

ABSTRACT
Latent membrane protein 1 (LMP1) is a major oncogene essential for primary B cell transformation by Epstein-Barr virus (EBV). Previous studies suggested that some transcription factors, such as PU.1, RBP-Jκ, NF-κB, and STAT, are involved in this expression, but the underlying mechanism is unclear. Here, we identified binding sites for PAX5, AP-2, and EBF in the proximal LMP1 promoter (ED-L1p). We first confirmed the significance of PU.1 and POU domain transcription factor binding for activation of the promoter in latency III. We then focused on the transcription factors AP-2 and early B cell factor (EBF). Interestingly, among the three AP-2-binding sites in the LMP1 promoter, two motifs were also bound by EBF. Overexpression, knockdown, and mutagenesis in the context of the viral genome indicated that AP-2 plays an important role in LMP1 expression in latency II in epithelial cells. In latency III B cells, on the other hand, the B cell-specific transcription factor EBF binds to the ED-L1p and activates LMP1 transcription from the promoter.
IMPORTANCE Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) is crucial for B cell transformation and oncogenesis of other EBV-related malignancies, such as nasopharyngeal carcinoma and T/NK lymphoma. Its expression is largely dependent on the cell type or condition, and some transcription factors have been implicated in its regulation. However, these previous reports evaluated the significance of specific factors mostly by reporter assay. In this study, we prepared point-mutated EBV at the binding sites of such transcription factors and confirmed the importance of AP-2, EBF, PU.1, and POU domain factors. Our results will provide insight into the transcriptional regulation of the major oncogene LMP1.
INTRODUCTION
The Epstein-Barr virus (EBV) is a human gammaherpesvirus that mainly infects and establishes latent infection in B lymphocytes, but it can also infect other types of cells, including NK, T, and epithelial cells. EBV infection has been implicated as a causal factor in a variety of malignancies, and the expression pattern of viral latent genes varies depending on the tissue of origin and the state of the tumors. Neoplasms such as Burkitt lymphomas or gastric carcinomas express only EBV-encoded small RNA (EBER) and EBV nuclear antigen 1 (EBNA1) (type I latency), whereas some Hodgkin lymphomas, nasopharyngeal carcinomas (NPC), and NK/T lymphomas express EBER, EBNA1, latent membrane protein 1 (LMP1), and LMP2 genes (type II latency). In addition to the type II genes, EBNA2, EBNA3, and EBNA-LP are also expressed in immunosuppression-related lymphomas or lymphoblastoid cell lines (LCLs; type III latency). LMP1 constitutively activates cellular signaling through NF-κB, mitogen-activated protein, JAK/STAT, and AKT and is believed to be a major oncogene encoded by EBV (1–11).
Two promoters regulate LMP1 gene transcription, with mechanisms that differ between type II and type III infection. In latency III in B lymphocytes, LMP1 transcription from the proximal ED-L1 promoter is activated by EBNA2 (12–14). Although EBNA2 shows no DNA-binding activity, it enhances LMP1 promoter activity by functioning as a cofactor. It associates with cellular transcriptional factors, including the recombination signal binding protein Jκ (RBP-Jκ) (14–16) and PU-box 1 (PU.1) (12, 13, 17, 18), which are then recruited onto the LMP1 promoter for transactivation. Viral factors, including EBNA-LP and EBNA3C, also associate with the complex and further modify the activation process (19–22).
On the other hand, LMP1 is expressed in an EBNA2-independent manner in type II latency, since EBNA2 is not available in this state. Cytokines, such as interleukin-4 (IL-4), IL-6, IL-10, IL-13, and IL-21, have been frequently reported to activate the JAK/STAT pathway, thereby inducing LMP1 gene expression through STAT (23–28). In certain latency II infected cells, including NPC cells (29), LMP1 transcription originates from a STAT regulated upstream promoter, termed TR-L1p, located within the terminal repeats (TRs), in addition to the proximal ED-L1p (23, 24, 27, 30, 31). We previously identified a CCAAT enhancer-binding protein (C/EBP) family transcription factor that augments both proximal and distal promoter activation of LMP1 in type II latency by binding to a sequence motif in the proximal promoter (32).
Elsewhere, the involvement of transcriptional factors, such as NF-κB (33, 34), AP-2 (35), POU domain protein (17), ATF/CREB (36), Sp1/3 (37), and IRF7 (38), has been observed. Type I interferons were also reported to upregulate LMP1 expression, presumably through NF-κB, PKC, and JNK in Burkitt lymphoma cells (39).
Despite the presence of these well-targeted, focused reports, functional testing of the cis (and trans) elements in the context of virus genomes has not received sufficient attention since most mutagenesis studies have analyzed the importance of transcription factor binding sites in reporter assays.
AP-2 is a family of transcription factors containing a helix-span-helix motif for DNA binding at the carboxyl terminus with possible roles in development, the control of apoptosis and cell cycling, and oncogenesis (40, 41). Its members are clearly distinct from AP-1 family transcription factors, homo/heterodimers composed of c-Fos, c-Jun, or ATF, which share a b-Zip motif for dimerization and DNA binding. Moreover, AP-2 proteins can bind to G/C-rich elements, such as 5′-[G/C]CCN(3,4)GG[G/C]-3′ (41, 42).
Early B cell factor (EBF) is a transcription factor that contains a helix-loop-helix motif, which binds to the G/C-rich motif, 5′-CCCNNGGG-3′. It is expressed in B cell lineages and is a master regulator of early B cell differentiation (43, 44).
In the present study, we applied small interfering RNA (siRNA)-mediated knockdown and/or overexpression and showed that AP-2 and EBF play important roles in EBNA2-independent and -dependent LMP1 expression, respectively. The introduction of mutations into the AP-2/EBF binding sites in the promoter of recombinant EBV inhibited B cell transformation efficiency. Taken together, we observed a crucial role of AP-2 and EBF in LMP1 expression in both type II and type III latency.
MATERIALS AND METHODS
Cell culture and reagents.
HEK293EBV-BAC and HeLa-CR2/GFP-EBV (32) cells were maintained in Dulbecco modified Eagle medium (Sigma) supplemented with 10% fetal bovine serum. Akata(−), C666-1 (45), and LCLs were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. Antibodies against SP1 and PAX5 were obtained from Santa Cruz Biotechnology. Anti-AP-2α, -FLAG, -myc, and -tubulin antibodies were purchased from Abcam, Sigma, MBL, and Cell Signaling Technology, respectively. The anti-LMP1 and EBNA2 antibodies have been described previously (46). Horseradish peroxidase-linked goat antibodies to mouse/rabbit IgG were obtained from Amersham Biosciences. The expression vectors, pcDNAFLAGhTFAP-2α (47), pcDNAPAX5 (48), and pcDNAmycEBF1 (49) were provided by K. Miyazono, F. Hayakawa, and M. Sigvardsson, respectively.
Genetic manipulation of EBV-BAC DNA and cloning of HEK293 cells with EBV-BAC.
EBV-BAC DNA was provided by W. Hammerschmidt (50). Homologous recombination was carried out in Escherichia coli as described previously (32, 51).
To prepare EBV-BAC mutants, a transfer DNA fragment for the first recombination was generated by PCR using rpsL-neo (Gene Bridges) as the template, with Neo/stFor (TGCCGCCAACGACCTCCCAACGTTGCGCGCCCCGCGCCTCTTTGTGCAGATTACACTGCCGGCCTGGTGATGATGGCGGGATC) and Neo/stRev (CAGTGTGAGAGGCTTATGTAGGGCGGCTACGTCAGAGTAACGCGTGTTTCTTGGGATGTATCAGAAGAACTCGTCAAGAAGG) primers. After recombination, kanamycin-resistant colonies were selected and checked by colony PCR using the primers TAGTCCTGCCTTTCCATTTCCTG and GTCTCAGAAGGGGGAGTGCGTAG to generate intermediate DNA. The Neo/st cassette in the intermediate DNA was then replaced using the next transfer vector DNA, containing each mutation in the LMP1 promoter. The AP-2 binding motif at −75 (CCCCCCGGGCCTAC) was modified to CCCCCCTTTCCTAC. Motifs TGCCTCCGGCAGA (−100) and GCCCCCCGGGGACCCGC (−205) were edited to TGCCTAATTCAGA and GCCCAAATGGGACCCGC, respectively. Electroporation of E. coli was performed using a Gene Pulser III (Bio-Rad), and purification of EBV-BAC DNA was achieved with NucleoBond Bac100 (Macherey-Nagel). Recombination was confirmed with PCR products of the promoter region, by electrophoresis of the BamHI-digested viral genome and sequencing analysis. EBV-BAC DNA was transfected into HEK293 cells using Lipofectamine 2000 reagent (Invitrogen), followed by culture on 10-cm dishes with 100 to 150 μg of hygromycin B/ml for 10 to 15 days to clone green fluorescent protein (GFP)-positive cell colonies as described previously (51). Briefly, for each recombinant virus, we picked up more than 10 hygromycin-resistant, GFP-positive cell colonies to obtain at least three typical clones exhibiting minimal spontaneous expression of viral lytic proteins and significant induction of these proteins upon BZLF1 transfection.
Transfection and immunoblotting.
Transfections were carried out by lipofection using Lipofectamine 2000 reagent (Invitrogen) or by electroporation using a Microporator (Digital Bio). The total amount of plasmid DNA was standardized by the addition of an empty vector. Knockdown of AP-2 was performed by transfection of duplexes of 21-nucleotide siRNAs. The sense and antisense sequences for the siRNAs were as follows: si-control (GCAGAGCUGGUUUAGUGAAdTdT and UUCACUAAACCAGCUCUGCdTdT), si-AP-2α1 (CCGAAUUUCCUGCCAAAGCdTdT and GCUUUGGCAGGAAAUUCGGdTdT), and si-AP-2α2 (CGCCAAAAGCAGUGACAAAdTdT and UUUGUCACUGCUUUUGGCGdTdT). Immunoblotting was carried out as described previously (52).
Quantitative real-time RT-PCR (qRT-PCR).
