

Abstract
Immunosuppression is the typical measure to prevent rejection after heart transplantation. Although rejection is the usual cause of cardiac hypertrophy, numerous other factors warrant consideration. Calcineurin inhibitors rarely cause hypertrophic cardiomyopathy; the few relevant reports have described children after orthotopic kidney or liver transplantation. We present the case of a 73-year-old woman, an asymptomatic orthotopic heart transplantation patient, in whom chronic immunosuppression with prednisone and cyclosporine apparently caused a phenotype of hypertrophic cardiomyopathy. The natural course of her midapical hypertrophy was revealed by single-photon-emission computed tomography, positron-emission tomography, and 2-dimensional echocardiography.
Clinicians and radiographers should be alert to progressive left ventricular hypertrophy and various perfusion patterns in heart transplantation patients even in the absence of underlying coronary artery disease. Toward this end, we recommend that advanced imaging methods be used to their fullest extent.
Keywords: Cardiomyopathy, hypertrophic/chemically induced/diagnosis/drug therapy; cyclosporine/therapeutic use; heart transplantation; heart ventricles/physiopathology; immunosuppressive agents/adverse effects; positron-emission tomography; prednisone/therapeutic use; time factors; tomography, emission-computed, single-photon
Hypertrophic cardiomyopathy (HCM) is associated with sarcomere disarray and pathologic hypertrophy of the myocardium. Ischemic or fixed perfusion abnormalities in patients who have HCM and no substantial coronary artery disease (CAD) can indicate small-vessel disease or microvascular dysfunction. Acquired variants resembling the HCM phenotype have typically been seen in the presence of hypertensive heart disease, particularly in elderly patients.
Years after an elderly woman had undergone orthotopic heart transplantation (OHT), we detected the natural development of an apical-variant HCM phenotype by analyzing images from single-photon-emission computed tomography (SPECT), 2-dimensional echocardiography (TTE), and positron-emission tomography (PET). We describe the possible contribution of long-term cyclosporine and steroidal use to the patient's acquired hypertrophy, and we review typical causes.
Case Report
A 73-year-old woman who had undergone OHT in 1990 was referred to our nuclear cardiology stress laboratory in July 2011. She was asymptomatic. This annual evaluation for transplant vasculopathy included regadenoson rubidium82 PET rest–stress imaging (an off-label use in cardiac PET). The PET scan revealed substantial, reversible perfusion defects in the mid-to-distal anterior and inferior left ventricular (LV) walls and apex; transient ischemic dilation was also confirmed (Fig. 1). The pattern on the rest images—normal perfusion along with preferential, marked uptake of radioisotope medium in the apical and mid ventricle—suggested midapical LV hypertrophy (LVH).
Fig. 1.
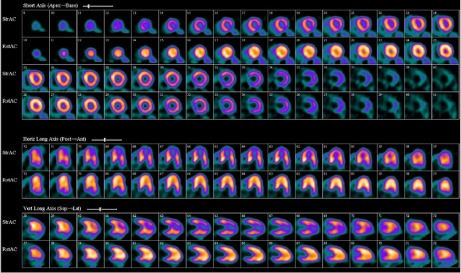
In July 2011, regadenoson rubidium82 rest–stress positron-emission tomographic images show a large, severe perfusion defect involving the mid-to-distal anterior, inferior, and apical left ventricular walls. The rest perfusion images (bottom row) notably show a pattern of enhanced radiotracer uptake in the mid-to-distal anterior and inferior walls and in the apex, similar to the spade shape on 2-dimensional echocardiograms and ventriculograms in patients who have apical-variant hypertrophic cardiomyopathy.
We reviewed the patient's medical records since 1991. No donor-specific antibodies, biopsy results indicating cellular or humoral rejection, or substantial epicardial CAD were ever noted. She had undergone TTE annually after OHT. Her first TTE, in 1991, showed no LVH; mild LVH was first reported in 1997. An electrocardiogram performed in 1997 showed right bundle branch block, LVH, and deep T-wave inversions in a pattern typical of apical-variant HCM (Fig. 2).
