4507. doi: 10.1128/JVI.78.9.4498-4507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sambucetti LC, Cherrington JM, Wilkinson GW, Mocarski ES. 1989. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J 8:4251–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasparian A, Burkhart C, Purmal A, Brodsky L, Pal M, Saranadasa M, Bosykh D, Commane M, Guryanova O, Pal S, Safina A, Sviridov S, Koman I, Veith J, Komar A, Gudkov A, Gurova K. 2011. Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Transl Med 3:95ra74. doi: 10.1126/scitranslmed.3002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Connor CM, Shenk T. 2012. Human cytomegalovirus pUL78 G protein-coupled receptor homologue is required for timely cell entry in epithelial cells but not fibroblasts. J Virol 86:11425–11433. doi: 10.1128/JVI.05900-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Umashankar M, Goodrum F. 2014. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol Biol 1119:99–112. doi: 10.1007/978-1-62703-788-4_7. [DOI] [PubMed] [Google Scholar]
- 20.Huang JY, Chen WH, Chang YL, Wang HT, Chuang WT, Lee SC. 2006. Modulation of nucleosome-binding activity of FACT by poly(ADP-ribosyl)ation. Nucleic Acids Res 34:2398–2407. doi: 10.1093/nar/gkl241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuevas-Bennett C, Shenk T. 2008. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J Virol 82:9525–9536. doi: 10.1128/JVI.00946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]