

ABSTRACT
Human cytomegalovirus (HCMV) resides latently in hematopoietic progenitor cells (HPCs). During latency, only a subset of HCMV genes is transcribed, including one of the four virus-encoded G protein-coupled receptors (GPCRs), US28. Although US28 is a multifunctional lytic protein, its function during latency has remained undefined. We generated a panel of US28 recombinant viruses in the bacterial artificial chromosome (BAC)-derived clinical HCMV strain TB40/E-mCherry. We deleted the entire US28 open reading frame (ORF), deleted all four of the viral GPCR ORFs, or deleted three of the HCMV GPCRs but not the US28 wild-type protein. Using these recombinant viruses, we assessed the requirement for US28 during latency in the Kasumi-3 in vitro latency model system and in primary ex vivo-cultured CD34+ HPCs. Our data suggest that US28 is required for latency as infection with viruses lacking the US28 ORF alone or in combination with the remaining HCMV-encoded GPCR results in transcription from the major immediate early promoter, the production of extracellular virions, and the production of infectious virus capable of infecting naive fibroblasts. The other HCMV GPCRs are not required for this phenotype as a virus expressing only US28 but not the remaining virus-encoded GPCRs is phenotypically similar to that of wild-type latent infection. Finally, we found that US28 copurifies with mature virions and is expressed in HPCs upon virus entry although its expression at the time of infection does not complement the US28 deletion latency phenotype. This work suggests that US28 protein functions to promote a latent state within hematopoietic progenitor cells.
IMPORTANCE Human cytomegalovirus (HCMV) is a widespread pathogen that, once acquired, remains with its host for life. HCMV remains latent, or quiescent, in cells of the hematopoietic compartment and upon immune challenge can reactivate to cause disease. HCMV-encoded US28 is one of several genes expressed during latency although its biological function during this phase of infection has remained undefined. Here, we show that US28 aids in promoting experimental latency in tissue culture.
INTRODUCTION
The betaherpesvirus human cytomegalovirus (HCMV) is a ubiquitous pathogen that, once acquired, remains with its host for life. HCMV establishes a latent infection in CD34+ hematopoietic progenitor cells (HPCs), and individuals with a competent immune system are, for the most part, asymptomatic for disease. Under weakened immune conditions, however, the virus can reactivate, causing severe morbidity and often mortality. In developed countries, such as the United States, approximately 80% of the population is HCMV positive by 40 years of age (reviewed in reference 1). In adults, HCMV-associated disease is due mostly to reactivation of latent infection, whereas primary infections rarely cause significant health burdens in this population (reviewed in reference 1). Current treatments administered in clinical settings to sequester HCMV infection include those that predominantly target late stages of viral lytic replication although by this point of infection, HCMV-associated disease has often already manifested. Furthermore, CMV drug-resistant strains have emerged, thus underscoring the need for additional therapies (2). Therefore, understanding the factors that influence viral latency and reactivation may lead to the identification of novel therapeutics aimed at targeting a stage of viral infection prior to the manifestation of disease.
While alpha- and gammaherpesvirus latency model systems are well developed, having existed for decades, HCMV latency studies are in their infancy, and our understanding of the control of HCMV latency and reactivation is incomplete. This is due, at least in part, to the lack of an animal model system with which to interrogate this human virus and, until recently (i.e., within the last decade), the absence of suitable ex vivo or in vitro HCMV model systems. HCMV latency culture systems that utilize ex vivo primary hematopoietic cells (3–14) and in vitro model systems (15–27) are gaining momentum and being used more widely, and thus we are learning more about these stages of infection. Repressive chromatin marks are critical in HCMV genomic silencing during latency, and both histone deacetylases and methyltransferases function to aid in this repression (reviewed in reference 28). The major immediate early promoter (MIEP) contains multiple transcription factor binding sites, and these are also modulated by chromatinization and associated with repressive marks during latency (reviewed in reference 28). Although chromatinization plays a critical role in latency and reactivation, it is clear that other viral and cellular factors are involved. For example, viral proteins including UL138 (29, 30), pp71 (13), LUNA (31), UL144 (32), and viral interleukin-10 (latency-associated HCMV homolog of IL-10 [LAcmvIL-10]) (33–36) contribute to successful latency and reactivation in culture. HCMV has co-opted cellular factors as well, such as cellular microRNAs (miRNAs) (36–38), transcription factors (32, 38), and cell signaling (38, 39). It is clear that HCMV latency and reactivation are multifaceted processes and thus likely that our full understanding of these stages of infection remains incomplete.
