

Abstract
Background
With ever increasing numbers of closely related virus genomes being sequenced, it has become desirable to be able to compare two genomes at a level more detailed than gene content because two strains of an organism may share the same set of predicted genes but still differ in their pathogenicity profiles. For example, detailed comparison of multiple isolates of the smallpox virus genome (each approximately 200 kb, with 200 genes) is not feasible without new bioinformatics tools.
Results
A software package, Base-By-Base, has been developed that provides visualization tools to enable researchers to 1) rapidly identify and correct alignment errors in large, multiple genome alignments; and 2) generate tabular and graphical output of differences between the genomes at the nucleotide level. Base-By-Base uses detailed annotation information about the aligned genomes and can list each predicted gene with nucleotide differences, display whether variations occur within promoter regions or coding regions and whether these changes result in amino acid substitutions. Base-By-Base can connect to our mySQL database (Virus Orthologous Clusters; VOCs) to retrieve detailed annotation information about the aligned genomes or use information from text files.
Conclusion
Base-By-Base enables users to quickly and easily compare large viral genomes; it highlights small differences that may be responsible for important phenotypic differences such as virulence. It is available via the Internet using Java Web Start and runs on Macintosh, PC and Linux operating systems with the Java 1.4 virtual machine.
Background
The recent advances in large-scale DNA sequencing technologies has significantly reduced the cost of this work, and it has become feasible to determine the sequence of multiple isolates of a number of viruses and examine smaller and smaller differences between them. The availability of this type of data permits novel hypotheses to be tested, but it requires new bioinformatics tools. For a number of years, our laboratory has been designing software specifically to manage and analyze large numbers of the bigger virus genomes such as those of poxviruses. Poxvirus genomes range in size from 150–380 kb and encode several hundred proteins [1]; there are now more than 30 completely sequenced poxvirus genomes available from GenBank. Coronaviruses provide another important example, with the largest genome (~30 kb) of all RNA viruses and 99 isolates of the SARS virus completely sequenced. This wealth of sequence data provides excellent opportunities to study new aspects of virus virulence and evolution. The Poxvirus Orthologous Clusters (POCs; [2]) mySQL database and query tools, now expanded to Virus Orthologous Clusters (VOCs), was constructed to organize all the genomes of one virus family into a single database and to group orthologous genes into families. A variety of tools are integrated into the database for rapid comparison of not only individual genes and proteins but also entire gene families if required. These tools include BLAST [3,4], CLUSTALW [5,6], T-coffee [7], hydrophobicity plots (using Kyte-Doolittle, Hopp-Woods and Parker-Guo-Hodges scales of hydrophobicity), DNA/protein dotplots and NAP [8] to align protein to DNA sequences. Gene maps of complete virus genomes are also generated automatically in VOCs. Continuing on the theme of working with gene families and complete genomes, the Viral Genome Organizer (VGO; [9]) was designed to display multiple whole genomes in a single working window with zooming capabilities and showing a variety of information such as ORFs, start/stop codons and AT%. VGO acts as a graphical portal to a variety of data that is stored in VOCs (amino acid composition, nucleotide composition, presence of orthologs in other genomes and pre-processed BLAST searches) or generated on-the-fly; for example clicking on gene X in one genome will cause the orthologous gene to be highlighted in a related genome. This software has the ability to easily and quickly answer a database query such as display genes present in variola virus that are absent from vaccinia virus. However, no software existed that could display all nucleotide differences between variola virus strain 1 and variola virus strain 2 and provide a summary of implications of these base changes; therefore the development of Base-By-Base (BBB) was initiated. This level of detail is required by current sequencing projects that aim to identify the genetic determinants responsible for phenotypes such as mortality rate/virulence, host tropism or tissue tropism displayed by different isolates of a single virus species. BBB allows the user to quickly display all nucleotide differences between genomes, and provides a detailed summary of the effect that each change produces in the relevant genes.
