

ABSTRACT
The type I interferon (IFN) response is an important aspect of innate antiviral defense, and the transcription factor IRF3 plays an important role in its induction. Membrane perturbation during fusion, a necessary step for enveloped virus particle entry, appears sufficient to induce transcription of a subset of IFN-stimulated genes (ISGs) in an IRF3-dependent, IFN-independent fashion. IRF3 is emerging as a central node in host cell stress responses, although it remains unclear how different forms of stress activate IRF3. Here, we investigated the minimum number of Sendai virus (SeV) and human cytomegalovirus (HCMV) particles required to activate IRF3 and trigger an antiviral response. We found that Ca2+ signaling associated with membrane perturbation and recognition of incoming viral genomes by cytosolic nucleic acid receptors are required to activate IRF3 in response to fewer than 13 particles of SeV and 84 particles of HCMV per cell. Moreover, it appears that Ca2+ signaling is important for activation of STING and IRF3 following HCMV particle entry, suggesting that Ca2+ signaling sensitizes cells to recognize genomes within incoming virus particles. To our knowledge, this is the first evidence that cytosolic nucleic acid sensors recognize genomes within incoming virus particles prior to virus replication. These studies highlight the exquisite sensitivity of the cellular response to low-level stimuli and suggest that virus particle entry is sensed as a stress signal.
IMPORTANCE The mechanism by which replicating viruses trigger IRF3 activation and type I IFN induction through the generation and accumulation of viral pathogen-associated molecular patterns has been well characterized. However, the mechanism by which enveloped virus particle entry mediates a stress response, leading to IRF3 activation and the IFN-independent response, remained elusive. Here, we find that Ca2+ signaling associated with membrane perturbation appears to sensitize cells to recognize genomes within incoming virus particles. To our knowledge, this is the first study to show that cytosolic receptors recognize genomes within incoming virus particles prior to virus replication. These findings not only highlight the sensitivity of cellular responses to low-level virus particle stimulation, but provide important insights into how nonreplicating virus vectors or synthetic lipid-based carriers used as clinical delivery vehicles activate innate immune responses.
INTRODUCTION
Cells defend themselves from viral infection by producing antiviral proteins, which cumulatively make them nonpermissive to virus replication (1). Large sets of antiviral proteins are induced by type I interferons (IFNs), and this response is critical for defense against viral infection (2, 3). IFN-β has no direct antiviral activity but signals induction of a set of IFN-stimulated genes (ISGs) encoding proteins with antiviral activity (4, 5). IFN-β is produced by a wide array of cells, and as it is the first IFN subtype produced in response to virus infection, many subsequent immune responses hinge on this initial signal (6–8). Following viral recognition, the transcription factors NF-κB, ATF2/c-Jun, and IFN-regulatory factor 3 (IRF3) are activated and form an enhanceosome on the IFN-β promoter, which is critical for its induction (1, 9).
While certain stimuli activate the IFN pathway to induce ISGs, low-level infection with enveloped virus particles is sufficient to directly induce a subset of ISGs in the absence of IFN (10, 11). Unlike IFN-β production, which can occur in an IRF3-independent fashion (12–14), IFN-independent induction of ISGs by virus particles occurs in an IRF3-dependent, NF-κB-independent manner (15, 16). Based on our observations that the threshold for activation of IRF3 is lower than that of NF-κB (15), we previously proposed a model in which the IFN-independent antiviral response serves to efficiently and quietly induce a localized and primarily intracellular protective response to low-level virus stimulation without inducing unwanted or unnecessary immune activity (15). The ability of IRF3 to function independently of the IFN-β enhanceosome and in the absence of traditional markers of activation (16, 17) suggests a means of IRF3 activation distinct from the canonical virus-activated signaling pathway.
Canonical activation of IRF3 by virus infection requires recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) (18, 19). The IFN-independent response is associated with the entry of enveloped virus particles (10, 11, 16, 20). All enveloped viruses must fuse with cell membranes during entry, and reports suggest membrane fusion itself is sufficient to induce ISGs. Enveloped virus particles and lipoprotein complexes containing purified reovirus fusion-associated small transmembrane (p14-FAST) protein directly induce ISGs in primary fibroblasts (16, 21), while in immune cells, virus-like particles (VLPs) and fusogenic liposomes induce type I IFN (22). Interestingly, membrane fusion by p14 lipoplexes induces the same noncanonical IRF3 activation and ISG subset as virus particles. A number of stress pathways have been associated with noncanonical IRF3 activation, leading us to speculate that, like cell stress, membrane perturbation is sensed as a danger signal of infection (23, 24).
One cellular pathway associated with both stress and innate signaling is the Ca2+ signaling pathway (23, 25). Ca2+ influx to the cytoplasm acts as a second messenger and can originate from outside the cell or from endoplasmic reticulum (ER)-associated stores (26). Signaling often takes the form of spikes or oscillations in cytoplasmic Ca2+ and utilizes localization and/or oscillation frequency to confer signaling specificity (26, 27). Entry of herpes simplex virus (HSV) or VLPs causes rapid Ca2+ influx from intracellular stores (22, 28, 29), providing evidence of a link between Ca2+ signaling and membrane perturbation. Furthermore, Ca2+-mobilized ER stress is sufficient to activate IRF3 and enhance induction of certain ISGs, while other pathways require Ca2+ signaling for IRF3 activation and full ISG induction (30, 31). These results suggest Ca2+ signaling acts as a danger signal, priming the response to viral infection.
To explore the hypothesis that entry of low levels of enveloped virus particles is detected as a danger signal prior to virus replication and a prototypic PAMP response, we set out to investigate the cellular pathways induced by membrane perturbation and entry of low levels of enveloped virus particles. We found that recognition of low-level enveloped virus particle entry involves sensing both membrane perturbation and incoming viral genomes and that Ca2+ signaling plays a central role.
