

ABSTRACT
Parainfluenza viruses are known to inhibit type I interferon (IFN) production; however, there is a lack of information regarding the type III IFN response during infection. Type III IFNs signal through a unique heterodimeric receptor, IFN-λR1/interleukin-10R2 (IL-10R2), which is primarily expressed by epithelial cells. Parainfluenza virus 3 (PIV-3) infection is highly restricted to the airway epithelium. We therefore sought to examine type III IFN signaling pathways during PIV-3 infection of epithelial cells. We used three strains of PIV-3: human PIV-3 (HPIV-3), bovine PIV-3 (BPIV-3), and dolphin PIV-1 (Tursiops truncatus PIV-1, or TtPIV-1). Here, we show that message levels of IL-29 are significantly increased during PIV-3 infection, yet downstream antiviral signaling molecules are not upregulated to levels similar to those of the positive control. Furthermore, in Vero cells infected with PIV-3, stimulation with recombinant IL-29/-28A/-28B does not cause upregulation of downstream antiviral molecules, suggesting that PIV-3 interferes with the JAK/STAT pathway downstream of the IFN-λR1/IL-10R2 receptor. We used Western blotting to examine the phosphorylation of Stat1 and Stat2 in Vero cells and the bronchial epithelial cell line BEAS-2B. In Vero cells, we observed reduced phosphorylation of the serine 727 (S727) site on Stat1, while in BEAS-2B cells Stat1 phosphorylation was decreased at the tyrosine 701 (Y701) site during PIV-3 infection. PIV-3 therefore interferes with the phosphorylation of Stat1 downstream of the type III IFN receptor. These data provide new evidence regarding strategies employed by parainfluenza viruses to effectively circumvent respiratory epithelial cell-specific antiviral immunity.
IMPORTANCE Parainfluenza virus (PIV) in humans is associated with bronchiolitis and pneumonia and can be especially problematic in infants and the elderly. Also seen in cattle, bovine PIV-3 causes respiratory infections in young calves. In addition, PIV-3 is one of a number of pathogens that contribute to the bovine respiratory disease complex (BRDC). As their name suggests, interferons (IFNs) are produced by cells to interfere with viral replication. Paramyxoviruses have previously been shown to block production and downstream signaling of type I IFNs. For the first time, it is shown here that PIV-3 can induce protective type III IFNs in epithelial cells, the primary site of PIV-3 infection. However, we found that PIV-3 modulates signaling pathways downstream of the type III IFN receptor to block production of several specific molecules that aid in a productive antiviral response. Importantly, this work expands our understanding of how PIV-3 effectively evades host innate immunity.
INTRODUCTION
Parainfluenza virus 3 (PIV-3) causes a prominent respiratory infection in both cattle and humans. The CDC reports that, in humans, most children have antibodies against human PIV-3 (HPIV-3) by 5 years of age (http://www.cdc.gov/parainfluenza/hcp/clinical.html). There is currently no vaccine available for control of HPIV-3 infection; however, a few studies have examined the use of an attenuated bovine PIV-3 (BPIV-3) to protect against HPIV-3 because of the homology between bovine and human strains (1–3). Given the lack of an efficacious vaccine for HPIV-3, there is a critical need to understand the mechanisms of HPIV-3-induced disease and the molecular pathways associated with viral modulation of the host antiviral defenses.
Paramyoxviruses are negative-sense single-stranded RNA viruses which are part of the Paramyxoviridae family and Paramyxovirinae subfamily (4). PIV-3 is found within the genus Respirovirus. PIV-3 is a respiratory virus that primarily infects the epithelial cells of the lung. Symptoms of HPIV-3 infection include bronchiolitis and pneumonia, and HPIV-3 is especially problematic in infants (4). Airway epithelial cells recognize viral infection through pattern recognition receptors (PRRs): RIG-I like receptors (RLRs) and Toll-like receptors (TLRs) (5). Viral RNA binds to RLRs and TLRs, creating a signaling cascade to produce proinflammatory cytokines and interferons (IFNs). Type I and type III IFNs are important in mounting a robust antiviral response by inducing various IFN-stimulating genes (ISGs) (6).
Type I IFNs, particularly IFN-α and -β, are known for their antiviral role; however, many viruses have been shown to prevent type I IFN production and responses. Paramyxoviruses in particular are known to block type I IFN production and downstream signaling pathways (7–16). A newly described class of IFNs, the type III IFNs or IFN-λs, were identified independently by two groups and published in the same issue of Nature Immunology (17, 18). The type III IFNs were termed interleukin-29 (IL-29)/IL-28A/IL-28B or IFN-λ1/IFN-λ2/IFN-λ3, respectively. IFN-λs bind a novel heterodimeric class II cytokine receptor, with IFN-λR1/IL-28Rα and IL-10R2/IL-10Rβ subunits (19). In some infections, including hepatitis C, type I IFNs are used for treatment. Nevertheless, giving IFN-α to humans is problematic in itself because of the lengthy list of adverse side effects (http://www.hepatitis.va.gov/provider/reviews/treatment-side-effects.asp). IFN-λs may be especially beneficial during respiratory infections because the IFN-λR1 is more restricted to epithelial cells (20). Type III IFNs may therefore be a useful treatment in HPIV-3 viral infection until an efficacious vaccine is developed.
Like type I IFNs, the IFN-λs bind their distinct receptor to induce Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways (17, 18). The JAK/STAT pathway activated by either type I or III IFNs can turn on many ISGs to control viral infection (6). The antiviral regulator protein kinase R (PKR) is responsible for phosphorylating eukaryotic initiation factor 2α (eIF2α) to halt protein synthesis. OAS (2′-5′ oligoadenylate synthetase) activates RNase L, which, as the name suggests, degrades viral RNA. The GTPase MxA (myxovirus resistance protein 1) mediates antiviral activity by controlling vesicle budding, organogenesis, and cytokinesis (6). Finally, a multitude of ISGs exist as antiviral mediators. Specifically, ISG56 has been shown to be responsible for inhibiting protein synthesis during PIV infection (21).
Here, we examined type III IFN signaling during a PIV-3 infection. BEAS-2B cells, a bronchial epithelial cell line, were used because the epithelial cells of the lung are the natural site of infection for PIV-3. Vero cells were used as a tool to study the IFN-λ response independent of that of IFN-α/β because they are unable to produce type I IFNs (22–24). Here, we demonstrate that IL-29 message is upregulated during PIV-3 infection in both Vero and BEAS-2B cells. Surprisingly, however, expression of the downstream antiviral molecules PKR, OAS, MxA, and ISG56 is not induced to levels near those of the positive control in Vero cells. This suggested that PIV-3 infection may be blocking downstream signaling. Both the tyrosine 701 (Y701) and serine 727 (S727) phosphorylation sites on Stat1 are necessary for full activation of downstream antiviral molecules (25–27). A Western blot showed that phosphorylation of S727 on Stat1 was reduced in the PIV-3-infected Vero cells stimulated with type III IFNs, while Y701 was phosphorylated. The reverse was true in the BEAS-2B cells: phospho-Stat1 (pStat1) S727 was phosphorylated normally, and phosphorylation of Y701 was decreased during PIV-3 infection. These results suggest that PIV-3 reduces production of antiviral molecules downstream of the type III IFN receptor by specifically causing reduced phosphorylation of Stat1.