A second problem associated with the alignment of closely related, large virus genomes, is that the available software such as Dialign2 [10], used to produce acceptably fast, global alignments of complete genomes, invariably makes occasional small errors, often around the positioning of multiple small gaps. These small alignment errors pose a serious problem with fine analysis of genomes because the insertion of erroneous gaps results in an increase in the number of apparent, but not real, nucleotide substitutions. BBB provides a visual display to help the molecular biologist recognize these errors and quickly correct them using the built in sequence editor. Accurate large-scale alignments are essential for determining the evolutionary relationship of closely related viruses (such as isolates of a single strain), and in such alignments insertions and deletions themselves are very useful indicators of evolutionary lineage. Thus BBB will be very useful for phenotype/genotype analysis and for epidemiology.
Implementation
Design rationale
BBB was coded in Java to simplify support for multiple platforms including Mac OS X, MS Windows and LINUX. A user initially accesses the application (client) from a web page using Java Web Start, which also automatically downloads the application from the host server computer whenever a new version is available. This approach greatly simplifies the distribution of updates and ensures users are taking advantage of the latest version of the software; it has worked very well for the distribution of VOCs and VGO.
Although BBB was primarily designed to be used as an editor for large DNA sequences, the software also works with protein sequences and is used as the multiple alignment interface in VOCs. Both FASTA and ClustalW (.aln) formatted text files can be loaded into the program; multiple sequences may be placed in a single file and additional sequences can also be added at any time. Similarly, alignments may be exported in either of these formats. The native file format of BBB is, however, based on the Bioinformatics Sequence Markup Language (BSML) [11] standard that is itself an Extensible Markup Language (XML) dedicated to the needs of bioinformatics; it aims to provide an open language definition for distributing sequence data. The BBB file format stores the sequence alignment, gene features and other user-defined annotations for the sequences; because of the highly modular nature of XML, it is easy to incorporate new information in the alignment while maintaining compatibility with previous versions. XML is also highly interoperable since it has as its root a plain text file, allowing BBB and BSML files to be easily integrated with other software packages.
Since BBB provides access to ClustalW [5,6] and T-coffee [7] software, specific regions or entire genome sequences can be selected and aligned within the program itself, but in normal operation it is expected that the user would import large sequences that had already been aligned by a more appropriate program such as DIALIGN2 [10]. ClustalW and T-coffee are not distributed in the BBB jar file and are run instead on the remote BBB server. The user selects regions of the sequences to be re-aligned, and these are formatted by BBB, submitted to the server and subsequently received back in a new BBB window.
The annotations for viral genomes in a BBB alignment are read into BBB from a GenBank file or from a VOCs database. Currently, poxviruses [12] and coronaviruses [13] are available in our VOCs databases although herpesviruses, baculoviruses and adenoviruses will be available in the near future [14]. The gene annotations are required by BBB to generate the tabular summary of the differences between genomes that details the effect of nucleotide changes on the genes and predicted proteins. These annotations are not required to produce a visual summary of sequence alignments, although we find it useful to have the genes displayed for one of the genomes in a large alignment.
Results and Discussion
Editing and display of sequence differences in real-time
BBB is primarily intended to be a sequence analysis tool focused on genome sequence comparisons, but the program also provides similar functionality to other alignment viewers and editors such as GeneDoc [15] and BioEdit [16]. BBB gives users the ability to load, correct and save alignments. Alignments loaded into BBB are easily edited by dragging sequences across the screen to insert and remove gaps; multiple sequences may be selected and edited as one. One of many unique features of BBB is that the main window displays the DNA or protein sequences together with flags that show all the nucleotide or amino acid differences that are present between the sequences shown in the window. This information provides very important visual cues as to the position of major and minor differences between the sequences in an alignment and is updated in real-time as the user makes edits to the alignments (Figure 1). BBB provides a button to allow the user to skip through a sequence from one flagged nucleotide difference to the next, alleviating the need to scroll through an entire genome and detect the differences by eye. The sequence substitutions, insertions and deletions are flagged and color-coded in a single row between the pairs of sequences (Figure 1). The program preferences can be set so that the difference data represents differences between each adjacent pair of sequences or to a consensus sequence calculated by BBB. In pairwise comparison mode, the first sequence is compared to the second, second to the third and so on, making BBB especially useful for visualizing evolution of sequences. When comparing to the consensus, the sequence differences indicated on the screen are those differences between each sequence and the consensus sequence, this allows for a global view of the alignment and easily identifies conserved/variable regions in genomes which is useful information in drug design and vaccine development. The consensus sequence generated from an alignment can also be saved to a file (FASTA format) for analysis with other software.
