

ABSTRACT
The alphaviral 6k gene region encodes the two structural proteins 6K protein and, due to a ribosomal frameshift event, the transframe protein (TF). Here, we characterized the role of the 6k proteins in the arthritogenic alphavirus Ross River virus (RRV) in infected cells and in mice, using a novel 6k in-frame deletion mutant. Comprehensive microscopic analysis revealed that the 6k proteins were predominantly localized at the endoplasmic reticulum of RRV-infected cells. RRV virions that lack the 6k proteins 6K and TF [RRV-(Δ6K)] were more vulnerable to changes in pH, and the corresponding virus had increased sensitivity to a higher temperature. While the 6k deletion did not reduce RRV particle production in BHK-21 cells, it affected virion release from the host cell. Subsequent in vivo studies demonstrated that RRV-(Δ6K) caused a milder disease than wild-type virus, with viral titers being reduced in infected mice. Immunization of mice with RRV-(Δ6K) resulted in a reduced viral load and accelerated viral elimination upon secondary infection with wild-type RRV or another alphavirus, chikungunya virus (CHIKV). Our results show that the 6k proteins may contribute to alphaviral disease manifestations and suggest that manipulation of the 6k gene may be a potential strategy to facilitate viral vaccine development.
IMPORTANCE Arthritogenic alphaviruses, such as chikungunya virus (CHIKV) and Ross River virus (RRV), cause epidemics of debilitating rheumatic disease in areas where they are endemic and can emerge in new regions worldwide. RRV is of considerable medical significance in Australia, where it is the leading cause of arboviral disease. The mechanisms by which alphaviruses persist and cause disease in the host are ill defined. This paper describes the phenotypic properties of an RRV 6k deletion mutant. The absence of the 6k gene reduced virion release from infected cells and also reduced the severity of disease and viral titers in infected mice. Immunization with the mutant virus protected mice against viremia not only upon exposure to RRV but also upon challenge with CHIKV. These findings could lead to the development of safer and more immunogenic alphavirus vectors for vaccine delivery.
INTRODUCTION
Alphaviruses like Ross River virus (RRV), Barmah Forest virus (BFV), Semliki Forest virus (SFV), Sindbis virus (SINV), chikungunya virus (CHIKV), and O'nyong-nyong virus (ONNV) are small, enveloped RNA viruses of the family Togaviridae. They frequently cause human diseases manifesting with signs and symptoms such as encephalitis, acute or persistent arthritis, fever, arthralgia, and myalgia (1). RRV is a prevalent disease-causing alphavirus in the Australasian region and a major health hazard, with 5,000 cases of Ross River virus disease (RRVD) reported annually (2). Patients infected with RRV can develop a febrile illness, rash, polyarthritis, and myalgia (3). A number of patients also experience chronic arthralgia and debilitating joint pain that can last for months. Despite this, the pathogenic determinants of RRV have not been extensively studied.
The alphaviral genome comprises the 5′ region, encoding the nonstructural proteins, and the 3′ region, encoding the structural proteins. The structural proteins, capsid protein (C), glycoproteins E1, E2, and E3, 6K protein, and transframe protein (TF), are derived from progressive cleavage of the major polyproteins, C-E3-E2-6K-E1, and minor polyproteins, C-E3-E2-TF (1). TF shares the same N-terminal sequence as the 6K protein but contains an extended C terminus as a result of ribosomal frameshifting that occurs during the translation of the 6k gene (2). C, E2, and E1 are the obligatory components of mature virions that morphologically appear as a nucleocapsid enveloped by a lipid bilayer that is studded with E1/E2 trimers.
Although the 6K and TF 6k proteins are translated alongside the other structural proteins, they are generally present at very low levels within virus particles (4–6). Both 6k proteins are membrane-associated proteins, containing hydrophobic (supposed transmembrane) domains and numerous acylated amino acid residues (4, 7). The role of the 6k proteins in alphaviral disease remains unclear, although they have been shown to be involved in processes of viral replication, assembly, and release. For example, mutations of SINV 6k resulting from either an in-frame insertion of 15 amino acids in the 6k gene (8) or a deletion of 21 amino acids from the 6k gene (9) led to defective glycoprotein processing, trafficking, and viral budding. These findings are in line with a previous report demonstrating the involvement of an SFV 6k protein sequence in the insertion of E1 into the endoplasmic reticulum (ER) membrane (10). Interestingly, complete deletion of the SFV 6k gene had little effect on glycoprotein processing and transport but significantly impaired the release of virus particles, suggesting a role for the 6k proteins in viral assembly or budding (5, 11). Similarly, disruption of SINV TF production reduced infectious particle release during infection (6). The severity of disease experienced by mice infected with SINV TF mutants was also greatly reduced relative to the severity of disease experienced by mice infected with the wild-type virus in a model of SINV neuropathogenesis (6). The size, hydrophobicity, and structural characteristics of the alphaviral 6k proteins led to the hypothesis that they could function as viral ion channels, or viroporins. In this model, the 6k proteins are thought to be responsible for increasing host cell plasma membrane permeability, a common phenomenon of cytolytic viral infections (12–14). A number of studies also supported a role for the alphaviral 6K protein in enhancing membrane permeability through alternative mechanisms (15–18).
The 6k region is not highly conserved among the alphaviruses. In particular, the relevance of the 6k proteins for the replication and pathogenesis of RRV has not been assessed, despite the importance of this virus. Hence, we have constructed a 6K/TF deletion mutant of RRV [RRV-(Δ6K)] containing an in-frame deletion of the entire 6k gene region, thereby ensuring that the translation of proteins encoded by sequences on either side of the 6k gene region remains unaffected. Therefore, the potential caveats associated with partial gene deletions or insertions, as were done previously for SINV 6k (8, 9), are avoided. Using this mutant, we demonstrate that the 6k proteins are involved in virus release in BHK-21 cells. Furthermore, we show that RRV-(Δ6K) induces only mild disease in mice compared to the severity of the disease induced by its wild-type (WT) counterpart and that the viral load is reduced. RRV-(Δ6K)-immunized mice possessed the capacity to significantly reduce viremia upon alphavirus challenge. These results demonstrate the major role of the 6k proteins in RRV pathogenesis and the potential of 6k deletion as a vaccination strategy against RRV.