Total cell RNA was purified using the TriPure isolation reagent (Roche) and subjected to reverse transcription and real-time PCRs by using a One-Step SYBR PrimeScript RT-PCR Kit II (TaKaRa) and a real-time PCR System 7300, as described previously (53), except that the 40-s extension period at 60oC was extended to 70 s for detecting long species of LMP1 mRNA expressed from the TR-L1 promoter. The primers used for the qRT-PCR of the GAPDH, BZLF1, and EBNA2 genes were as follows: GAPDH, (TGCACCACCAACTGCTTAGC and GGCATGGACTGTGGTCATGAG), BZLF1 (AACAGCCAGAATCGCTGGAG and GGCACATCTGCTTCAACAGG), and EBNA2 (TTAGAGAGTGGCTGCTACGCATT and TCACAAATCACCTGGCTAAG). The primers used to distinguish distal (TR-L1) and proximal (ED-L1) primers were as follows: TR-L1 (C666-1), TACGGTTACCCCACAGCCTT and TGAGTAGGAGGGTGATCATC; TR-L1+ED-L1 (C666-1), CTATTCCTTTGCTCTCATGC and TGAGTAGGAGGGTGATCATC; TR-L1 (B95-8), TACGGTTACCCCACAGCCTT and TGAGCAGGAGGGTGATCATC; and TR-L1+ED-L1 (B95-8), CTATTCCTTTGCTCTCATGC and TGAGCAGGAGGGTGATCATC.
EMSA and chromatin immunoprecipitation (ChIP).
Electromobility shift assay (EMSA) was carried out as described previously (54). PAX5, FLAG-tagged AP-2α, and myc-tagged EBF proteins were produced using the TNT Quick-Coupled transcription/translation system (Promega) according to the manufacturer's instructions. Probe DNAs were prepared by hybridization of the sense and antisense oligonucleotides listed below. Because the DNAs have 5′ protruding ends, they could be labeled by 3′-end labeling using the Klenow fragment (Toyobo) and [32P]dCTP (Institute of Isotopes Co., Hungary). Unincorporated deoxynucleotide triphosphates were removed with Chromaspin-10 columns (Clontech). The in vitro-translated FLAG-tagged AP-2α protein and labeled DNA sequences were incubated in the EMSA binding buffer [20 mM Tris-HCl (pH 7.6), 1 mM EDTA, 1 mM dithiothreitol, 5% glycerol, 80 mM NaCl, and 0.5 mg of poly(dI-dC)/ml] at room temperature for 30 min. The composition of the EMSA binding buffer for PAX5 was as follows: 20 mM Tris-HCl (pH 7.9), 0.5 mM EDTA, 0.5 mM dithiothreitol, 10% glycerol, 6.25 mM MgCl2, 0.5 mg of poly(dI-dC)/ml, and 0.01% NP-40. The composition of the EMSA buffer for EBF was as follows: 10 mM HEPES (pH 7.9), 70 mM KCl, 1 mM dithiothreitol, 1 mM EDTA, 2.5 mM MgCl2, 10% glycerol, and 0.5 mg of poly(dI-dC)/ml. The samples were then separated in a 4% nondenaturing polyacrylamide gel in 0.5×Tris-borate-EDTA buffer, and the radioactivity was visualized. The sense and antisense sequences of oligonucleotide probes I to V were as follows: I, TGAATCCGCCACCTCATTCTGAAATTCCCATATCCGCCGTCTGCTGCTTCGTCACCCGCCGACCCTTAGCCCTCTTAGCCGCCTCACCCGCCTCCCCTACGGTTACCCCACAGCCTTGCCTCACCTGAAC and GGGGGTTCAGGTGAGGCAAGGCTGTGGGGTAACCGTAGGGGAGGCGGGTGAGGCGGCTAAGAGGGCTAAGGGTCGGCGGGTGACGAAGCAGCAGACGGCGGATATGGGAATTTCAGAATGAGGTGGCGGAT; II, CCTGAACCCCCCTAAAGCACGGCCTCCCGCCTGCCGCCAACGACCTCCCAACGTTGCGCGCCCCGCGCCTCTTTGTGCAGATTACACTGCCGCTTCCCACAACACTACGCACTCCCCCTTCTGATTGCCGCACTG and GGCAGTGCGGCAATCAGAAGGGGGAGTGCGTAGTGTTGTGGGAAGCGGCAGTGTAATCTGCACAAAGAGGCGCGGGGCGCGCAACGTTGGGAGGTCGTTGGCGGCAGGCGGGAGGCCGTGCTTTAGGGGG; III, CCGCACTGCCTTTCCATTTCCTGTTGCACTTGGCCACCGCATTCCCACAGCTTGCCCCCCGGGGACCCGCTTTTCTAACACAAACACACGCTTTCTACTTCCCCTTTCTACGCTTACATGCACACACA and GGTGTGTGTGTGCATGTAAGCGTAGAAAGGGGAAGTAGAAAGCGTGTGTTTGTGTTAGAAAAGCGGGTCCCCGGGGGGCAAGCTGTGGGAATGCGGTGGCCAAGTGCAACAGGAAATGGAAAGGCAGTG; IV, CACACACACACCGCCGCTTTCGGGAAATCTGTACCCGTACTGCCTCCGGCAGACCCCGCAAATCCCCCCGGGCCTACATCCCAAGAAACACGCGTTACTCTGACGTAGCCGCCCTACATAAGCCTCTCACACTG and GAGCAGTGTGAGAGGCTTATGTAGGGCGGCTACGTCAGAGTAACGCGTGTTTCTTGGGATGTAGGCCCGGGGGGATTTGCGGGGTCTGCCGGAGGCAGTACGGGTACAGATTTCCCGAAAGCGGCGGTGT; and V, CACACTGCTCTGCCCCCTTCTTTCCTCAACTGCCTTGCTCCTGACACACTGCCCTGAGGATGGAACACGACCTTGAGAGGGGCCCACCGGGCCCGCGACGGCCCCCTCGAGGACCCCCCCTCTCCTCTTCCCTAGG and GGCCTAGGGAAGAGGAGAGGGGGGGTCCTCGAGGGGGCCGTCGCGGGCCCGGTGGGCCCCTCTCAAGGTCGTGTTCCATCCTCAGGGCAGTGTGTCAGGAGCAAGGCAGTTGAGGAAAGAAGGGGGCAGAG. The sequences for probes III-1 to III-4 and probe iv were as follows: III-1, CCGCACTGCCTTTCCATTTCCTGTTGCACTTGGCCAC and GCGGTGGCCAAGTGCAACAGGAAATGGAAAGGCAGTGCGG; III-2, TGGCCACCGCATTCCCACAGCTTGCCCCCCGGGGACCCG and AGCGGGTCCCCGGGGGGCAAGCTGTGGGAATGCGGTGG; III-3, GGGACCCGCTTTTCTAACACAAACACACGCTTTCTACTT and GGAAGTAGAAAGCGTGTGTTTGTGTTAGAAAAGCGGGTC; III-4, TTCTACTTCCCCTTTCTACGCTTACATGCACACA and TGTGTGTGTGCATGTAAGCGTAGAAAGGGGAAGTAGAA; and iv, TACCCGTACTGCCTCCGGCAGACCCCGCAAATCCCCCCGGGCCTACATCCCAAGAAACA and GCGTGTTTCTTGGGATGTAGGCCCGGGGGGATTTGCGGGGTCTGCCGGAGGCAGTACGGGTA. ChIP assays were carried out as described previously (32).
B cell transformation assay.
First, wild-type or mutant EBVs were collected from wild-type or mutant HEK293EBV-BAC cell supernatants. Virus titers in the media were determined by infecting Akata(−) cells, followed by counting the percentage of enhanced GFP (EGFP)-positive cells using flow cytometry (FACSCalibur; Becton Dickinson). Titers were normalized according to the percentages by adding control media. Peripheral blood monocytes (PBMCs) were infected with 10-fold dilutions of adjusted culture supernatant media obtained from wild-type or mutant HEK293EBV-BAC cells and seeded onto 96-well plates at 105 cells. For PBMCs, blood samples were obtained from healthy adult donors who provided written informed consent according to protocols approved by the Institutional Review Board of Aichi Cancer Center and Nagoya University. Cells were cultured in the presence of cyclosporine. Half of the medium was exchanged once a week with fresh medium containing cyclosporine. After 3 weeks, 50% transforming doses were calculated.
RESULTS
Preparation of mutant EBVs in the proximal LMP1 promoter.
Despite numerous reports on transcriptional activators of the major EBV oncogene LMP1, the significance of cis-acting binding sites for such factors has been analyzed mostly using reporter assays. Since these assays do not necessarily or proportionally reflect the actual transcriptional levels in the genome, the confirmation of cis elements in the viral genome is important. Therefore, we first prepared recombinant EBVs carrying mutations in the proximal ED-L1 LMP1 promoter, as shown in Fig. 1. We constructed mutants of NF-κB, RBP-Jκ, C/EBP, PU.1, and POU domain factor (Table 1) (12, 32–34). As shown in Fig. 1A, a part of the LMP1 ED-L1 promoter sequence (−360 to −11), containing the cis-acting binding sites of reported transcription factors, was first replaced with a marker cassette (Neo/st), which was then exchanged with each sequence with a mutation (marked by an “X” in Fig. 1A). Sequencing analysis confirmed that each of the EBV-BACmt DNA sequences contained the intended mutations. Integrity of the BAC DNA was checked by BamHI digestion, followed by electrophoresis to confirm that the recombinant viruses did not carry obvious deletions or insertions (Fig. 1B). Recombinant EBV-BAC DNA was introduced into a virus-producing cell line, HEK293, followed by hygromycin selection to establish cell lines in which recombinant viruses were maintained as episomes.
FIG 1.

Construction of recombinant EBV featuring a mutation in the transactivator binding site of the LMP1 promoter. (A) Schematic arrangement of the recombination of the EBV genome using the tandemly arranged neomycin resistance and streptomycin sensitivity genes (Neo/st). Sequences of the B95-8 ED-L1 LMP1 promoter (−360 to −11) were first replaced with the Neo/st cassette, which was then replaced with mutated sequences (ringed “X”) to construct EBV-BAC mt. (B) Electrophoresis of the recombinant virus genomes. Recombinant EBV genomes were digested with BamHI and separated in an agarose gel.