MATERIALS AND METHODS
Cells and reagents.
Human embryonic lung (HEL) fibroblasts (American Type Culture Collection [ATCC]) were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Murine embryonic fibroblasts (MEFs) were obtained from wild-type, STING−/− Golden-ticket (Jackson) and MyD88−/− MAVS−/− Toll-like receptor 3−/− (TLR3−/−) triple-knockout (TKO) mice and maintained in minimal essential medium with alpha modification (α-MEM) supplemented with 12% FBS. All media were supplemented with 1% l-glutamine. The Ca2+ inhibitors 2-aminoethyl diphenylborinate (2-APB) and BAPTA-AM (Life Technologies) were reconstituted in methanol or dimethyl sulfoxide (DMSO), respectively, and diluted in serum-free medium to a working concentration of 200 μM or 10 μM. The cells were pretreated with 2-APB for 60 min or with BAPTA-AM for 30 min prior to treatment, and the inhibitor was present for the duration of the experiment. The synthetic double-stranded RNA (dsRNA) mimetic poly(I · C) was resuspended in phosphate-buffered saline (PBS) and diluted in serum-free medium to a working concentration of 20 μM. Antibodies against Sendai virus (SeV) (a kind gift from Yoshiyuki Nagai) and human cytomegalovirus (HCMV) IE1 (Rumbaugh-Goodwin Institute) were used for Western immunoblotting.
Viruses and p14 lipoplexes.
Herpes simplex virus 1 (HSV-1) (KOS strain) and vesicular stomatitis virus expressing green fluorescent protein (VSV-GFP) (Indiana strain) were grown, and their titers were determined on Vero cells; HCMV (strain Ad169) was grown, and its titer was determined on HEL fibroblasts; SeV (strain Cantell) was purchased from Charles River Laboratories, and its titer was determined on CV-1 cells with 1 μg/ml TPCK (tosylsulfonyl phenylalanyl chloromethyl ketone)-treated trypsin overlay; and replication-deficient adenovirus (AdV E1/E3) was grown, and its titer was determined on HEK 293 cells. HSV-1 was inactivated with 575 mJ/cm2 UV and used at a multiplicity of infection (MOI) of 10 PFU/ml, while HCMV and SeV were inactivated with 800 mJ/cm2 and used at an MOI of 0.02 PFU/cell and 0.14 PFU/cell, respectively, unless otherwise stated. UV inactivation was performed using a CL-1000 UV cross-linker (UVP). All infections were performed for 1 h in minimal medium at 37°C, except VSV-GFP infections, which were done in 40 min. p14 lipoplexes were created by diluting 4 μg of purified p14 (a kind gift from Roy Duncan) and 3 μl of Lipofectamine 2000 in PBS and nuclease-free water, respectively, and incubating them separately for 5 min before mixing.
VSV-GFP plaque reduction assay.
Cells were conditioned with virus particles or p14 lipoplexes and later challenged with VSV-GFP, and F11 overlay medium containing 1% FBS and 1% methyl-cellulose was added to restrict plaques. Green fluorescence from VSV-GFP was measured using a Typhoon laser scanner (GE Healthcare) and quantified using ImageQuant software. The fluorescence was then expressed as a percentage of that of unconditioned cells challenged with VSV-GFP.
Particle counting.
Particles of SeV or HCMV were counted by tunable resistive pulse sensing (TRPS), based on the Coulter principle, using a qViro-X particle counter (Izon). Virus stocks were diluted in filtered pH 7.4 formulation buffer containing 10 mM HEPES, 150 mM NaCl and 4% sucrose and sonicated briefly before measurement. Diluted samples were run through an ∼400-nm-diameter pore at constant stretch, pressure, and voltage. The amplitude and frequency of disruptions in the current signal trace corresponded to the size and concentration of particles, while beads of known size and concentration were run to calibrate the measurements and give quantitative data. All measurement and analysis were done using Izon protocols and software.
Calcium microscopy.
HEL cells were grown on glass bottom petri plates for fluorimetry, and changes in the Ca2+ concentration were monitored using Oregon Green (Invitrogen, USA), a Ca2+ indicator dye. A stock solution of Oregon Green was prepared in DMSO and 20% pluronic acid. Cells were incubated with Oregon Green (5 μM) and sulfobromophthalein (100 μM) for 30 min at 37°C and then treated with different particles for another 30 min. The cells were infected with UV-inactivated HSV (HSV-UV), HCMV (HCMV-UV), or SeV (SeV-UV) at 10, 0.04, or 0.14 PFU/cell, respectively, to visualize Ca2+ signaling under conditions where IFN is not induced. The cells were then placed in a Plexiglas recording chamber and perfused with Hanks balanced salt solution (HBSS) for a period of 15 to 30 min prior to experimentation to allow complete dye hydrolysis. Confocal microscopy was then performed at room temperature (21 to 23°C) using a custom-built apparatus based on an inverted Nikon Eclipse TE2000-4 microscope (32); the recording rate was generally 1 frame/s. Picture frames were stored in TIF stacks of several hundred frames on a local hard drive using image acquisition software (Video Savant 4.0; IO Industries, London, ON, Canada). The image files were then analyzed using ImageJ software.
Quantitative reverse transcription (RT)-PCR.
