

ABSTRACT
Bacterial genomes encode numerous homologs of Cas9, the effector protein of the type II CRISPR-Cas systems. The homology region includes the arginine-rich helix and the HNH nuclease domain that is inserted into the RuvC-like nuclease domain. These genes, however, are not linked to cas genes or CRISPR. Here, we show that Cas9 homologs represent a distinct group of nonautonomous transposons, which we denote ISC (insertion sequences Cas9-like). We identify many diverse families of full-length ISC transposons and demonstrate that their terminal sequences (particularly 3′ termini) are similar to those of IS605 superfamily transposons that are mobilized by the Y1 tyrosine transposase encoded by the TnpA gene and often also encode the TnpB protein containing the RuvC-like endonuclease domain. The terminal regions of the ISC and IS605 transposons contain palindromic structures that are likely recognized by the Y1 transposase. The transposons from these two groups are inserted either exactly in the middle or upstream of specific 4-bp target sites, without target site duplication. We also identify autonomous ISC transposons that encode TnpA-like Y1 transposases. Thus, the nonautonomous ISC transposons could be mobilized in trans either by Y1 transposases of other, autonomous ISC transposons or by Y1 transposases of the more abundant IS605 transposons. These findings imply an evolutionary scenario in which the ISC transposons evolved from IS605 family transposons, possibly via insertion of a mobile group II intron encoding the HNH domain, and Cas9 subsequently evolved via immobilization of an ISC transposon.
IMPORTANCE Cas9 endonucleases, the effectors of type II CRISPR-Cas systems, represent the new generation of genome-engineering tools. Here, we describe in detail a novel family of transposable elements that encode the likely ancestors of Cas9 and outline the evolutionary scenario connecting different varieties of these transposons and Cas9.
INTRODUCTION
Cas9 (CRISPR-associated protein 9) is the effector protein of the type II CRISPR-Cas systems, which represent about 10% of the CRISPR-Cas adaptive-immunity systems and, unlike other CRISPR-Cas types, are present only in bacteria (1). The Cas9 protein has a complex domain architecture, with a RuvC-like endonuclease domain; an HNH endonuclease domain that is inserted within the RuvC-like domain; and a large, α-helical recognition lobe for which no homologs have been identified in other proteins (1–4). Cas9 is an RNA-guided dual nuclease that recognizes and cleaves DNA of invading phages and plasmids. More specifically, Cas9 binds mature crRNAs, the products of CRISPR array expression followed by transcript processing, and employs the approximately 20-nucleotide unique portions of these RNAs (corresponding to the spacers in the CRISPR array) as guides to recognize and cleave DNA molecules that contain a sequence complementary to the guide adjacent to a short protospacer-associated motif (PAM). When the crRNA complexed with Cas9 forms a heteroduplex with the cDNA, the HNH domain of Cas9 cleaves the heteroduplex DNA strand, whereas the RuvC-like domain cleaves the second, unpaired DNA strand (3, 4). In addition to the recognition and cleavage of the target DNA at the interference stage of the CRISPR response, Cas9 is involved in PAM recognition during the adaptation stage, where short foreign DNA sequences are excised and inserted as spacers between the type II CRISPR repeats (5). The spacers, which are transcribed and incorporated into crRNAs, embody the bacterial immune memory, the key feature of CRISPR-Cas adaptive immunity.
In addition to the bona fide Cas9 genes that are lodged within type II CRISPR-Cas loci, numerous more distant, standalone homologs of Cas9 have been identified outside the CRISPR-Cas systems (1, 6). These proteins possess sequence features that are shared with Cas9, including the RuvC-like and HNH nuclease domains and a characteristic arginine-rich region (1, 6). The RuvC-like nuclease domain is also present in the widespread family of TnpB proteins that are often encoded by transposons of the IS605 superfamily (1). TnpB, often encoded as a standalone gene, is not required for transposition and, moreover, has been shown to downregulate transposition (7–9). Thus, the insertion elements that encode TnpB proteins alone are considered to be nonautonomous transposons.
It has been noticed that some bacterial genomes encode multiple Cas9 homologs (1, 6), but none of these proteins has been experimentally studied, and no specific evidence that the proteins are encoded in transposable elements (TEs) has been presented. Here, we show that these Cas9 homologs are encoded by a novel group of mostly nonautonomous transposons that we denote ISC (insertion sequences Cas9-like). Based on their structural features and the predicted target site specificity, the ISC elements form a distinct group within the IS605/IS200 superfamily of bacterial and archaeal transposons that are mobilized by the Y1 tyrosine transposase (7, 9). The ISC transposon-encoded two nuclease domain-containing proteins are the likely ancestors of the CRISPR-associated Cas9.
MATERIALS AND METHODS
A set of 2,751 complete bacterial and archaeal genomes was retrieved from the RefSeq database (February 2014) (10), and additional draft genomes were retrieved from GenBank. For detection of homologous proteins, the PSI-BLAST program (11) was used with default parameters to search the NCBI NR database. The same program was used to assign proteins from complete genomes to profiles of known protein families from the CDD database (12), with an E value threshold of 10−4 and low-complexity filtering turned off. Protein secondary structure was predicted using Jpred 4 (13). The HHpred program was used with default parameters for detection of remote sequence similarity (14). The BLASTCLUST program was used to cluster sequences with at least 90% length coverage and 90% sequence identity (15). Multiple-protein-sequence alignments for phylogenetic analysis were constructed using the MUSCLE program (16). Phylogenetic analysis was performed using the FastTree program with the WAG evolutionary model and the discrete gamma model with 20 rate categories (17) or using the PhyML program with automatic model selection (18).