HCMV is a large virus, containing over 200 open reading frames (ORFs) (40–43). However, during latency only a small subset of genes is expressed (5, 44). US28 is one of four HCMV-encoded G-protein coupled receptor (GPCR) homologs and is expressed during both the latent (5, 32, 44, 45) and lytic (46, 47) cycles. Although many studies have focused on understanding US28's functions during lytic replication (reviewed in reference 48), there is little known about the role US28 plays during latency although it is one of only a few genes associated with latent transcription. US28 transcripts have been detected both during natural latency (32, 45) and during ex vivo latent infection studies (4–6, 44, 49). To begin to elucidate the role of US28 during latency, we have utilized the Kasumi-3 model for HCMV latency and reactivation (23). The Kasumi-3 cell line is a CD34+ hematopoietic progenitor cell (HPC) line that shares many of the same cell surface markers described for the ex vivo systems utilizing primary CD34+ HPCs isolated from either bone marrow or cord blood (50). We have previously shown that the Kasumi-3 cell line supports all of the hallmarks of HCMV latency, including reactivation resulting in the production of infectious virus (23). Using this in vitro model for HCMV latency and a panel of viral recombinants, we show that US28 aids in promoting successful latent infection. Additionally, we found that this phenotype also occurs during ex vivo infection of primary CD34+ HPCs. Together, our findings reveal that US28 plays a role in successful latent infection of HPCs.
MATERIALS AND METHODS
Cells and viruses.
Kasumi-3 cells (ATCC CRL-2725) were cultured in RPMI 1640 medium (ATCC 30-2001) supplemented with 20% fetal bovine serum (FBS), 100 U/ml each of penicillin and streptomycin, and 100 μg/ml gentamicin at a density of 3 × 105 to 3 × 106 cells/ml. Primary newborn human foreskin fibroblasts (NuFF-1 cells; GlobalStem) were maintained in Dulbecco's modified Eagle medium (DMEM), supplemented with 10% FBS, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 10 mM HEPES, and 100 U/ml each of penicillin and streptomycin. Irradiated stromal cells (1:1 mixture of S1/S1 and MG3 cells) were a kind gift from Felicia D. Goodrum (University of Arizona) and were thawed directly into human CD34+ long-term culture medium (hLTCM) consisting of MyeloCult H5100 (Stem Cell Technologies) supplemented with 1 μM hydrocortisone and 100 U/ml each of penicillin and streptomycin. Primary CD34+ hematopoietic progenitor cells (HPCs) were isolated from deidentified cord blood samples by magnetic separation, as described elsewhere (4, 5, 51). Cells were immediately infected after isolation (see below). All cells were maintained at 37°C with 5% CO2. Isolation and culture conditions for primary CD34+ cells are described in the next section.
HCMV bacterial artificial chromosome (BAC)-derived strain TB40/E (clone 4) (52) was used in this study. We previously engineered this strain to express mCherry (TB40/E-mCherry) (53). TB40/E-mCherry-US28Δ (US28Δ), in which the entire US28 open reading frame (ORF) is deleted, and TB40/E-mCherry-allΔ (allΔ), in which all four HCMV-encoded GPCR ORFs (UL33, UL78, US27, and US28) are deleted, have been described previously (54). Two independently generated clones were constructed in which the ORFs for UL33, UL78, and US27 were excised, while the US28 ORF remained. This virus, TB40/E-mCherry-US28wt (US28wt; where wt indicates the wild-type US28 ORF) was generated using galK recombineering techniques, as described previously (53, 55), using the primer sets shown in Table 1. Each US28wt clone was verified by sequence analysis. All viral stocks were grown on primary fibroblasts (NuFF-1) and harvested by ultracentrifugation through a 20% sorbitol cushion after 100% cytopathic effect (CPE) was observed. Viral pellets were resuspended in X-VIVO15 medium (Lonza) supplemented with 1.5% bovine serum albumin. Aliquots were flash frozen in liquid nitrogen prior to long-term storage at −80°C. Titers of all viral stocks were determined by 50% tissue culture infective dose (TCID50) assays on primary fibroblasts.
TABLE 1.