RNA was extracted using TRIzol reagent (Invitrogen) and treated with DNase (Ambion) according to the manufacturer's instructions. Five hundred nanograms of RNA was reverse transcribed using SuperScript II reverse transcriptase (Invitrogen) and a random-hexamer primer according to the manufacturer's instructions. The abundances of cDNAs were measured using specific TaqMan probes and reagents on a StepOnePlus Q-PCR instrument (Applied Biosystems) according to the manufacturer's instructions. Threshold cycle (CT) values were calculated, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous control to calculate individual ΔΔCT values. The ΔΔCT values of samples were compared with those of mock-treated samples to calculate the fold change. Specific probes for human GAPDH (Hs02758991_g1), ISG56 (Hs03027069_s1), STING (Hs00736958_m1), and cGAS (Hs00403553_m1) or murine GAPDH (Mm99999915_g1) and ISG56 (Mm00515153_m1) from ThermoFisher were used.
siRNA knockdown.
Cells at 50% confluence were transfected with pooled Stealth small interfering RNA (siRNA) sequences directed against STING or cGAS (HSS139156-58 and HSS132955-57; purchased from Life Technologies). Specific or nontargeting control siRNAs were diluted in Opti-MEM medium (Life Technologies), combined with RNAiMax Lipofectamine (Life Technologies), and added to the cells according to the manufacturer's instructions. The media were changed to reduced-serum DMEM 6 h after the addition of siRNA, and experiments were carried out at 72 h posttransfection, corresponding to optimal knockdown.
Immunofluorescence.
Fibroblasts were seeded on acid-washed glass coverslips to reach approximately 50% confluence. Five hours postinfection, the cells were formalin fixed, permeabilized in 0.1% Triton-X PBS, and blocked in 3% FBS, 3% goat serum in PBS. Anti-IRF3 (Santa Cruz; FL-425), anti-STING (Abcam; EPR13130), and anti-rabbit AlexaFluor 488 (Life Technologies) were diluted in blocking buffer prior to use, and Hoechst dye was diluted in PBS. A Leica DM IRE2 microscope was used, and IRF3-positive nuclei were counted using OpenLab software and calculated as a percentage of the total nuclei.
RESULTS
Low numbers of enveloped virus particles are sufficient for IFN-independent ISG induction.
We previously proposed that sensing membrane perturbation plays an important role in the first line of defense against enveloped viruses (16, 21). Membrane perturbation and low-level infection with enveloped virus particles induce similar IFN-independent responses (11, 15, 21). However, it remains unclear what signals are required to overcome the activation threshold of IRF3. To define low-level infection, we previously described the number of PFU sufficient to induce an IFN-independent antiviral response (15). Titers based on replication competency, however, rarely correspond to the number of virus particles, and our studies indicate that replication is not required for IRF3 activation. To understand how many physical virus particles cells are exposed to, we used TRPS to measure particle numbers, with SeV and HCMV used as representative RNA and DNA viruses, respectively. We measured the size distribution (Fig. 1A), as well as the particle concentration (Fig. 1B), of our stocks. We determined that as few as 13 particles from the SeV preparation or 84 particles from the HCMV preparation per cell are sufficient for full antiviral protection in HEL cells. We further measured the number of particles in extracts from uninfected cells, purified similarly to HCMV, and found particles (<1 log unit in abundance) of similar size to HCMV that on their own do not contribute to antiviral protection (data not shown). Therefore, while we cannot determine the exact number of virus particles required to elicit a response, it is likely lower than we have estimated. Unlike virus particles, p14 lipoplexes adopt ill-defined sizes and shapes and thus cannot be counted using this technique.
FIG 1.

Low particle multiplicity is sufficient to induce an antiviral response. Virus stocks of SeV or HCMV were diluted, and their properties were measured using tunable resistive pulse sensing. The size distribution (A) and particle concentration, with standard error (B), of virus stocks were calculated by comparing the amplitude and frequency of pore obstructions to calibration beads of known size and concentration. PFU were converted to total virus particles when determining how many particles are sufficient to induce an IFN-independent response.
Ca2+ oscillations are associated with lipid-based particle stimulation.
To investigate whether Ca2+ signaling is an inherent feature of envelope fusion with the cell, we measured cytosolic Ca2+ following addition of virus particles. The minimum numbers of UV-inactivated SeV, HCMV, or HSV-1 particles necessary for an antiviral response were added, while 500 particles/cell of nonreplicating AdV were added as a control. We found that all UV-inactivated enveloped virus particles, as well as p14 lipoplexes, induced Ca2+ oscillations (Fig. 2). Although the amplitude and frequency of these oscillations varied between experiments and between cells, we reproducibly detected similar oscillation patterns with all of the lipid-based particles relative to mock-treated cells. In the discussion of our results below, we use the term Ca2+ signaling to refer to the oscillation patterns observed. Nonreplicating adenovirus with E1/E3 deleted did not induce calcium oscillations. The variety of lipid-based particles that induce Ca2+ oscillations suggests that Ca2+ signaling is associated with entry of enveloped particles.
FIG 2.

Ca2+ oscillations are associated with lipid-based particles. HEL fibroblasts were loaded with Ca2+-sensitive dye and mock infected; infected with UV-inactivated HSV (≤178 particles/cell), SeV (≤13 particles/cell), or HCMV (≤84 particles/cell) or live AdV (≤500 particles/cell); or treated with p14 lipoplexes. The fluorescence (F510) of representative cells, beginning 45 min after the addition of treatment, is plotted.
Ca2+ oscillations are required for the response to membrane perturbation and enveloped virus particle entry.
To determine the role of Ca2+ signaling in the antiviral response to membrane perturbation, we used the inhibitor 2-APB, which broadly disrupts Ca2+ signaling (33, 34). Treatment with 2-APB was sufficient to completely abolish Ca2+ signaling following HSV-UV infection (Fig. 3, top). ***, P < 0.001.
FIG 3.