MATERIALS AND METHODS
Cells.
Baby hamster kidney (BHK-21; ATCC CCL-10) cells were maintained at 37°C in F15 medium supplemented with nonessential amino acids, 5% fetal calf serum (FCS), 10% tryptose phosphate broth, and 2 mM glutamine. Vero cells (ATCC CCL-81) were cultured in Opti-MEM medium supplemented with 10% FCS. Human osteosarcoma (HOS) cells (ATCC CRL-1543) were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% FCS, and HOS cells stably transfected with enhanced green fluorescent protein (EGFP)-tagged LC3 (19) were kept in DMEM with 10% FCS and 1 g/liter G418.
Construction of an RRV 6k deletion mutant.
The fragment of pRR64, an RRV T48 infectious cDNA clone, spanning from the RsrII to the SmaI restriction site was cloned into pKS+ (Stratagene, Germany). The sequence corresponding to the 6k region was removed by polymerase incomplete primer extension PCR (20) using oligonucleotides with the sequences 5′-TGC TCG TAT GCG TTC GCC CTC GGT GCG CAG C-3′ and 5′-GAA CGC ATA CGA GCA CAC AGC CAC AAT TCC GAA TGT GG-3′ as the primers and Phusion DNA polymerase (Thermo Scientific, USA) as the enzyme. The fragment containing the correct deletion was used to replace the corresponding fragment of pRR64. The sequence of the obtained clone, designated pRR64-(Δ6K), was verified by sequencing.
Viruses.
Infectious RRV RNA transcripts were generated as described by Kuhn et al. (21). Briefly, pRR64 and pRR64-(Δ6K) were linearized with SacI and transcribed using SP6 RNA polymerase, to generate full-length transcripts that were 5′ capped by the inclusion of m7G(5′)ppp(5′)A in the transcription reaction. BHK-21 cells were transfected with these RNAs by electroporation, and both WT RRV and the 6k deletion mutant [RRV-(Δ6K)] were harvested at 24 to 48 h posttransfection. The deletion of the 6k region was confirmed by RT-PCR, and the lack of 6K protein expression was verified by immunofluorescence. CHIKV (Bali isolate) was kindly provided by Linda Hueston (Centre for Infectious Diseases & Microbiology Laboratory Services, Westmead Hospital, Sydney, Australia) and propagated in Vero cells.
Infection and replication in cell culture.
Cells were infected with viruses in Hanks' buffered salt solution (HBSS) at a multiplicity of infection (MOI) of 0.1 PFU/cell. Following adsorption of virus for 1 h at 37°C, cell monolayers were washed and fresh growth medium was added. The culture medium was assayed for the presence of infectious particles at various time points. To determine the amount of cell-associated virus, cell monolayers were washed and lysed by osmotic shock and four freeze-thaw cycles to release internal virus particles. The cell lysate was then assayed for infectious particles by a plaque assay or for viral RNA by reverse transcription-quantitative PCR (RT-qPCR).
Temperature sensitivity.
Plaque assays were performed on Vero cells incubated at 37°C, 39°C, or 41°C. Cells were incubated with WT RRV or RRV-(Δ6K) for 1 h, the virus inoculum was removed, and an overlay of semisolid medium (0.16% agarose in DMEM with 2% FCS) was added to the cells. The cells were incubated at the temperatures specified below for 48 h prior to staining with crystal violet. To assay the thermal stability of WT RRV or RRV-(Δ6K) particles, 100 PFU of each virus was incubated at 52°C or 56°C for 0, 6, or 12 min and cooled on ice. BHK-21 cells were incubated with virus for 1 h at 37°C and overlaid with semisolid medium (2% carboxymethyl cellulose [CMC] in Glasgow minimum essential medium [GMEM]). At 72 h postinfection (p.i.), the cells were fixed with 10% formaldehyde for 20 min and stained with crystal violet. The experiments were repeated three times.
pH sensitivity assessment.
WT RRV or RRV-(Δ6K) (100 PFU) was treated at room temperature with 0.3 M morpholinoethanesulfonic acid (MES) at pH 5.5 for 0, 10, 20, 30, 40, 50, or 60 s and neutralized with 1 M NaOH. BHK-21 cells were incubated with virus for 1 h at 37°C and overlaid with semisolid medium (2% CMC in GMEM). At 72 h p.i., the cells were fixed with 10% formaldehyde for 20 min and stained with crystal violet. The experiments were repeated three times.
Virus quantification. (i) Infectious viral particle quantification by plaque assay.
All plaque assays on Vero cells were performed as described previously (22).
(ii) Viral load quantification by RT-qPCR.
RNA extraction was performed using the TRIzol reagent (Life Technologies) following the manufacturer's instructions. Extracted RNA was reverse transcribed using random nonamers and Moloney murine leukemia virus reverse transcriptase (Sigma-Aldrich) according to the manufacturer's instructions. A standard curve was generated using serial dilutions of pRR64 as described previously (23). Quantification of the viral load was performed using SsoAdvanced universal probes supermix (Bio-Rad) in a 12.5-μl reaction volume to detect the nsP3 region (23). Primers nsp3 Forward (5′-CCG TGG CGG GTA TTA TCA AT-3′) and nsp3 Reverse (5′-AAC ACT CCC GTC GAC AAC AGA-3′) and fluorogenic probe RRV nsp3 (5′-ATT AAG AGT GTA GCC ATC C-3′) were used All reactions were performed using a Bio-Rad CFX96 Touch real-time PCR detection system in 96-well plates. Cycler conditions were as follows: (i) a PCR initial activation step of 95°C for 15 min for 1 cycle and (ii) 3-step cycling at 94°C for 15 s, followed by 55°C for 30 s and 72°C for 30 s for 40 cycles. A dissociation curve was acquired using CFX Manager software to determine the specificity of the amplified products. A standard curve was plotted, and the copy numbers of the amplified products were interpolated from the standard curve using GraphPad Prism software to determine the viral load.