Primers used for generating TB40/E-mCherry-US28wt (US28wt)b
| Recombinant virus (TB40/E-mCherry) | Primer descriptiona | Primer sequence (5′–3′) |
|---|---|---|
| US27Δ-galK insertion | FOR | GTGTAATGCTTTTTACAGGACCGTTCAACAGGTGATACTACCTGCAAGGTACCTGTTGACAATTAATCATCGGCA |
| REV | CGTGCAATTAGCAAAAATAGATGTGCGGCGGACGCGTGAGAGAGGATCGAATCAGCACTGTCCTGCTCCTT | |
| US27Δ | DS oligonucleotide | TTTACAGGACCGTTCAACAGGTGATACTACCTGCAAGGTATTCGATCCTCTCTCACGCGTCCGCCGCACATCTATTTTTG |
| UL33Δ-galK insertion | FOR | TTCCGCCCAGACCCGCAACAACACTCCTCCGCACATCAATGACACTTGCAACCCTGTTGACAATTAATCATCGGCA |
| REV | GGGGAAATGGCGACGGGTTCTGGTGCTTTCTGAATAAAGTAACAGGAAAGCTCAGCACTGTCCTGCTCCTT | |
| UL33Δ | DS oligonucleotide | CCGCAACAACACTCCTCCGCACATCAATGACACTTGCAACGCTTTCCTGTTACTTTATTCAGAAAGCACCAGAACCCGTC |
| UL78Δ-galK insertion | FOR | GTCCCCGGAGAGGGTATATTCGTTCGGCGAGAGCGGGCGGCGGTGGTGGGTCCTGTTGACAATTAATCATCGGCA |
| REV | TAACGTGATTTATCTGCCACTTTTCTCCCCGCTGCCGTACAGCGCCGCCGCTCAGCACTGTCCTGCTCCTT | |
| UL78Δ | DS oligonucleotide | GGGTATATTCGTTCGGCGAGAGCGGGCGGCGGTGGTGGGTGCGGCGGCGCTGTACGGCAGCGGGGAGAAAAGTGGCAGAT |
FOR, forward; REV, reverse; DS, double-stranded.
Boldface sequences are complementary to the galK template plasmid.
Infection of fibroblasts, Kasumi-3 cells, and CD34+ HPCs.
For viral growth comparative analyses on fibroblasts, 2.5 × 105 NuFF-1 cells were infected at a multiplicity of infection of 0.5 TCID50/cell or 2.0 TCID50s/cell for 1 h at 37°C. After adsorption, the inoculum was removed, cells were washed three times with phosphate-buffered saline (PBS), and then cultures were replenished with fresh medium. Viral supernatants were collected every 24 h over a 120-h time course. Viral titers were determined by TCID50 assay.
Kasumi-3 cells were infected as described previously (23, 37). In brief, cells were starved in X-VIVO15 medium (Lonza) for 48 h prior to infection and then infected with each virus at a multiplicity of infection of 1.0 TCID50/cell by centrifugal enhancement at 1,000 × g for 30 min at room temperature in X-VIVO15 medium (Lonza). The following day, cells were treated with trypsin to remove any virus that had not entered the cell and then cushioned onto Ficoll-Pacque (GE Healthcare) to remove residual virus and debris. Infected cells were washed with PBS and then replated in X-VIVO15 medium (Lonza) and harvested as indicated in the text. To stimulate viral lytic transcription, 20 nM 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma) was added for an additional 2 days, as previously described (23, 37).
Where indicated in the text and in Fig. 8, viral inoculum was inactivated by UV treatment at maximum energy (0.9999 J/cm2) two times using an HL-2000 HybriLinker UV cross-linker (UVP). Equal multiplicities (1.0 PFU/cell) of UV-inactivated virus and untreated virus were subsequently used to coinfect Kasumi-3 cells, as described above. As controls, UV- and non-UV-treated inoculum was qualitatively and quantitatively measured by IE1 immunofluorescence (55) and TCID50 assay in highly permissive fibroblasts, respectively.
FIG 8.

Virion-associated US28 fails to complement the US28Δ latency phenotype in Kasumi-3 cells. Kasumi-3 cells were coinfected with WT and WT-UV, WT and US28Δ-UV, US28Δ and WT-UV, or US28Δ and US28Δ-UV, as indicated, at a multiplicity of infection of 1.0 PFU/cell each. Each coinfection was assessed for its ability to produce infectious virus capable of infecting naive fibroblasts by coculture (top panels), as well as for UL123 transcription (bottom panels). Fibroblast-associated plaques (mCherry-positive) were visualized by fluorescence microscopy. RT-qPCR was used to quantify UL123 transcripts relative to cellular GAPDH transcripts. For both WT and US28Δ coinfections with US28Δ-UV, UL123/GAPDH values are shown relative to those of the coinfections with WT-UV. All samples were analyzed in triplicate. Inoculum from each virus (left) was qualitatively assessed by immunofluorescence for IE1-positive nuclei using a monoclonal antibody directed at IE1 (clone 1B12) (56) (C) or quantitatively assessed by TCID50 assay (D). Samples for the experiment shown in panel D were analyzed in triplicate. The dashed line represents the level of detection.