Figure 1.
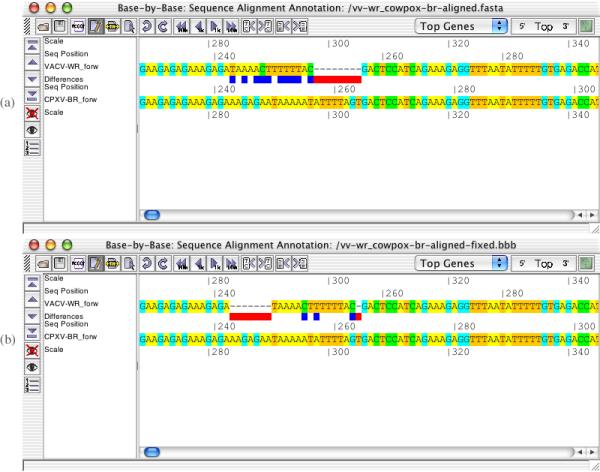
Use of BBB to identify and correct small mis-aligned regions within alignments of large virus genomes. (a) Region of vaccinia virus (strain WR) and cowpox virus (strain Brighton Red) genomes aligned by DIALIGN2 (b) Manually corrected version of region shown in (a). Gaps and mismatched nucleotides are shown as red and navy blue boxes or bars in between the 2 sequences, respectively.
During development, it was discovered that it was important to have the flags that highlight nucleotide differences updated in real-time as the user manually modifies the alignment. These flags are created for the displayed area only, and are then updated and augmented when needed, such as when the user changes the alignment, by inserting or deleting gaps, or changes the view, by scrolling or setting the display area. This enables BBB to work with multiple large genome sequences and keep an acceptable refresh rate for displayed data. A Block Glue option facilitates the movement of long sequence blocks that are bigger than the editing window by permitting the dragging of complete blocks without introduction of new gaps within the alignment. Often alignment errors are obvious to an experienced molecular biologist and the alignment can be manually corrected, simply by dragging the appropriate nucleotides into new positions. However, if there are doubts about the DIALIGN2 alignment or if there are a large number of sequences in the alignment, the user may choose to perform a local re-alignment of selected regions of the sequences using the ClustalW or T-coffee module of BBB. After the server completes an alignment, a new BBB window displays the new local alignment and the user is offered the option to import this new local alignment back into the original complete genome alignment. If required, forward and backward 3-frame translations of the DNA sequences can be displayed in the main sequence alignment window to help with alignment decisions. Methionines (green) and stop codons (red) are highlighted on the 3-frame translations, as are the genes that are associated with individual sequences through annotations in the BBB file. Figure 1 shows an example of a correction of a small mismatched region of two poxvirus genomes; breaking the single gap and moving the two resulting gaps reduces the number of mismatched nucleotides (not including those opposite a gap) from 10 to 3.
Sequence filtering and display customization
Since Base-By-Base has been designed for users to manipulate alignments of complete poxvirus genomes (150–300 kb), features to simplify and enhance the user-interface and to speed up the program's manipulation of the sequences have been incorporated where possible. However, some speed has been sacrificed by using Java, which was chosen to provide cross-platform functionality. BBB allows users to filter their view of the data in two ways, 1) complete genome sequences can be sorted and reordered in the main window or placed in the background, completely hidden from view to make analysis of dozens of sequences possible; 2) long genome alignments can be masked from the 5' and/or 3' ends to allow a user to focus on any particular region within the genome; this is especially useful for visually tracking the differences between individual orthologous genes in several different genomes. These user selected viewing options also apply to the functions for generating visual or tabular reports; only the genomes shown in the main window and the unmasked regions are evaluated by these report routines. From the preferences window, users can also toggle on/off the display of the sequence difference flags, sequence-numbering scales, user annotations, and there is a button in the main window to toggle on/off the display of 3-frame translation of DNA sequences (Figure 1). These features are especially useful for alignments of 30 sequences or more, where screen real estate precludes the display of all sequences or all features associated with the sequences.