Immunofluorescence.
Cells were grown on glass coverslips and infected with virus as described above. Nonpermeabilized cells were fixed on ice for 30 min with 2% paraformaldehyde in phosphate-buffered saline (PBS), or, alternatively, cells were permeabilized and fixed with 2% paraformaldehyde containing 0.1% Triton X-100 in PBS, followed by blocking in 5% horse serum in PBS at 4°C for at least 16 h. RRV 6k proteins and E2 were labeled using rabbit polyclonal antibodies raised against the RRV 6K N-terminal region or the RRV E2 protein; mouse monoclonal antibodies against double-stranded RNA (monoclonal antibody J2; English and Scientific Consulting), calnexin (monoclonal antibody AF18; catalog number MA3027; Pierce/Thermo), GM130 (catalog number 610822; Becton Dickinson), EEA1 (catalog number 610456; Becton Dickinson), and LAMP1 (catalog number ab25630; Abcam) were employed to stain replication complexes, the ER, cis-Golgi network, early endosomes, and lysosomes, respectively. Human cells had to be used for this analysis because not all these organelle marker antibodies reacted with the hamster proteins in BHK-21 cells (data not shown). Autophagosomes were detected by fluorescence of EGFP-tagged LC3 in HOS cells stably transfected with this marker (19). Antibodies were diluted in 5% horse serum-PBS; incubation was performed for 2 to 5 h at 21°C, followed by washing in PBS; and secondary antibodies were applied for 30 min at 21°C; suitable fluorescein isothiocyanate (FITC)-, Alexa Fluor 488-, or Alexa Fluor 555-labeled anti-rabbit and anti-mouse immunoglobulin secondary antibodies plus DRAQ5 (Thermo) were used to stain the nuclei. Samples were imaged with either an Olympus FV1000 confocal microscope or a Leica TCS SP5 X confocal microscope equipped with a supercontinuum pulsed white laser.
Mouse studies.
The mouse model of RRV infection was established as described previously by Lidbury et al. (22). To assess the pathogenic potentials of WT RRV and RRV-(Δ6K), 17-day-old outbred Swiss mice were infected intraperitoneally (i.p.) with 103 PFU of each virus and the clinical manifestations were monitored for 20 days postinfection. Mock-inoculated animals were injected with the diluent alone. Clinical scores and body weights were recorded as previously described (24). To evaluate the prophylactic potential of RRV-(Δ6K), 21-day-old C57BL/6 mice were infected with 104 PFU RRV-(Δ6K). At day 30 postinfection, the C57BL/6 mice were challenged with 106 PFU RRV T48 or CHIKV. Viral titers in sera were evaluated by plaque assay at days 1, 2, and 3 postchallenge. All experiments were approved by the animal ethics committees of the University of Canberra and The Australian National University.
Statistical analysis.
The statistical significance between experimental groups was determined by the Student's unpaired t test (P < 0.05). Data are presented as the mean ± standard error of the mean (SEM). For disease scores, experimental results were analyzed by the Mann-Whitney test. Statistical analyses were performed using GraphPad Prism software (version 4.0b; GraphPad Software Inc.). Differences in means were considered significant at a P value of <0.05.
RESULTS
Subcellular localization of the 6k proteins in RRV-infected cells.
We first characterized the distribution and localization of the RRV 6k proteins in RRV-infected BHK-21 cells at 12 h postinfection (p.i.) by immunofluorescence. When infected cells were processed by fixation, permeabilization, and immunostaining for either the RRV 6k proteins or RRV E2, we readily detected these proteins (Fig. 1A, right), while nonpermeabilized infected cells were positive for E2 but not the 6k proteins (Fig. 1A, left). This suggests that the RRV 6k proteins were not exposed to the extracellular space. When examined with a higher magnification, both the 6k proteins and E2 were predominantly present at the periphery of the nuclei with punctate appearances (Fig. 1B), reminiscent of localization to the vesicular organelles.
FIG 1.

Cellular distribution of the RRV 6k and E2 proteins in WT RRV-infected BHK-21 cells. BHK-21 cell monolayers were infected with WT RRV at an MOI of 0.1 for 1 h, placed in fresh growth medium, and incubated for 12 h. (A) Cells were either fixed with paraformaldehyde (nonpermeabilized) or permeabilized and fixed with paraformaldehyde containing 0.1% Triton X-100. The cellular location of the 6k proteins and the E2 protein was visualized using rabbit polyclonal antibodies raised against the RRV 6K N-terminal region and the RRV E2 protein, respectively, followed by FITC-conjugated secondary antibodies. Cells were examined using fluorescence (top row) and light (middle row) microscopy, and merged images (bottom row) were generated. Bar = 40 μm. (B) RRV 6k proteins and the E2 protein exhibit a punctate distribution in BHK-21 cells. BHK-21 cell monolayers were infected with WT RRV, fixed, and permeabilized at 12 h p.i. and stained as described in the legend to panel A. Bar = 20 μm.
We then performed higher-resolution confocal microscopy to determine the subcellular localization of the 6k proteins (Fig. 2). A punctate intracellular distribution of the 6k proteins without discernible accumulation at the plasma membrane was observed. These intracellular foci did not represent replication complexes, as evidenced by costaining for the replication intermediate double-stranded RNA (Fig. 2A). The lack of 6k signals in cells infected with RRV-(Δ6K) demonstrates the specificity of the staining (Fig. 2B). Further, colocalization analysis with organelle markers showed that the 6k protein foci in cells infected with WT RRV extensively colocalized with calnexin, a marker for the ER (Fig. 2C), while the 6k proteins did not colocalize to any considerable extent with markers for the cis-Golgi network (Fig. 2D), early endosomes (Fig. 2E), or lysosomes (Fig. 2F). Also, the 6k proteins were not targeted to autophagosomes (Fig. 2G), which accumulate in alphavirus-infected cells (25). Thus, this in-depth microscopic analysis shows that the 6k proteins were predominantly localized to the ER in RRV-infected cells.