Fig. 2.
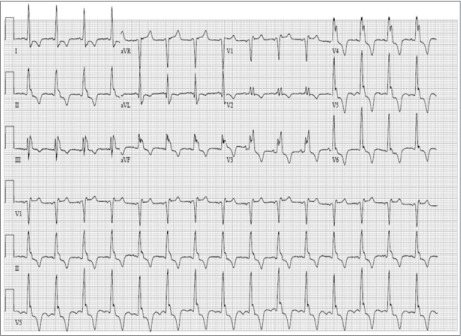
In 1997, this 12-lead electrocardiogram showed sinus rhythm, complete right bundle branch block, and deep T-wave inversions in the anterolateral precordial leads.
The patient's earlier SPECT scans (from 2004, 2005, and 2008) showed normal perfusion with enhanced isotope uptake in the mid apex of the LV; this suggested apical or midapical hypertrophy. The SPECT study performed in 2009 revealed a subtle, reversible, distal, anteroapical perfusion defect (Fig. 3). To clarify this finding, the patient had undergone dobutamine stress echocardiography in August 2010. Those results included preserved systolic function, no wall-motion abnormality, and midapical hypertrophy in a spade-shaped LV (Fig. 4).
Fig. 3.
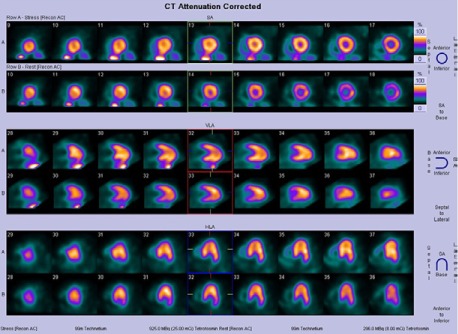
In 2009, rest–stress single-photon-emission computed tomographic images showed a subtle reversible defect in the anterior and apical walls and a midapical hypertrophic pattern with enhanced isotope uptake.
Fig. 4.
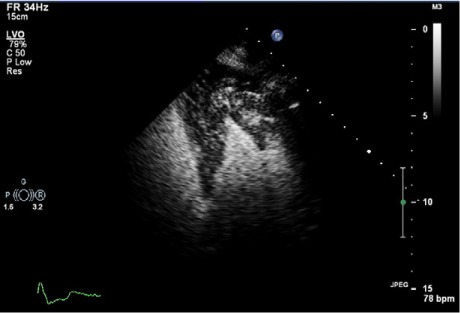
In 2010, this 2-dimensional contrast echocardiogram (4-chamber view) showed a spade-shaped left ventricle with preferential severe hypertrophy of the apex and mid ventricle.
The patient had developed hypertension soon after transplantation. In 1991, she was started on enalapril; in 1996, bumetanide was added. In 2003, the enalapril was changed to candesartan, and carvedilol was added to the antihypertensive regimen in 2004. Over several years, the patient developed calcineurin-induced nephropathy, so the candesartan was replaced with isosorbide dinitrate and hydralazine in 2006.
Coronary angiograms from 1991 through 2000 had shown no significant transplant vasculopathy or obstructive CAD, so the patient had been treated medically. Her initial maintenance immunosuppressive therapy included cyclosporine, azathioprine, and prednisone (1990–1998). From 1998 on, the patient had complied with an immunosuppressive regimen of cyclosporine, mycophenolate mofetil, and prednisone. She had remained completely asymptomatic, so her transplant physicians had recommended annual monitoring with the use of SPECT, PET, and contrast TTE.
Discussion
Cyclosporine and steroids, widely used for immunosuppression, have dramatically improved the outcomes of transplant patients; however, these drugs might have contributed to this patient's midapical-variant HCM.