The Censor program was used for the identification of similar nucleotide sequences and mapping the detected sequences on the genome (19). Each family of transposons was characterized either by its consensus sequences or by a single, apparently intact copy (when the family consisted of fewer than 3 copies >90% identical to each other). Transposon sequences with >75% pairwise nucleotide sequence identity were classified as members of the same family. Multiple alignments of DNA sequences were constructed using MAFFT (20). The consensus sequences were derived from multiple alignments using the “cons” routine of the EMBOSS package (21). For local sequence manipulations, programs from the EMBOSS and ALNPACK packages were used (19, 20).
In order to precisely identify the transposon termini, positions of insertions, and putative modifications of the target sites, two approaches were applied. Under the first strategy, transposons inserted into copies of some other TEs or unclassified repeats were identified. Based on pairwise alignments of these inserted copies of transposons with the consensus sequences, the exact termini of the inserted elements and target sites were identified.
Given that only a small fraction of the analyzed transposons are inserted into copies of other transposons or repetitive elements, we found useful the second strategy, which was based on the detection of polymorphic transposon insertions where one of the compared genomes contained an insert whereas the second genome lacked it. In the genome of a particular species, each transposon copy with intact ends was extended by 70 to 210 bp in each direction. Then, the transposon sequence was excised, and the 3′ and 5′ flanking sequences were concatenated and used as queries in BLASTN searches against the bacterial and archaeal NR, Genomes, and WGS sections of GenBank. Among all the significant hits with >85% identity to the query, those that did not contain insertions longer than 5 bp were selected. These hits corresponded to “empty target sites” in syntenic regions in the genomes of other strains or species closely related to the query species. Each empty syntenic region was aligned with the query sequence containing the transposon copy using the aaln2 program (“−mon” alignment option for logarithmic gap penalty) from Censor-ALNPACK. Based on manual inspection of these pairwise alignments, the exact termini of transposons and the positions of their insertions into the target sites were reevaluated.
RESULTS
Cas9 homologs in bacteria and archaea.
It has been shown previously that Cas9 homologs belong to two major families. One family contains the HNH nuclease domain insertion within the RuvC-like domain, whereas in the other, the RuvC-like domain is continuous (1, 6). The two families are jointly represented in the Pfam database as pfam14239, RRXRR proteins; this Pfam entry includes the N-terminal portion of the proteins, which encompasses the first RuvC motif and the arginine-rich region. In order to avoid multiple false-positive hits to numerous HNH nucleases, we used representatives of the second, RuvC-only family as a query for a PSI-BLAST search. A PSI-BLAST search against the NCBI NR database initiated with the WP_044523571.1 protein from Nostoc sp. strain PCC 7120 identified 1,227 Cas9 homologs after 6 iterations (see Table S1 in the supplemental material). We then generated clusters of highly similar sequences using the BLASTCLUST program in order to identify potential recent expansions of the family (see Table S1 in the supplemental material).
As expected from previous observations (1), we identified many cases in which multiple, highly similar copies of Cas9 homologs were present in the same organism, e.g., 30 copies in the euryarchaeon Methanosarcina lacustris, 15 in the cyanobacterium Microcystis aeruginosa, 13 in the bacterium Lactobacillus delbrueckii, and 8 in the bacterium Ktedonobacter racemifer (see Table S1 in the supplemental material). We then compared the distributions and abundances of this family of Cas9 homologs, Cas9 and TnpB, within the set of 2,751 complete bacterial and archaeal genomes (see Table S2 in the supplemental material). The TnpB proteins are extremely widespread in both bacteria and archaea (see Table S2 in the supplemental material). In contrast, Cas9 (the component of type II CRISPR-Cas loci) and Cas9 homologs show relatively narrow distributions that are limited to several bacterial phyla (see Table S2 in the supplemental material). The Cas9 homologs are especially abundant in cyanobacteria (31% of the available genomes encode at least one protein of the family). They are rarely present in the same genome with Cas9. Both Cas9 and Cas9 homologs are extremely rare in archaea, especially thermophiles (see Table S2 in the supplemental material).
The results of HHpred searches show that Cas9 homologs are much more similar to Cas9 than to the TnpB family, which, despite the presence of the readily detectable RuvC-like domain, is not even detected in HHpred searches starting from either Cas9 or Cas9 homologs, at least not with a probability of >50% (see Table S3 in the supplemental material). The multiple-sequence alignment of the RuvC-like domain and the arginine-rich helix region for the selected representatives of Cas9, Cas9 homologs, and TnpB proteins also clearly demonstrates the pronounced similarity between Cas9 and its homologs, especially in the arginine-rich helix and the RuvC-like motif III (Fig. 1). Taken together with the presence of the HNH domain inserted into the RuvC domain, a unique derived shared character, these findings are indicative of a direct evolutionary relationship between Cas9 and the family of Cas9 homologs.
FIG 1.

Multiple-sequence alignment of conserved motifs in selected representatives of IscB, TnpB, and Cas9 families. The catalytic residues are shaded black; conserved hydrophobic residues are highlighted in yellow; conserved small residues are highlighted in green; in the bridge helix alignment, positively charged residues are in red. Secondary-structure prediction is shown below the aligned sequences: H denotes α-helix, and E denotes extended conformation (β-strand). The poorly conserved spacers between the alignment blocks are shown by numbers. The bottom sequence shows the RuvC nuclease from Thermus thermophilus (Protein Data Bank [PDB] ID 4EP5), with the catalytic amino acid residues denoted.
Cas9 homologs are encoded by transposable elements.
We sought to characterize the genes encoding Cas9 homologs and in particular, given the distant relationship with TnpB, the possibility that these proteins are encoded by a distinct family of TEs. The key feature of TEs is the presence of unique terminal structures, often terminal inverted repeats (TIR), and/or target site duplication. For the identification of these diagnostic features, it is essential to compare multiple copies of closely related TEs. For initial analysis, we selected one of the largest tight clusters of genes encoding Cas9 homologs, which was detected in the genome of K. racemifer DSM 493 (9 sequences altogether, 2 of which are identical). Using the multiple alignment of the corresponding DNA sequences that were expanded by 2 kb in each direction, we derived a 1,594-bp consensus sequence that showed, on average, 96% identity to the individual copies (the DNA and protein sequences of transposons are shown in Fig. S1 in the supplemental material). All the copies contained the perfectly conserved terminal sequences 5′-GTCA and ATGG-3′. The presence of these specific termini suggests that the genes encoding Cas9 homologs are copies of a TE rather than products of segmental duplications. Moreover, as shown below, some copies of the transposon are inserted into different unclassified repetitive elements.