Ca2+ signaling is required for the antiviral response to membrane perturbation. (Top) HEL fibroblasts were infected with UV-inactivated HSV (≤178 particles/cell) in the presence of the inhibitor 2-APB or MeOH alone, and the fluorescence (F510) of representative cells, beginning 45 min after infection, was plotted. (Bottom) HEL fibroblasts were treated with p14 lipoplexes or Lipofectamine 2000 alone (LF) in the presence of 2-APB or MeOH alone and challenged with VSV-GFP in a plaque reduction assay 12 h later. GFP fluorescence was quantified from plate scans and plotted as a percentage of that of unconditioned VSV-GFP-infected cells. The average and standard error of 3 biological replicates were determined, and significance was calculated by an unpaired t test. ***, P < 0.001.
Membrane perturbation by p14 lipoplexes induces ISGs and antiviral protection in the absence of nucleic acid (21), making it the simplest lipid-based particle for examining pathways leading to activation of the key node protein IRF3. We treated HEL fibroblasts with p14 lipoplexes in the presence or absence of 2-APB, and measured antiviral protection. Disruption of Ca2+ signaling completely prevented the antiviral response to p14 lipoplexes, suggesting that Ca2+ is necessary for the antiviral response to membrane perturbation mediated by this stimulus (Fig. 3, bottom).
Next, we examined the roles of membrane perturbation and Ca2+ signaling in the response to enveloped virus particles. Disrupting Ca2+ with 2-APB during SeV-UV or HCMV-UV infection significantly reduced antiviral protection (Fig. 4A). 2-APB also limited ISG induction under these conditions (Fig. 4B). In addition, we observed that specifically chelating intracellular Ca2+ with BAPTA-AM significantly reduced the antiviral response to SeV-UV and HCMV-UV (Fig. 4C). Similar experiments chelating extracellular Ca2+ with EGTA showed no effect (data not shown). Additionally, inhibition of Ca2+ signaling had no effect on the antiviral protection of poly(I · C), which robustly produces IFN under the experimental conditions used. Finally, we probed protein extracts with virus-specific antibodies and saw that Ca2+ chelation had little to no effect on entry and early gene expression during live-virus infection (Fig. 4D).
FIG 4.

Ca2+ signaling is involved in the antiviral response to enveloped virus particles. (A and B) HEL cells were infected with SeV-UV (≤13 particles/cell) or HCMV-UV (≤84 particles/cell) in the presence of 2-APB or MeOH alone and challenged with VSV-GFP in a plaque reduction assay 12 h later (A) or ISG56 induction was measured by quantitative RT-PCR, as described in Materials and Methods, after 6 h (B). (C) Cells similarly infected in the presence of BAPTA-AM or DMSO alone were challenged with VSV-GFP in a plaque reduction assay 7 h after initial infection. GFP fluorescence is plotted as a percentage of that of mock-treated VSV-GFP-infected cells. (D) Protein extracts from cells infected with live SeV (≤13 particles/cell) or HCMV (≤84 particles/cell) in the presence of BAPTA-AM or DMSO were probed with virus-specific antibodies to measure viral entry and gene expression. All the graphs report the average and standard error of 3 biological replicates. Significance was calculated by two-way analysis of variance (ANOVA) and Bonferroni posttests. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Sensing of packaged genomic nucleic acid during low-level enveloped virus particle infection is required for the antiviral response.
While disruption of Ca2+ signaling completely blocked the antiviral activity of p14 lipoplexes, only a partial, but statistically significant, block was observed in response to enveloped virus particles, suggesting that additional pathways are involved in particle recognition. Although cytoplasmic PRRs recognize viral nucleic acids that accumulate during viral replication, it is not known how sensitive these PRRs are to incoming packaged genomes. To determine whether nucleic acid sensing plays a role in response to low-level enveloped particle treatment, we asked whether known PRR signaling pathways contribute to IRF3 activation and subsequent ISG induction. We used primary MEFs lacking essential components of either RNA- or DNA-sensing pathways. TKO MEFs deficient in TLR3, MyD88, and MAVS were used to investigate the role of RNA sensing, while MEFs derived from Golden-ticket mice encoding a null mutation in STING were used to investigate the role of DNA sensing (35). Unlike wild-type MEFs, treatment of TKO MEFs with SeV-UV did not induce antiviral protection (Fig. 5A) or upregulation of ISG56 (Fig. 5B). Similarly, STING KO MEFs had an impaired response to HCMV-UV (Fig. 5A and C). As expected, the absence of STING did not impair the response to SeV-UV, and TKO MEFs responded fully to HCMV-UV (Fig. 5A).
FIG 5.