FIG 2.

Subcellular localization of the RRV 6k proteins. (A and B) BHK-21 cell monolayers were infected with WT RRV (A) or RRV-(Δ6K) (B) at an MOI of 0.1 for 1 h, placed in fresh growth medium, and incubated for 12 h. Cells were permeabilized and fixed with paraformaldehyde containing 0.1% Triton X-100. The subcellular localization of the 6k proteins was visualized using rabbit polyclonal antibodies raised against the RRV 6K N-terminal region (pseudocolored red), combined with staining of the indicated organelles (green). dsRNA, double-stranded RNA. Bar = 10 μm. (C to G) HOS cells were infected with WT RRV as indicated and fixed and permeabilized, followed by costaining for the 6k proteins (red) and the indicated organelle marker (green). Mouse monoclonal antibodies against calnexin, GM130, EEA1, and LAMP1 were used to stain the ER (C), cis-Golgi network (D), early endosomes (E), and lysosomes (F), respectively. (G) Autophagosomes were detected by fluorescence of EGFP-tagged LC3 in HOS cells stably transfected with this marker.
Effect of the 6k deletion on RRV temperature and pH sensitivity.
We then went on to assess whether the 6k proteins affect the sensitivity of RRV virions to variations in temperature or pH, as such a role has been suggested in the context of other alphavirus 6k mutations (26). To assess the thermostability of virions, WT RRV and RRV-(Δ6K) particles were treated at elevated temperatures (52 or 56°C) for 0, 6 and 12 min, before being cooled to 4°C and assayed by plaque assay. WT RRV and RRV-(Δ6K) were both completely inactivated following incubation at 56°C, and no difference in the amount of infectious virus after incubation at 52°C was found (data not shown), indicating that there was no difference in the thermal stability of RRV-(Δ6K) and WT virus particles. To examine the temperature sensitivity of RRV-(Δ6K) during replication, 10-fold dilutions of either WT RRV or RRV-(Δ6K) were overlaid onto Vero cells and incubated at different temperatures. Variations in temperature did not differentially affect the infectivity of the WT and mutant viruses at 37°C and 39°C, where similar plaque numbers were counted, while no plaques were detected at 41°C (Fig. 3A and B). The plaque size of RRV-(Δ6K) was, however, significantly reduced compared to that of WT RRV at 39°C but not at 37°C (Fig. 3C). Thus, unlike the stability of virions and their ability to infect cells, a subsequent step of the RRV replication cycle was clearly affected by the deletion of the 6k region.
FIG 3.

RRV-(Δ6K) temperature and pH sensitivity. (A) Representative plaque phenotypes of WT RRV and RRV-(Δ6K) at the specified temperatures. (B) Average plaque count for WT RRV and RRV-(Δ6K) at 37°C and 39°C. The plaque count was zero at 41°C. The data represent the means ± SEMs from three independent determinations. (C) Average plaque diameter for WT RRV and RRV-(Δ6K) at 37°C and 39°C. (D) Relative amount of infectious WT RRV and RRV-(Δ6K) after exposure to pH 5.5 for the indicated times normalized to the amount at time zero (100%). The data represent the means ± SEMs from three independent determinations. P values were determined by Student's unpaired t test. **, P < 0.01; ***, P < 0.001.
To examine the effect of deletion of the 6k proteins on the pH sensitivity of virions, WT RRV and RRV-(Δ6K) particles were incubated at pH 5.5 for various times prior to neutralization and subsequent plaque assay. WT RRV was stable at pH 5.5 with no reduction of infectivity (Fig. 3D). In contrast, RRV-(Δ6K) virions were sensitive to low pH, as their infectivity was significantly reduced following incubation at pH 5.5 for 60 s compared to that of WT RRV. In summary, we observed a tendency for RRV to have a higher vulnerability to temperature and pH when it lacked the 6k proteins.
6k deletion does not affect virus particle production but affects the release of virus particles from BHK-21 cells.
Next, we asked whether deletion of the 6k proteins affected RRV replication. To this end, BHK-21 cells were infected with either WT RRV or RRV-(Δ6K), and the supernatants were harvested at various times postinfection and titrated by plaque assay. At early times postinfection (0 to 16 h), no significant difference in the amount of infectious cell-associated WT RRV or RRV-(Δ6K) was observed, suggesting that deletion of the 6k gene did not affect the rate of replication of RRV (Fig. 4A). However, at late times (>24 h) postinfection, the amount of cell-associated RRV-(Δ6K) significantly increased compared to the amount of WT RRV, indicating that virions of RRV-(Δ6K) were not efficiently released and accumulated within and/or at the surface of infected cells. Additionally, the significant increase in the amount of WT RRV released from BHK-21 cells compared to the amount of RRV-(Δ6K) released, which was especially evident at 16 h p.i. and later time points, also supports the conclusion that the release of RRV-(Δ6K) was compromised (Fig. 4B). Complementary RT-qPCR analysis of culture supernatants and lysates containing cell-associated virus also revealed evidence of the disturbed release of RRV-(Δ6K); the cell-associated viral genome copy numbers of RRV-(Δ6K) significantly increased at 24 h p.i. with respect to the cell-associated viral genome copy numbers of WT RRV (Fig. 4C), a finding which correlates with the larger amount of infectious cell-associated RRV-(Δ6K) detected (Fig. 4A). The genome copy numbers of RRV-(Δ6K) were significantly reduced in culture supernatants at 16 h p.i. though not at 24 h p.i. (Fig. 4D). This may result from increased survival of RRV-(Δ6K)-infected cells, allowing a prolonged or more efficient synthesis of viral RNA. To rule out the possibility that this effect may be caused by a reduced genome copy number/PFU ratio of RRV-(Δ6K), such ratios were determined for both WT RRV and RRV-(Δ6K) (data not shown). No difference was found, indicating that the high genome copy number of RRV-(Δ6K) in the cell culture supernatant at 24 h p.i. may be due to the release of virus RNA-containing cell debris into the culture medium. Together these data suggest that, although virus production was not reduced, deletion of 6k inhibited the release of RRV from host cells.