The consensus sequence of this transposon, here denoted ISC2-1_KR (insertion sequences encoding Cas9 homolog of family 2, subfamily 1, from Ktedonobacter racemifer), encodes a single protein, a Cas9 homolog, which we here denote IscB by analogy with TnpB. The subfamily of IscB proteins that contain the HNH insertion into the RuvC-like domain is referred to here as IscB2 (and the respective TE as ISC2), and the family with the RuvC-like domain only is referred to as IscB1 (and the respective TE as ISC1).
To identify the target sites of ISC2-1_KR, we searched for insertions of the transposon into other repetitive elements and identified three copies of ISC2-1_KR inserted into unclassified repeats Rep1, Rep2, and Rep3 (most likely, these repeats, identified by Censor, belong to unclassified TEs). Using pairwise alignments of the repeat copies containing insertions of ISC2-1_KR with the other copies of these repeats that were free of the insertions, we found that the actual termini of ISC2-1_KR are 5′-CA and AT-3′ and that the ISC2-1_KR copies are specifically inserted into GTGG target sites. Apparently, the insertions occurred precisely between GT and GG dinucleotides (GT|GG), without generating any target site duplications (see Fig. S2 in the supplemental material).
By similar analysis of other IscB clusters in the K. racemifer genome consisting of 2 or 3 elements each, we identified three additional families of TEs related to ISC2-1_KR and one to ISC1-1_KR. All these transposons are short (1,341 to 1,707 bp) and encode only the IscB2 or IscB1 protein, respectively (Fig. 2A). Even from this analysis of several TE families in a single species, it was clear that these transposons are highly diverse, with the pairwise protein identity in a range from 30% to 63%. Despite the high interfamily sequence divergence, transposons from different families of ISC2_KR elements (and the single ISC1 element) show notable sequence conservation at the 5′ termini and are inserted specifically between the GT and GR dinucleotides of the target sites without generating target site duplications (Fig. 2B).
FIG 2.

Structures of nonautonomous ISC transposons. (A) Organizations of ISC transposons. Green rectangles, ORFs encoding IscB; RuvC and HNH, nuclease domains; RR, arginine-rich region. The bacterial hosts and the length of each transposon are indicated on the left and right of the transposon schematics, respectively. In the names of transposons, the KR, GS, CC, YF, and SM suffixes denote K. racemifer DSM 44963, Geitlerinema sp. PCC 7105, C. chthonoplastes PCC 7420, Y. fragilis 232.1, and S. mucosus DSM 16094. (B) Termini and target site specificity among diverse groups of IscB transposons. Target sites are shown in green. The conserved nucleotides at the 5′ and 3′ termini are shown in boldface. The unusual nucleotides are shown in red. Subterminal inverted repeats forming imperfect hairpins recognized by the Y1 transposase are marked by arrows above the corresponding sequences.
We then sought to determine whether the identified features of the ISC2 and ISC1 transposons from K. racemifer (Fig. 2) are shared by transposons from other prokaryotes. For comparison, we selected several additional families of putative ISC elements that were detected in other bacteria, namely, Firmicutes (Youngiibacter fragilis 232.1, Anoxybacillus flavithermus TNO-09.006, Mahella australiensis, Anaerostipes hadrus, and Halobacteroides halobius), Cyanobacteria (Geitlerinema sp. strain PCC 7105, Microcystis aeruginosa NIES-843, and Coleofasciculus chthonoplastes PCC 7420), Proteobacteria (Salipiger mucosus DSM 16094, Marinobacter sp. strain ELB17, and Halomonas), and a methanogenic archaeon (M. lacustris Z-7289). Most of the TEs belong to the ISC2 group, with the exception of two ISC1 transposons from the cyanobacteria C. chthonoplastes and M. aeruginosa. Analysis of these TE families using the approach described above confirmed that the key features of the IscB-encoding transposons initially identified in the K. racemifer genome are shared by transposons populating the genomes of diverse bacteria and some archaea (Fig. 2 and Table 1).
TABLE 1.