MATERIALS AND METHODS
Cell culture and medium recipes.
BEAS-2B cells (ATCC, CRL-9609) were grown as per ATCC's recommendations. Briefly, flasks were coated in 0.01 mg/ml fibronectin (356008; Corning), 0.03 mg/ml bovine collagen type I (5005-B; Advanced Biomatrix), and 0.01 mg/ml bovine serum albumin (BSA) (A3059; Sigma). No antibiotics or antimycotics were used in the bronchial epithelial cell growth medium (BEGM), but BEGM was supplemented with multiple growth factors (CC-3170; Lonza). Vero cells (ATCC, CCL-81) were grown in Eagle's minimum essential medium (EMEM) (ATCC, 30-2003) supplemented with 1× antibiotic-antimycotic (A5955; Sigma) and 10% heat-inactivated fetal bovine serum (FBS) (A11-265; PAA). Both cell lines were grown at 37°C in 5% CO2.
Viruses.
The bovine parainfluenza virus 3 strain SF-4 was purchased from ATCC (VR-281). The dolphin isolate of parainfluenza virus (Tursiops truncatus PIV-1, or TtPIV-1) was obtained from the Marine Mammal Health Program at the University of Florida (28). Both BPIV-3 and TtPIV-1 were propagated in primary bovine turbinate (BT) cells. The human parainfluenza virus 3 C243 strain used was purchased from ATCC (VR-93). HPIV-3 was grown in Vero cells. The HPIV-3 and BPIV-3 used here are commonly studied strains, which were both isolated in the 1950s. The TtPIV-1 isolate is a more recently described clinical strain that Eberle et al. concluded is actually a BPIV-3 (29). Therefore, all viruses used throughout the manuscript are considered PIV-3 viruses.
Recombinants and antibodies.
The following recombinant cytokines were purchased from R&D Systems: IL-29 (catalog number 1598-IL), IL-28A (1587-IL), and IL-28B (5259-IL). Anti-human IL-29 (hIL-29)/IFN-λ1 antibody was used for blocking experiments (AF1598; R&D Systems). Western blotting was performed with the following antibodies from Cell Signaling: Stat1 (catalog number 9172), Stat2 (4594), phospho-Stat1 (Ser727) (9177), phospho-Stat1 (Tyr701) 58D6 (9167), and phospho-Stat2 (Tyr690) (4441). Anti-STAT2 (phosphorylated at Y690) antibody from Abcam (ab53132) was used for the BEAS-2B cells. β-Actin (ab82270-50; Abcam) was used as an internal control antibody. Goat anti-rabbit IgG(H+L), horseradish peroxidase (HRP) conjugate (31460; Thermo Fisher), was used as a secondary antibody for all Western blots.
Time course for real-time PCR.
Virus was added to cells at a multiplicity of infection (MOI) of 2 and left to incubate for 6, 12, 24, 48, or 72 h. A TLR3 agonist, poly(I·C) (P1530; Sigma), was used at 20 μg/ml. A 10-min Lipofectamine 3000 (L3000-015; Invitrogen) treatment with poly(I·C) was necessary in the BEAS-2B cells to see a positive cytokine response (9). Recombinant cytokine treatment with either IFN-α (11200-2; PBL) and IFN-β (11415-1; PBL) at 500 U/ml or with IL-29/-28A/-28B at 1 μg/ml was also used as a positive control. Recombinants were incubated for 24 h. At harvest, samples were collected in RLT lysis buffer (1015762; Qiagen) and 2-beta-mercaptoethanol (2-ME) (BP176; Sigma).
RNA extraction and cDNA synthesis.
RNA extraction and cDNA synthesis have been described previously by Eberle et al. (29). Samples were collected in RLT lysis buffer (1015762; Qiagen) and 2-beta-mercaptoethanol (2-ME) (BP176; Sigma). Total RNA was isolated using an RNeasy Mini RNA Isolation kit (74106; Qiagen) per the manufacturer's protocol. DNase digestion was achieved using a Qiagen RNase-Free DNase set (79254; Qiagen). After elution, RNaseOUT (10777; Invitrogen) was added to each sample. Eluted RNA was transcribed using random primers (48190; Invitrogen) and deoxynucleoside triphosphate (dNTP) mix (18427; Invitrogen), followed by 5× First-Strand Buffer, 0.1 M dithiothreitol (DTT), and Superscript III reverse transcriptase (18080; Invitrogen) per the manufacturer's instructions.
Real-time PCR.
Real-time PCR was described previously by Eberle et al. (29). Briefly, Power SYBR green PCR master mix (4367659; Invitrogen), 10 μM both the forward and reverse primer (Integrated DNA Technologies), distilled H2O (dH2O), and cDNA template were combined. The amplification conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min, with a final dissociation step. Plates were run on an Applied Biosystems 7300 real-time PCR system. Relative gene expression was determined using the 2−ΔΔCT (where CT is threshold cycle) method (30). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference gene. Primer sequences used are listed in Table 1.
TABLE 1.
Forward and reverse primer sequences used in real-time PCR assays
| Target (cell line) | Forward primer | Reverse primer | Reference |
|---|---|---|---|
| IL-29 | GAA GCC TCA GGT CCC AAT TC | GAA GCC TCA GGT CCC AAT TC | 45 |
| IL-28 | ACT GCA GCC ACT CCC | CTC CAG AAC CTT CAG CGT CAG | 45 |
| IFN-α1 | CAG AGT CAC CCA TCT CAG CA | CAC CAC CAG GAC CAT CAG TA | 34 |
| IFN-α2 | CTG GCA CAA ATG GGA AGA AT | CTT GAG CCT TCT GGA ACT GG | 34 |
| IFN-β | AAG GCC AAG GAG TAC AGT C | ATC TTC AGT TTC GGA GGT AA | 45 |
| PKR | ACT TTT TCC TGG CTC ATC TC | ACA TGC CTG TAA TCC AGC TA | 46 |
| OAS | AGA AGG CAG CTC ACG AAA CC | CCA CCA CCC AAG TTT CCT GTA | 47 |
| MxA (Vero) | TTC AGC ACC TGA TGG CCT ATC | TGG ATG ATC AAA GGG ATG TGG | 48 |
| MxA (BEAS-2B) | ACC ACA GAG GCT CTC AGC AT | CTC AGC TGG TCC TGG ATC TC | 49 |
| ISG56 | CAG CAA CCA TGA GTA CAA AT | AAG TGA CAT CTC AAT TGC TC | 45 |
| GAPDH | ACT TTG GTA TCG TGG AAG GAC T | GTA GAG GCA GGG ATG ATG TTC T | 50 |
Recombinant cytokine treatment and blocking.
Vero cells were preincubated with 1 μg/ml of IL-29/IL-28A/IL-28B for 24 h. For the blocking experiments, anti-IL-29 antibody was incubated with recombinant IL-29 for 1 h at room temperature before being added to the cells for 24 h. After 24 h with the recombinant, virus was added on top of the IFN treatment and allowed to incubate for an additional 24 h. At the time of harvest, plates were placed at −80°C for a single freeze-thaw cycle. This allowed cell lysis and viral release. Serial dilutions in serum-free EMEM were made of lysates and transferred to Vero cell microtiter plates. The microtiter plates were read after 6 to 7 days at 37°C in 5% CO2. An endpoint titer in units of 50% tissue culture infective doses (TCID50)/ml was calculated using the Reed-Muench method (31).