TABLE 1.
Wild-type and mutated sequences of EBV-BAC
| Protein | Sequence (5′–3′)a |
|
|---|---|---|
| Wild type | Mutation | |
| C/EBP | tctgATTGCCGCACtgcc | tctgAGACTAGTCCtgcc |
| RBP-Jκ | ccgcTTCCCACAacac | ccgcTTCCAGAAacac |
| POU, PAX5 | ttacATGCACACacac | ttacACTAGAACacac |
| NF-κB | ccccGCAAATCCCCccgg | ccccGCGGGTCCCcccgg |
| PU.1 | ctacTTCCCCTttct | ctacGCGTCCTttct |
| AP2,EBF(−75) | CCCCCCGGGcctac | CCCCCCTTTcctac |
| AP2(−100) | tGCCTCCGGCaga | tGCCTAATTCaga |
| AP2,EBF(−205) | gcCCCCCGGGGacccgc | gcCCAAATGGGacccgc |
Mutations are indicated by boldface latters. Uppercase and lowercase letters indicate binding motifs and the surrounding sequences, respectively.
Attenuation of transformation efficiency by mutations in the POU factors and PU.1 binding sites.
After preparing HEK293 cell clones with mutant EBVs, we explored whether mutations could affect the expression of LMP1. Since EBNA2 is not produced in HEK293EBV-BAC cells (32), it is clear that the virus produces LMP1 in an EBNA2-independent manner in HEK293 cells. Levels of LMP1 protein were comparable overall (Fig. 2A). We next examined the effect of mutations in the LMP1 promoter of EBV during type III latency when LMP1 is produced in an EBNA2-dependent manner. To accomplish this, B cells were infected with mutant viruses. Prior to infection, we measured viral titers in supernatant solutions using Akata(−) cells to adjust the infectious virus particle numbers per milliliter. After adjustment, viruses in the media were cocultured with PBMC B cells in the presence of cyclosporine for 3 weeks. The wild-type EBV-BAC virus could produce 3.9 × 102 clumps per ml (Fig. 2B). Unexpectedly, all mutant viruses could transform B cells almost as efficiently as the wild-type virus, except for relatively lower efficiencies with the POU factor binding site and PU.1 binding site mutants (2.0 × 102 and 1.6 × 102 per ml, respectively) (Fig. 2B). However, we assume that the actual effect of PU.1 mutation and POU factor mutation is more significant than the calculated result of severalfold repression of the transformation unit (Fig. 2B), because the sizes of the cell clumps formed by the PU.1 or POU mutant virus were markedly smaller than in other cases (data not shown). To test this hypothesis, the growth properties of LCL clones were determined (Fig. 2C). We examined two clones of each mutant, but only one clone was tested for the PU.1 mutant because we could not obtain more than one clone, probably due to the low growth rate of the strain. Compared to the wild type (Fig. 2C, black circles), two POU mutant clones and one PU.1 clone grew significantly slower (Fig. 2C, diamonds and an asterisk). In addition, we analyzed the levels of LMP1 in the LCLs by Western blotting (Fig. 2D). We did not observe a marked difference in LMP1 levels in the LCL clones shown here, but mutation of POU domain factors and PU.1 might result in a mild decrease (Fig. 2D). These results implied that binding of PU.1 and POU domain factors to the proximal LMP1 promoter plays a role in EBNA2-dependent LMP1 expression in B cells. However, this does not mean that NF-κB, RBP-Jκ, and C/EBP are not important because we could disrupt only one “major” site according to reporter assays for each factor, and more than one binding site may exist in the LMP1 promoter.
FIG 2.

Effect of mutations in HEK293 and LCLs. (A) LMP1 protein levels in HEK293 cells with mutant EBV-BACs in the transactivator binding site of the LMP1 promoter. Immunoblotting was performed using anti-LMP1 and -tubulin antibodies. Independent cell clones that latently maintain EBV-BAC were obtained by transfection of each mutant DNA, and the LMP1 levels of two or three typical clones were examined. (B) Transformation efficiency of recombinant EBVs carrying mutations in the transactivator binding site of the LMP1 promoter. Viruses obtained from different clones of wild-type or mutant HEK293EBV-BACs were normalized based on the data of EGFP-positive Akata ratios and infected with PBMCs in the presence of cyclosporine. Three weeks later, transformation units were determined. The mean and standard deviation (SD) values of three independent assays are shown. A Student t test was performed, but statistical significances between WT and any of the mutants are not indicated. (C) Growth properties of LCLs. LCLs (35 × 104 cells/ml) prepared in panel B were seeded and, after 3 and 8 days, the cell numbers were counted. (D) Levels of LMP1 in LCL clones. Independent one or two LCL clones obtained in panel B were subjected to immunoblotting with anti-LMP1 and -tubulin antibodies.
A previous report (17) showed that, in addition to the POU domain factor (termed Dα1), unidentified host factor (termed Dα2) also binds to the POU domain site within the ED-L1 promoter. Thus, we searched for this unknown factor and found that a paired box family transcription factor, PAX5 (or B-cell lineage-specific activator protein [BSAP]) binds to this motif (Fig. 3). To identify the binding site in the proximal LMP1 promoter (ED-L1p), the 635-bp region was divided into five overlapping nucleotide sequences and used as probes (Fig. 3A, probes I to V) for EMSA; probe III was targeted most efficiently by PAX5 (Fig. 3B, leftmost panel, white arrowhead). Addition of an antibody against PAX5 removed the PAX5-probeIII band, indicating that binding between the two is specific (Fig. 3B, second panel from the left). Probe III was further divided into four fragments (Fig. 3A, probes III-1 to III-4), and PAX5 was confirmed to bind to probe III-4 (Fig. 3B, third panel, white arrowhead). When the same point mutation introduced into the POU domain factor was introduced into the probe III-4 (Table 1 and Fig. 1 and 2), PAX5 binding was diminished (Fig. 3B, rightmost panel). This POU site (TGTGCATG [antisense]) contains a sequence similar to the PAX5 consensus sequence (GC[A,G]TG). Therefore, it is highly likely that the previously unidentified host factor in B cell lysate that binds to the POU domain factor site in ED-L1p (Dα2) is PAX5. Interestingly, multiple copies of PAX5 reportedly target the TR of EBV and negatively regulate LMP1 transcription in B cells (55, 56). This is in agreement with previous speculation that Dα2 is a negative regulator of LMP1 expression (17). When these reports and our results are taken into consideration, regardless of whether ED-L1p or TR-L1p, PAX5 binding to the LMP1 promoter region may negatively regulate LMP1 transcription. However, in our mutagenesis experiment, the positive effect of POU factor binding might be greater than the negative effect of PAX5 binding to the same motif (Fig. 2B to D).
FIG 3.

Binding of PAX5 to the LMP1 promoter. (A) Schematic illustration of the LMP1 promoter and the probes (I to V and III-1 to III-4) used in EMSA. (B) EMSA was carried out as described in Materials and Methods. PAX5 protein was produced in vitro and incubated with 32P-labeled probes. Probes I to V cover sequences from positions −514 to −381, −391 to −255, −264 to −133, −141 to −8, and −13 to +121 of the LMP1 promoter, relative to the transcription start, respectively. Samples were then separated in a 4% polyacrylamide gel and analyzed by autoradiography. Supershift analysis was performed using a mouse anti-PAX5 monoclonal antibody (+α-PAX5, second panel from the left). Addition of the antibody caused the band to disappear but not to supershift probably because binding of the antibody influenced the DNA-binding activity of PAX5. Further fragmentation of the probe III was performed (III-1 to III-4), and the resulting fragments were used in an EMSA (third panel). Lastly, a mutant probe for POU binding was assessed (mPOU, rightmost panel). White arrowheads indicate bands specific for DNA-PAX5.
Effect of AP-2 on LMP1 production.
We then applied the knockdown method to examine the importance of specific transcription factors for LMP1 expression in infected cells. We first tried to ablate NF-κB, RBP-Jκ, and PU.1, which proved to be difficult, probably because these factors play essential roles in cell fate. Instead, we focused on the AP-2α protein, since Rymo's group suggested the involvement of AP-2 (or an unknown host protein that binds to the AP-2 motif in ED-L1p) by mutagenesis of ED-L1p using reporter assay systems in EBNA2-dependent LMP1 expression in B cells (35). To further characterize the role of AP-2 in LMP1 expression, we explored whether the transcription factor could induce LMP1 in an epithelial NPC cell line C666-1, in which LMP1 is expressed in the absence of EBNA2 (Fig. 4A to D). When AP-2α, a typical member of the AP-2 family (40), was exogenously overexpressed, mRNA levels of LMP1 were induced, as expected in the NPC cell line (Fig. 4A). We examined mRNA but not protein levels of LMP1, since LMP1 protein is not detectable in this cell line (32). The results of qRT-PCR showed that exogenous expression of AP-2α did not affect the level of LMP1 transcription from TR-L1p, but it resulted in a 3.2-fold induction of LMP1 (Fig. 4B, TR+ED).
FIG 4.

Activation of LMP1 expression by AP-2α in C666-1 and LCLs. (A and B) A nasopharyngeal carcinoma cell line C666-1 was transfected with empty vector or the FLAG-tagged AP-2α expression vector (flag AP-2α). Three days after transfection, the cells were harvested and subjected to RT-PCR for detection of LMP1 and GAPDH gene expression and for immunoblotting with anti-FLAG and -tubulin antibodies (A). Parts of the RNA samples were subjected to qRT-PCR to examine promoter usage (B). (C and D) C666-1 cells were transfected with control siRNA (si control) or the siRNA against AP-2α (si AP2α-1,2). Three days after transfection, the cells were harvested and subjected to RT-PCR for LMP1 and GAPDH (C) and immunoblotting with anti-AP-2α and -tubulin antibodies (C). Parts of the RNA samples were subjected to qRT-PCR to examine promoter usage (D). (E and F) LCLs were transfected with empty vector or the FLAG-tagged AP-2α expression vector (flag AP2α). Three days after transfection, the cells were harvested and subjected to immunoblotting using anti-LMP1, -FLAG, and -tubulin antibodies (E) and qRT-PCR to examine promoter usage (F). Three independent samples were assayed and Student t test was performed. *, P < 0.05; **, P < 0.02.