ISC transposons in bacteria and archaea
| Transposon family | Species | No. of copies (% identity)a | IS605 transposon(s) with similar terminus (no. of copies, % identity)a,b | Target sitec | Taxonomy |
|---|---|---|---|---|---|
| ISC2-1_KR | Ktedonobacter racemifer DSM 493 | 7 (99) | IS605B-1_KR (2, 99) | GT|GGd (GT|GG) | Chloroflexi |
| ISC2-2_KR | 3 (99) | IS605B-2_KR (4, 99) | GT|GA (CT|AA) | Chloroflexi | |
| ISC2-3_KR | 2 (91) | IS605B-3_KR (6, 99) | GT|GA (AGT|GA) | Chloroflexi | |
| ISC2-4_KR | 2 (97) | IS605B-4_KR | GT|GA | Chloroflexi | |
| ISC1-1_KR | 3 (97) | GT|GT | Chloroflexi | ||
| ISCY2-1_KR | 2 (99) | IS605B-1_ME (6, 94) | ? | Chloroflexi | |
| ISC1-1_CC | Coleofasciculus chthonoplastes PCC 7420 | 4 (100) | TT|GG | Cyanobacteria | |
| ISC1-2_CC | 2 (99) | IS605-1_CC | TT|GA | Cyanobacteria | |
| ISC1-1_MA | Microcystis aeruginosa NIES-843 | 4 (97) | IS605B-1_MA (46, 96) | N|ATGAd (N|ATGA)d | Cyanobacteria |
| ISC2-1_GS | Geitlerinema sp. PCC 7105 | 10 (98) | GT|GG | Cyanobacteria | |
| ISC2-2_GS | 2 (91) | GT|GG | Cyanobacteria | ||
| ISC2-1_AH | Anaerostipes hadrus strain PEL 85 | 2 (97) | IS605B-1_AH, IS605B-2_AH | GT|AA (GT|AA) | Firmicutes |
| ISC2-1_HH | Halobacteroides halobius DSM 5150 | 2 (98) | AA|GG ? | Firmicutes | |
| ISC2-1_AF | Anoxybacillus flavithermus TNO-09.006 | 6 (97) | IS605B-1_AF (3, 99) | GT|GAd (AT|GA)d | Firmicutes |
| ISC2-1_YF | Youngiibacter fragilis 232.1 | 6 (96) | GT|GA | Firmicutes | |
| ISC2Y-1_CH | Clostridium haemolyticum NCTC 9693 | 1 | GT|AAd | Firmicutes | |
| ISC2-1_SM | Salipiger mucosus DSM 16094 | 4 (98) | CC|CG | Proteobacteria | |
| ISC2-1_HM | Halomonas meridiana strain R1t3 | 1 | IS605B-1_OS (3, 97), IS605-1_OS, IS605B-1_HM | N|ATGAd (N|ATGA) | Proteobacteria |
| ISC2-1_MS | Marinobacter sp. ELB17 | 5 (94) | IS605B-1_MS | N|ATGAd (N|ATNA)d | Proteobacteria |
| ISC2-1_NH | Nitrosococcus halophilus Nc 4 | 4 (99) | ? | Proteobacteria | |
| ISC2-1_ML | Methanosarcina lacustris Z-7289 | 12 (96) | GT|GAd | Archaea | |
| ISC2-2_ML | 10 (97) | IS605B-1_ML (7, 99) | GT|GA | Archaea |
For each transposon, the number of copies is accompanied by the mean percent identity of the copies to the respective consensus sequence (the pairwise identity for transposons represented by two copies).
IS605 and IS605B transposons that share common terminal regions with the corresponding ISC transposons.
Target sites of the corresponding ISC and IS605 (in parentheses) transposons. ?, target site is not defined.
Target site verified based on the identification of “empty” orthologous loci or repeats including empty copies and a copy harboring an ISC transposon (see Fig. S2, S4, and S6 in the supplemental material).
The ISC transposons share termini with IS605-like transposons encoding TnpB proteins.
During the analysis of the consensus sequence of the ISC transposons in the K. racemifer genome, we identified two ∼1,360-bp elements that were ∼95% identical to each other, contained termini similar to those of ISC2-1_KR, and encoded the TnpB protein in their antisense strands (Fig. 3). Here, we refer to the “top” 5′-to-3′ DNA strand of a transposon as the sense strand, whereas the complementary strand, also from 5′ to 3′, is denoted antisense. Thus, the elements that encode TnpB alone appear to be copies of a nonautonomous transposon, here called IS605B-1_KR, which most likely is mobilized by the Y1 tyrosine transposase (TnpA) of autonomous transposons. These autonomous transposons encode either both TnpA and TnpB (IS605 family) or TnpA alone (IS200 family) (7–9). Here, we use “IS605 superfamily” as an encompassing term that also includes the nonautonomous IS1341 and PATE (palindrome-associated transposable elements) (7, 22, 23). For simplicity, we refer to IS1341-like, TnpB-only transposons as IS605B. The same mode of transposition dependent on Y1 transposases supplied in trans can be predicted for the ISC transposons. Indeed, the vast majority of the genomes that contain ISC transposons carry at least one Y1 family transposase, whereas serine transposases are much less common (Table 2).
FIG 3.

ISC2-1_KR and IS605B-1_KR transposons share similar termini. (A) Map of ISC2-1_KR copies in the K. racemifer DSM 44963 genome obtained by using Censor. The first three columns from the left contain the GenBank accession numbers and coordinates of GenBank DNA sequences similar to the query sequence (column 4); columns 5 and 6 give the first and last positions of a region in the query sequence that is similar to the corresponding GenBank sequence; column 7 shows the orientation of the GenBank sequence (d, direct; c, complementary); column 8 shows DNA identity (0 to 1). Two 34-bp sequences similar to the 5′ terminus of ISC2-1_KR are shaded in light brown. These two sequences are the 5′ termini of two 95% identical copies of the IS605B-1_KR transposon. The 1,357-bp IS605B-1_KR transposon encodes the TnpB protein in the antisense orientation. The two terminal hairpins are rendered in black and blue. (B) Pairwise alignment of the 5′ and 3′ termini in both transposons; the terminal hairpins are formed by inverted repeats (marked by arrows). The identical GTGG target sites are shown in green.
TABLE 2.
Cooccurrence of IscB and TnpB with Y1 tyrosine and serine transposases (S_T) in complete genomesa
| Y1/S_T presence | No. of genomes with IscB/TnpB presence |
|||
|---|---|---|---|---|
| ISC (−)/ TnpB (−) |
ISC (−)/ TnpB (+) |
ISC (+)/ TnpB (−) |
ISC (+)/ TnpB (+) |
|
| Y1 (−)/S_T (−) | 1,296 | 235 | 0 | 4 |
| Y1 (−)/S_T (+) | 81 | 133 | 0 | 4 |
| Y1 (+)/S_T (−) | 532 | 285 | 1 | 24 |
| Y1 (+)/S_T (+) | 34 | 88 | 0 | 34 |
−, absent; +, present.