Since individual researchers frequently are interested in different genome features that may not be annotated in GenBank files, a tool was created in BBB that allows the user to add comments to different regions of one or more sequences in an alignment. These comments can be color coded, labeled with text and hidden or viewed in the main window as required; they are saved within the native .bbb alignment file. Again, since the aligned sequences are frequently very long, BBB provides a button to skip through the sequences from one comment to the next.
To provide the user with additional information about the sequence alignments, BBB provides several different coloring styles for viewing the alignments in the main window. These include the default character-identity based scheme in which each nucleotide or amino acid is colored based on which nucleotide or amino acid it represents and a simple percent identity style which uses shades to indicate the frequency of each nucleotide or amino acid at each position in the alignment. Protein sequence alignments may also be viewed with similarity-matrix based (BLOSUM62 or PAM250) shading for which residues "similar" to the most frequently occurring amino acid are also colored. Lastly, a hydrophobicity coloring scheme shades amino acids based on the hydrophobicity score of each residue.
Reporting sequence differences and effects of nucleotide changes
Another of the unique and primary features of BBB is its ability to summarize the differences between two large closely related virus genomes; without such bioinformatics tools, this type of large-scale analysis is not feasible. Using the genome annotation information imported from a GenBank file, or from our own VOCs database, BBB is able to determine which pairs of genes have nucleotide differences and what, if any, effect these nucleotide substitutions have on the predicted proteins from each gene. Basic gene information is listed in a table, including start/stop positions, strand and length, followed by: 1) the number of nucleotide differences counted and categorized as substitutions, deletions and insertions; 2) calculation of the percent difference; 3) changes in promoter regions; 4) nucleotide substitutions categorized as silent, or listed with amino acid change (Figure 2). In addition, BBB displays the size of deletions and insertions to guide the user in comparison of percent differences since a single deletion event removing 12 nucleotides creates larger effect in the percent difference table than 6 single substitution events that could affect 6 different amino acids. The summary table, which can be very large for the poxviruses, can be sorted using the data in any column by simply clicking on any column header.
Figure 2.
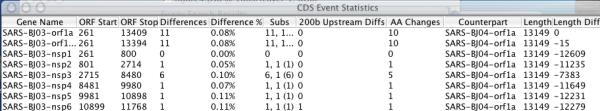
Section of a gene feature report comparing SARS coronavirus strains BJ03 and BJ04. The Base-By-Base table header describes the listed data. Since the non-structural proteins (NSP) that we have annotated in VOCs are fragments of the ORF1a and ORF1ab polyproteins, the length differences appear as negative numbers
The percent difference between genes is useful for spotting regions that may be mis-aligned by highlighting unusually large differences between particular genes. A region of 200 nucleotides upstream of each gene is also analyzed for differences; in poxviruses, promoters are small and almost all are within 200 nucleotides of the initiating ATG [17,18]. Thus, with very similar genomes it is very simple to determine which few genes have differences in the promoter regions and select these for further analysis. This type of analysis of promoter regions is very important when looking at different isolates of a single virus strain since modulation of gene expression may occur rapidly. Changes of one or two nucleotides in the small promoters of poxviruses can have a drastic effect on transcription rate and ultimately on protein expression [17,18]. This type of adaptation of a virus to a particular environment/host occurs at a much higher frequency than the acquisition of novel genes/promoters derived from the host or other viruses. These differences can easily be identified and viewed in BBB.
Comparison of very closely related genomes is the primary purpose for which BBB was designed and there is usually a simple one-to-one correspondence of complete genes between such genomes. However, BBB also handles situations where fragmented genes may exist in one genome; it chooses counterpart genes in the other genome by determining the greatest portion of overlap between the gene on the first genome and the gene on the second genome. The tabular report includes raw information on the genes, such as the position and length of the genes (Figure 2).