Immediately following isolation, CD34+ HPCs were infected at a multiplicity of infection of 2.0 TCID50s/cell in infection medium, as described elsewhere (51). CD34+ HPC infection medium contains Iscove's modified Dulbecco's medium (IMDM; Corning) supplemented with 10% BIT9500 serum substitute (Stem Cell Technologies), 2 mM l-glutamine, 20 ng/ml low-density lipoproteins, and 50 μM 2-mercaptoethanol. The following day, cultures were washed three times in PBS and replated in transwells over stromal cells in hLTCM, as described previously (51). At 5 days postinfection (p.i.), cells were washed three times in PBS, and half of each infected culture was returned to the transwell culture in hLTCM, while the remaining half was shifted to reactivation medium (51) containing alpha modification of Eagle's medium (α-MEM) supplemented with 20% FBS, 1 μM hydrocortisone, 0.02 mM folic acid, 0.2 mM i-inositol, 0.1 mM 2-mercaptoethanol, 2 mM l-glutamine, 100 U/ml each of penicillin and streptomycin, and 15 ng/ml of each of the following cytokines: IL-6, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-3 (R&D Systems). The infected cells were cultured an additional 2 days prior to harvest for reverse transcription-quantitative PCR (RT-qPCR) analyses.
RNA and DNA analyses.
Total RNA was isolated from cells as described previously (55). Briefly, RNA was isolated with TRI reagent (Sigma) according to the manufacturer's instructions. Next, cDNA (1 μg/sample) was reverse transcribed with a TaqMan RT reagent kit (Applied Biosystems), according to the manufacturer's protocol. Transcripts were quantified by using equal volumes of cDNA template for RT-qPCR using SYBR green PCR mix (Applied Biosystems). All samples were analyzed in triplicate and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All primers are listed in Table 2.
TABLE 2.
Primers used for DNA and RNA analyses
| Gene name | Primer directiona | Primer sequence (5′–3′) |
|---|---|---|
| UL123 | FOR | GCCTTCCCTAAGACCACCAAT |
| REV | ATTTTCTGGGCATAAGCCATAATC | |
| UL44 | FOR | TACAACAGCGTGTCGTGCTCCG |
| REV | GGCGTGAAAAACATGCGTATCAAC | |
| UL99 | FOR | GTGTCCCATTCCCGACTCG |
| REV | TTCACAACGTCCACCCACC | |
| UL138 | FOR | GGTTCATCGTCTTCGTCGTC |
| REV | CACGGGTTTCAACAGATCG | |
| US28 | FOR | CCAGAATCGTTGCGGTGTCTCAGT |
| REV | CGTGTCCACAAACAGCGTCAGGT | |
| US27 | FOR | CCGTATGGTGCGGTTTATCATTA |
| REV | CTAAAAATAGCGCCAGGTTGAAAGG | |
| GAPDH | FOR | ACCCACTCCTCCACCTTTGAC |
| REV | CTGTTGCTGTAGCCAAATTCGT |
FOR, forward; REV, reverse.
Extracellular viral genomes were quantified by qPCR as described previously (37). Viral genomes were detected using primers directed at UL123 (Table 2). All samples were normalized to input and analyzed in triplicate.
Kasumi-3 cell immunofluorescence.