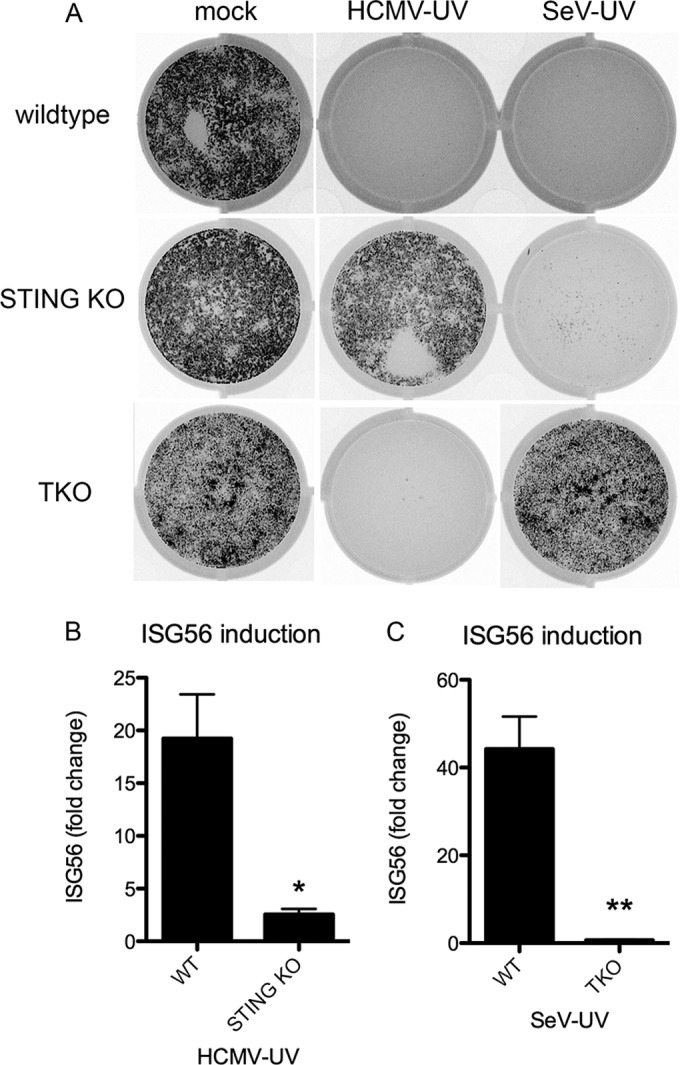
Nucleic acid-sensing pathways are necessary for the antiviral response to low-level incoming virus particles. (A to C) Wild-type, STING KO, or TKO MEFs lacking MAVS, TLR3, and MyD88 were mock infected or infected with SeV-UV (≤7 particles/cell) or HCMV-UV (≤84 particles/cell). The MEFs were then challenged with VSV-GFP in a plaque reduction assay, and a representative plate scan was included (A), or ISG56 induction was measured by quantitative RT-PCR after 12 h and plotted relative to mock-infected cells (B and C). The fold change from quantitative RT-PCR was calculated as described in Materials and Methods and graphed with the average and standard deviation of 3 biological replicates. Significance was calculated by unpaired t test. *, P < 0.05, **, P < 0.01.
While STING has been characterized as a critical adaptor in DNA sensing (36), additional roles for STING have been suggested, including recognition of envelope-membrane fusion (22, 30, 37). Although the ability of SeV-UV particles to induce full antiviral protection in STING KO MEFs suggests that STING participates in sensing DNA genomes and not envelope-membrane fusion per se, we asked whether the associated cytoplasmic DNA sensor cGAS is required for the antiviral response to HCMV-UV. We first depleted STING or cGAS in HEL fibroblasts using pooled siRNA sequences and then infected the cells with HCMV-UV. STING and cGAS mRNAs were reduced by more than 90% for up to 72 h posttransfection prior to experiments (Fig. 6A). Both STING and cGAS knockdown similarly reduced antiviral protection and ISG56 induction in response to incoming HCMV-UV particles (Fig. 6B and C) but had no effect on the response to SeV-UV particles (data not shown). Taken together, these data suggest that sensing of incoming packaged genomes is an essential component of the host response to enveloped virus particle entry, even with stimulation with as few as 7 (SeV) or 84 (HCMV) nonreplicating virus particles per cell.
FIG 6.
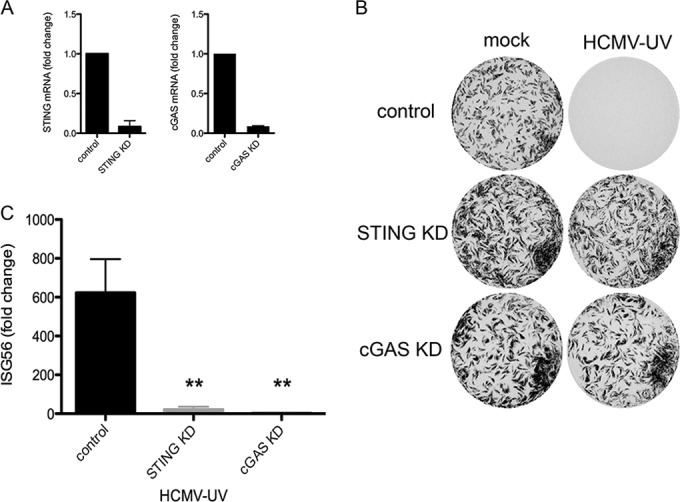
The cytoplasmic DNA sensor cGAS is necessary for the antiviral response to incoming HCMV particles. (A) STING and cGAS were knocked down (KD) in HEL fibroblasts, and transcript levels of STING and cGAS were measured by quantitative RT-PCR after 72 h and plotted relative to control siRNA transfection. (B and C) After knockdown, the cells were mock infected or infected with HCMV-UV (≤84 particles/cell) and then challenged with VSV-GFP in a plaque reduction assay, and a representative plate scan was included (B), or ISG56 induction was measured by quantitative RT-PCR and plotted relative to that of mock-infected cells (C). The fold change from quantitative RT-PCR was calculated as described in Materials and Methods and graphed with the average and standard error of 3 biological replicates. Significance was calculated by two-way ANOVA and Bonferroni posttests. **, P < 0.01.
Ca2+ signaling is upstream of STING and IRF3 in HCMV particle recognition.
The host response to enveloped virus particles appears to require multiple signals, including both Ca2+ signaling and viral genome recognition, prompting the question of where these signals converge upstream of antiviral gene induction. In this experiment, we used the lowest number of HCMV-UV particles that elicited clear activation of both STING and IRF3. Upon activation, STING translocates from the ER to cytoplasmic vesicles (38, 39), where it acts as a scaffold for the activation of IRF3, which subsequently translocates to the nucleus. Both translocation events can be visualized using immunofluorescence microscopy. Preventing Ca2+ signaling with 2-APB reduced the relocation of STING to cytoplasmic foci (Fig. 7A), as well as nuclear translocation of IRF3 following infection with HCMV-UV (Fig. 7B), suggesting Ca2+ signaling lies upstream of STING activation in the response to HCMV-UV.