Knockdown experiments were performed using two siRNAs for AP-2α (si AP-2α-1,2). Either of the siRNAs clearly ablated protein levels of AP-2α in the C666-1 cell line (Fig. 4C), and the reductions were correlated with decreases in LMP1 expression levels (Fig. 4C and D). The effect of si AP-2α-1 on LMP1 expression was less potent than si AP-2α-2, for unknown reasons. Notably, ablation of AP-2α from the NPC cells caused a significant loss of LMP1 transcription from TR-L1p (Fig. 4D), which suggested that an ectopic excess supply of AP-2 most obviously activates proximal ED-L1p, but not TR-L1p (Fig. 4B), although endogenous levels of the transcription factor activate TR-L1p (Fig. 4D). These results indicated that AP-2 is a crucial determinant of type II LMP1 production and that natural levels of AP-2 activate the distal LMP1 promoter, at least in NPC cells latently infected with EBV.
Although previous study reported that AP-2 may not play a central role in LMP1 expression in latency III B cells because B cells express very low levels of AP-2 (35), we tested the effect of overexpression of AP-2 on LMP1 expression in LCLs. Ectopic expression of FLAG-AP-2α was lower than that in C666-1, but it could also induce LMP1 protein in the latency III B cells (Fig. 4E). Quantitation of LMP1 mRNAs indicated that the ED-L1, but not the TR-L1 promoter, was activated by exogenous expression of FLAG-AP-2α, although the enhancement was less prominent in LCLs (Fig. 4F).
Identification of AP-2-binding elements in the LMP1 promoter.
After confirming the importance of AP-2 for LMP1 expression, we used EMSA to examine whether the AP-2α protein could bind to motifs in the LMP1 promoter (Fig. 5). Here, we used the same probes (probes I to V) as in Fig. 3. The addition of FLAG-AP-2α did not produce any DNA-AP-2 complexes in the case of probes I and II, and a very weak signal may have occurred for probe V (Fig. 5B). On the other hand, probe III yielded a prominent band of the AP-2α-nucleotide complex (Fig. 5B, white arrowhead), and the band shifted up in the presence of the anti-Flag antibody (Fig. 5B). Probe IV yielded two discernible bands (Fig. 5B, black arrowheads), both of which were supershifted by specific antibody addition, indicative of binding of AP-2α to two possible elements. To locate the AP-2 binding sites, we searched the promoter region for G/C-rich sequences similar to the AP-2 consensus motif ([G/C]CCN(3,4)GG[G/C]) and identified one such element in probe III (−205, CCCCCGGGG) and two in probe IV (−75, CCCCCCGGG; −100, GCCTCCGGC). The introduction of mutations in the binding sites of the probes (Table 1) diminished binding to AP-2α (Fig. 5C, III′, and IV′), indicating that these are the actual AP-2 binding sites in ED-L1p.
FIG 5.
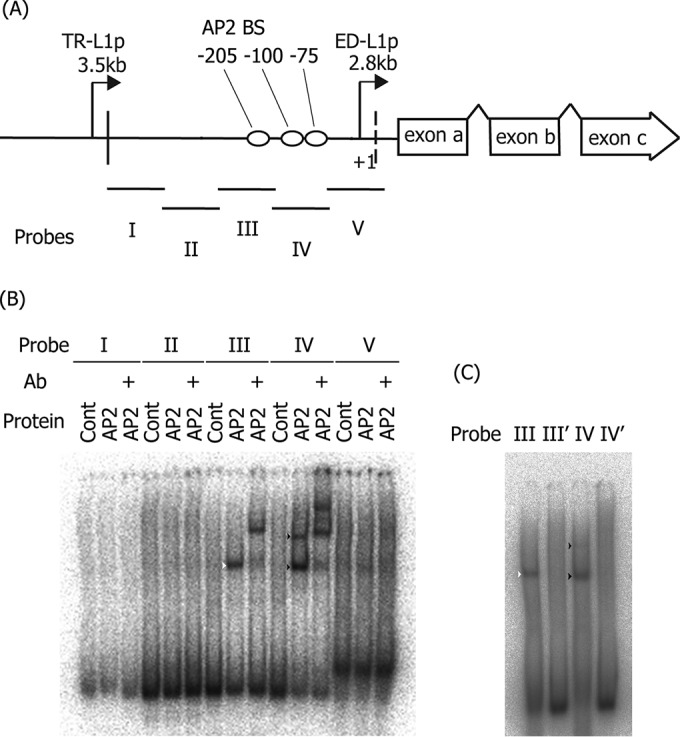
Binding of AP-2α to the LMP1 promoter. (A) Schematic illustration of the LMP1 promoter and the probes (I to V) used in EMSA. (B) EMSA was carried out as described in Materials and Methods. FLAG-tagged AP-2α was produced in vitro and incubated with 32P-labeled probes. Supershift analysis was performed using mouse anti-FLAG monoclonal antibody (+Ab). Samples were then separated in a 4% polyacrylamide gel and analyzed by autoradiography. (C) EMSA was carried out as described in panel B, except the AP-2α-binding motif in probes III and two motifs in IV were mutated to make the III′ and IV′ probes, respectively. White and black arrowheads indicate bands for the probe III-AP-2 and probe IV-AP-2 complexes, respectively.
Mutation at −75 in the LMP1 promoter had little effect on LMP1 expression.
To confirm the significance of these cis-acting binding motifs, we prepared recombinant EBV carrying mutations in the proximal ED-L1p. We first mutated AP-2 motif at −75, since reporter assays previously showed the importance of the site for LMP1 expression (33). Part of the LMP1 ED-L1p sequence (−360 to −11), containing the cis-acting binding sites of AP-2, was replaced with a marker cassette (Neo/st), which after was exchanged with the sequence containing a mutation (ringed “X” in Fig. 6A). Sequencing analysis confirmed that the EBV-BAC AP2(−75)mt DNA sequences contained the intended mutations. Integrity of the BAC DNA was examined based on BamHI digestion, followed by electrophoresis to confirm that the recombinant viruses did not carry obvious deletions or insertions (Fig. 6B). Recombinant EBV-BAC DNA was introduced into a virus-producing cell line, HEK293, followed by hygromycin selection to establish cell lines in which recombinant viruses were maintained as episomes.
FIG 6.

Effect of the mutation in the AP-2α-binding site (−75) of the LMP1 promoter. (A) Schematic arrangement of the recombination of the EBV genome using the tandemly arranged neomycin-resistance and streptomycin-sensitivity genes (Neo/st). Sequences of the ED-L1 LMP1 promoter (−360 to −11) were first replaced with the Neo/st cassette, which was then replaced with mutated sequences (ringed “X”) to construct EBV-BAC AP2(−75)mt. (B) Electrophoresis of the recombinant viruses. Recombinant EBV genomes were digested with BamHI and separated on an agarose gel. (C) HEK293 cell clones latently maintaining EBV-BAC wild-type (WT) or AP2(−75)mt were subjected to immunoblotting with anti-LMP1 and -tubulin antibodies. Independent cell clones that latently maintain EBV-BAC were obtained by transfection of each mutant DNA, and LMP1 levels of three typical clones were examined. (D) Effect of the mutation in the AP-2α-binding site (−75) of the LMP1 promoter on B cell transformation. Viruses obtained from WT or the mutant HEK293 EBV-BAC cells were normalized based on data of EGFP-positive Akata ratios and infected with PBMCs in the presence of cyclosporine. Twenty days later, transformation units were determined. The mean and SD values of three independent assays are shown. A Student t test was performed, but the statistical significance between WT and the mutant is not indicated. (E) Growth properties of LCLs. LCLs (20 × 104 cells/ml) prepared in panel D were seeded and, after 4 and 8 days, the cell numbers were counted. (F) Levels of LMP1 in LCL clones. Two independent LCL clones obtained in panel D were subjected to immunoblotting with anti-LMP1 and -tubulin antibodies.
After preparing HEK293 cell clones with wild-type and mutant EBV, we explored whether mutations could affect the expression of LMP1. Because EBNA2 is not produced in HEK293EBV-BAC cells, the virus produces LMP1 in an EBNA2-independent manner in HEK293 cells (32). The levels of LMP1 protein were comparable overall in HEK293 cells (Fig. 6C), indicating that the AP-2 binding motif at −75 does not play a major role in LMP1 expression in the cell line. B cells of human PBMCs were then infected with the wild-type and AP2(−75)mt viruses, and the transformation activity of the viruses was determined (Fig. 6D). The mutant virus showed the same degree of B cell immortalization efficiency as the wild-type virus. After the development of LCLs, the growth behavior (Fig. 6E) and the LMP1 protein (Fig. 6F) were examined, but no obvious differences were observed between the lines. Therefore, the AP-2 binding motif at position −75 of ED-L1p may not be required for the production of LMP1 either in latency II or III, unlike the previous report shown by reporter assays (33).
Mutation at positions −75, 100, and 205 in the LMP1 promoter caused a loss of LMP1 production.
Next, we prepared the mutant virus, in which all three AP-2-binding sites were mutated (−75, −100, and −205), and another virus, in which two sites (−100 and −205) were modified (Fig. 7A and B). We observed fluctuation to some extent, but the expression of LMP1 in wild type, AP2(−100,205)mt, and AP2(−75,100,205)mt was comparable in HEK293 (Fig. 7C). Infection of AP2(−100,205)mt EBV to human primary B cells for 21 days caused minor decreases in transformation compared with the wild type, and triple mutation (−75, −100, and −205) caused statistically significant reduction in transformation efficiency of about 1 order (Fig. 7D). In fact, when the triple-mutant virus was infected, the size of the cell clumps was markedly smaller compared to the wild type or even the double mutant (−100 and −205); thus, the effect of triple mutation was more profound than 1 order of magnitude. In agreement with this assumption, we could not further amplify and develop LCLs infected with the triple mutant, whereas we could readily prepare LCLs infected with wild-type or double-mutant EBVs. We speculate that the wild-type LMP1 promoter could resist forceful pressure of epigenetic gene silencing after 21 days because of transcriptional activation through AP-2 sites. On the other hand, the triple mutant could express LMP1 to some extent, but the loss of AP-2 binding sites caused silencing of LMP1 gene expression and an arrest in cell growth. Because LCLs with the triple mutation could not be obtained, we compared LCLs infected with the wild type and the double mutant, AP2(−100,205)mt (Fig. 7E and F). Cell growth of the double mutant was slightly slower (Fig. 7E), but LMP1 production of the mutant did not seem decreased, due to the fluctuation in wild-type samples (Fig. 7F).