In the K. racemifer genome, we identified 8 short elements (62 bp) with terminal sequences 78 to 83% identical to the 65-bp 3′ terminus of the ISC2-2_KR transposon (see Fig. S3 in the supplemental material). Examination of pairwise and multiple DNA alignments of these elements expanded 5 kb in both directions showed that they constituted the 3′ termini of >95% identical copies of another, 1,832-bp transposon, IS605B-2_KR. Analogous to IS605B-1_KR, this transposon encodes only one protein, a distinct variant of TnpB that showed the closest similarity (23% identity; E = 8e−33; BLASTP) to the TnpB protein encoded by the ISCbt transposon, a typical autonomous IS605 transposon encoding both TnpA and TnpB (24).
In both examples described above, an ISC transposon was accompanied by an IS605 or IS605B transposon, with its 3′-terminal sequence similar to that of the 3′ terminus of the ISC transposon. Compared to the iscB genes, the tnpB genes in both IS605B transposons had the opposite orientation, i.e., were encoded by the antisense DNA strands. Surprisingly, this pattern of gene orientation holds among the other 9 families of ISC transposons and their IS605B or, less often, IS605 counterparts (Table 1; see Fig. S1, S3, and S4A and B in the supplemental material).
Flexibility among the target site cutting positions.
In addition to the several families of ISC transposons that are inserted precisely between the dinucleotides of the GTGG and GTGA target sites (see Fig. S2 and S5 in the supplemental material), transposons from several other ISC families from Cyanobacteria and Proteobacteria are inserted upstream of the ATGA target sites. For example, in the genome of M. aeruginosa NIES-843, we identified 4 copies of the ISC1-1_MA transposon that are ∼97% identical to their consensus sequence (see Fig. S4 in the supplemental material). The consensus sequence contained the 5′-GTCA and CGATGA-3′ termini. Therefore, based on the previously described target site specificity, we assumed that the terminal GT and GA dinucleotides belonged to the GT|GA target site (“|” shows the exact position of the transposon insertions) rather than to the termini of the transposon. To verify this assumption, we examined the cross-genome comparison aiming to identify the counterpart loci free of the transposon copies and found that the M. aeruginosa 9717 genome contains one such locus (see Fig. S4C in the supplemental material). Pairwise alignment of this locus with the corresponding ISC1-1_MA-containing locus of M. aeruginosa NIES-843 showed that the 5′-GT dinucleotide is the end of the transposon, whereas ATGA-3′ is the target site. In agreement with this observation, we found that two full-length copies of ISC1-1_MA in the M. aeruginosa 9717 genome were inserted at the loci syntenic to those in M. aeruginosa NIES-843 that did not contain the transposon insertion (see Fig. S4D in the supplemental material). For both loci, pairwise alignment of their DNA sequences with the sequences of their transposon-free orthologs confirmed that ISC1-1_MA has the 5′-GT and CG-3′ termini and is inserted in the N|ATGA target site.
As mentioned above, ISC1-1_MA and IS605B-1MA transposons share a 3′ terminus (see Fig. S4A and B in the supplemental material), which in IS605 family transposons is recognized by the Y1 transposase (25). Importantly, these two transposons share not only the Y1-binding sites but also the same target site specificity. In contrast to ISC1-1_MA (4 copies), IS605B-1_MA is much more prolific and is represented in M. aeruginosa NIES-843 by 46 full-length copies. For 12 loci harboring these transposons, we identified transposon-free counterparts (empty target sites) in M. aeruginosa 9717. Examination of all 12 pairwise alignments of the transposon-carrying and transposon-free loci indicated that the IS605B-1_MA transposon was inserted precisely in the N|ATGA target sites and contained the 5′-TT and CG-3′ termini (Fig. S4E in the supplemental material). A comparison of the IS605B-1_HM transposon-carrying locus of Halomonas meridiana strain R1t3 and the corresponding transposon-free locus of Halomonas sp. strain TD01 identified the same target site, N|ATGA (see Fig. S6E in the supplemental material). The target site specificity was further confirmed by analysis of the Marinobacter sp. ELB17 genome, which contains 5 copies of ISC2-1_MS (see Fig. S6A and B in the supplemental material), as well as 3 copies of ISC2-2_MS (see Fig. S6C and D in the supplemental material), that all share identical target sites.
The IS605B-1_MS transposon is inserted precisely upstream of the ATCA and ATAA target sites, resembling the ATGA target site of ISC2-1_MS and ISC2-2_MS (see Fig. S6G in the supplemental material). Taken together, these findings strongly suggest that the transposition of ISC and IS605 transposons is catalyzed by the same or related Y1 transposases.
ISC transposons encoding TnpA-like tyrosine transposases.
Through analysis of the IscB cluster from K. racemifer, which consists of only two sequences (NCBI accession numbers ZP_06974220 and ZP_06973729), we identified ∼99% identical copies of an autonomous ISC2Y-1_KR transposon that, in addition to IscB, encodes a TnpA homolog, a Y1 tyrosine transposase (Fig. 4) (9, 26). In the ISfinder database of bacterial and archaeal transposons (24), the Y1 sequences most similar to the ISC2Y-1_KR transposase are those of the archaeal IS605 family transposons ISMa19, ISMba17, and ISMac7 (50%, 49%, and 45% identity, respectively) and bacterial IS605 (40%). In these transposons, TnpA is encoded in the antisense strand, whereas TnpB is encoded in the sense strand. In contrast, in the ISC2Y-1_KR transposon, the iscA (tnpA) gene is located downstream of the iscB gene in the sense strand so that the two genes are transcribed codirectionally; furthermore, the coding regions of IscA and IscB overlap by 82 nucleotides.
FIG 4.