Western blotting.
Protein was run on 10% Tris-glycine protein gels (EC6078BOX; Novex), followed by transfer to nitrocellulose using iBlot transfer stacks (IB3010-02). Antibody mixtures were made in 5% BSA and 0.1% Tween 20 in Tris-buffered saline (TBS) as per the recommendations of Cell Signaling for phospho-antibody use. After samples were blocked for 30 min at room temperature with StartingBlock (TBS) blocking buffer (37542; Thermo Fisher), antibodies were left to incubate at 4°C with constant shaking overnight. After vigorous washing with 0.1% Tween 20 in TBS, the secondary HRP antibody was added for 1 h at room temperature. The blots were washed as before and developed with SuperSignal West Dura Chemiluminescent Substrate (34076; Thermo Fisher). Images were developed using the medical film processor SRX-101A by Konica Minolta Medical and Graphic, Inc.
RESULTS
Real-time PCR of type I and type III IFNs in BEAS-2B cells infected with PIV-3.
The epithelial cells of the lung are the primary site of PIV-3 infection. The type III IFN response during PIV-3 infection has not been previously described. We therefore initially sought to determine if type III IFNs were produced during PIV-3 infection in the human bronchial epithelial cell line BEAS-2B. BEAS-2B cells were infected at an MOI of 2 with TtPIV-1, BPIV-3, or HPIV-3, and expression levels of the type III IFNs (IL-28 and IL-29) were measured by real-time PCR at multiple time points after infection. In BEAS-2B cells, IL-29 message was induced to levels similar to those of the positive control, poly(I·C), by 24 h and continued through 48 h (Fig. 1). IL-28 expression was not significantly upregulated during PIV-3 infection. IFN-β expression in the BEAS-2B cells was significantly increased during HPIV-3 infection at 24 and 48 h (Fig. 1). IFN-α was not significantly upregulated during PIV-3 infection in BEAS-2B cells.
FIG 1.

Real-time PCR of type I and type III IFNs in BEAS-2B cells infected with PIV-3. BEAS-2B cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 6, 12, 24, or 48 h. Cells treated with 20 μg/ml poly(I·C) and 0.6% Lipofectamine 3000 for 10 min were used as a positive control (+). IL-28, IL-29, IFN-α1 (IFNa.1), IFN-α2 (IFNa.2), and IFN-β (IFNb) message levels were measured by real-time PCR. The relative quantification (RQ) was calculated by normalization first to the GAPDH level to give a ΔCT value, followed by normalization to the mock level at a given time point (ΔΔCT). The RQ or fold change was then calculated as 2−ΔΔCT. A significant change in levels from those in uninfected controls at a particular time point is denoted as follows: *, P < 0.05; #, P < 0.01; +, P < 0.001.
ELISA of type I and type III IFNs in BEAS-2B cells infected with PIV-3.
Enzyme-linked immunosorbent assays (ELISAs) were run on cell culture supernatants to confirm our real-time PCR results (Fig. 2). BEAS-2B cells produced significant levels of IFN-β protein by 48 and 72 h postinfection with HPIV-3. Concentrations of IL-29 protein were also significantly upregulated by 72 h in response to BPIV-3 and HPIV-3 infection but not to TtPIV-1 infection.
FIG 2.
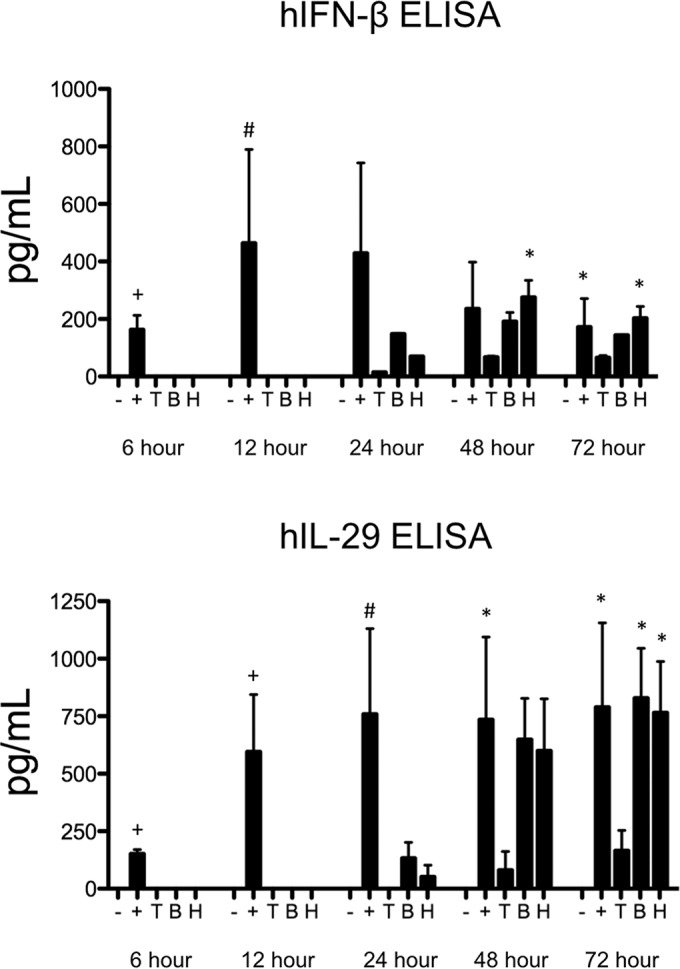
ELISA of type I and type III IFNs in BEAS-2B cells infected with PIV-3. BEAS-2B cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 6, 12, 24, or 48 h. Cells treated with 20 μg/ml poly(I·C) and 0.6% Lipofectamine 3000 for 10 min were used as a positive control (+). ELISAs were conducted on the supernatants to measure human IFN-β (hIFN-β) and IL-29 (hIL-29). A significant change in levels from those in uninfected controls at a particular time point is denoted as follows: *, P < 0.05; #, P < 0.01.
Real-time PCR of antiviral molecules downstream of the IFN receptor in BEAS-2B cells infected with PIV-3.
PIV-3 infection induced IL-29 production by BEAS-2B cells. Therefore, we next wanted to confirm that the expression of downstream antiviral molecules was induced as one would expect based on previous reports (Fig. 3). In BEAS-2B cells, expression of OAS and ISG56 message increased significantly after infection with all of the PIV-3s as early as 24 h postinfection. However, PKR and MxA message levels were not upregulated, even by 72 h postinfection.
FIG 3.