FIG 7.

Effect of the mutations in the AP-2α-binding sites (−75, −100, and −205) of the LMP1 promoter. (A) Schematic arrangement of the recombination of the EBV genome using the tandemly arranged neomycin resistance and streptomycin sensitivity genes (Neo/st). The sequences of the ED-L1 LMP1 promoter (−360 to −11) were first replaced with the Neo/st cassette, which was then replaced with mutated sequences (ringed “X”) to construct EBV-BAC AP2(−100,205)mt and AP2(−75,100,205)mt. (B) Electrophoresis of recombinant viruses. The recombinant EBV genomes were digested with BamHI and separated on an agarose gel. (C) HEK293 cell clones latently maintaining EBV-BAC WT, AP2(−100,205)mt, or AP2(−75,100,205)mt were subjected to immunoblotting with anti-LMP1 and -tubulin antibodies. Independent cell clones that latently maintain EBV-BAC were obtained by transfection of each mutant, and LMP1 levels of three typical clones were examined. (D) Effect of the mutation in the AP-2α-binding sites (−100,205 or −75,100,205) of the LMP1 promoter on B cell transformation. Viruses obtained from WT or the mutant HEK293 EBV-BAC cells were normalized based on the data of EGFP-positive Akata ratios and infected with PBMCs in the presence of cyclosporine. Twenty days later, transformation units were determined, and the mean and SD values are shown. Three independent infections were assayed, and a Student t test was performed. *, P < 0.05. (E) Growth properties of LCLs. LCLs (20 × 104 cells/ml) prepared in (D) were seeded, and after 4 and 8 days, cell numbers were counted. (F) Two independent LCL clones obtained in panel D were subjected to immunoblotting with anti-LMP1 and -tubulin antibodies.
Since LCLs infected with the triple mutant could not be developed (Fig. 7E and F), cells were harvested at earlier time points and LMP1 expression levels were analyzed (Fig. 8A). At 2 days after the infection of PBMC B cells, LMP1 could not be detected in either the wild type or the triple mutant (Fig. 8A). This result is in accordance with a previous report that LMP1 expression is highly restricted for about 1 week after primary B cell infection (57). LMP1 expression from cells infected with the wild-type virus increased dramatically by day 13, and LMP1 mRNA levels were markedly lower in the case of the triple mutant, AP2(−75,100,205)mt (Fig. 8A). EBNA2 mRNA levels were relatively higher from 2 days after infection (Fig. 8B), which can be explained by a previous study (57). The immediate-early gene of the lytic infection cycle, BZLF1, was highest on day 2, which likely is a reflection of the prelatent abortive lytic phase (57, 58), and was silenced later (Fig. 8C). Importantly, levels of EBNA2 and BZLF1 expressed in wild-type samples on day 2 were almost equal to those in triple-mutant samples, indicating that the loss of LMP1 expression in the triple mutant (Fig. 8A) was not attributable to the difference in the multiplicity of infection of the infected viruses. We also confirmed these results in the double mutant virus (Fig. 8D and E). Viruses were prepared from HEK293 cells containing wild-type EBV-BAC (WT) or two independent HEK293 cell clones with the EBV-BAC triple mutant [AP2(−75,100,205)mt] or the double mutant [AP2(−75,100,205)mt]. PBMC B cells were infected with these viruses and RNA was harvested for qRT-PCR 7 days after infection. At 7 days, LMP1 mRNA levels were significantly reduced by the double mutation and were further decreased in the triple mutant (Fig. 8D); however, EBNA2 mRNA levels were comparable (Fig. 8E).
FIG 8.

Effect of the mutations in the AP-2α-binding sites (−75, −100, and −205) of the LMP1 promoter on primary B cell infection. (A to C) PBMC B cells were mock infected or infected with EBV-BAC WT or AP2(−75,100,205)mt, as described in Fig. 7D. Cellular RNA was collected at 2, 7, and 13 days after infection and subjected to qRT-PCR to detect the LMP1, EBNA2, BZLF1, and GAPDH genes. Relative mRNA levels were shown after normalization to GAPDH. (D and E) Likewise, B cells were infected with WT EBV-BAC or EBV-BAC viruses produced from two independent HEK293 clones of AP2(−75,100,205)mt and AP2(−100,205)mt. Cellular RNA was obtained on day 7 and subjected to qRT-PCR for detection of the LMP1, EBNA2, and GAPDH genes. Relative mRNA levels are shown after normalization to GAPDH. Three independent infections were assayed, and a Student t test was performed. **, P < 0.02.
These results indicated that the three AP-2 binding motifs act together to induce LMP1 expression and thereby immortalize B cells. Among the three binding sites, the distal two (−100 and −205) seemed to be more important for LMP1 expression than the most proximal one (−75, Fig. 6).
Mutation at −100 or 205 in the LMP1 promoter had little effect.
Having confirmed the importance of three AP-2 binding sites in the LMP1 promoter for LMP1 expression, particularly the distal two sites (−100 and −205), we next mutated the two sites separately, as shown in Fig. 9A and B. We did not observe a significant difference in LMP1 levels among the wild type, AP2(−100)mt, and AP2(−205)mt in HEK293 cells (Fig. 9C). In infected B cells, the levels of immortalization (Fig. 9D), cell proliferation (Fig. 8E), and LMP1 protein of the LCLs (Fig. 9F) were similar between the wild type and the mutants, although the cell growth rate may have been slightly slower in LCLs infected with AP2(−205)mt. Taken together, the transcription factor AP-2 is crucial for LMP1 expression in LCLs, and three AP-2 binding sites in the promoter contribute additively to LMP1 induction.
FIG 9.

Effect of mutations in the AP-2α-binding sites (−100 or −205) of the LMP1 promoter. (A) Schematic arrangement of the recombination of the EBV genome using the tandemly arranged neomycin resistance and streptomycin sensitivity genes (Neo/st). Sequences of the ED-L1 LMP1 promoter (−360 to −11) were first replaced with the Neo/st cassette, which was then replaced with mutated sequences (ringed X) to construct EBV-BAC AP2(−100)mt and AP2(−205)mt. (B) Electrophoresis of the recombinant viruses. The recombinant EBV genomes were digested with BamHI and separated on an agarose gel. (C) HEK293 cell clones latently maintaining EBV-BAC WT, AP2(−100)mt, or AP2(−205)mt were subjected to immunoblotting with anti-LMP1 and -tubulin antibodies. Independent cell clones that latently maintain EBV-BAC were obtained by transfection of each mutant, and LMP1 levels of three typical clones were examined. (D) Effect of the mutation in the AP-2α-binding sites (−100 or −205) of the LMP1 promoter on B cell transformation. Viruses obtained from WT or mutant HEK293 EBV-BAC cells were normalized based on the EGFP-positive Akata ratios and infected with PBMCs in the presence of cyclosporine. Twenty days later, transformation units were determined. The mean and SD values of three independent assays are shown. A Student t test was performed, but the statistical significance between WT and the mutant is not indicated. (E) Growth properties of LCLs. LCLs (20 × 104 cells/ml) prepared in panel D were seeded and, after 4 and 8 days, the cell numbers were counted. (F) Two independent LCL clones obtained in panel D were subjected to immunoblotting with anti-LMP1 and -tubulin antibodies.
Binding of EBF to the AP-2 motifs in ED-L1p.
Although the expression of AP-2α is weak in B lymphocytes, including Akata, P3HR1, and LCLs (data not shown) (35), the simultaneous mutation of three AP-2 binding sites clearly diminished LMP1 expression in LCLs (Fig. 7 and 8). Johannsen et al. reported that one of the G/C-rich AP-2 binding sites (−205 motif in the present study) was predicted to be bound by an unknown factor (termed LBF7) in B cell lysate (12). Therefore, we searched for this factor and found that two of the AP-2 binding sites in ED-L1p could also be targeted by the B cell-specific transcription factor EBF (Fig. 10). EBF bound to probes III and IV (Fig. 10A and B, left panel, white and black arrowheads), and this association was supershifted by the addition of anti-myc antibody (Fig. 10B, second panel, white and black arrowheads). Mutation of the −205 AP-2 motif in probe III (III′) or the −75 and −100 motifs in probe IV (IV′) prevented the binding between EBV and the DNA probes (Fig. 10B, third panel). Additional mutagenesis demonstrated that EBF binds to the −75 but not the −100 motif (Fig. 10B, rightmost panel). This is expected because the sequences of the −75 (CCCCCGGG) and −205 (CCCCCGGGG) motifs, but not of the −100 AP-2 motif (GCCTCCGGC), coincide with the EBF consensus sequence (CCCNNGGG). Indeed, Zhao et al. reported that the −205 motif in ED-L1p was targeted by EBF (59). To examine the importance of EBF, an expression vector harboring myc-tagged EBF was transfected into LCLs (Fig. 10C and D). The expression of myc-EBF increased the LMP1 protein level (Fig. 10C). Moreover, qRT-PCR analysis revealed that the proximal ED-L1 promoter, but not the distal TR-L1 promoter, was activated by exogenous EBF production, although the induction was modest (Fig. 10D). Most recently, Lu and others have shown that knockdown of EBF1 leads to a loss of LMP1 protein expression in LCLs (77). These results suggest that EBF plays an important role for transcription of LMP1 in B cells.
FIG 10.