FIG 4.

Deletion of RRV 6k has no effect on RRV particle production in BHK-21 cells but inhibits particle release. BHK-21 cells were infected with either WT RRV or RRV-(Δ6K) at an MOI of 0.1 for 1 h and placed in growth medium. (A and B) Quantification of infectious cell-associated virus (A) and virus released into the cell culture supernatant (B) by plaque assay. The limit of detection for the plaque assay was 10 PFU/ml. (C and D) Determination of viral genome copy numbers by RT-qPCR at the indicated times p.i. for cell-associated virus (C) and virus in the culture supernatant (D). P values were determined by Student's unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
RRV-(Δ6K)-infected mice show a reduced severity and duration of RRV disease.
Several in vivo studies examining the role of the alphavirus structural proteins have outlined the importance of the envelope glycoproteins in viral pathogenesis (27, 28). However, little is known about the effect of the 6k proteins on alphaviral virulence. To examine this for RRV, Swiss mice were mock infected or infected with 103 PFU of WT RRV or RRV-(Δ6K) and monitored for disease signs and body weight. WT RRV-infected mice developed severe disease signs, including a loss of hind limb grip strength and dragging of the hind limbs. These disease signs peaked at day 10 p.i. and resolved by day 20 (Fig. 5A). Furthermore, WT RRV-infected mice failed to gain weight, showing signs of wastage, which also peaked at day 10 p.i. (Fig. 5B). In contrast, RRV-(Δ6K)-infected mice did not present the same severity of disease as WT RRV-infected mice, showing only mild hind limb weakness and a low level of lethargy. This mild disease peaked at day 6 and resolved by day 12 p.i. (Fig. 5A). RRV-(Δ6K)-infected mice also started to gain weight by day 8 (Fig. 5B). In addition, a significant decrease in the viral load was found in both the quadriceps muscle (Fig. 5C) and the ankle joints (Fig. 5D) of RRV-(Δ6K)-infected mice. While peak viral titers in WT RRV-infected mice were observed at day 5 p.i., RRV-(Δ6K) titers in the quadriceps muscle peaked at day 3 and were markedly lower than those of WT RRV. A significant decrease in the RRV-(Δ6K) titer in comparison to the WT RRV titer was observed in both quadriceps muscle and ankle joint tissues from day 5 p.i. RRV-(Δ6K) was cleared from the ankle joints of infected mice at day 8 p.i. In addition, we detected viral RNA in the ankle joints of mice up to 1 month p.i. with WT RRV but not with RRV-(Δ6K) (Fig. 5E), indicating the persistence of WT RRV but not the 6k mutant.
FIG 5.

RRV-induced disease in mice infected with WT RRV and RRV-(Δ6K). Seventeen-day-old outbred Swiss mice were infected with 103 PFU of WT RRV or RRV-(Δ6K) by intraperitoneal injection. Mock-inoculated mice were injected with PBS alone. (A) Mice were scored for lethargy and hind limb dysfunction on the basis of the following scale: 0, no disease signs; 1, ruffled fur; 2, very mild hind limb weakness; 3, mild hind limb weakness; 4, moderate hind limb weakness and dragging of hind limbs; 5, severe dragging; 6, complete loss of hind limb function; 7, moribund state. Data for disease scores were analyzed by the Mann-Whitney test. *, P < 0.05; **, P < 0.01. (B) Mice were monitored daily for changes in weight. Values are the means ± SEMs for 8 mice per group. *, P < 0.05. (C and D) On days 1, 3, 5, and 8 p.i., the quadriceps muscle (C) and ankle joints (D) were collected and the amount of infectious virus was determined by plaque assay. The limit of detection for the plaque assay was 10 PFU/ml. The data represent the means ± SEMs for 4 mice per group and are representative of those from two separate experiments. P values were determined by Student's unpaired t test.*, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) RT-PCR analysis of the E2 region of viral RNA in the ankle joint tissue of WT RRV- or RRV-(Δ6K)-infected mice at different times after infection. The hypoxanthine-guanine phosphoribosyltransferase (HPRT) housekeeping gene was used as a loading control. Lanes 1, day 5 p.i.; lanes 2, day 10 p.i.; lanes 3, day 15 p.i.; lanes 4, day 30 p.i.
RRV-(Δ6K) immunization results in a reduced viral burden during alphaviral infection.
As RRV-(Δ6K) showed significantly reduced virulence in the host, we tested its prophylactic efficacy. Mice were immunized with 104 PFU of RRV-(Δ6K) or mock immunized with PBS and at day 30 p.i. were challenged with a high dose (106 PFU) of WT RRV or CHIKV. As shown in Fig. 6, RRV-(Δ6K)-immunized mice showed an overall reduced viral load in serum after infection with RRV (Fig. 6A) or CHIKV (Fig. 6B). Immunization with RRV-(Δ6K) resulted in the effective reduction of the RRV load already by day 1 p.i. (Fig. 6A). When the RRV-(Δ6K)-immunized mice were challenged with CHIKV, viral titers at day 2 were reduced 10-fold compared to those in mock-immunized mice (Fig. 6B), indicating cross protection. By day 3 postchallenge, infectious virus was cleared from the serum of RRV-(Δ6K)-immunized mice but not control mice in both RRV- and CHIKV-challenged mice (Fig. 6). These findings suggest that RRV-(Δ6K) immunization offers protection against reinfection with the same or even a closely related alphavirus by reducing the viral burden in the host.