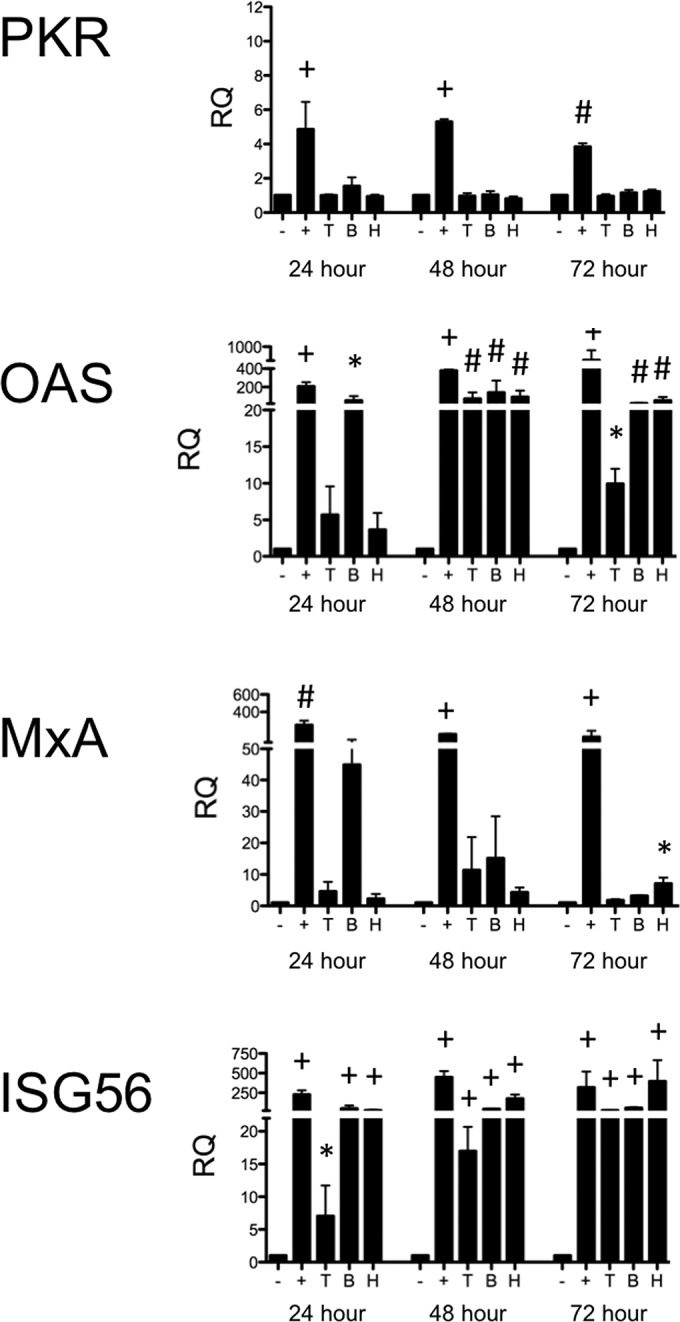
Real-time PCR of antiviral molecules downstream of the IFN receptor in BEAS-2B cells infected with PIV-3. BEAS-2B cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 24, or 48, or 72 h. Cells treated with 20 μg/ml poly(I·C) and 0.6% Lipofectamine 3000 for 10 min were used as a positive control (+). Levels of downstream antiviral molecules (PKR, OAS, MxA, and ISG56) were measured by real-time PCR. The relative quantification (RQ) was calculated by normalization first to the GAPDH level to give a ΔCT value, followed by normalization to the mock level at a given time point (ΔΔCT). The RQ or fold change was then calculated as 2−ΔΔCT. A significant change in levels from those in uninfected controls at a particular time point is denoted as follows: *, P < 0.05; #, P < 0.01; +, P < 0.001.
Examining type III IFN signaling in BEAS-2B cells using Western blotting.
Type III IFNs signal through a unique receptor but are thought to utilize the same JAK/STAT signaling pathways as type I IFNs (17, 18). Failure of the BEAS-2B cells to upregulate expression of PKR and MxA in response to PIV-3 infection suggested a blockade in the signaling pathways downstream of the type I or type III receptors. We therefore wanted to determine the effect of PIV-3 infection on the JAK/STAT signaling pathway. BEAS-2B cells were infected with TtPIV-1, BPIV-3, or HPIV-3 at an MOI of 2 for 24 h. After 24 h, the cells were treated with 1 μg/ml of recombinant IL-29/-28A/-28B for 30 min. Western blotting was then used to examine the phosphorylation of Stat during PIV-3 infection and subsequent recombinant cytokine treatment. Stat1 has two critical phosphorylation sites: Y701 and S727. Both sites are required to mediate downstream signaling (25–27). Representative blots are depicted in Fig. 4A; densitometry quantification is shown in Fig. 4B. As seen in Fig. 4, phosphorylation at Y701 on Stat1 is significantly reduced during PIV-3 infection compared to the level in cells treated with IFN-λ only, while phosphorylation of the S727 site is unaffected. Importantly, calculations to normalize values to those of Stat1 were based on the active form, Stat1-α because Stat1-β lacks the 38-amino-acid transcriptional activation domain (32). In addition, during HPIV-3 infection, IFN-λ-induced phosphorylation at the Y690 site of Stat2 is significantly reduced. Phosphorylation of Stat2 Y690 also appears to be decreased during BPIV-3 and TtPIV-1 infection, but these changes are not significant.
FIG 4.

Examining type III IFN signaling in BEAS-2B cells using Western blotting. Vero cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 24 h. At 24 h postinfection, Vero cells were stimulated with recombinant IL-29/-28A/-28B (1 μg/ml) for 30 min to drive signaling downstream of the type III IFN receptor. (A) Western blotting was used to examine Stat1 and Stat2 phosphorylation during PIV-3 infection. β-Actin was used as a loading control. (B) Densitometry quantification from Western blotting in BEAS-2B cells was calculated. The relative quantification from band intensity was calculated by its expression relative to that of the IFN-λ-stimulated positive control (striped bars). This RQ was then normalized to the appropriate total Stat1 or Stat2 to give the percent pStat1 to total Stat1 or the percent pStat2 to total Stat2 value. A significant change in the level of phosphorylation from that in the IFN-λ-treated positive control is denoted as follows: *, P < 0.05; #, P < 0.01.
Because BEAS-2B cells have the capacity to secrete both type I and type III IFNs, it is difficult to dissect the effect of PIV-3 infection on IL-29 signaling, independent of IFN-α/β. In preliminary experiments, we sought to knock down the expression of type I IFN receptor (IFNAR1/IFNAR2) by small interfering RNA (siRNA) treatment. After siRNA knockdown and restimulation with recombinant IFN-α/β, there was a 60 to 70% reduction in expression levels of OAS, MxA, and ISG56 (data not shown). However, Lipofectamine treatment alone caused untoward changes in the JAK/STAT signaling pathway that would affect any conclusions. Jensen et al. showed a similar phenomenon in that multiple transfection reagents artificially upregulated IFIT1, which is an antiviral molecule downstream of the IFN receptor (33).
Real-time PCR of type III IFNs in Vero cells infected with PIV-3.
We had observed intriguing results about the type III IFN response in BEAS-2B cells during PIV-3 infection; however, conclusions made in the BEAS-2B cells are complicated by the presence of IFN-α/β. We therefore chose to repeat these experiments in a model independent of type I IFN. Vero cells were specifically used because several previous studies have shown that they are not able to produce type I IFNs (22–24). As in the BEAS-2B cells, Vero cells were infected at an MOI of 2 with TtPIV-1, BPIV-3, or HPIV-3, and expression levels of the type III IFNs (IL-28 and IL-29) were measured by real-time PCR at multiple time points after infection. As observed with the BEAS-2B cells, IL-29 message was significantly increased in Vero cells by 48 h postinfection in response to all virus strains (Fig. 5). Treatment with recombinant IFN-α/β was not able to significantly upregulate IL-29 or IL-28 message in Vero cells. IL-28 expression was not detectable in Vero cells during PIV-3 infection; however, we were unable to establish a positive control to confirm the capacity of these cells to produce IL-28.