Kasumi-3 cells were infected as described above at a multiplicity of infection of 1.0 PFU/ml. Cells were prepared for immunofluorescence as described previously (23). In brief, Kasumi-3 cells were washed with PBS twice and fixed in 4% formaldehyde (Thermo Scientific) for 10 min at room temperature, and then the fixative was diluted with 5 times the volume of permeabilization buffer (1% bovine serum albumin [BSA], 2 mM EDTA, 0.1% sodium azide, 0.1% saponin in PBS), after which the cells were pelleted at 0.5 × g for 7 min. This washing step was repeated two additional times. Cells were then incubated for 20 min with an IE1 monoclonal antibody (clone 1B12) (56), diluted 1:10 in permeabilization buffer. Next, cells were diluted and washed as described above and then incubated with Alexa Fluor 488 anti-mouse secondary antibody (Molecular Probes), diluted 1:1,000 in permeabilization buffer, for 15 min in the dark at room temperature. Cells were then diluted and washed as described above, washed an additional two times in permeabilization buffer lacking saponin, resuspended in SlowFade Gold Antifade Reagent containing 4′,6′-diamidino-2-phenylindole (DAPI; Molecular Probes), and then mounted onto Superfrost Plus microscope slides (Fisher). Images were collected on a Leica DM IRB inverted fluorescence microscope using a 40× objective.
Protein analyses.
For fibroblast (NuFF-1)-associated US28 expression, 5 × 106 NuFF-1 cells were infected with TB40/E-mCherry-US28-3×F (US28-3×F) carrying a triple FLAG tag (3×F) (54) at a multiplicity of infection of 1.0 TCID50/cell for 1 h. Inoculum was then removed, and cells were washed three times with PBS and then cultured for 96 h in fresh medium. For Kasumi-3 cell-associated US28 expression, cells were first starved in X-VIVO15 medium (Lonza) for 48 h. Then, 1.65 × 105 cells per condition were pretreated with or without cycloheximide (CHX; 100 μg/ml) for 1 h prior to mock infection or infection with US28-3×F at a multiplicity of infection of 50 TCID50s/cell (55, 57) by centrifugal enhancement (1,000 × g for 30 min at room temperature). Following adsorption, inoculum was removed, and the cells were washed three times with PBS. All mock- or virus-infected Kasumi-3 cell cultures were treated with trypsin (100 μg/ml) at 37°C for 15 min to remove residual, extracellular virions. Soybean trypsin inhibitor (100 μg/ml; Sigma) was then added (55, 57). For both fibroblast and Kasumi-3 cell cultures, total cell lysates were collected and lysed in radioimmunoprecipitation assay (RIPA) buffer, and equal protein amounts (15 μg) were analyzed by Western blotting.
To assess the presence of US28 in mature viral particles, NuFF-1 cells were infected at a multiplicity of infection of 1.0 TCID50/cell. When 100% cytopathic effect was observed, the infectious medium was collected and precleared of cellular debris two times by centrifugation (3,000 × g for 5 min at room temperature). The virus-containing medium was then purified through a 20% sorbitol cushion. Viral particles were concentrated 100× in 10 mM Tris-Cl, pH 8.0, 400 mM NaCl, and 10 mM EDTA, and one-fifth of the concentrated particles were then analyzed by Western blotting.
All proteins were denatured for 10 min at 42°C prior to SDS-PAGE separation. Antibodies used in the analyses included the following: anit-FLAG M2 (Sigma) diluted 1:7,500; anti-IE2 (clone 3A9) (58) and anti-pp65 (clone 8A8) (59), each diluted at 1:100; anti-tubulin (Sigma) diluted at 1:5,000; and horseradish peroxidase (HRP)-conjugated goat-anti-mouse secondary antibody (Jackson Immuno Research Labs) diluted at 1:10,000.
RESULTS
Generation of viral recombinants.
To begin to understand how US28 influences a latent infection, we generated a panel of viral mutants utilizing the galK system for bacterial recombineering (60). We constructed each recombinant in the wild-type (WT) background of the bacterial artificial chromosome (BAC)-derived clinical isolate TB40/E-mCherry (55). We have previously described the mutants in which we deleted either the entire US28 ORF (US28Δ) (reference (54) or all four GPCRs (allΔ) (54). For this study, we created an additional recombinant in which US28 was the only GPCR expressed (US28wt) (Fig. 1). At multiplicities of infection of both 0.5 TCID50/cell (Fig. 2A) and 2.0 TCID50s/cell (Fig. 2B), allΔ infections of fibroblasts resulted in low titers at 72 h p.i. (hpi) that eventually grew to WT titer levels by 120 hpi; US28wt infection resulted in titers comparable to those of the WT virus at each time point assessed in lytically infected fibroblasts and in a slight growth advantage at late times at each multiplicity of infection (Fig. 2). As expected, US28 is dispensable for lytic replication in fibroblasts (Fig. 2), as we along with others have previously reported (46, 54, 61–63). Importantly, although titers for US28wt at 96 hpi were significantly higher than those of the other three viruses, none of these viral recombinants displayed a significant growth advantage in lytically infected fibroblasts compared to growth of the WT at 120 hpi (Fig. 2), and each of the two independent clones for all of the constructs displayed similar phenotypes, suggesting that the growth properties we observed during infections that favor latency (see below) are not due to defects in lytic replication.