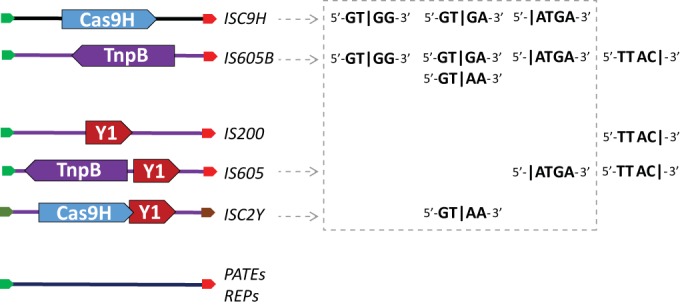
Organizations of the major groups of transposons of the IS605 superfamily. Transposition of nonautonomous transposons encoding IscB and TnpB is predicted to be catalyzed by the Y1 tyrosine transposase encoded by autonomous transposons IS200, IS605, and ISC2Y. The Y1 transposase binds to the transposon termini, which contain imperfect hairpins formed by the subterminal inverted repeats (green and red arrows). Short nonautonomous TEs transposed by the IS605-like Y1 transposase that do not encode any proteins are known as PATEs and REPs. Four types of specific 4-bp target sites are listed for transposons from different groups in the 4 columns on the right. In the target site sequences, the vertical lines indicate the exact positions of transposon insertion. Previously reported target sites and the transposon insertion positions are shown in the last column. The dashed box denotes new types of target sites described in this work.
In addition to ISC2Y-1_KR, the K. racemifer genome contains four ∼98% identical copies of an ISC2Y-2_KR transposon. These two families of ISC2Y transposons are close to each other (∼74% DNA identity). However, in the second transposon, both open reading frames (ORFs) coding for the IscA and IscB proteins are corrupted by identical mutations in all copies. As a result, the ISC2Y-2_KR transposon codes only for the C-truncated proteins and should be classified as a nonautonomous transposon. Although the termini of IS605-1_KR and IS605-2_KR were not similar to the termini of any of the ISC transposons, we found that the 35-bp 5′ termini were similar to the 35-bp 5′ terminus of an IS605B-1_ME TnpB-encoding transposon from the archaeon Methanohalobium evestigatum Z-7303.
To investigate the distribution and evolution of the ISC2Y transposons, we used the homologs of the IscA and IscB proteins from ISC2Y-1_KR as queries in a search of the nonredundant protein database and identified 5 additional ISC2Y transposons in the firmicutes Clostridium haemolyticum NCTC 9693 (plasmid p1Ch9693), Enterococcus cecorum DSM 20682 (ATCC 43198), Anaeromusa acidaminophila DSM 3853, and Coprobacillus sp. strain 3_3_56FAA and the chlorobi Microscilla marina ATCC 23134 (see Fig. S1 in the supplemental material). A TBLASTN search with the same queries against all bacterial and archaeal WGS GenBank DNA sequences failed to identify any additional ISC2Y transposons, except for many strains of the same species.
In all these transposons, ORF1 and ORF2, which encode IscB and IscA, respectively, are located in the same order in the sense strand and overlap by 12 to 39 nucleotides. The considerable diversity of the IscA and IscB protein sequences in the ISC2Y transposons (39 to 69% and 33 to 65% pairwise identity, respectively) implies that ISC2Y transposons are an old TE family. However, ISC2Y transposons are quite rare, with full-length elements detected in only 14 bacterial and archaeal genomes, typically as a single copy (Table 1). Only one genome (K. racemifer) harbors both ISC and ISC2Y transposons. Thus, in contrast to autonomous transposons of the ISC605 superfamily, ISC2Y transposons might be deleterious to the hosts and are not often fixed in the course of evolution. Apparently, the great majority of the ISC transposons rely on Y1 transposases encoded in TEs of other families for their transposition.
Using the IscA protein sequences encoded by ISC2Y-1_KR, ISC2Y-1_CH, and ISC2Y-1_EC as queries in BLASTP searches against the NR database, we extracted ∼150 sequences that were highly similar to the queries. After elimination of sequences >65% identical to each other, they were reduced to a group of 10 protein sequences >40% identical to the ISC2Y transposases. Each of these 10 transposases is encoded by an IS605 transposon that also carries a tnpB gene separated from the transposase gene by less than 1 kb.
Unexpectedly, a phylogenetic tree of the 6 identified ISC2Y transposases and 10 diverse IS605 transposases contains two distinct, strongly supported branches, each of which joins ISC2Y (IscA) and IS605 (TnpA) transposases (Fig. 5; see Fig. S7 in the supplemental material). Given that in all ISC2Y transposons the IscB and IscA ORFs are codirectional and overlapping, whereas in all but one of the closest IS605 elements the two genes do not overlap and are divergently transcribed (Fig. 5), it is highly unlikely that these two groups of ISC2Y transposons evolved independently from different IS605 transposons. Thus, the opposite direction of evolution, namely, that ISC2Y transposons “gave birth” independently to at least two groups of IS605 elements, in each case yielding a chimeric IscA-TnpB transposon, appears most parsimonious (Fig. 5).
FIG 5.

Phylogenetic tree of Y1 transposases encoded by ISC2Y and IS605 transposons (IscA and TnpA, respectively). In ISC2Y transposons, IscA (red arrows) and IscB (blue arrows) ORFs overlap by 12 to 82 nucleotides. In IS605 transposons, TnpA and TnpB are depicted as red and black arrows. The right and left arrows indicate ORFs encoded by the sense and antisense strands, respectively. The light-green and brown shading highlights the two distinct clades, each of which combines IscA and TnpA. The tree was obtained using the PhyML program (automatic model selection): LG model, discrete gamma model with 6 categories, and estimated gamma shape parameter. The support for internal branches is indicated by Bayes approximation values above 95%. Species abbreviations: KR, K. racemifer DSM 44963; CS, Coprobacillus sp. 3_3_56FAA; EC, E. cecorum DSM 20682 (ATCC 43198); AA, A. acidaminophila DSM 3853; CH, C. haemolyticum NCTC 9693; MA, M. marina ATCC 23134; VB, Vibrio breoganii; BC, Bacteroides coprophilus; MM, Methanosarcina mazei; MZ, Methanosalsum zhilinae; EH, Eubacterium hallii DSM 3353; BSp, Butyrivibrio sp. strain MB2005; BMT2, Bacillus sp. strain MT2; FP, Francisella philomiragia; HP, Helicobacter pylori Hp H-16; RI, Roseburia inulinivorans.