FIG 5.
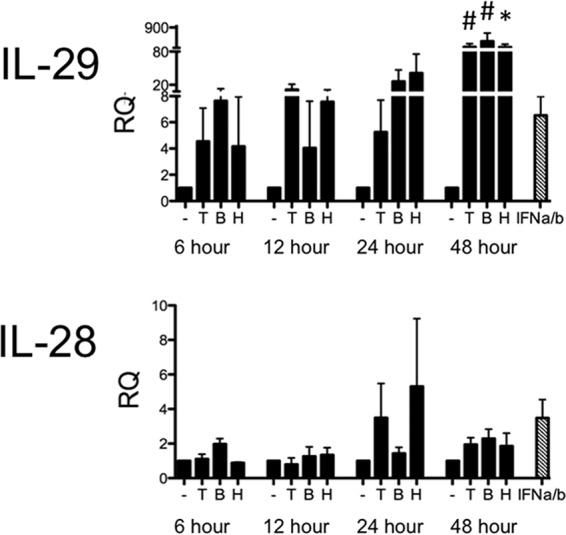
Real-time PCR of type III IFNs in Vero cells infected with PIV-3. Vero cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 6, 12, 24, or 48 h. Cells were incubated with recombinant IFN-α/β at 500 U/ml for 24 h. IL-28 or -29 message levels were measured by real-time PCR. The relative quantification (RQ) was calculated by normalization first to the GAPDH level to give a ΔCT value, followed by normalization to the mock level at a given time point (ΔΔCT). The RQ or fold change was then calculated as 2−ΔΔCT. A significant change from uninfected controls at a particular time point is denoted as follows: *, P < 0.05; #, P < 0.01. Striped bars, positive controls.
Real-time PCR of antiviral molecules downstream of the IFN receptor in Vero cells infected with PIV-3 and stimulated with recombinant type III IFNs.
We again wanted to examine downstream antiviral molecules that may be upregulated in the Vero cells during PIV-3 infection (Fig. 6). Cells were infected with TtPIV-1, BPIV-3, or HPIV-3, as described for the experiment shown in Fig. 5, and expression of PKR, OAS, MxA, and ISG56 was measured by real-time PCR at multiple time points after infection. As shown by the data in Fig. 6, expression of OAS was upregulated in response to all three viruses within 48 h postinfection. ISG56 expression was also upregulated by 48 h following HPIV-3 infection and by 72 h following BPIV-3 and TtPIV-1 infection. However, PKR and MxA expression levels were not significantly induced in response to any of the virus strains. Importantly, none of the downstream signaling molecules were expressed at levels near those of the recombinant type III or I IFN treatments used as positive controls (Fig. 6, striped bars), suggesting an effect of PIV-3 on downstream signaling pathways.
FIG 6.
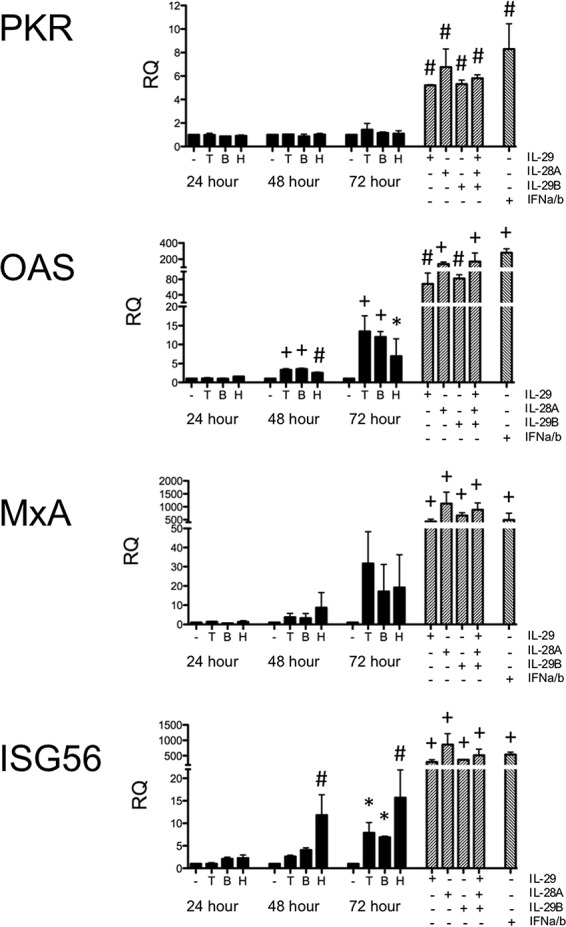
Real-time PCR of antiviral molecules downstream of the IFN receptor in Vero cells infected with PIV-3. Vero cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 24, or 48, or 72 h. Cells were incubated with recombinant IFN-α/β (500 U/ml) or IL-29/-28A/-28B (1 μg/ml) for 24 h. Levels of downstream antiviral molecules (PKR, OAS, MxA, and ISG56) were measured by real-time PCR. The relative quantification (RQ) was calculated by normalization first to the GAPDH level to give a ΔCT value, followed by normalization to the mock level at a given time point (ΔΔCT). The RQ or fold change was then calculated as 2−ΔΔCT. A significant change in levels from those in the uninfected controls at a particular time point is denoted as follows: *, P < 0.05; #, P < 0.01; +, P < 0.001. Striped bars, positive controls.
We sought to further determine if treatment with recombinant type III IFN could overcome the blockade resulting from PIV-3 infection and induce signaling downstream of IFN-λR1/IL-10R2. Therefore, Vero cells were infected with PIV-3 and 24 h postinfection treated with recombinant type III IFNs for 2 or 6 h. As seen from the data in Fig. 7, downstream signaling molecules were still not upregulated to levels near those of uninfected, IFN-λ-treated positive-control cells. Because recombinant type III IFN treatment could not rescue the effect of virus infection on downstream antiviral molecules, we hypothesized that PIV-3 infection directly affects the JAK/STAT signaling pathway that is downstream of IFN-λR1/IL-10R2.
FIG 7.