FIG 7.

Ca2+ signaling is important for activation of STING and IRF3 following entry of HCMV particles. (A and B) HEL fibroblasts were infected with HCMV-UV (≤840 particles/cell) in the presence of 2-APB or MeOH alone, and STING relocalization was visualized by immunofluorescence 4 h postinfection (A) or IRF3 translocation was visualized 5 h postinfection (≤168 particles/cell) (B). (C) IRF3 translocation was graphed as the percentage of IRF3-positive nuclei with standard errors from 4 biological replicates. Significance was calculated by unpaired t test. ***, P < 0.001.
DISCUSSION
We previously reported that enveloped virus particle entry or membrane perturbation by p14 lipoplexes induces an ISG subset through an IFN-independent, IRF3-dependent pathway (16, 21). However, since the known pathways upstream of IRF3 involve nucleic acid sensing or TLR signaling through TRIF, neither of which is necessary for the IFN-independent induction of ISGs (15, 20, 21), the mechanisms by which low-level enveloped virus particle exposure activates IRF3 remained elusive. Of interest, IRF3 is also activated in response to different forms of cellular stress, such as redox and ER stresses and cytoskeleton disruption (30, 40–42). Consistent with IRF3 being increasingly recognized as a key mediator of diverse host stress responses, it is likely that the activation profile of IRF3 is equally diverse. While activation of IRF3 following virus replication and accumulation of viral PAMPs can lead to common modifications, such as Ser385 and Ser396 hyperphosphorylation, the host response to more subtle stimuli, such as enveloped virus particle entry, often lacks canonical, or indeed any, IRF3 activation markers (17). Accordingly, we proposed that membrane perturbation serves as a stress or danger signal prior to, or in the absence of, a prototypic pathogen-sensing response (23, 24).
An intriguing pathway to investigate was the Ca2+ signaling pathway, as HSV-1 infection or VLP treatment induces rapid Ca2+ fluxes from intracellular stores (22, 28, 29), and Ca2+ signaling is associated with stress responses, homeostatic regulation, and innate signaling (23, 25). The Ca2+ activity we observed in primary fibroblasts appears to be a common feature following treatment with diverse lipid-based particles, but not nonenveloped viruses, despite the assumption that nonenveloped virus particles would need to perturb a cellular membrane in some capacity to access the interior of a susceptible cell. Enveloped viruses enter either at the cell membrane or from within endosomes, and thus, any signal triggered during entry could originate from either membrane.
Our investigation has not uncovered how membrane perturbation is sensed and signals the release of Ca2+ from intracellular stores. The phosphatidylinositol 3-kinase (PI3K) pathway commonly lies upstream of Ca2+ store-mediated signaling, and we and others have shown that the PI3K inhibitor LY294002 (LY) blocks the antiviral response to enveloped virus particles (22, 43). However, the exact means by which LY blocks antiviral gene induction is unknown. LY does not inhibit IRF3 nuclear translocation following entry of enveloped virus particles but instead appears to modulate a pathway downstream of IRF3 activation (43). Moreover, the prototypic p85/p110 PI3K complex is not involved in the IFN-independent antiviral response (43). Thus, the relationship between Ca2+-mediated and PI3K-mediated signaling during this response is unclear and requires further investigation.
Although a common Ca2+-dependent pathway exists for enveloped particle recognition, we found that recognition of incoming genomes by cytoplasmic DNA- and RNA-sensing pathways significantly contributed to the IFN-independent response. While previous findings suggested that neither virus replication nor TLR signaling is required (15, 16, 20), the contributions of incoming viral genomes or cytoplasmic nucleic acid-sensing pathways were not fully evaluated. While diverse enveloped virus particles induce the IFN-independent antiviral response (16), we used SeV and HCMV particles as representative RNA and DNA virus particles, respectively. SeV belongs to the paramyxovirus family of nonsegmented negative-strand RNA viruses and releases its genome into the cytoplasm following entry and uncoating. RIG-I recognizes genomes of SeV and other negative-strand RNA viruses when transfected into the cell and activates the signaling adaptor MAVS to induce IFN (44, 45). However, the ability of RIG-I and MAVS to recognize incoming virus particles has not been described, especially in the range of 7 to 13 particles/cell used here. HCMV belongs to the herpesvirus family of dsDNA viruses, and following entry, capsids translocate to the nucleus and inject their genomes through the nuclear pore (46). HCMV DNA can be recognized by DAI (ZBP2) in fibroblasts (47) and, additionally, IFI16 in macrophages (48), but to our knowledge, no role for the cytoplasmic sensor cGAS has been shown. Conceptually, while incoming HCMV genomes should not be exposed within the cytoplasm, virus entry is an imperfect process, and HCMV capsid degradation by the proteasome, and thus viral DNA release, has been demonstrated in macrophages (48).
Unlike herpesviruses, AdV capsids uncoat in the cytoplasm at the nuclear pore prior to import, which leaves AdV genomes exposed at this bottleneck (49). The cGAS/STING/TBK-1 pathway is capable of sensing cytoplasmic AdV DNA, leading to IRF3 activation (50, 51). In our studies, however, infection with 1,000 PFU/cell of AdV particles failed to elicit a response (data not shown), while infection with 0.02 PFU/cell of HCMV particles efficiently induced a response. This represents greater than 25,000 particles/cell of AdV versus fewer than 100 particles/cell of HCMV. Thus, the simplest interpretation is that in human fibroblasts, DNA sensing of incoming viral genomes alone is insufficient to activate IRF3 and requires additional signals, such as Ca2+ oscillations. However, the nature, timing, or amplitude of the Ca2+ signal is likely important (26, 27); indeed, Ca2+ mobilization induced by ionomycin is insufficient to activate IRF3 (31). Consistent with these findings, preliminary experiments combining ionomycin treatment with AdV infection failed to elicit IRF3 activation and ISG induction (data not shown).