DISCUSSION
The findings described here show that IscB proteins, the closest homologs of Cas9 not linked to CRISPR-Cas systems, are encoded by a distinct group of transposons, which we denote ISC. The extensive mobility of ISC transposons is supported by the discovery of numerous empty target sites in pairs of closely related genomes, one of which contains a transposon.
Virtually all types of DNA transposons are defined by their relatively short terminal regions, which are recognized by the respective transposases. The ISC transposons share similar terminal sequences containing characteristic imperfect palindromes with the transposons of the IS605B and IS200/IS605 (super)families but not with any known transposons from other (super)families with terminal sequences significantly similar to those of the ISCs. Terminal sequences with this structure are typical of the so-called HUH Y1 transposons, so named after the conserved metal-binding motif (histidine-hydrophobic residue-histidine) in their tyrosine transposases (7, 26). Most of the ISC transposons do not encode a transposase and, accordingly, are nonautonomous elements that are most likely mobilized in trans by Y1 transposases (TnpA proteins) encoded by other transposons of the IS605 superfamily (23). Indeed, most of the genomes that contain ISC transposons also encompass at least one TnpA-encoding transposon. We identified several transposons that encode both IscB and TnpA (ISC2Y transposons), which is obviously compatible with the involvement of TnpA in ISC transposition. However, ISC2Y transposons are rare and apparently do not typically serve as helpers to nonautonomous ISC elements. The Y1 (TnpA) transposase forms a dimer and contains hybrid active sites, with an HUH motif in one monomer and the catalytic tyrosine in the other monomer (26); these catalytic residues are also conserved in the Y1 transposase (IscA) that is encoded by the ISCY transposons. Overall, the identified ISC elements extend the group of numerous nonautonomous IS200/IS605 transposons, including so-called REP and IStron elements (27).
In contrast to many other transposons, ISC and IS605 elements lack terminal inverted repeats that are typically recognized by diverse DDE or cut-and-paste transposases (7, 23). Instead, they contain subterminal imperfect hairpins that are recognized by the Y1 transposase (25, 26). All transposons that employ Y1 transposases are target site specific and integrate into the target sites without generating target site duplications. Unlike many other bacterial DNA transposons that are often inserted into random sites, the IS605 superfamily transposons are inserted 3′ of a specific 4- or 5-bp target site (7, 23). As shown here, the IS605 target site specificity is much more diverse than previously suspected and can be extended to include GT|GG, GT|RA, and N|ATGA target sites (Fig. 4 and Table 1), with transposons inserting either inside the specific target site or 5′ of it. Considering all these observations, the ISC transposons appear to represent a distinct family within the IS200/IS605 superfamily.
The phylogenetic tree of the IscB family consists of two distinct clades, IscB1 and IscB2 (Fig. 6; see Fig. S8 in the supplemental material). Although the tree could not be rooted by TnpB proteins due to the insufficient number of reliably aligned positions between IscB and TnpB, this topology clearly is consistent with the origin of IscB2 from IscB1 through insertion of the HNH nuclease-coding sequence into the ancestral iscB1 gene. The HNH nuclease could originate from a group II intron. Indeed, we detected insertion of group II introns encoding a reverse transcriptase-HNH fusion protein into IS605 transposons in several bacterial genomes (e.g., Anabaena cylindrica PCC 7122 and Nostoc sp. strain PCC 7107) (see Fig. S9 in the supplemental material).
FIG 6.

Phylogenetic tree of IscB proteins encoded by the ISC transposons. The unrooted maximum-likelihood phylogenetic tree was constructed using the FastTree program from a multiple-sequence alignment built for a nonredundant set of 443 full-size IscB sequences and containing 207 informative positions. The tree is shown schematically; the complete tree is available in Fig. S8 in the supplemental material. The ISC transposons described in this work are mapped to the respective collapsed branches. The FastTree program was also used to compute bootstrap values indicated for the branches with more than 70% support.
In more than 15 identified examples of highly diverse ISC transposons with terminal sequences similar to those of IS605B or IS605 transposons (Table 1; see Fig. S1, S3, and S4A and B in the supplemental material), the IscB and TnpB proteins are always encoded in the sense and antisense strands, respectively. The simplest explanation of this unusually stable orientation of the tnpB and iscB genes could be a tight association of the promoters of these genes with the 3′-subterminal regions of the respective transposons.
Remarkable features of TnpB are its high abundance and wide spread among bacteria and archaea, which are surprising because, as shown in several experimental studies on IS605 and IS607 transposons, TnpB is not essential for transposition and is thought to be involved in the regulation of transposase expression and activity (7, 8, 28, 29). The major function of the RuvC nuclease in bacteria is resolution of the Holliday junctions during recombination and repair of DNA breaks (30, 31). Transposition can lead to a substantial increase in the number of DNA breaks, which could be deleterious to both the host and the transposons. The TnpB proteins could be directly involved in DNA break repair via the activity of the RuvC-like endonuclease domain. As a result, the TnpB-encoding transposons could be more successful than transposons devoid of TnpB in terms of long-term coevolution with the hosts due to minimization of the damage to the host caused by transposition. However, more complicated mechanisms of action of TnpB and IscB proteins cannot be ruled out, in particular those that would involve RNA binding via the arginine-rich helices of these proteins. Such a possibility seems particularly plausible for the IscB2 proteins, which might function analogously to Cas9 proteins, which employ the HNH nuclease domain to cleave the DNA strand paired with the CRISPR RNA and the RuvC-like domain to cleave the opposite strand (3, 32).