Several mechanisms might underlie the development of cardiac hypertrophy after OHT. One is systemic, hypertension-induced LVH after OHT. Another is immune injury and cytokines produced by the transplanted heart.1 A third is the independent contribution of immunosuppressive agents.
We think that our patient's HCM resulted from a variety of factors, including chronic exposure to immunosuppressants for at least 12 years. Calcineurin inhibitors are known to cause side effects such as renal impairment, disturbances of glucose metabolism, and neurologic sequelae. A less frequent side effect is cardiotoxicity, including the development of symmetric HCM in children years after kidney and liver transplantation and subsequent immunosuppression with tacrolimus.2–5 As part of immunosuppressive regimens in patients who were given bone marrow to suppress graft-versus-host disease, chronic cyclosporine therapy has been associated with LVH and increased LV mass.6
Cyclosporine-induced LVH has been associated with systemic hypertension. Our patient's hypertension was managed with 2 blood pressure medications. Nevertheless, cyclosporine might have a direct vasoconstrictive effect on both the renal and systemic circulations.7,8 Postulated mechanisms include changes in prostaglandin synthesis and activation of the renin-angiotensin and sympathetic nervous systems. In addition, steroids can cause HCM that typically resolves after steroidal therapy is discontinued.9
Stetson and colleagues1 have proposed that LVH in OHT patients might be due to a persistent expression of tumor necrosis factor-α (TNF-α), which induces the expression of angiotensin II—a known mediator of fibrosis and cardiac hypertrophy. Levels of total collagen, collagen I, and collagen III were also increased in OHT patients who had elevated TNF-α levels. In association with those changes in collagen content, the investigators postulated that the hypertrophy was secondary to increased cell size and the extracellular matrix deposition of collagen. In that study, patients with high myocardial TNF-α content still had preserved systolic function.1 On the basis of previous evidence in intact hearts of TNF-α's role in increasing protein synthesis and inducing angiotensin II,10,11 one could extend this concept to the phenomenon of induced LVH after OHT.
In our patient, multimodal images clearly showed the progression of LVH from mild to severe. An apical-variant HCM phenotype appeared as a midapical, increased perfusion pattern on SPECT, and PET revealed the progressive development of a dramatic ischemic pattern. Ward and colleagues12 noted a “solar polar” SPECT pattern at the apex during their evaluation of HCM, particularly the apical variant.11 However, to our knowledge, no investigators have described the role of SPECT or PET in evaluating acquired HCM of a midapical variant in OHT patients who were taking calcineurin inhibitors and steroids.
Although multiple mechanisms might have been involved, our theory—on the basis of the prior evidence—is that the calcineurin inhibitors and steroids contributed to our patient's LVH. The classic distribution of increased radiotracer uptake with normal perfusion patterns in the mid apex of the LV on the 3 earliest SPECT scans, the subtle anteroapical perfusion defect apparent on the 2009 SPECT scan, and thereafter the substantial worsening of the ischemic area in the same hypertrophied region as seen on PET, all probably indicate profound microvascular flow-reserve abnormalities precipitated by vasodilator stress. The dobutamine stress echocardiogram showed no pressure gradient.
Echocardiography, SPECT, and PET all helped to clarify the course of the HCM-like phenotype and noteworthy complications in our OHT patient. Perfusion imaging can reveal the progressive LVH and ischemic patterns that can appear in OHT patients even in the absence of underlying CAD. The numerous possible causes of hypertrophic response should alert clinicians caring for OHT patients to use multimodal imaging to its fullest extent.
Footnotes
From: Heart & Vascular Institute (Drs. Ananthasubramaniam and Williams) and Department of Internal Medicine (Dr. Garikapati), Henry Ford Hospital, Detroit, Michigan 48202
Dr. Ananthasubramaniam receives research grants from Astellas Pharma US, Inc.