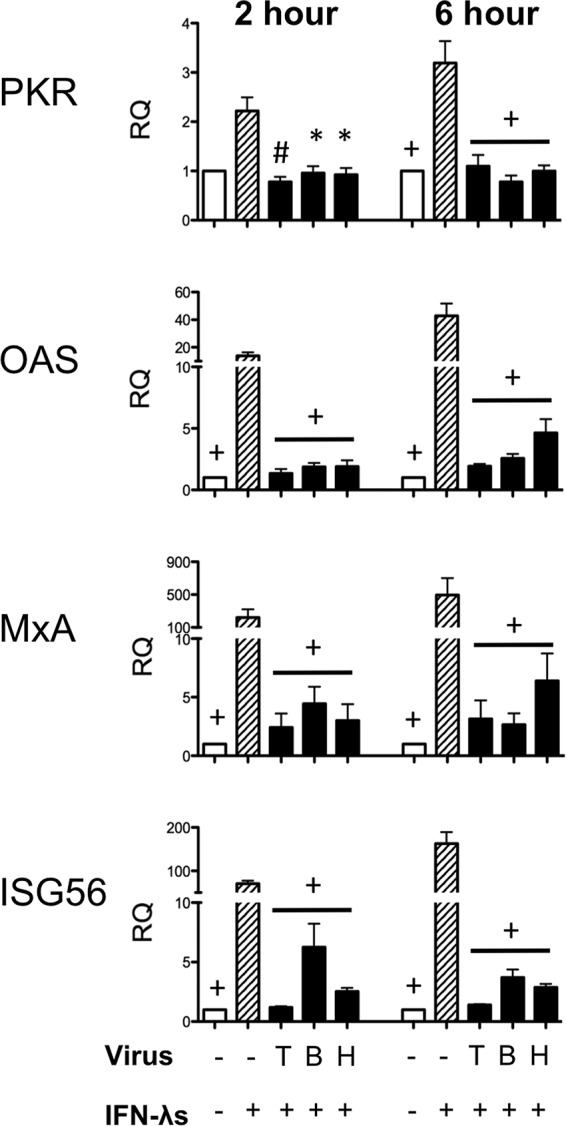
Real-time PCR of antiviral molecules downstream of the IFN receptor in Vero cells stimulated with recombinant type III IFNs after PIV-3 infection. Vero cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 24 h. At 24 h postinfection, Vero cells were stimulated with recombinant IL-29/-28A/-28B (1 μg/ml) for 2 or 6 hours. Levels of downstream antiviral molecules (PKR, OAS, MxA, and ISG56) were measured by real-time PCR. The relative quantification (RQ) was calculated by normalization first to the GAPDH level to give a ΔCT value, followed by normalization to the mock level at a given time point (ΔΔCT). The RQ or fold change was then calculated as 2−ΔΔCT. A significant change in levels from those in the IFN-λ-treated positive control at a particular time point is denoted as follows: *, P < 0.05; #, P < 0.01; +, P < 0.001.
Examining type III IFN signaling in Vero cells using Western blotting.
IL-29 expression was upregulated in Vero cells infected with PIV-3 (Fig. 5), while downstream antiviral signaling molecules were not induced (Fig. 6). In addition, recombinant type III IFN treatment of PIV-3-infected Vero cells failed to induce expression of downstream antiviral molecules to levels observed in uninfected controls (Fig. 7). This suggested that, through an unknown mechanism, PIV-3 inhibits the JAK/STAT signaling pathway downstream of IFN-λR1/IL-10R2. Similarly to previous experiments, Vero cells were infected with TtPIV-1, BPIV-3, or HPIV-3 at an MOI of 2 for 24 h. After 24 h, the cells were treated with recombinant type III IFN to drive signaling downstream of the type III IFN receptor. In BEAS-2B cells we observed, by Western blotting, reduced phosphorylation of pStat1 Y701 during PIV-3 infection. In Fig. 8, we show that, in Vero cells, phosphorylation is decreased at the S727 site on Stat1 during PIV-3 infection. This reduced phosphorylation is specific to the S727 site because the Y701 site of Stat1 is unaffected. As depicted by densitometric quantitation in Fig. 8B, Stat1 phosphorylation at S727 is significantly reduced during PIV-3 infection while phosphorylation at the Y701 site significantly increased. In addition, levels of total Stat1 and Stat2 and of phosphorylation at the tyrosine 690 site on Stat2 (pStat2 Y690) are not affected. To calculate the relative quantification (RQ) of band intensity, the level of pStat1 was normalized to that of Stat1-α as previously described (32).
FIG 8.

Examining type III IFN signaling in Vero cells using Western blotting. Vero cells were infected with dolphin (T), bovine (B), or human (H) PIV-3 for 24 h. At 24 h postinfection, Vero cells were stimulated with recombinant IL-29/-28A/-28B (1 μg/ml) for 30 min to drive signaling downstream of the type III IFN receptor. (A) Western blotting was used to examine Stat1 and Stat2 phosphorylation during PIV-3 infection. β-Actin was used as a loading control. (B) Densitometry quantification from Western blotting in Vero cells was calculated. Relative quantification from band intensity was calculated by its expression relative to the level of the IFN-λ-stimulated positive control. This RQ was then normalized to the level of the appropriate total Stat1 or Stat2 to give the percent pStat1 to total Stat1 or percent pStat2 to total Stat2 value. A significant change in phosphorylation levels from those in the IFN-λ-treated positive control is indicated (*, P < 0.05).
Prophylactic treatment of Vero cells with type III IFNs before PIV-3 viral infection.
Because PIV-3 blocks signaling downstream of the type III IFN receptor, therapeutic treatment with type III IFNs would not be useful. However, prophylactic treatment with IL-29, IL-28A, and/or IL-28B in Vero cells does appear effective against PIV-3 infection (Fig. 9, left panels). Vero cells were treated with the type III IFNs, singly or pooled, for 24 h before PIV-3 infection, and then the virus titer was measured 24 h after PIV-3 infection. As seen in Fig. 9, recombinant type III IFN treatment resulted in significantly reduced viral titers. These results are evidently independent of MOI in that MOIs of 2 and 0.2 were used. This antiviral response was specific to the recombinant type III interferon added because 80 μg/ml blocking antibody was able to reverse the titer repression (Fig. 9, right panels).
FIG 9.

Prophylactic treatment of Vero cells with type III IFNs before PIV-3 viral infection. Vero cells were prophylactically treated with recombinant IL-29/-28A/-28B, alone or pooled, at 1 μg/ml for 24 h before TtPIV-1, BPIV-3, or HPIV-3 infection at an MOI of 2 or 0.2 for an additional 24 h. After 48 h, titer plates were used to determine the effect of type III IFNs on virus growth. In the experiment shown in the right panels, IL-29 blocking antibody was added in increasing doses to show specificity of the viral inhibition by type III IFNs. At the highest blocking antibody dose of 80 μg/ml, a log(titer) similar to that of the control was observed. A significant reduction in log(titer) compared to that of the untreated control sample is denoted as follows: *, P < 0.05; #, P < 0.01; +, P < 0.001.
DISCUSSION
Type I and type III IFNs are induced by viral sensors including RLRs and TLRs. IFNs bind their respective receptors to activate JAK/STAT signaling and produce downstream antiviral molecules, including PKR, OAS, MxA, and ISGs. Parainfluenza virus predominantly infects the airway epithelium. Because the type III IFN receptor is primarily located on epithelial cells, we chose to examine the IFN-λ response to PIV-3 infection. While many respiratory viruses, including respiratory syncytial virus (RSV), rhinovirus, and influenza virus, have been shown to induce IFN-λs (33–37), this is the first report, to our knowledge, of PIV-3 inducing expression of type III IFNs and modulating the downstream antiviral signaling pathways.