Binding of EBF to the LMP1 promoter. (A) Schematic illustration of the LMP1 promoter and the probes (I to V and iv) used in EMSA. (B) EMSA was carried out as described in Materials and Methods. myc-tagged EBF protein was produced in vitro and incubated with 32P-labeled probes. Supershift analysis was performed using mouse anti-myc monoclonal antibody (α-myc, second panel). As shown in the third panel, EMSA was carried out likewise, except that the AP-2α-binding motif in probe III and two motifs in probe IV were mutated to produce the III′ and IV′ probes, respectively (third panel). Lastly, using probe iv (shorter than probe IV but covers both motifs −75 and −100), motifs were mutated one by one (rightmost panel). (C and D) Activation of LMP1 expression by EBF in LCLs. LCLs were transfected with empty vector or the myc-tagged EBF expression vector (myc EBF). Three days after transfection, the cells were harvested and subjected to immunoblotting with anti-LMP1, -myc, and -tubulin antibodies (C) and to qRT-PCR to examine promoter usage (D). Three independent samples were assayed, and a Student t test was performed. *, P < 0.05. (E) SP1 does not bind to AP-2 motifs. HEK293 cell clones latently maintaining EBV-BAC WT or AP2(−75,100,205)mt were subjected to ChIP assays using anti-AP2 and -SP1 antibodies. Levels of the LMP1 proximal promoter region precipitated were determined by qPCR and are shown as the percentage of input. Three independent samples were assayed, and a Student t test was performed. *, P < 0.05.
Because AP-2 binds to G/C-rich elements, reminiscent of SP1-binding motifs, we lastly determined whether these sites are bound by the transcription factor. ChIP assays further confirmed that AP-2 binding was inhibited in the triple mutant, whereas SP1 binding was unaffected (Fig. 10E).
DISCUSSION
In this study, we first explored the role of AP-2 based on exogenous overexpression and/or knockdown (Fig. 4). We then identified three AP-2 binding sites (−75, −100, and −205) in the proximal (ED-L1) LMP1 promoter (Fig. 5) and evaluated their significance in the context of the EBV-BAC system (Fig. 6 to 9). The results documented here show involvement of AP-2 binding sites in the upregulation of the LMP1 gene in both latency II and III. Interestingly, two of the AP-2 binding sites were bound by the B cell transcription factor EBF, too.
Among the three AP-2 sites in ED-L1p, Rymo's group predicted binding of AP-2 to two motifs (−75 and −100) in 2007 and confirmed binding of in vitro translated AP-2 to one of the motifs (−100) (35). These authors further showed that introduction of mutations into the AP-2 site (−100) markedly reduced EBNA2-mediated transcriptional activation of LMP1 as determined by luciferase assays. Nevertheless, they speculated that an unknown host factor other than AP-2 binds to the motif and mediates EBNA2-dependent expression of LMP1 since AP-2 protein levels are low in B cells. Next, Demetriades and Mosialos showed in 2009 that introduction of a point mutation into another AP-2 site (−75) significantly decreased proximal LMP1 promoter activity in latency III B95-8 cells, as determined by luciferase assays (33). Here, we confirmed the binding of AP-2 to two of the sites (−75 and −100) and identified an additional motif (−205) in ED-L1p. Introduction of point mutations into the three motifs simultaneously or individually in the context of the virus demonstrated the importance of all of these sites for LMP1 expression in latency III. However, the expression of the AP-2 protein is low in B cells. We examined binding of other transcription factors expressed in B cells and found that the B cell-specific transcription factor EBF can bind to at least two of the three AP-2 sites (−75 and −205). Indeed, this transcription factor has been shown to bind to the proximal LMP1 promoter by ChIP-seq (56), and more detailed analysis by EMSA by Zhao et al. demonstrated that EBF binds to the −205 AP-2 motif (59). Coenrichment of EBNA2 and EBF sites in LCLs in the ChIP-seq analysis and the luciferase assays further indicated the importance of EBF for EBNA2-dependent LMP1 expression in latency III B cells (56, 59). EBF binding to the −75 motif has not been reported to date, and the physiological role of these binding sites has not been analyzed using recombinant virus. Our results, in conjunction with previous reports (60–62), confirm the central role of EBF in EBNA2-dependent activation of the ED-L1 promoter in B cells.
In this study, we also confirmed the involvement of PU.1 and POU domain factors in LMP1 transcription (Fig. 2). On the other hand, mutations in the major binding site of RBP-Jκ, which have been more extensively studied with regard to EBNA2-dependent LMP1 expression, did not affect the B cell transformation efficiency (Fig. 2). We cannot preclude involvement of RBP-Jκ in EBNA2-mediated LMP1 expression since binding sites may be redundant for RBP-Jκ and a mutation in only one major motif may not be sufficient to inhibit expression. Likewise, contributions of NF-κB to type III LMP1 expression cannot be ruled out based on our data. Because PU.1 is a lymphocyte-specific transcription factor, whereas RBP-Jκ and NF-κB are ubiquitously expressed in various types of cells and tissues, PU.1 may account for the B cell specificity of latency III LMP1 expression (63, 64). In accordance with our result, Johannsen et al. reported that PU.1 plays a role in EBNA2-mediated LMP1 expression but also demonstrated, based on mutational analysis, that EBNA2 activation of the LMP1 promoter in B cells is partially dependent on the interaction with RBP-Jκ and is completely dependent on the interaction with PU.1 protein (12). Our functional library screening also identified PU.1 as a transcriptional activator of LMP1 (32). Zhao and Sample reported a role for the PU.1 binding site in the LMP1 promoter for promoter activation in the presence of EBNA2 (20). Therefore, it can be assumed that PU.1 protein and its binding site in the LMP1 promoter are very important for the EBNA2-dependent production of the EBV oncogene LMP1 in type III latency.
POU domain factors include Oct-1 and Oct-2. The expression of Oct-1 is ubiquitous, whereas that of Oct-2 is B cell specific. Because disruption of the POU domain binding motif within the LMP1 promoter inhibited LMP1 expression (Fig. 2), Oct-2 may also contribute to latency III LMP1 induction in B cells. However, Rymo's group showed that an unidentified factor (Dα1) belonging to the POU domain family, but distinct from Oct-1 and Oct-2, binds to the POU site in ED-L1p, because an antibody against POU domain proteins, but not antibodies against Oct-1 or Oct-2, supershifted the Dα1 band in EMSA (17). In addition to a POU factor (Dα1), these researchers also demonstrated that a negative factor binding to the POU motif, or in the vicinity thereof, within ED-L1p is expressed in B cells (17). We found here that PAX5, a master regulator of B cell function, development, and leukemogenesis, also binds to the POU site in the ED-L1 promoter. PAX5 has been shown to negatively regulate LMP1 transcription through binding to TR (55, 56). Knockdown of PAX5 in LCLs increased transcription of LMP1, indicating that PAX5 serves to suppress LMP1 expression. We speculate that the virus fine-tunes the expression of LMP1 by activating the promoter, on the one hand, and delicately suppressing it, on the other hand, because this oncogene might be toxic to cells when produced in excess (11).
It is interesting that mutations of a single binding site for either transcription factor that we tested had almost no effect (NF-κB, RBP-Jκ, and C/EBP) or only a moderate effect (PU.1 and POU) on LMP1 transcription. No single transcription factor's binding site in the ED-L1p was essential for LMP1 expression and B cell transformation, indicating robustness and redundancy of the LMP1 promoter.
Regarding latency II, we found that LMP1 levels were increased by AP-2 in NPC C666-1 (Fig. 4A to D) and HeLa-EBV cells (data not shown), which is convincing, since AP-2 proteins are abundantly expressed in these epithelial cells. In addition to C666-1 and HeLa cells, we confirmed that levels of AP-2α are high in HEK293 cells (data not shown), but the effect of AP-2 was weak in this cell line (Fig. 7C). In addition, levels of AP-2α in the SNK6 NK cell lymphoma line, in which LMP1 is highly expressed in an EBNA2-independent manner (65), were low (not shown). Therefore, levels of AP-2α do not necessarily correlate with LMP1 expression. We speculate that other transcription factors, such as other members of the AP-2, STAT (23–28), C/EBP (32), or E-box-binding proteins such as MAD and MAX (66) may account for this inconsistency. Several reports indicate that activation of the JAK/STAT pathway by some cytokines is of major importance in latency II (23–28).
The reasons for the low expression of LMP1 protein in C666-1 cells remain unclear. LMP1 protein in the cell may be unstable and easily degraded. Notably, LMP1 is reported to be degraded rapidly through the ubiquitin/proteasome-dependent pathway (67), and LMP1 degradation is specifically regulated in NPC cells (45). Furthermore, since LMP1 mRNA levels are low in C666-1 cells, LMP1 may be regulated prior to its translation. LMP1 mRNA levels are reportedly downregulated by EBV-encoded microRNAs, BARTs, which are abundantly expressed in C666-1 cells (68).
The activity of AP-2 proteins can be controlled not only based on protein abundance but also at the posttranslational level, such as through protein kinase A-mediated phosphorylation (69). Carcinogens, including nitrosamines in salted fish, have been reported to aggravate NPC (70, 71), and nitrosamines were found to activate PKA (72), probably inducing LMP1 in NPC. Other factors, such as growth factors or cytokines, are also feasible candidate modulators of PKA activity.
Overall, we confirmed the importance of AP-2 and EBF for LMP1 expression in latency II and III. Because LMP1 plays a major role in immortalization, development, metastasis, and malignancy of NPC (73–75), inhibition of AP-2 and EBF may offer an avenue to treat these cancers. A search for small molecules that inhibit LMP1 expression is under way (76).
ACKNOWLEDGMENTS
We thank W. Hammerschmidt, H. J. Delecluse, and S. W. Tsao for providing the EBV-BAC system, HEK293 cells, and C666-1 cells. We also are grateful to K. Miyazono, F. Hayakawa, and Sigvardsson for the AP-2α, PAX5, and EBF expression vectors.
This study was supported by grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (to T.T. [23390118 and 23114512], T.M. [15K08494], Y.N. [14J01982], and H.K. [25293109]); the Ministry of Health, Labor, and Welfare (to T.T.); the Japan Agency for Medical Research and Development (to H.K. [Practical Research Project for Rare/Intractable Diseases, 15ek0109098]); and partly by the Uehara Memorial Research Fund, the Takeda Science Foundation (to T.T. and T.M.), the Senshin Medical Research Foundation, the Kanae Foundation for the Promotion of Medical Science, the General Assembly of the Japanese Association of Medical Sciences (to T.M.), and a Grant for the Joint Research Program of the Institute for Genetic Medicine, Hokkaido University (to T.T., T.K., and H.Y.).