FIG 1.

Schematic of recombinant viruses. (A) BAC-derived TB40/E-mCherry was used to generate TB40/E-mCherry-US28-3×F (US28-3×F), which contains three tandem FLAG epitopes at the C terminus of the US28 ORF (54). TB40/E-mCherry-US28Δ (US28Δ) was generated by deleting the entire US28 epitope-tagged ORF (54). (B) To generate a recombinant in which US28 is the only intact HCMV GPCR ORF, we performed a series of recombination events whereby the entire coding regions for UL33, UL78, and US27 were excised and repaired by linear recombination. This resulted in a virus termed TB40/E-mCherry-US28wt (US28wt). Finally, using US28wt, we deleted US28 in its entirety, yielding TB40/E-mCherry-allΔ (allΔ) (54). TRL and TRS, long and short terminal repeat, respectively; UL and US, long and short unique region, respectively; IRL and IRS, long and short internal repeat, respectively.
FIG 2.

US28 is dispensable for lytic grown in fibroblasts. Primary fibroblasts (NuFF-1) were infected with the viruses indicated at a multiplicity of infection of 0.5 TCID50/cell (A) or 2.0 TCID50s/cell (B). Supernatants were harvested over a time course of 120 h, and then titers for each time point were determined by TCID50 assays. Samples were analyzed in triplicate.
US28 is expressed in infected Kasumi-3 cells and primary CD34+ HPCs under conditions that promote latency.
Investigators have previously reported that US28 is expressed during latency in natural (32, 45) and experimental (4–6, 44, 49) infections. To confirm these findings in our culture systems, we examined the expression of US28 transcripts in both in vitro-cultured CD34+ Kasumi-3 cells and ex vivo-cultured primary cord blood-derived CD34+ HPCs. To this end, we latently infected either Kasumi-3 cells (23) or primary CD34+ HPCs (4, 5) with TB40/E-mCherry. Following 18 days of latent infection of each cell type, we induced lytic reactivation in parallel cultures (4, 5, 23). To induce lytic reactivation, Kasumi-3 cells were treated with the phorbol ester TPA, whereas the primary CD34+ HPCs were cultured in reactivation medium (see Materials and Methods). Utilizing RT-qPCR, we assessed the expression of US28 cDNA relative to that of UL123. Evaluating the ratio of US28 to UL123 is critical since US28 is expressed during both lytic and latent HCMV replication. The UL123 transcript, however, is highly expressed during lytic replication yet silenced during viral latency. Thus, during latency, the ratio of US28/UL123 transcripts should be increased, whereas during active/lytic transcription when both US28 and UL123 are expressed, the expression levels should be closer to a 1:1 ratio. We found that US28 mRNA is expressed in the Kasumi-3 model for in vitro latency (Fig. 3, first panel) and confirmed previously published findings (4, 5, 44) showing that US28 RNA is expressed during ex vivo latency in primary CD34+ cells (Fig. 3, first panel). As a control, we assessed the expression of the latency-associated transcript UL138 during latent infection and following reactivation in Kasumi-3 cells (Fig. 3, second panel) and CD34+ cells (Fig. 3, second panel). We also assessed the viral early lytic gene UL44 and the late lytic gene UL99, both of which are associated with lytic transcription, in each cell type as controls for successful latent infection and subsequent reactivation (Fig. 3, third and fourth panels, respectively). Finally, we confirmed that while the viral GPCR US28 is associated with latent transcription, another HCMV GPCR, US27, is expressed during lytic, rather than latent, infection in both Kasumi-3 cells and CD34+ cells (Fig. 3, last panel). Taken together, these data confirm that, similar to findings in latently infected primary CD34+ HPCs, the US28 transcript is actively transcribed in Kasumi-3 cells cultured under conditions shown to support latency. Thus, these cells are a suitable platform with which to begin to investigate US28-associated phenotypes during latency and/or reactivation.
FIG 3.