The Visual Overview Summary creates a pictorial representation of the differences (default) or similarities between each sequence in the main alignment window and the consensus sequence. This summary window is not a static picture, but rather it communicates with the alignment window (Figure 3). Examples of this communication activity are: 1) information showing the length of the sequence displayed in the alignment window is superimposed on the summary picture, and clicking in another area of the summary window causes that new region to be displayed in the main window; this feature also provides a very rapid way for navigating through large genomes; 2) when edits are made to the underlying sequences in the main sequence window, the consensus is automatically recalculated and the summary display is updated. Changes to the main window are polled approximately once per second, so that the system is not inundated with consensus refresh requests that can, in some cases, be quite time consuming. The default color scheme used in this window is identical to that of the main alignment display and nucleotide differences (or regions of mostly substitutions) are displayed as blue ticks or bars, and insertions and deletions are displayed in green and red, respectively. Similarity shading styles, scored with BLOSUM62 and PAM250 matrices, are available for analyzing protein sequences that might not be similar enough for the default parameters (Figure 3) and has proven to be very useful for the analysis of gene orthologs and protein families.
Figure 3.

Scored similarity visual summary of an alignment of coronavirus spike glycoproteins. PAM250 substitution matrix was used to shade the figure; a grey scale indicates similarity (black-perfect match; white-low) and gaps are shown as dashes.
As discussed above, BBB stores the sequence alignment, gene features and user annotation in its own XML format. It can, however, export the alignment text in FASTA and ClustalW (.aln) formats for convenient transfer to other programs. To capture a graphical view of the main window alignment, the user may choose to export either a full alignment or a particular sequence range to an image file in JPEG or PNG file formats. This permits the user to view and print a full alignment by wrapping the single row, at a user-specified width, which is normally displayed on the screen. All graphics, showing sequence differences, nucleotide translations and user-added annotations are preserved in the picture. For publication purposes, however, features such as user comments can be easily hidden from view by changing BBB preferences (Edit menu).
Other methods of summarizing the information from a multiple sequence alignment in BBB are by using the Phylogeny tools or the Alignment Info tool. Trees are calculated from the alignment and drawn by routines from the Phylogenetic Analysis Library (PAL) [19]. The Alignment Info tool generates a tabular report of the percent identity between all the pairs of sequences in the alignment; this data can be exported as a tab-delimited file for convenient importing into a spreadsheet or table in a word processor. Since the percent identity is only calculated on the region of the alignment set in Display area, this provides a useful tool to calculate conservation in different regions of an alignment.
Regular expression and fuzzy motif searches
It is frequently necessary to search through viral genome sequences for restriction sites, primer sequences and other patterns. For most restriction sites a regular expression search suffices, however, to look for less well-defined motifs such as promoters or other regulatory sequences, allowance for a significant element of degeneracy is desirable. This is a tedious process using regular expressions; therefore, a fuzzy motif search capability was also implemented in BBB that allows a user-selected number of mismatches and ignores gaps in sequence alignments. The search results are returned in two formats (Figure 4); they are presented in table format in a new window together with information indicating the percent match to the query motif and are also indicated directly on the main sequence alignment window. To facilitate searches of long sequences or those that return a large number of matches, the search results table can be sorted by location of the match within the genome or by percent match (Figure 4). Users can also rapidly move from hit to hit along each of the sequences searched by simply clicking on the icon representing the search hit in the search results table. Fuzzy search expressions follow a simple grammar that is explained in the user documentation http://www.virology.ca/pbr/bbb/manual, and the regular expression searching capability is provided by the Jakarta-ORO text-processing package. BBB uses the Perl 5 regular expressions from the ORO package with case insensitive searching; this function searches both strands of the nucleotide sequence.
Figure 4.