FIG 6.
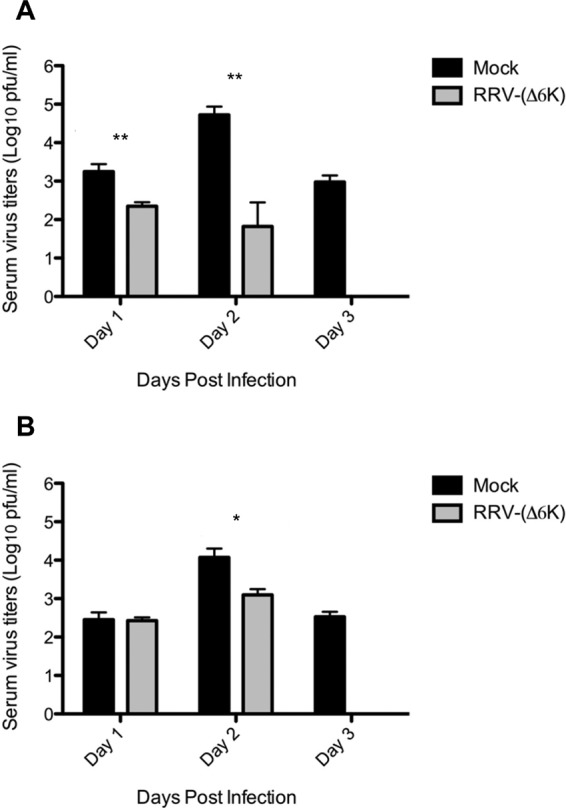
Primary RRV-(Δ6K) infection reduces the viral burden upon secondary exposure to alphavirus in mice. C57BL/6 mice were immunized with 104 PFU RRV-(Δ6K) or mock immunized with PBS alone. At day 30 p.i., the mice were challenged with 106 PFU WT RRV (A) and CHIKV (B). Viral titers in sera were evaluated by the plaque assay at days 1, 2, and 3 postchallenge. The limit of detection for the plaque assay was 10 PFU/ml. Each data point represents the mean ± SEM for 4 mice per group, and the data are representative of those from two separate experiments. P values were determined by Student's unpaired t test. *, P < 0.05; **, P < 0.01.
DISCUSSION
Little is known about the importance and role of the RRV 6k proteins, 6K and TF, during virus infection and replication. A number of previous studies have used partial gene deletions or insertions to investigate the role of the alphaviral 6K protein (8, 9). However, in such mutants, it is not known whether protein function is completely abolished. Also, it is not known whether these approaches result in the expression of a novel protein with different properties and what effects that this might have on processes such as viral replication and host cell metabolism (29, 30). Furthermore, the recent discovery of TF, the result of a translational frameshift in the 6k gene, brings added uncertainty to the validity of these approaches (2, 31). Additionally, the 6k gene is not highly conserved between alphaviruses, making it difficult to compare findings in different alphaviruses. In this study, an RRV mutant containing an in-frame deletion of the entire 6k gene region was constructed, thereby ensuring that the translation of proteins encoded by sequences on either side of the 6k gene region remained unaffected. This mutant is directly analogous to the alphavirus 6k deletion mutant of SFV described previously (11).
Immunofluorescence detection of the 6k proteins showed a punctate perinuclear distribution, suggesting that these proteins were largely retained within the cell. The 6k proteins were not detectable in RRV-infected nonpermeabilized cells by immunofluorescence, indicating that the epitope for the 6k antibody, the N-terminal end of the 6k proteins, was not detectably exposed at the cell surface in infected cells. The predominant localization of the RRV 6k proteins was at the ER, in line with the findings of previous studies on SINV (4) and SFV (7) 6k proteins. At the ER, the alphaviral 6k proteins may function as ion channels, thereby disrupting calcium homeostasis by the sustained release of calcium from the ER store (13, 31). The sustained release of calcium from the ER store leads to increased cytoplasmic calcium levels, triggering cell shrinkage and cell permeabilization, which are likely to play a role in the later stages of alphavirus replication, such as viral budding and release (15). The subcellular localization of the 6k proteins was, however, not completely congruent with the ER, implying some shuttling of 6K/TF to other vesicular organelles and/or the plasma membrane.
The reduced infectious RRV-(Δ6K) titers for released but not cell-associated virus suggest that the RRV 6k proteins are required for efficient viral dissociation from the cell. The inefficient cell dissociation of RRV-(Δ6K) virions may be linked to the formation of ion channels by the 6k proteins (16). Membrane permeabilization by ion channel formation leads to changes in the local membrane potential, creating an environment thought to promote alphaviral budding (9, 32). This defect in cell dissociation was most likely enhanced at 39°C, as at this temperature RRV-(Δ6K) made the same number of plaques as WT RRV, but the plaques were smaller than those of WT RRV. With the 6K protein of SFV thought to play a role in the organization and stability of the heterodimeric E1-E2 glycoprotein complex, it is possible that deletion of the RRV 6k proteins may influence the downstream processing and/or trafficking of the RRV glycoproteins and ultimately affect virus assembly and replication (31, 33). Notably, in contrast to RRV-(Δ6K), the SFV 6k deletion mutant demonstrated a dramatic decrease in the rate of viral replication (5, 11). In addition, a 6k deletion mutant of salmonid alphavirus (SAV) was found to be nonviable (34), outlining the functional divergence of the 6k proteins in different alphaviruses.