US28 is expressed in hematopoietic progenitor cells during latent infection. To confirm US28 expression during latency, we infected Kasumi-3 cells or primary CD34+ cells under conditions favoring a latent infection. Following 18 days in culture, each cell subset was cultured for an additional 2 days under conditions favoring either latency, using Kasumi-3 cells without TPA (23) or CD34+ cells in human long-term culture medium (51), or lytic reactivation, using Kasumi-3 cells with TPA (23) or CD34+ cells in reactivation medium (51). Cells were then harvested for RNA isolation, and the ratio of cDNA expression of US28 to UL123 was assessed by RT-qPCR in triplicate. Under latent conditions, the ratio of US28/UL123 is high, suggesting that US28 is expressed while UL123 is repressed. However, the US28/UL123 ratio nears 1 upon reactivation as both of these genes are highly expressed during lytic replication. The latent transcript UL138, the early lytic transcript UL44, the late lytic transcript UL99, and an additional HCMV GPCR, US27, are also shown as controls. *, P < 0.001. AU, arbitrary units.
US28 is required for latent infection in Kasumi-3 cells.
To determine the contribution of US28 toward a successful latent infection of Kasumi-3 cells, we assessed the production of extracellular virions over a 5-day time course following latent infection of Kasumi-3 cells and assessed viral DNA using qPCR (37). The cells infected with either the US28Δ or allΔ virus yielded an increase in extracellular viral genomes (Fig. 4A). It is important to note that due to the lytic-like phenotype we observed for the viruses lacking US28, we restricted our analyses to a 5-day time course as we noted an increase in cytopathic effect (CPE) of US28Δ- and allΔ-infected Kasumi-3 cells at later times postinfection (data not shown) concomitant with virus production, as shown in Fig. 4. Importantly, we were able to induce transcription from the MIEP by culturing a portion of each group of infected cells with TPA (Fig. 4B). These data confirm that both WT and US28wt infections reactivate from viral latency. Furthermore, not only is an increase in extracellular viral genomes detected in the infected cells lacking US28, but these cells also produce infectious virus capable of establishing a lytic infection within fibroblasts (Fig. 4C) in a coculture assay, suggesting that viruses that lack US28 favor a lytic rather than latent infection. Consistent with this finding, the infected Kasumi-3 cells are positive for IE1 protein by immunofluorescence (Fig. 5). Taken together, these data argue that US28 is critical to ensuring either the establishment or the maintenance of viral latency during in vitro infection of Kasumi-3 cells.
FIG 4.

US28 is important for a successful latent infection in Kasumi-3 cells. (A) Kasumi-3 cells were infected with each of the indicated viruses. For each infected cell population, extracellular DNA was collected over a 5-day time course and quantified by qPCR using primers directed at UL123. Each sample was analyzed in triplicate. *, P < 0.001 relative to the WT value at 5 days p.i. (B) Parallel cultures from the experiment shown in panel A were treated with TPA to induce lytic transcription for an additional 2 days. Extracellular viral genomes were quantified as described for panel A. *, P < 0.001. (C) To confirm that the extracellular virions produced in the experiment shown in panel A were infectious, the infected Kasumi-3 cell subsets were each cocultured with naive fibroblasts for 7 days. Viral plaques (mCherry-positive) were visualized by fluorescence microscopy.
FIG 5.
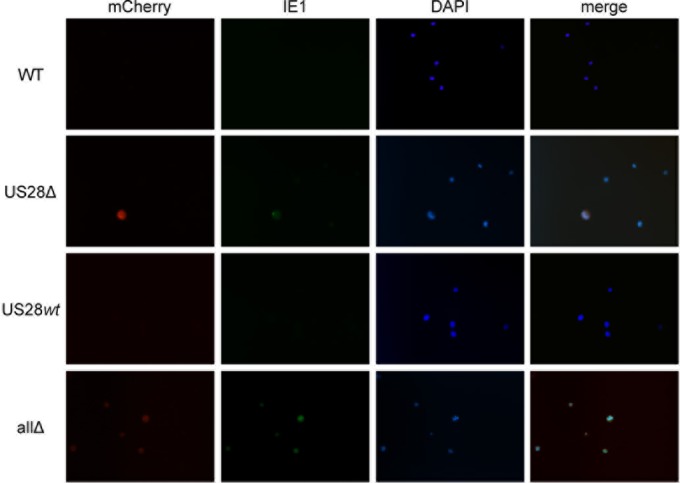
US28Δ- and allΔ-infected Kasumi-3 cells result in IE1-positive infected cells. Kasumi-3 cells infected with each of the indicated viruses under latent conditions as described in the legend of Fig. 4 were harvested for immunofluorescence assay. Cells were stained with a monoclonal antibody directed at IE1 (clone 1B12) (56), shown in green. mCherry (red) is a marker of lytic infection; DAPI (blue) is shown as a nuclear marker. Images were collected using a 40× objective and depict representative fields for each infection.