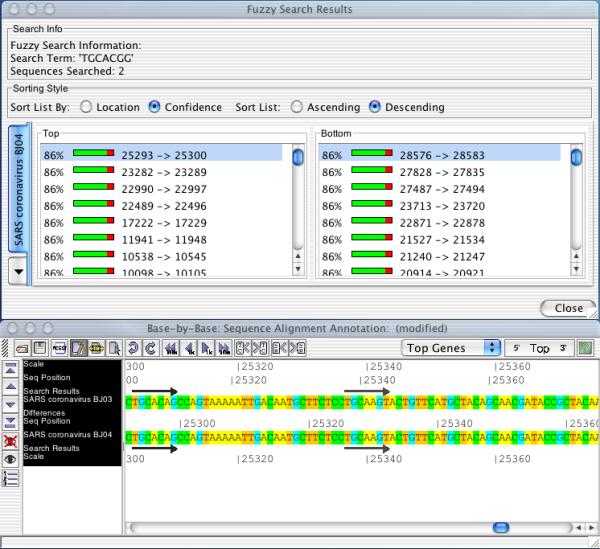
Results of a fuzzy search. The query sequence was 'TGCACGG', allowing 2 mismatches (user option), in 2 SARS coronavirus genomes. Upper panel, search results window with hits sorted by confidence (percent match) and the location of the hit within the genome; lower panel, hits displayed as black arrows in sequence alignment window.
Data connectivity
BBB has been designed to accommodate large virus genomes and therefore works with sequences in the order of 300 kb. Initially, alignments are read into BBB from FASTA or ClustalW format alignment files which are then converted to the native BBB file format. By their XML nature, BBB files are simple text files that research labs can easily post on their websites, which are then accessible from inside the program. Users of the program have found it convenient to maintain a series of BBB alignment files, each containing a multiple alignment of one group of closely related virus genomes, which serve as founder alignments for users; for example, a large alignment of all SARS genomes is maintained at http://athena.bioc.uvic.ca/sars/bbb. These files may then be edited by users who can delete the genome sequences they are not interested in and save the new files to their local computer. If, by deleting a series of genomes from the multiple alignment, there are then some positions that have gaps in all remaining genomes, BBB asks the user if these empty columns should be removed from the alignment.
Once a genome sequence alignment has been corrected, one of the most frequent requests is to view the visual summary of the alignment. Therefore an interface to generate and view these BBB summary files directly from a WWW page has been developed. Figure 5 shows a visual summary of 11 SARS genomes with an ORF map and substitutions, insertions and deletions color-coded. The WWW package allows the user to select an alignment file and view either the entire sequence alignment or a selected region of the genome sequences. If the BBB alignment file contains gene feature information, the position of genes will be superimposed on the alignment summary. Since the views are produced on-the-fly, the site maintainer only has to save the BBB file to the appropriate WWW directory and the users can be sure that they have access to the latest information. This feature also provides access to the summary display without the need to download BBB or learn how to run the software.
Figure 5.
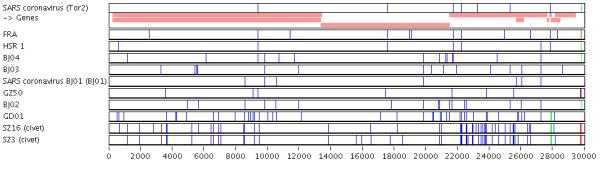
Web interface of a Base-By-Base summary. Display of 11 SARS coronavirus genomes. Pink boxes at the top of the figure represent genes; annotations are derived from the VOCs database. After comparison to the consensus, nucleotide differences are displayed as vertical blue ticks or bars, and insertions and deletions are displayed in green and red, respectively.
Conclusions
The goal of this project was to produce a tool to facilitate the comparison of closely related large viral genomes such as isolates of a single virus strain. To this end, a new software package called Base-By-Base has been developed; it uses a graphical interface to highlight differences between genomes and includes a multiple alignment editor so that the user can manually correct the errors made by programs making global alignments of complete genomes. When combined with our gene feature database, the Viral Orthologous Clusters system, or a GenBank file BBB is able to map gene features onto whole genome alignments, thereby giving users the ability to manipulate their alignment within the context of the annotated genes. Graphical summaries of multiple genome alignments are available from BBB in several formats. Furthermore, by using genome annotations, BBB is able to create tables that summarize all of the nucleotide differences between genomes and the implication of these changes on proteins encoded by the viral genes. Both coding and intergenic (e.g. promoter) sequences are analyzed. We believe that BBB will significantly enhance the analysis of a growing set of sequence data, namely the accumulation of multiple closely related virus genomes. Correlations between sequence and phenotype can be analyzed and hypotheses developed for testing. Conserved and variable regions can be viewed for phylogenetic relationships or vaccine or drug development. BBB is written in Java and has been tested on Linux, Mac OS X, and Windows. It is freely available for use under the terms of the GNU General Public License (GPL) at http://www.virology.ca/pbr/bbb/.