In animal studies, RRV-(Δ6K)-infected mice did not show disease of the same severity as that in WT RRV-infected mice. RRV-(Δ6K) induced mild disease, which culminated and resolved 4 and 8 days earlier, respectively, than WT RRV disease. RRV-(Δ6K)-infected mice continued to gain weight, in contrast to WT RRV-infected mice. These observations are in line with those of previous studies into the role of SINV TF during infection in vivo, which found that mice infected with an SINV TF deletion mutant developed a disease less severe than that in mice infected with WT SINV (6). The attenuated disease in mice infected with the SINV TF deletion mutant was associated with a reduced viral burden in the target organs (6), much like the reduced viral titers observed here in the quadriceps muscle and ankle joint of RRV-(Δ6K)-infected mice. It is therefore likely that the inefficient release of RRV-(Δ6K) from the host cell, observed in vitro, contributed to the reduced viral burden in RRV-(Δ6K)-infected mice and, subsequently, the reduced inflammation and disease. In this regard, no RRV RNA was detectable within joint tissue of RRV-(Δ6K)-infected mice at 30 days postinfection.
The attenuated nature of infection with RRV-(Δ6K) provides the possibility for using this virus as a vaccine vector, avoiding severe RRV disease. Similarly, Hallengärd and colleagues reported that CHIKV with a 6k deletion was also attenuated in vivo, induced high levels of neutralizing antibodies, and protected animals against viremia and joint swelling upon challenge with WT CHIKV (35). Thus, the deletion of 6k has the potential to generally attenuate alphaviruses and may be used as a potential vaccination strategy against RRV. Importantly, our results show that immunization with RRV-(Δ6K) also protected against disease upon challenge with the related arthritogenic alphavirus CHIKV. This is evidence for cross protection, a phenomenon reported for the respective wild-type viruses and may be caused by the induction of broadly neutralizing antialphavirus antibodies (36, 37). Thus, vaccination with a 6k deletion virus has the potential to protect against disease by several related alphaviruses.
In conclusion, we describe the phenotypic properties of an RRV 6k deletion mutant, whereby the 6k gene locus likely plays a key role in the enhanced pathogenicity exhibited by RRV. Understanding the viral genetic loci that govern pathogenesis could ultimately lead to the development of safer and more immunogenic alphavirus vectors for vaccine delivery. In this regard, manipulating the 6k gene locus could be a potential strategy for future viral vector-based vaccine development.
ACKNOWLEDGMENTS
We thank Pontus Aspenström, Karin Loré, and Lisa Westerberg (Karolinska Institutet, Stockholm, Sweden) for the generous provision of reagents.
Part of this work was supported by a National Health and Medical Research Council grant to S.M. (grant 399700) and a Swedish Research Council grant (621-2014-4718) to G.M.M. A.T. is the recipient of a National Health and Medical Research Council early career research fellowship (1073108). L.J.H. is the recipient of an Australian Research Council Discovery early career researcher award (140101493). S.M. is the recipient of a National Health and Medical Research Council senior research fellowship (1059167).
With great sadness we note that both Julian V. Melton and Peter W. Gage passed away before completion of this research study. The other authors dedicate this paper to these two outstanding researchers.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58:491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell RC. 2002. Ross River virus: ecology and distribution. Annu Rev Entomol 47:1–31. doi: 10.1146/annurev.ento.47.091201.145100. [DOI] [PubMed] [Google Scholar]
- 3.Harley D, Sleigh A, Ritchie S. 2001. Ross River virus transmission, infection, and disease: a cross-disciplinary review. Clin Microbiol Rev 14:909–932. doi: 10.1128/CMR.14.4.909-932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaedigk-Nitschko K, Schlesinger MJ. 1990. The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology 175:274–281. doi: 10.1016/0042-6822(90)90209-A. [DOI] [PubMed] [Google Scholar]
- 5.Loewy A, Smyth J, von Bonsdorff CH, Liljestrom P, Schlesinger MJ. 1995. The 6-kilodalton membrane protein of Semliki Forest virus is involved in the budding process. J Virol 69:469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snyder JE, Kulcsar KA, Schultz KL, Riley CP, Neary JT, Marr S, Jose J, Griffin DE, Kuhn RJ. 2013. Functional characterization of the alphavirus TF protein. J Virol 87:8511–8523. doi: 10.1128/JVI.00449-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welch WJ, Sefton BM. 1980. Characterization of a small, nonstructural viral polypeptide present late during infection of BHK cells by Semliki Forest virus. J Virol 33:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlesinger MJ, London SD, Ryan C. 1993. An in-frame insertion into the Sindbis virus 6K gene leads to defective proteolytic processing of the virus glycoproteins, a trans-dominant negative inhibition of normal virus formation, and interference in virus shut off of host-cell protein synthesis. Virology 193:424–432. doi: 10.1006/viro.1993.1139. [DOI] [PubMed] [Google Scholar]
- 9.Sanz MA, Carrasco L. 2001. Sindbis virus variant with a deletion in the 6K gene shows defects in glycoprotein processing and trafficking: lack of complementation by a wild-type 6K gene in trans. J Virol 75:7778–7784. doi: 10.1128/JVI.75.16.7778-7784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liljestrom P, Garoff H. 1991. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J Virol 65:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liljestrom P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol 65:4107–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrasco L. 1995. Modification of membrane permeability by animal viruses. Adv Virus Res 45:61–112. doi: 10.1016/S0065-3527(08)60058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrasco L. 1977. The inhibition of cell functions after viral infection. A proposed general mechanism. FEBS Lett 76:11–15. [DOI] [PubMed] [Google Scholar]