References
- 1.Stetson SJ, Perez-Verdia A, Mazur W, Farmer JA, Koerner MM, Weilbaecher DG et al. Cardiac hypertrophy after transplantation is associated with persistent expression of tumor necrosis factor-alpha. Circulation. 2001;104(6):676–81. doi: 10.1161/hc3101.093765. [DOI] [PubMed] [Google Scholar]
- 2.Dehghani SM, Haghighat M, Imanieh MH, Zahmatkeshan M, Borzooei M, Amoozegar H et al. Tacrolimus related hypertrophic cardiomyopathy in liver transplant recipients. Arch Iran Med. 2010;13(2):116–9. [PubMed] [Google Scholar]
- 3.Jarzembowski TM, John E, Panaro F, Manzelli A, Cabrera A, Greco A et al. Reversal of tacrolimus-related hypertrophic obstructive cardiomyopathy 5 years after kidney transplant in a 6-year-old recipient. Pediatr Transplant. 2005;9(1):117–21. doi: 10.1111/j.1399-3046.2005.00260.x. [DOI] [PubMed] [Google Scholar]
- 4.Atkison P, Joubert G, Barron A, Grant D, Paradis K, Seidman E et al. Hypertrophic cardiomyopathy associated with tacrolimus in paediatric transplant patients. Lancet. 1995;345(8954):894–6. doi: 10.1016/s0140-6736(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 5.Turska-Kmiec A, Jankowska I, Pawlowska J, Kalicinski P, Kawalec W, Tomyn M et al. Reversal of tacrolimus-related hypertrophic cardiomyopathy after conversion to rapamycin in a pediatric liver transplant recipient. Pediatr Transplant. 2007;11(3):319–23. doi: 10.1111/j.1399-3046.2006.00633.x. [DOI] [PubMed] [Google Scholar]
- 6.Espino G, Denney J, Furlong T, Fitzsimmons W, Nash RA. Assessment of myocardial hypertrophy by echocardiography in adult patients receiving tacrolimus or cyclosporine therapy for prevention of acute GVHD. Bone Marrow Transplant. 2001;28(12):1097–103. doi: 10.1038/sj.bmt.1703304. [DOI] [PubMed] [Google Scholar]
- 7.Antunes ML, Spotnitz HM, Clark MB, Steinhardt MJ, Marboe CC, Smith CR et al. Long-term function of human cardiac allografts assessed by two-dimensional echocardiography. J Thorac Cardiovasc Surg. 1989;98(2):275–84. [PubMed] [Google Scholar]
- 8.Lipkin GW, Tucker B, Giles M, Raine AE. Ambulatory blood pressure and left ventricular mass in cyclosporin- and non-cyclosporin-treated renal transplant recipients. J Hypertens. 1993;11(4):439–42. doi: 10.1097/00004872-199304000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Vimala J, Prabhu A, Pavithran S, Kumar RN. Hydrocortisone induced hypertrophic cardiomyopathy. Int J Cardiol. 2011;150(3):e94–5. doi: 10.1016/j.ijcard.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Kapadia S, Torre-Amione G, Yokoyama T, Mann DL. Soluble TNF binding proteins modulate the negative inotropic properties of TNF-alpha in vitro. Am J Physiol. 1995;268(2 Pt 2):H517–25. doi: 10.1152/ajpheart.1995.268.2.H517. [DOI] [PubMed] [Google Scholar]
- 11.Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation. 1997;95(5):1247–52. doi: 10.1161/01.cir.95.5.1247. [DOI] [PubMed] [Google Scholar]
- 12.Ward RP, Pokharna HK, Lang RM, Williams KA. Resting “solar polar” map pattern and reduced apical flow reserve: characteristics of apical hypertrophic cardiomyopathy on SPECT myocardial perfusion imaging. J Nucl Cardiol. 2003;10(5):506–12. doi: 10.1016/s1071-3581(03)00455-0. [DOI] [PubMed] [Google Scholar]