REFERENCES
- 1.Soni V, Cahir-McFarland E, Kieff E. 2007. LMP1 TRAFficking activates growth and survival pathways. Adv Exp Med Biol 597:173–187. doi: 10.1007/978-0-387-70630-6_14. [DOI] [PubMed] [Google Scholar]
- 2.Lam N, Sugden B. 2003. CD40 and its viral mimic, LMP1: similar means to different ends. Cell Signal 15:9–16. doi: 10.1016/S0898-6568(02)00083-9. [DOI] [PubMed] [Google Scholar]
- 3.Shair KH, Bendt KM, Edwards RH, Bedford EC, Nielsen JN, Raab-Traub N. 2007. EBV latent membrane protein 1 activates Akt, NFκB, and Stat3 in B cell lymphomas. PLoS Pathog 3:e166. doi: 10.1371/journal.ppat.0030166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kulwichit W, Edwards RH, Davenport EM, Baskar JF, Godfrey V, Raab-Traub N. 1998. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc Natl Acad Sci U S A 95:11963–11968. doi: 10.1073/pnas.95.20.11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lam N, Sugden B. 2003. LMP1, a viral relative of the TNF receptor family, signals principally from intracellular compartments. EMBO J 22:3027–3038. doi: 10.1093/emboj/cdg284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dirmeier U, Hoffmann R, Kilger E, Schultheiss U, Briseno C, Gires O, Kieser A, Eick D, Sugden B, Hammerschmidt W. 2005. Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 24:1711–1717. doi: 10.1038/sj.onc.1208367. [DOI] [PubMed] [Google Scholar]
- 7.Kilger E, Kieser A, Baumann M, Hammerschmidt W. 1998. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J 17:1700–1709. doi: 10.1093/emboj/17.6.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kieser A, Kaiser C, Hammerschmidt W. 1999. LMP1 signal transduction differs substantially from TNF receptor 1 signaling in the molecular functions of TRADD and TRAF2. EMBO J 18:2511–2521. doi: 10.1093/emboj/18.9.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paine E, Scheinman RI, Baldwin AS Jr, Raab-Traub N. 1995. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-κB/Rel family proteins. J Virol 69:4572–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, Kikutani H. 1999. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 286:300–303. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- 11.Ito T, Kawazu H, Murata T, Iwata S, Arakawa S, Sato Y, Kuzushima K, Goshima F, Kimura H. 2014. Role of latent membrane protein 1 in chronic active Epstein-Barr virus infection-derived T/NK-cell proliferation. Cancer Med 3:787–795. doi: 10.1002/cam4.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman SR. 1995. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by Jκ and PU.1. J Virol 69:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laux G, Adam B, Strobl LJ, Moreau-Gachelin F. 1994. The Spi-1/PU.1 and Spi-B ets family transcription factors and the recombination signal binding protein RBP-Jκ interact with an Epstein-Barr virus nuclear antigen 2 responsive cis-element. EMBO J 13:5624–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the Jκ recombination signal binding protein. Proc Natl Acad Sci U S A 91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waltzer L, Logeat F, Brou C, Israel A, Sergeant A, Manet E. 1994. The human Jκ recombination signal sequence binding protein (RBP-Jκ) targets the Epstein-Barr virus EBNA2 protein to its DNA responsive elements. EMBO J 13:5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henkel T, Ling PD, Hayward SD, Peterson MG. 1994. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein Jκ. Science 265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- 17.Sjoblom A, Jansson A, Yang W, Lain S, Nilsson T, Rymo L. 1995. PU box-binding transcription factors and a POU domain protein cooperate in the Epstein-Barr virus (EBV) nuclear antigen 2-induced transactivation of the EBV latent membrane protein 1 promoter. J Gen Virol 76(Pt 11):2679–2692. [DOI] [PubMed] [Google Scholar]
- 18.Sjoblom A, Nerstedt A, Jansson A, Rymo L. 1995. Domains of the Epstein-Barr virus nuclear antigen 2 (EBNA2) involved in the transactivation of the latent membrane protein 1 and the EBNA Cp promoters. J Gen Virol 76(Pt 11):2669–2678. [DOI] [PubMed] [Google Scholar]
- 19.Harada S, Kieff E. 1997. Epstein-Barr virus nuclear protein LP stimulates EBNA-2 acidic domain-mediated transcriptional activation. J Virol 71:6611–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao B, Sample CE. 2000. Epstein-barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an spi-1/Spi-B binding site. J Virol 74:5151–5160. doi: 10.1128/JVI.74.11.5151-5160.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin J, Johannsen E, Robertson E, Kieff E. 2002. Epstein-Barr virus nuclear antigen 3C putative repression domain mediates coactivation of the LMP1 promoter with EBNA-2. J Virol 76:232–242. doi: 10.1128/JVI.76.1.232-242.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nitsche F, Bell A, Rickinson A. 1997. Epstein-Barr virus leader protein enhances EBNA-2-mediated transactivation of latent membrane protein 1 expression: a role for the W1W2 repeat domain. J Virol 71:6619–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kis LL, Gerasimcik N, Salamon D, Persson EK, Nagy N, Klein G, Severinson E, Klein E. 2011. STAT6 signaling pathway activated by the cytokines IL-4 and IL-13 induces expression of the Epstein-Barr virus-encoded protein LMP-1 in absence of EBNA-2: implications for the type II EBV latent gene expression in Hodgkin lymphoma. Blood 117:165–174. doi: 10.1182/blood-2010-01-265272. [DOI] [PubMed] [Google Scholar]
- 24.Kis LL, Salamon D, Persson EK, Nagy N, Scheeren FA, Spits H, Klein G, Klein E. 2010. IL-21 imposes a type II EBV gene expression on type III and type I B cells by the repression of C- and activation of LMP-1-promoter. Proc Natl Acad Sci U S A 107:872–877. doi: 10.1073/pnas.0912920107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kis LL, Takahara M, Nagy N, Klein G, Klein E. 2006. IL-10 can induce the expression of EBV-encoded latent membrane protein-1 (LMP-1) in the absence of EBNA-2 in B lymphocytes and in Burkitt lymphoma- and NK lymphoma-derived cell lines. Blood 107:2928–2935. doi: 10.1182/blood-2005-06-2569. [DOI] [PubMed] [Google Scholar]
- 26.Konforte D, Simard N, Paige CJ. 2008. Interleukin-21 regulates expression of key Epstein-Barr virus oncoproteins, EBNA2 and LMP1, in infected human B cells. Virology 374:100–113. doi: 10.1016/j.virol.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 27.Chen H, Lee JM, Zong Y, Borowitz M, Ng MH, Ambinder RF, Hayward SD. 2001. Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J Virol 75:2929–2937. doi: 10.1128/JVI.75.6.2929-2937.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Hutt-Fletcher L, Cao L, Hayward SD. 2003. A positive autoregulatory loop of LMP1 expression and STAT activation in epithelial cells latently infected with Epstein-Barr virus. J Virol 77:4139–4148. doi: 10.1128/JVI.77.7.4139-4148.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsao SW, Tramoutanis G, Dawson CW, Lo AK, Huang DP. 2002. The significance of LMP1 expression in nasopharyngeal carcinoma. Semin Cancer Biol 12:473–487. doi: 10.1016/S1044579X02000901. [DOI] [PubMed] [Google Scholar]
- 30.Sadler RH, Raab-Traub N. 1995. The Epstein-Barr virus 3.5-kilobase latent membrane protein 1 mRNA initiates from a TATA-less promoter within the first terminal repeat. J Virol 69:4577–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsiao JR, Chang KC, Chen CW, Wu SY, Su IJ, Hsu MC, Jin YT, Tsai ST, Takada K, Chang Y. 2009. Endoplasmic reticulum stress triggers XBP-1-mediated up-regulation of an EBV oncoprotein in nasopharyngeal carcinoma. Cancer Res 69:4461–4467. doi: 10.1158/0008-5472.CAN-09-0277. [DOI] [PubMed] [Google Scholar]
- 32.Noda C, Murata T, Kanda T, Yoshiyama H, Sugimoto A, Kawashima D, Saito S, Isomura H, Tsurumi T. 2011. Identification and characterization of CCAAT enhancer-binding protein (C/EBP) as a transcriptional activator for Epstein-Barr virus oncogene latent membrane protein 1. J Biol Chem 286:42524–42533. doi: 10.1074/jbc.M111.271734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demetriades C, Mosialos G. 2009. The LMP1 promoter can be transactivated directly by NF-κB. J Virol 83:5269–5277. doi: 10.1128/JVI.00097-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansson P, Jansson A, Ruetschi U, Rymo L. 2009. Nuclear factor-κB binds to the Epstein-Barr virus LMP1 promoter and upregulates its expression. J Virol 83:1393–1401. doi: 10.1128/JVI.01637-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansson A, Johansson P, Yang W, Palmqvist L, Sjoblom-Hallen A, Rymo L. 2007. Role of a consensus AP-2 regulatory sequence within the Epstein-Barr virus LMP1 promoter in EBNA2 mediated transactivation. Virus Genes 35:203–214. doi: 10.1007/s11262-007-0116-x. [DOI] [PubMed] [Google Scholar]
- 36.Sjoblom A, Yang W, Palmqvist L, Jansson A, Rymo L. 1998. An ATF/CRE element mediates both EBNA2-dependent and EBNA2-independent activation of the Epstein-Barr virus LMP1 gene promoter. J Virol 72:1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai CN, Lee CM, Chien CK, Kuo SC, Chang YS. 1999. Additive effect of Sp1 and Sp3 in regulation of the ED-L1E promoter of the EBV LMP 1 gene in human epithelial cells. Virology 261:288–294. doi: 10.1006/viro.1999.9851. [DOI] [PubMed] [Google Scholar]