Here, we show that Ca2+ signaling plays a role upstream of both STING and IRF3 activation following HCMV-UV infection. Rather than converging on the activation of IRF3, as we had hypothesized, Ca2+ signaling associated with membrane perturbation appears to be necessary for activation of STING. While no role for Ca2+ in STING activation had been observed previously, the crystal structures of STING show an important Ca2+-binding pocket at the interface of 2 STING dimers (52, 53). While these results suggest that Ca2+ signaling is similarly upstream of MAVS activation following stimulation with enveloped RNA virus particles, we were unable to reproducibly detect MAVS activation in primary fibroblasts to test this hypothesis.
p14 lipoplexes do not contain nucleic acid, and the sensor upstream of Ca2+ is currently unknown. While one explanation is that the p14 protein is sensed by an unknown receptor, it is unlikely that p14 serves as a PAMP due to its size and structural dissimilarity with enveloped virus fusion proteins (54). It is intriguing that while we routinely detect ISG and antiviral state induction in response to envelope virus particle entry within 6 to 8 h in fibroblasts, the response to p14-lipoprotein complexes requires ∼12 h, despite induction of the same subset of ISGs (11, 21). Studies are currently ongoing to elucidate the mechanism(s) by which p14-lipoprotein complexes and Ca2+ signaling lead to IRF3 activation.
Although we found that viral nucleic acid sensing was required for the antiviral response to incoming virus particles, we did not observe typical markers of IRF3 activation, such as phosphorylation of S386 or S396. While this could be explained by simple limitations of detection when using so few particles, there are clear differences in IRF3 activation following infection with different live or inactivated virus preparations, despite equal induction of ISGs (17), suggesting that a linear relationship between canonical activation of IRF3 and ISG induction does not exist. Accumulating data instead suggest that noncanonical and undefined modifications additionally play roles in the activation of IRF3.
Together with previous reports, our study underscores the complexity of the host intrinsic/innate response and how different cell types and species uniquely respond to incoming stimuli. For example, VLPs and fusogenic liposomes efficiently induce type I IFN and ISGs in human immune cells (22). Similarly, AdV activates the cGAS/STING pathway and elicits IRF3 activation in murine macrophages (50, 51), while AdV infection of primary mouse lung fibroblasts leads to type I IFN induction (55). Our studies also highlight the exquisite sensitivity of cells to incoming low-level virus particle entry.
Little is known about how membrane perturbation leads to IRF3 activation and ISG induction, but our data suggest Ca2+ plays an important role in this process. Both Ca2+ signaling and recognition of packaged viral genomes contribute to IRF3 activation following low-level enveloped virus particle entry. However, we know very little about the interaction between pathways or how membrane perturbation alone leads to IRF3 activation. There is emerging cross talk between cell stress responses and intrinsic/innate immunity (23, 24), and further work examining the roles of Ca2+ and related pathways in innate signaling will help uncover these pathways.
REFERENCES
- 1.Fensterl V, Sen GC. 2009. Interferons and viral infections. Biofactors 35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 2.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 3.Sancho-Shimizu V, Perez de Diego R, Jouanguy E, Zhang SY, Casanova JL. 2011. Inborn errors of anti-viral interferon immunity in humans. Curr Opin Virol 1:487–496. doi: 10.1016/j.coviro.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fensterl V, Sen GC. 2015. Interferon-induced Ifit proteins: their role in viral pathogenesis. J Virol 89:2462–2468. doi: 10.1128/JVI.02744-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Crouse J, Kalinke U, Oxenius A. 2015. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol 15:231–242. doi: 10.1038/nri3806. [DOI] [PubMed] [Google Scholar]
- 8.Swiecki M, Colonna M. 2011. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr Opin Virol 1:463–475. doi: 10.1016/j.coviro.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thanos D, Maniatis T. 1995. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 10.Preston CM, Harman AN, Nicholl MJ. 2001. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J Virol 75:8909–8916. doi: 10.1128/JVI.75.19.8909-8916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J Virol 75:750–758. doi: 10.1128/JVI.75.2.750-758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeWitte-Orr SJ, Mehta DR, Collins SE, Suthar MS, Gale M Jr, Mossman KL. 2009. Long double-stranded RNA induces an antiviral response independent of IFN regulatory factor 3, IFN-beta promoter stimulator 1, and IFN. J Immunol 183:6545–6553. doi: 10.4049/jimmunol.0900867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548. doi: 10.1016/S1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 14.Takaki H, Honda K, Atarashi K, Kobayashi F, Ebihara T, Oshiumi H, Matsumoto M, Shingai M, Seya T. 2014. MAVS-dependent IRF3/7 bypass of interferon beta-induction restricts the response to measles infection in CD150Tg mouse bone marrow-derived dendritic cells. Mol Immunol 57:100–110. doi: 10.1016/j.molimm.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Paladino P, Cummings DT, Noyce RS, Mossman KL. 2006. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol 177:8008–8016. doi: 10.4049/jimmunol.177.11.8008. [DOI] [PubMed] [Google Scholar]