Availability and requirements
Project name: Base-By-Base
Project home page: http://www.virology.ca/pbr/bbb/
Operating system(s): Platform independent
Programming language: Java
Other requirements: Java 1.4 or higher, this requires at least system 10.2.8 on the Macintosh.
License: GNU General Public License
Any restrictions to use by non-academics: Contact authors
Authors' contributions
RB and AJS were principle programmers of the BBB software. RLR, VT and CU contributed ideas for features and display requirements, and tested the program.
Supplementary Material
This file contains 3 SARS coronavirus genomes, aligned with annotations, in Base-By-Base format. After downloading this file, the alignment can be opened from 1) the BBB file menu; open alignment.... From your local file; 2) Load sequences icon (open folder) after selecting the BBB file format from the menu.
Acknowledgments
Acknowledgements
This work was funded by NIAID/DARPA grant U01 AI48653-02 and Canadian NSERC Strategic Grant STPGP 269665-03. We would like to thank: Angelika Ehlers for systems administration and Melissa Da Silva for beta-testing and critical review of the manuscript.
Contributor Information
Ryan Brodie, Email: ryan@ryanbrodie.com.
Alex J Smith, Email: ajsmith@uvic.ca.
Rachel L Roper, Email: roperr@mail.ecu.edu.
Vasily Tcherepanov, Email: vasilyt@uvic.ca.
Chris Upton, Email: cupton@uvic.ca.
References
- Moss B. Poxviruses. In: Knipe DM and Howley P M, editor. Fields Virology. Vol. 2. Philadelphia, Lippincott Williams & Wilkins; 2001. pp. 2849–2884. [Google Scholar]
- Ehlers A, Osborne J, Slack S, Roper RL, Upton C. Poxvirus Orthologous Clusters (POCs) Bioinformatics. 2002;18:1544–1545. doi: 10.1093/bioinformatics/18.11.1544. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. JMolBiol. 1990;215:403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Huang X, Zhang J. Methods for comparing a DNA sequence with a protein sequence. Comput Appl Biosci. 1996;12:497–506. doi: 10.1093/bioinformatics/12.6.497. [DOI] [PubMed] [Google Scholar]
- Upton C, Hogg D, Perrin D, Boone M, Harris NL. Viral genome organizer: a system for analyzing complete viral genomes. Virus Res. 2000;70:55–64. doi: 10.1016/S0168-1702(00)00210-0. [DOI] [PubMed] [Google Scholar]
- Morgenstern B. DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics. 1999;15:211–218. doi: 10.1093/bioinformatics/15.3.211. [DOI] [PubMed] [Google Scholar]
- Bioinformatics Sequence Markup Language http://www.bsml.org
- Poxvirus Bioinformatics Resource http://www.poxvirus.org
- Coronavirus Bioinformatics Resource http://www.sarsresearch.ca
- The Virus Bioinformatics Resource http://www.virology.ca
- GeneDoc http://www.psc.edu/biomed/genedoc
- BioEdit http://www.mbio.ncsu.edu/BioEdit/bioedit.html
- Davison AJ, Moss B. Structure of vaccinia virus late promoters. J Mol Biol. 1989;210:771–784. doi: 10.1016/0022-2836(89)90108-3. [DOI] [PubMed] [Google Scholar]
- Davison AJ, Moss B. Structure of vaccinia virus early promoters. JMolBiol. 1989;210:749–769. doi: 10.1016/0022-2836(89)90107-1. [DOI] [PubMed] [Google Scholar]
- PAL: Phylogenetic Analysis Library http://www.cebl.auckland.ac.nz/pal-project
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains 3 SARS coronavirus genomes, aligned with annotations, in Base-By-Base format. After downloading this file, the alignment can be opened from 1) the BBB file menu; open alignment.... From your local file; 2) Load sequences icon (open folder) after selecting the BBB file format from the menu.