- 14.Carrasco L, Perez L, Irurzun A, Lama J, Martinez-Abarca F, Rodrigez P, Guinea R, Castrillo JL, Sanz MA, Ayala MJ. 1993. Modification of membrane permeability by animal viruses, p 283–305. In Carrasco L, Sonenberg N, Wimmer E (ed), Regulation of gene expression in animal viruses. Plenum Publishing Corp, New York, NY. [Google Scholar]
- 15.Antoine A-F, Montpellier C, Cailliau K, Browaeys-Poly E, Vilain J-P, Dubuisson J. 2007. The alphavirus 6K protein activates endogenous ionic conductances when expressed in Xenopus oocytes. J Membr Biol 215:37–48. doi: 10.1007/s00232-007-9003-6. [DOI] [PubMed] [Google Scholar]
- 16.Melton JV, Ewart GD, Weir RC, Board PG, Lee E, Gage PW. 2002. Alphavirus 6K proteins form ion channels. J Biol Chem 277:46923–46931. doi: 10.1074/jbc.M207847200. [DOI] [PubMed] [Google Scholar]
- 17.Sanz MA, Perez L, Carrasco L. 1994. Semliki Forest virus 6K protein modifies membrane permeability after inducible expression in Escherichia coli cells. J Biol Chem 269:12106–12110. [PubMed] [Google Scholar]
- 18.Madan V, Redondo N, Carrasco L. 2010. Cell permeabilization by poliovirus 2B viroporin triggers bystander permeabilization in neighbouring cells through a mechanism involving gap junctions. Cell Microbiol 12:1144–1157. doi: 10.1111/j.1462-5822.2010.01460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eng KE, Panas MD, Hedestam GBK, McInerney GM. 2010. A novel quantitative flow cytometry-based assay for autophagy. Autophagy 6:634–641. doi: 10.4161/auto.6.5.12112. [DOI] [PubMed] [Google Scholar]
- 20.Klock HE, Koesema EJ, Knuth MW, Lesley SA. 2008. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71:982–994. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- 21.Kuhn RJ, Niesters HGM, Hong Z, Strauss JH. 1991. Infectious RNA transcripts from Ross River virus cDNA clones and the construction and characterization of defined chimeras with Sindbis virus. Virology 182:430–441. doi: 10.1016/0042-6822(91)90584-X. [DOI] [PubMed] [Google Scholar]
- 22.Lidbury BA, Simeonovic C, Maxwell GE, Marshall ID, Hapel AJ. 2000. Macrophage-induced muscle pathology results in morbidity and mortality for Ross River virus-infected mice. J Infect Dis 181:27–34. doi: 10.1086/315164. [DOI] [PubMed] [Google Scholar]
- 23.Shabman RS, Rogers KM, Heise MT. 2008. Ross River virus envelope glycans contribute to type I interferon production in myeloid dendritic cells. J Virol 82:12374–12383. doi: 10.1128/JVI.00985-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lidbury BA, Rulli NE, Suhrbier A, Smith PN, McColl SR, Cunningham AL, Tarkowski A, van Rooijen N, Fraser RJ, Mahalingam S. 2008. Macrophage-derived proinflammatory factors contribute to the development of arthritis and myositis after infection with an arthrogenic alphavirus. J Infect Dis 197:1585–1593. doi: 10.1086/587841. [DOI] [PubMed] [Google Scholar]
- 25.Eng KE, Panas MD, Murphy D, Hedestam GBK, McInerney GM. 2012. Accumulation of autophagosomes in Semliki Forest virus-infected cells is dependent on expression of the viral glycoproteins. J Virol 86:5674–5685. doi: 10.1128/JVI.06581-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McInerney GM, Smit JM, Liljestrom P, Wilschut J. 2004. Semliki Forest virus produced in the absence of the 6K protein has an altered spike structure as revealed by decreased membrane fusion capacity. Virology 325:200–206. doi: 10.1016/j.virol.2004.04.043. [DOI] [PubMed] [Google Scholar]
- 27.Santagati MG, Maatta JA, Itaranta PV, Salmi AA, Hinkkanen AE. 1995. The Semliki Forest virus E2 gene as a virulence determinant. J Gen Virol 76(Pt 1):47–52. [DOI] [PubMed] [Google Scholar]
- 28.Gorchakov R, Wang E, Leal G, Forrester NL, Plante K, Rossi SL, Partidos CD, Adams AP, Seymour RL, Weger J, Borland EM, Sherman MB, Powers AM, Osorio JE, Weaver SC. 2012. Attenuation of chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J Virol 86:6084–6096. doi: 10.1128/JVI.06449-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Firth AE, Chung BY, Fleeton MN, Atkins JF. 2008. Discovery of frameshifting in alphavirus 6K resolves a 20-year enigma. Virol J 5:108. doi: 10.1186/1743-422X-5-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung BY, Firth AE, Atkins JF. 2010. Frameshifting in alphaviruses: a diversity of 3′ stimulatory structures. J Mol Biol 397:448–456. doi: 10.1016/j.jmb.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 31.Yao JS, Strauss EG, Strauss JH. 1996. Interactions between PE2, E1, and 6K required for assembly of alphaviruses studied with chimeric viruses. J Virol 70:7910–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waite MR, Pfefferkorn ER. 1970. Inhibition of Sindbis virus production by media of low ionic strength: intracellular events and requirements for reversal. J Virol 5:60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lusa S, Garoff H, Liljestrom P. 1991. Fate of the 6k membrane-protein of Semliki Forest virus during virus assembly. Virology 185:843–846. doi: 10.1016/0042-6822(91)90556-Q. [DOI] [PubMed] [Google Scholar]
- 34.Guo TC, Johansson DX, Haugland O, Liljestrom P, Evensen O. 2014. A 6K-deletion variant of salmonid alphavirus is non-viable but can be rescued through RNA recombination. PLoS One 9:e100184. doi: 10.1371/journal.pone.0100184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hallengärd D, Kakoulidou M, Lulla A, Kummerer BM, Johansson DX, Mutso M, Lulla V, Fazakerley JK, Roques P, Le Grand R, Merits A, Liljestrom P. 2014. Novel attenuated chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. J Virol 88:2858–2866. doi: 10.1128/JVI.03453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A. 2010. Chikungunya virus arthritis in adult wild-type mice. J Virol 84:8021–8032. doi: 10.1128/JVI.02603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MK, Fong RH, Kahle KM, Smit JM, Jin J, Simmons G, Doranz BJ, Crowe JE Jr, Fremont DH, Rossmann MG, Diamond MS. 2015. Broadly neutralizing alphavirus antibodies bind an epitope on E2 and inhibit entry and egress. Cell 163:1095–1107. doi: 10.1016/j.cell.2015.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]