Here, we examined production of type III IFNs and antiviral molecules downstream of IFN-λR1/IL-10R2 during PIV-3 infection. IL-29 message levels were significantly increased in BEAS-2B cells as early as 24 h postinfection (Fig. 1). Other investigators have shown that type I IFNs are necessary to activate type III IFNs, but we did not observe the necessity of IFN-α/β for IL-29 production here (38). This can be seen from the data shown in Fig. 5, where IL-29 message was significantly upregulated by 48 h postinfection with PIV-3 in type I IFN-deficient Vero cells, and recombinant IFN-α/β alone (striped bars) did not stimulate IL-29 to a level similar to that observed during virus infection. We measured protein levels of IL-29 in cell culture supernatant from BEAS-2B cells, as shown in Fig. 2. Multiple IL-29 ELISAs were tested for use with Vero cell supernatants; however, we found that none of the antibodies were cross-reactive with Vero cell supernatants. It is therefore possible that PIV-3 could simply inhibit production of type III IFN protein by an unknown mechanism in Vero cells, which could, in turn, prevent induction of downstream antiviral molecules. A significant increase in OAS and ISG56 levels was seen in BEAS-2B cells infected with all three viruses, but this could be due to type I IFNs (Fig. 3). Furthermore, downstream antiviral molecules, including PKR, OAS, MxA, and ISG56, were not upregulated in the Vero cells infected with PIV-3 compared to levels in cells treated with recombinant type I or type III IFNs (Fig. 6, striped bars).
There is clear evidence that PIV-3 interferes with signaling downstream of the IFN-λR1/IL-10R2 receptor. Figure 7 showed that Vero cells stimulated with recombinant type III IFNs at 24 h postinfection with PIV-3 were unable to induce a robust antiviral response. This was a strong indication that PIV-3 interferes with the JAK/STAT signaling pathway downstream of the type III IFN receptor. It is well known that paramyxoviruses block type I IFN production (7–16). A few reports have studied the JAK/STAT downstream signaling response during PIV-3 infection, specifically in response to type I IFNs.
By examining the phosphorylation downstream of IFN-λR1/IL-10R2 through Western blotting, we specifically observed decreased phosphorylation of pStat1 Y701 in BEAS-2B cells (Fig. 4) and a decrease in phosphorylation at the S727 site on Stat1 in Vero cells (Fig. 8) during PIV-3 infection. It is well known that both the tyrosine and serine sites of Stat1 are necessary for full transcriptional activation (26). Specifically, phosphorylation at the Y701 site has been shown to be vital because of its role in hetero- or homodimerization of Stat1 (26, 32). On the other hand, the S727 site must be phosphorylated to allow binding of transcriptional coactivators such as CBP/p300 and MCM5 (26, 27, 39, 40). Phosphorylation of the serine and tyrosine sites on Stat1 has been shown to be independent of each other (41); here, we observed reduced phosphorylation of Stat1 specific to S727 or Y701 in the Vero or BEAS-2B cells, respectively. Because the Vero cells are naturally unable to produce type I IFNs, it may be most appropriate to base any conclusions on results obtained in the Vero cells. Other paramyxoviruses have been shown to decrease total Stat1 and Stat2 levels to reduce the type I IFN antiviral response (12, 15, 42). As was observed in the BEAS-2B cells (Fig. 4) but not seen in the Vero cells (Fig. 8), total Stat1-α and Stat2 may be slightly reduced during HPIV-3 infection. However, any total Stat1 or Stat2 reduction was accounted for during relative quantification performed for graphical analysis (Fig. 4B and 8B). It is of note that in the BEAS-2B cell line a species-specific decrease in phosphorylation of Stat2 was observed during infection with HPIV-3 that was not seen following infection with BPIV-3 or TtPIV-1 (Fig. 4).
We hypothesize that PIV-3 induces specific phosphatase activity to cause reduced phosphorylation of Stat1. The phosphatase SHP-2 has previously been shown to dephosphorylate Stat1 at both Y701 and S727 (43). In addition, it is plausible that alterations in tyrosine kinases (Jak1/Tyk2) could occur during PIV-3 infection that, in turn, cause reduced phosphorylation of Stat1 (44). Future experiments will need to be conducted to examine the specific mechanism of Stat1 dephosphorylation or reduced phosphorylation during PIV-3 infection.
Treatment of Vero cells with type III IFNs 24 h before PIV-3 infection was able to reduce viral titers (Fig. 9). Type III IFNs are therefore able to prophylactically induce downstream antiviral molecules to control viral growth in Vero cells. However, we show for the first time that PIV-3 induces type III IFNs and identify a novel mechanism by which PIV-3 inhibits production of the downstream antiviral molecules PKR, OAS, MxA, and ISG56. Downstream of the type III IFN receptor, PIV-3 causes reduced phosphorylation of Stat1 to negatively affect the JAK/STAT signaling pathway and reduce production of antiviral molecules.
ACKNOWLEDGMENTS
We acknowledge the excellent technical support of Theresa Waters and Tera Nyholm.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Karron RA, Thumar B, Schappell E, Surman S, Murphy BR, Collins PL, Schmidt AC. 2012. Evaluation of two chimeric bovine-human parainfluenza virus type 3 vaccines in infants and young children. Vaccine 30:3975–3981. doi: 10.1016/j.vaccine.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karron RA, Wright PF, Hall SL, Makhene M, Thompson J, Burns BA, Tollefson S, Steinhoff MC, Wilson MH, Harris DO. 1995. A live attenuated bovine parainfluenza virus type 3 vaccine is safe, infectious, immunogenic, and phenotypically stable in infants and children. J Infect Dis 171:1107–1114. doi: 10.1093/infdis/171.5.1107. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt AC, McAuliffe JM, Huang A, Surman SR, Bailly JE, Elkins WR, Collins PL, Murphy BR, Skiadopoulos MH. 2000. Bovine parainfluenza virus type 3 (BPIV3) fusion and hemagglutinin-neuraminidase glycoproteins make an important contribution to the restricted replication of BPIV3 in primates. J Virol 74:8922–8929. doi: 10.1128/JVI.74.19.8922-8929.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henrickson KJ. 2003. Parainfluenza viruses. Clin Microbiol Rev 16:242–264. doi: 10.1128/CMR.16.2.242-264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vareille M, Kieninger E, Edwards MR, Regamey N. 2011. The airway epithelium: soldier in the fight against respiratory viruses. Clin Microbiol Rev 24:210–229. doi: 10.1128/CMR.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossert B, Marozin S, Conzelmann KK. 2003. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol 77:8661–8668. doi: 10.1128/JVI.77.16.8661-8668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S. 2007. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 10.Childs KS, Andrejeva J, Randall RE, Goodbourn S. 2009. Mechanism of mda-5 Inhibition by paramyxovirus V proteins. J Virol 83:1465–1473. doi: 10.1128/JVI.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Childs K, Randall R, Goodbourn S. 2012. Paramyxovirus V proteins interact with the RNA helicase LGP2 to inhibit RIG-I-dependent interferon induction. J Virol 86:3411–3421. doi: 10.1128/JVI.06405-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, Buick R, Stevenson NJ, Touzelet O, Gadina M, Power UF, Johnston JA. 2007. Respiratory syncytial virus NS1 protein degrades STAT2 by using the elongin-cullin E3 ligase. J Virol 81:3428–3436. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitagawa Y, Yamaguchi M, Zhou M, Nishio M, Itoh M, Gotoh B. 2013. Human parainfluenza virus type 2 V protein inhibits TRAF6-mediated ubiquitination of IRF7 to prevent TLR7-and TLR9-dependent interferon induction. J Virol 87:7966–7976. doi: 10.1128/JVI.03525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu LL, Puri M, Horvath CM, Sen GC. 2008. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of κB kinase ε (IKKe)/TBK1. J Biol Chem 283:14269–14276. doi: 10.1074/jbc.M710089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. 2006. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 344:328–339. doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi M, Kitagawa Y, Zhou M, Itoh M, Gotoh B. 2014. An anti-interferon activity shared by paramyxovirus C proteins: inhibition of Toll-like receptor 7/9-dependent alpha interferon induction. FEBS Lett 588:28–34. doi: 10.1016/j.febslet.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 17.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. 2003. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 18.Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G, Clegg C, Foster D, Klucher KM. 2003. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 19.Miknis ZJ, Magracheva E, Li W, Zdanov A, Kotenko SV, Wlodawer A. 2010. Crystal structure of human interferon-λ1 in complex with its high-affinity receptor interferon-λR1. J Mol Biol 404:650–664. doi: 10.1016/j.jmb.2010.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog 4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrejeva J, Norsted H, Habjan M, Thiel V, Goodbourn S, Randall RE. 2013. ISG56/IFIT1 is primarily responsible for interferon-induced changes to patterns of parainfluenza virus type 5 transcription and protein synthesis. J Gen Virol 94:59–68. doi: 10.1099/vir.0.046797-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emeny JM, Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol 43:247–252. doi: 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- 23.Osada N, Kohara A, Yamaji T, Hirayama N, Kasai F, Sekizuka T, Kuroda M, Hanada K. 2014. The genome landscape of the African green monkey kidney-derived Vero cell line. DNA Res 21:673–683. doi: 10.1093/dnares/dsu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wathelet MG, Berr PM, Huez GA. 1992. Regulation of gene expression by cytokines and virus in human cells lacking the type-I interferon locus. Eur J Biochem 206:901–910. doi: 10.1111/j.1432-1033.1992.tb16999.x. [DOI] [PubMed] [Google Scholar]
- 25.Ooi EL, Chan ST, Cho NE, Wilkins C, Woodward J, Li M, Kikkawa U, Tellinghuisen T, Gale M, Saito T. 2014. Novel antiviral host factor, TNK1, regulates IFN signaling through serine phosphorylation of STAT1. Proc Natl Acad Sci U S A 111:1909–1914. doi: 10.1073/pnas.1314268111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen Z, Zhong Z, Darnell JE Jr. 1995. Maximal activation of transcription by Statl and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Takami K, Lo MS, Huang G, Yu Q, Roswit WT, Holtzman MJ. 2005. Modification of the Stat1 SH2 domain broadly improves interferon efficacy in proportion to p300/CREB-binding protein coactivator recruitment. J Biol Chem 280:34306–34315. doi: 10.1074/jbc.M503263200. [DOI] [PubMed] [Google Scholar]
- 28.Nollens HH, Wellehan JF, Saliki JT, Caseltine SL, Jensen ED, Van Bonn W, Venn-Watson S. 2008. Characterization of a parainfluenza virus isolated from a bottlenose dolphin (Tursiops truncatus). Vet Microbiol 128:231–242. doi: 10.1016/j.vetmic.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Eberle KC, Neill JD, Venn-Watson SK, McGill JL, Sacco RE. 2015. Novel Atlantic bottlenose dolphin parainfluenza virus TtPIV-1 clusters with bovine PIV-3 genotype B strains. Virus Genes 51:198–208. doi: 10.1007/s11262-015-1224-7. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Reed LJ, Muench J. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]
- 32.Horvath CM, Darnell JE. 1996. The antiviral state induced by alpha interferon and gamma interferon requires transcriptionally active Stat1 protein. J Virol 70:647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jensen K, Anderson JA, Glass EJ. 2014. Comparison of small interfering RNA (siRNA) delivery into bovine monocyte-derived macrophages by transfection and electroporation. Vet Immunol Immunopathol 158:224–232. doi: 10.1016/j.vetimm.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khaitov MR, Laza-Stanca V, Edwards MR, Walton RP, Rohde G, Contoli M, Papi A, Stanciu LA, Kotenko SV, Johnston SL. 2009. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy 64:375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 35.Okabayashi T, Kojima T, Masaki T, Yokota S-I, Imaizumi T, Tsutsumi H, Himi T, Fujii N, Sawada N. 2011. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res 160:360–366. doi: 10.1016/j.virusres.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Spann KM, Tran K-C, Chi B, Rabin RL, Collins PL. 2004. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J Virol 78:4363–4369. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ. 2009. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-1) in response to influenza A infection. J Immunol 182:1296–1304. doi: 10.4049/jimmunol.182.3.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H. 2008. An important role for type III interferon (IFN-λ/IL-28) in TLR-induced antiviral activity. J Immunol 180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- 39.Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. 2009. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J 28:948–958. doi: 10.1038/emboj.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang JJ, Zhao Y, Chait BT, Lathem WW, Ritzi M, Knippers R, Darnell JE. 1998. Ser727-dependent recruitment of MCM5 by Stat1α in IFN-γ-induced transcriptional activation. EMBO J 17:6963–6971. doi: 10.1093/emboj/17.23.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu X, Wen Z, Xu LZ, Darnell JE. 1997. Stat1 serine phosphorylation occurs independently of tyrosine phosphorylation and requires an activated Jak2 kinase. Mol Cell Biol 17:6618–6623. doi: 10.1128/MCB.17.11.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young DF, Didcock L, Goodbourn S, Randall RE. 2000. Paramyxoviridae use distinct virus-specific mechanisms to circumvent the interferon response. Virology 269:383–390. doi: 10.1006/viro.2000.0240. [DOI] [PubMed] [Google Scholar]
- 43.Wu TR, Hong YK, Wang XD, Ling MY, Dragoi AM, Chung AS, Campbell AG, Han ZY, Feng GS, Chin YE. 2002. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J Biol Chem 277:47572–47580. doi: 10.1074/jbc.M207536200. [DOI] [PubMed] [Google Scholar]
- 44.Senft AP, Taylor RH, Lei W, Campbell SA, Tipper JL, Martinez MJ, Witt TL, Clay CC, Harrod KS. 2010. Respiratory syncytial virus impairs macrophage IFN-α/β- and IFN-γ-stimulated transcription by distinct mechanisms. Am J Respir Cell Mol Biol 42:404–414. doi: 10.1165/rcmb.2008-0229OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. 2006. Lambda interferon (IFN-λ), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoon CH, Lee ES, Lim DS, Bae YS. 2009. PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc Natl Acad Sci U S A 106:7852–7857. doi: 10.1073/pnas.0812148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu MQ, Zhou DJ, Wang X, Zhou W, Ye L, Li JL, Wang YZ, Ho WZ. 2012. IFN-λ3 inhibits HIV infection of macrophages through the JAK-STAT pathway. PLoS ONE 7:e35902. doi: 10.1371/journal.pone.0035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J. Virol 81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuo TM, Hu CP, Chen YL, Hong MH, Jeng KS, Liang CC, Chen ML, Chang C. 2009. HBV replication is significantly reduced by IL-6. J Biomed Sci 16:41. doi: 10.1186/1423-0127-16-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hinzey A, Alexander J, Corry J, Adams KM, Claggett AM, Traylor ZP, Davis IC, Webster Marketon JI. 2011. Respiratory syncytial virus represses glucocorticoid receptor-mediated gene activation. Endocrinology 152:483–494. doi: 10.1210/en.2010-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]