The wide spread of TnpB in autonomous and nonautonomous transposons, which contrasts with the relatively narrow occurrence of both Cas9 (within type II CRISPR-Cas loci) and IscB (within ISC transposons), together with the simple domain organization of these proteins, suggests that TnpB is the ancestral form to both IscB and Cas9. The relatively high sequence similarity between Cas9 and IscB2 and, even more important, the shared presence of two nuclease domains indicate that these families have a common ancestor. The direction of evolution between IscB and Cas9 is difficult to infer with confidence, especially given that the IscB family shows considerably less sequence diversity than Cas9. Nevertheless, given the simpler domain architecture of IscB proteins and their inherent mobility as part of the ISC elements, a transposon ancestry of Cas9, with subsequent immobilization within the CRISPR-Cas loci, appears most likely. This scenario, together with the apparent origin of the adaptation modules of CRISPR-Cas systems from casposon-like TEs (33), highlights the major contribution made by mobile elements to the evolution of microbial defense systems, in particular adaptive immunity. The recent discovery of additional class 2 CRISPR-Cas systems, which encode distant homologs of TnpB containing only the RuvC-like nuclease domain (34, 35), is fully compatible with this conclusion.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yuri Wolf for invaluable technical help and Michael Chandler for useful discussions, particularly on TE nomenclature.
The research is supported by the intramural funds of the U.S. Department of Health and Human Services (to the National Library of Medicine).
V.V.K., K.S.M., and E.V.K. designed the research; V.V.K. and K.S.M. performed the research; V.V.K., K.S.M., and E.V.K. analyzed the results; V.V.K. and E.V.K. wrote the manuscript, which was edited and approved by all of us.
Funding Statement
The U.S. Department of Health and Human Services provided funding to Vladimir V. Kapitonov, Kira S. Makarova, and Eugene V. Koonin through the intramural funding program.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00783-15.
REFERENCES
- 1.Chylinski K, Makarova KS, Charpentier E, Koonin EV. 2014. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res 42:6091–6105. doi: 10.1093/nar/gku241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, Marraffini LA. 2015. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 519:199–202. doi: 10.1038/nature14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makarova KS, Aravind L, Wolf YI, Koonin EV. 2011. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct 6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siguier P, Gourbeyre E, Chandler M. 2014. Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol Rev 38:865–891. doi: 10.1111/1574-6976.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasternak C, Dulermo R, Ton-Hoang B, Debuchy R, Siguier P, Coste G, Chandler M, Sommer S. 2013. ISDra2 transposition in Deinococcus radiodurans is downregulated by TnpB. Mol Microbiol 88:443–455. doi: 10.1111/mmi.12194. [DOI] [PubMed] [Google Scholar]
- 9.He S, Guynet C, Siguier P, Hickman AB, Dyda F, Chandler M, Ton-Hoang B. 2013. IS200/IS605 family single-strand transposition: mechanism of IS608 strand transfer. Nucleic Acids Res 41:3302–3313. doi: 10.1093/nar/gkt014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41:D348–D352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drozdetskiy A, Cole C, Procter J, Barton GJ. 2015. JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43(W1):W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler D, Bhagwat M. 2007. BLAST QuickStart: example-driven web-based BLAST tutorial. Methods Mol Biol 395:149–176. doi: 10.1007/978-1-59745-514-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 19.Kohany O, Gentles AJ, Hankus L, Jurka J. 2006. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 22.Dyall-Smith ML, Pfeiffer F, Klee K, Palm P, Gross K, Schuster SC, Rampp M, Oesterhelt D. 2011. Haloquadratum walsbyi: limited diversity in a global pond. PLoS One 6:e20968. doi: 10.1371/journal.pone.0020968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siguier P, Gourbeyre E, Varani A, Ton-Hoang B, Chandler M. 2015. Everyman's guide to bacterial insertion sequences. Microbiol Spectr 3:MDNA3-0030-2014. doi: 10.1128/microbiolspec.MDNA3-0030-2014. [DOI] [PubMed] [Google Scholar]
- 24.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ronning DR, Guynet C, Ton-Hoang B, Perez ZN, Ghirlando R, Chandler M, Dyda F. 2005. Active site sharing and subterminal hairpin recognition in a new class of DNA transposases. Mol Cell 20:143–154. doi: 10.1016/j.molcel.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 26.Chandler M, de la Cruz F, Dyda F, Hickman AB, Moncalian G, Ton-Hoang B. 2013. Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat Rev Microbiol 11:525–538. doi: 10.1038/nrmicro3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He S, Corneloup A, Guynet C, Lavatine L, Caumont-Sarcos A, Siguier P, Marty B, Dyda F, Chandler M, Ton Hoang B. 2015. The IS200/IS605 family and “peel and paste” single-strand transposition mechanism. Microbiol Spectr 3. doi: 10.1128/microbiolspec.MDNA3-0039-2014. [DOI] [PubMed] [Google Scholar]
- 28.Boocock MR, Rice PA. 2013. A proposed mechanism for IS607-family serine transposases. Mob DNA 4:24. doi: 10.1186/1759-8753-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kersulyte D, Mukhopadhyay AK, Shirai M, Nakazawa T, Berg DE. 2000. Functional organization and insertion specificity of IS607, a chimeric element of Helicobacter pylori. J Bacteriol 182:5300–5308. doi: 10.1128/JB.182.19.5300-5308.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ariyoshi M, Vassylyev DG, Iwasaki H, Nakamura H, Shinagawa H, Morikawa K. 1994. Atomic structure of the RuvC resolvase: a Holliday junction-specific endonuclease from E. coli. Cell 78:1063–1072. doi: 10.1016/0092-8674(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 31.Lilley DM, White MF. 2001. The junction-resolving enzymes. Nat Rev Mol Cell Biol 2:433–443. doi: 10.1038/35073057. [DOI] [PubMed] [Google Scholar]
- 32.Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA. 2014. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koonin EV, Krupovic M. 2015. Evolution of adaptive immunity from transposable elements combined with innate immune systems. Nat Rev Genet 16:184–192. doi: 10.1038/nrg3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. 2015. Cpf1 Is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. 2015. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell 60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.