- 38.Ning S, Hahn AM, Huye LE, Pagano JS. 2003. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: a regulatory circuit. J Virol 77:9359–9368. doi: 10.1128/JVI.77.17.9359-9368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salamon D, Adori M, Ujvari D, Wu L, Kis LL, Madapura HS, Nagy N, Klein G, Klein E. 2012. Latency-type dependent modulation of Epstein-Barr virus encoded latent membrane protein 1 expression by type I interferons in B cells. J Virol 86:4701–4707. doi: 10.1128/JVI.06829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eckert D, Buhl S, Weber S, Jager R, Schorle H. 2005. The AP-2 family of transcription factors. Genome Biol 6:246. doi: 10.1186/gb-2005-6-13-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hilger-Eversheim K, Moser M, Schorle H, Buettner R. 2000. Regulatory roles of AP-2 transcription factors in vertebrate development, apoptosis and cell-cycle control. Gene 260:1–12. doi: 10.1016/S0378-1119(00)00454-6. [DOI] [PubMed] [Google Scholar]
- 42.Mohibullah N, Donner A, Ippolito JA, Williams T. 1999. SELEX and missing phosphate contact analyses reveal flexibility within the AP-2[alpha] protein: DNA binding complex. Nucleic Acids Res 27:2760–2769. doi: 10.1093/nar/27.13.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welinder E, Ahsberg J, Sigvardsson M. 2011. B-lymphocyte commitment: identifying the point of no return. Semin Cancer Biol 23:335–340. doi: 10.1016/j.smim.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 44.Northrup DL, Allman D. 2008. Transcriptional regulation of early B cell development. Immunol Res 42:106–117. doi: 10.1007/s12026-008-8043-z. [DOI] [PubMed] [Google Scholar]
- 45.Hau PM, Tsang CM, Yip YL, Huen MS, Tsao SW. 2011. Id1 interacts and stabilizes the Epstein-Barr virus latent membrane protein 1 (LMP1) in nasopharyngeal epithelial cells. PLoS One 6:e21176. doi: 10.1371/journal.pone.0021176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K. 2004. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J Virol 78:7004–7015. doi: 10.1128/JVI.78.13.7004-7015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, Sunamura M, Imamura T, Miyazono K, Aburatani H. 2009. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol Cell Biol 29:172–186. doi: 10.1128/MCB.01038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurahashi S, Hayakawa F, Miyata Y, Yasuda T, Minami Y, Tsuzuki S, Abe A, Naoe T. 2011. PAX5-PML acts as a dual dominant-negative form of both PAX5 and PML. Oncogene 30:1822–1830. doi: 10.1038/onc.2010.554. [DOI] [PubMed] [Google Scholar]
- 49.Sigvardsson M. 2000. Overlapping expression of early B-cell factor and basic helix-loop-helix proteins as a mechanism to dictate B-lineage-specific activity of the lambda5 promoter. Mol Cell Biol 20:3640–3654. doi: 10.1128/MCB.20.10.3640-3654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A 95:8245–8250. doi: 10.1073/pnas.95.14.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murata T, Isomura H, Yamashita Y, Toyama S, Sato Y, Nakayama S, Kudoh A, Iwahori S, Kanda T, Tsurumi T. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81. doi: 10.1016/j.virol.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Murata T, Sato Y, Nakayama S, Kudoh A, Iwahori S, Isomura H, Tajima M, Hishiki T, Ohshima T, Hijikata M, Shimotohno K, Tsurumi T. 2009. TORC2, a coactivator of cAMP-response element-binding protein, promotes Epstein-Barr virus reactivation from latency through interaction with viral BZLF1 protein. J Biol Chem 284:8033–8041. doi: 10.1074/jbc.M808466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murata T, Kondo Y, Sugimoto A, Kawashima D, Saito S, Isomura H, Kanda T, Tsurumi T. 2012. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J Virol 86:4752–4761. doi: 10.1128/JVI.06768-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murata T, Noda C, Saito S, Kawashima D, Sugimoto A, Isomura H, Kanda T, Yokoyama KK, Tsurumi T. 2011. Involvement of Jun dimerization protein 2 (JDP2) in the maintenance of Epstein-Barr virus latency. J Biol Chem 286:22007–22016. doi: 10.1074/jbc.M110.199836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee N, Moss WN, Yario TA, Steitz JA. 2015. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell 160:607–618. doi: 10.1016/j.cell.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Price AM, Luftig MA. 2015. To be or not IIb: a multi-step process for Epstein-Barr virus latency establishment and consequences for B cell tumorigenesis. PLoS Pathog 11:e1004656. doi: 10.1371/journal.ppat.1004656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murata T, Tsurumi T. 2014. Switching of EBV cycles between latent and lytic states. Rev Med Virol 24:142–153. doi: 10.1002/rmv.1780. [DOI] [PubMed] [Google Scholar]
- 59.Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE, Kieff E. 2011. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc Natl Acad Sci U S A 108:14902–14907. doi: 10.1073/pnas.1108892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davies ML, Xu S, Lyons-Weiler J, Rosendorff A, Webber SA, Wasil LR, Metes D, Rowe DT. 2010. Cellular factors associated with latency and spontaneous Epstein-Barr virus reactivation in B-lymphoblastoid cell lines. Virology 400:53–67. doi: 10.1016/j.virol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 61.Portal D, Zhou H, Zhao B, Kharchenko PV, Lowry E, Wong L, Quackenbush J, Holloway D, Jiang S, Lu Y, Kieff E. 2013. Epstein-Barr virus nuclear antigen leader protein localizes to promoters and enhancers with cell transcription factors and EBNA2. Proc Natl Acad Sci U S A 110:18537–18542. doi: 10.1073/pnas.1317608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou H, Schmidt SC, Jiang S, Willox B, Bernhardt K, Liang J, Johannsen EC, Kharchenko P, Gewurz BE, Kieff E, Zhao B. 2015. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 17:205–216. doi: 10.1016/j.chom.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fahraeus R, Jansson A, Sjoblom A, Nilsson T, Klein G, Rymo L. 1993. Cell phenotype-dependent control of Epstein-Barr virus latent membrane protein 1 gene regulatory sequences. Virology 195:71–80. doi: 10.1006/viro.1993.1347. [DOI] [PubMed] [Google Scholar]
- 64.Wang F, Tsang SF, Kurilla MG, Cohen JI, Kieff E. 1990. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J Virol 64:3407–3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagata H, Konno A, Kimura N, Zhang Y, Kimura M, Demachi A, Sekine T, Yamamoto K, Shimizu N. 2001. Characterization of novel natural killer (NK)-cell and γδ T-cell lines established from primary lesions of nasal T/NK-cell lymphomas associated with the Epstein-Barr virus. Blood 97:708–713. doi: 10.1182/blood.V97.3.708. [DOI] [PubMed] [Google Scholar]
- 66.Sjoblom-Hallen A, Yang W, Jansson A, Rymo L. 1999. Silencing of the Epstein-Barr virus latent membrane protein 1 gene by the Max-Mad1-mSin3A modulator of chromatin structure. J Virol 73:2983–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aviel S, Winberg G, Massucci M, Ciechanover A. 2000. Degradation of the Epstein-Barr virus latent membrane protein 1 (LMP1) by the ubiquitin-proteasome pathway: targeting via ubiquitination of the N-terminal residue. J Biol Chem 275:23491–23499. doi: 10.1074/jbc.M002052200. [DOI] [PubMed] [Google Scholar]
- 68.Lo AK, To KF, Lo KW, Lung RW, Hui JW, Liao G, Hayward SD. 2007. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci U S A 104:16164–16169. doi: 10.1073/pnas.0702896104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garcia MA, Campillos M, Marina A, Valdivieso F, Vazquez J. 1999. Transcription factor AP-2 activity is modulated by protein kinase A-mediated phosphorylation. FEBS Lett 444:27–31. doi: 10.1016/S0014-5793(99)00021-6. [DOI] [PubMed] [Google Scholar]
- 70.Brennan B. 2006. Nasopharyngeal carcinoma. Orphanet J Rare Dis 1:23. doi: 10.1186/1750-1172-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liebowitz D. 1994. Nasopharyngeal carcinoma: the Epstein-Barr virus association. Semin Oncol 21:376–381. [PubMed] [Google Scholar]
- 72.Laag E, Majidi M, Cekanova M, Masi T, Takahashi T, Schuller HM. 2006. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int J Cancer 119:1547–1552. doi: 10.1002/ijc.21987. [DOI] [PubMed] [Google Scholar]
- 73.Kondo S, Wakisaka N, Muramatsu M, Zen Y, Endo K, Murono S, Sugimoto H, Yamaoka S, Pagano JS, Yoshizaki T. 2011. Epstein-Barr virus latent membrane protein 1 induces cancer stem/progenitor-like cells in nasopharyngeal epithelial cell lines. J Virol 85:11255–11264. doi: 10.1128/JVI.00188-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shair KH, Schnegg CI, Raab-Traub N. 2008. EBV latent membrane protein 1 effects on plakoglobin, cell growth, and migration. Cancer research 68:6997–7005. doi: 10.1158/0008-5472.CAN-08-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yip YL, Tsang CM, Deng W, Cheung PY, Jin Y, Cheung AL, Lung ML, Tsao SW. 2010. Expression of Epstein-Barr virus-encoded LMP1 and hTERT extends the life span and immortalizes primary cultures of nasopharyngeal epithelial cells. J Med Virol 82:1711–1723. doi: 10.1002/jmv.21875. [DOI] [PubMed] [Google Scholar]
- 76.Murata T, Iwata S, Siddiquey MN, Kanazawa T, Goshima F, Kawashima D, Kimura H, Tsurumi T. 2013. Heat shock protein 90 inhibitors repress latent membrane protein 1 (LMP1) expression and proliferation of Epstein-Barr virus-positive natural killer cell lymphoma. PLoS One 8:e63566. doi: 10.1371/journal.pone.0063566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu F, Chen HS, Kossenkov AV, DeWispeleare K, Won KJ, Lieberman PM. 2016. EBNA2 drives formation of new chromosome binding sites and target genes for B-cell master regulatory transcription factors RBP-jκ and EBF1. PLoS Pathog 12:e1005339. doi: 10.1371/journal.ppat.1005339. [DOI] [PMC free article] [PubMed] [Google Scholar]