- 16.Collins SE, Noyce RS, Mossman KL. 2004. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 78:1706–1717. doi: 10.1128/JVI.78.4.1706-1717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noyce RS, Collins SE, Mossman KL. 2009. Differential modification of interferon regulatory factor 3 following virus particle entry. J Virol 83:4013–4022. doi: 10.1128/JVI.02069-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int Rev Immunol 30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 19.Hiscott J. 2007. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem 282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 20.Tsitoura E, Thomas J, Cuchet D, Thoinet K, Mavromara P, Epstein AL. 2009. Infection with herpes simplex type 1-based amplicon vectors results in an IRF3/7-dependent, TLR-independent activation of the innate antiviral response in primary human fibroblasts. J Gen Virol 90:2209–2220. doi: 10.1099/vir.0.012203-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noyce RS, Taylor K, Ciechonska M, Collins SE, Duncan R, Mossman KL. 2011. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J Virol 85:10926–10931. doi: 10.1128/JVI.00862-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, Rixon FJ, Herold BC, Fitzgerald KA, Paludan SR. 2012. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol 13:737–743. doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins SE, Mossman KL. 2014. Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev 25:525–531. doi: 10.1016/j.cytogfr.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Hare D, Mossman KL. 2013. Novel paradigms of innate immune sensing of viral infections. Cytokine 63:219–224. doi: 10.1016/j.cyto.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Smith JA. 2014. A new paradigm: innate immune sensing of viruses via the unfolded protein response. Front Microbiol 5:222. doi: 10.3389/fmicb.2014.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berridge MJ, Bootman MD, Roderick HL. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 27.Dupont G, Combettes L, Bird GS, Putney JW. 2011. Calcium oscillations. Cold Spring Harb Perspect Biol 3:a004226. doi: 10.1101/cshperspect.a004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheshenko N, Liu W, Satlin LM, Herold BC. 2007. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol Biol Cell 18:3119–3130. doi: 10.1091/mbc.E07-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin LM, Herold BC. 2003. Herpes simplex virus triggers activation of calcium-signaling pathways. J Cell Biol 163:283–293. doi: 10.1083/jcb.200301084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu YP, Zeng L, Tian A, Bomkamp A, Rivera D, Gutman D, Barber GN, Olson JK, Smith JA. 2012. Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J Immunol 189:4630–4639. doi: 10.4049/jimmunol.1102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Yao M, Li N, Wang C, Zheng Y, Cao X. 2008. CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 112:4961–4970. doi: 10.1182/blood-2008-03-144022. [DOI] [PubMed] [Google Scholar]
- 32.Mukherjee S, Kolb MR, Duan F, Janssen LJ. 2012. Transforming growth factor-beta evokes Ca2+ waves and enhances gene expression in human pulmonary fibroblasts. Am J Respir Cell Mol Biol 46:757–764. doi: 10.1165/rcmb.2011-0223OC. [DOI] [PubMed] [Google Scholar]
- 33.Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2002. 2-Aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J 16:1145–1150. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- 34.Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT, Roderick HL. 2003. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 34:97–108. doi: 10.1016/S0143-4160(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 35.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE. 2011. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maringer K, Fernandez-Sesma A. 2014. Message in a bottle: lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev 25:669–679. doi: 10.1016/j.cytogfr.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. 2015. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukherjee A, Morosky SA, Shen L, Weber CR, Turner JR, Kim KS, Wang T, Coyne CB. 2009. Retinoic acid-induced gene-1 (RIG-I) associates with the actin cytoskeleton via caspase activation and recruitment domain-dependent interactions. J Biol Chem 284:6486–6494. doi: 10.1074/jbc.M807547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. 2009. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A 106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Servant MJ, Grandvaux N, Hiscott J. 2002. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol 64:985–992. doi: 10.1016/S0006-2952(02)01165-6. [DOI] [PubMed] [Google Scholar]
- 43.Noyce RS, Collins SE, Mossman KL. 2006. Identification of a novel pathway essential for the immediate-early, interferon-independent antiviral response to enveloped virions. J Virol 80:226–235. doi: 10.1128/JVI.80.1.226-235.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 45.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 46.Kobiler O, Drayman N, Butin-Israeli V, Oppenheim A. 2012. Virus strategies for passing the nuclear envelope barrier. Nucleus 3:526–539. doi: 10.4161/nucl.21979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeFilippis VR, Alvarado D, Sali T, Rothenburg S, Fruh K. 2010. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J Virol 84:585–598. doi: 10.1128/JVI.01748-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones E, Landolfo S, Damania B, Melchjorsen J, Bowie AG, Fitzgerald KA, Paludan SR. 2013. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol 190:2311–2319. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang IH, Suomalainen M, Andriasyan V, Kilcher S, Mercer J, Neef A, Luedtke NW, Greber UF. 2013. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 14:468–480. doi: 10.1016/j.chom.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 50.Lam E, Stein S, Falck-Pedersen E. 2014. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J Virol 88:974–981. doi: 10.1128/JVI.02702-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stein SC, Falck-Pedersen E. 2012. Sensing adenovirus infection: activation of interferon regulatory factor 3 in RAW 264.7 cells. J Virol 86:4527–4537. doi: 10.1128/JVI.07071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shu C, Yi G, Watts T, Kao CC, Li P. 2012. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol 19:722–724. doi: 10.1038/nsmb.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boutilier J, Duncan R. 2011. The reovirus fusion-associated small transmembrane (FAST) proteins: virus-encoded cellular fusogens. Curr Top Membr 68:107–140. doi: 10.1016/B978-0-12-385891-7.00005-2. [DOI] [PubMed] [Google Scholar]
- 55.Nociari M, Ocheretina O, Schoggins JW, Falck-Pedersen E. 2007. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J Virol 81:4145–4157. doi: 10.1128/JVI.02685-06. [DOI] [PMC free article] [PubMed] [Google Scholar]