US28 aids in promoting latency in primary CD34+ HPCs.
We next asked if we could extend our findings to ex vivo-cultured primary CD34+ HPCs. To this end, we infected primary CD34+ HPCs isolated from cord blood under conditions reported to promote latency (4, 5, 51) with each of the viruses described in the legend of Fig. 1. After 5 days, we divided each infected culture such that half of the cells remained under latent culture conditions, whereas the remaining cells were cultured in reactivation medium. Two days later, we harvested the cells and assessed IE RNA expression by RT-qPCR for UL123 (37). The latent cultures infected with the viral recombinants that lacked the US28 ORF (i.e., US28Δ and allΔ viruses) resulted in IE gene expression, indicative of an active transcriptional profile (Fig. 6A), whereas the US28wt viral infection resulted in suppressed IE gene expression that phenotypically resembled that of the WT (Fig. 6A). Importantly, all of the infected cells that were subsequently cultured in reactivation medium, which promotes the initiation of the lytic life cycle, displayed an increase in UL123 expression (Fig. 6B), confirming that WT virus and the US28wt recombinant virus retain the capacity to reactivate. Infections with viruses lacking US28 resulted in a further increase in UL123 expression following reactivation, perhaps due to the already high levels of UL123 under latent culture conditions for both US28Δ and allΔ infections (Fig. 6B). Additionally, the US28Δ- and allΔ-infected CD34+ HPCs also transferred infectious virus to fibroblasts when cocultured under latent conditions, whereas the WT and US28wt latently infected cells were unable to produce plaques in naive fibroblasts (Fig. 6C). Together, these data suggest that US28 aids in promoting a latent state without the assistance from the other virus-encoded GPCRs.
FIG 6.

US28Δ- and allΔ-infected primary CD34+ HPCs fail to suppress MIEP-driven transcription. (A) Cord blood-derived CD34+ cells were infected with the indicated viruses. Total RNA was isolated from each population at 5 days p.i., and IE gene expression was assessed by RT-qPCR with primers directed at UL123. Samples were normalized to cellular GAPDH levels and analyzed in triplicate. *, P < 0.001 relative to the WT level. (B) Parallel cultures from the experiment shown in panel A were treated with reactivation medium for an additional 2 days, and then RT-qPCR was performed as described for panel A. *, P < 0.001. (C) Latently infected cells from the experiment shown in panel A were cocultured with naive fibroblasts for 7 days. Fibroblast-associated viral plaques (mCherry-positive) were visualized by fluorescence microscopy.
US28 copurifies with mature virions and is expressed in Kasumi-3 cells upon entry.
Based on our findings that US28 is required for silencing MIEP-driven transcription, we hypothesized that US28 is present in HPCs prior to IE transcription and thus is included within the mature viral particle and delivered upon viral entry into a cell. To test this hypothesis, we pretreated Kasumi-3 cells with cycloheximide in order to inhibit de novo translation of viral proteins or left the cells untreated. We next infected these cells with TB40/E-mCherry-US28-3×F, a recombinant virus that includes a triple FLAG epitope tag on the C-terminal end of the US28 ORF (54), and harvested the cell lysates at 4 hpi. We observed US28 expression in Kasumi-3 cells, regardless of cycloheximide treatment, suggesting that de novo translation is not required for US28 following Kasumi-3 cell infection (Fig. 7). This finding also strengthens the hypothesis that US28 is incorporated into mature viral particles. To this end, we infected primary fibroblasts (NuFF-1) and, when cultures reached 100% CPE, purified extracellular virions by ultracentrifugation. When we assessed these particles by immunoblotting for the US28 C-terminal FLAG epitope, we found that US28 copurified with mature viral particles (Fig. 7). Although cell-associated US28 is not glycosylated (61), this viral GPCR is phosphorylated (64, 65), and we along with others have previously shown that US28 migrates as broad polypeptide species (54, 61, 66), consistent with the results shown in Fig. 7. Importantly, IE2, a non-virion-associated protein, and cellular tubulin were not detected in this same preparation, while the tegument protein pp65 was indeed present in the virion preparation as well as in the cell-associated preparations (Fig. 7). Together, these findings argue that US28 is incorporated into the mature viral particle and is expressed upon entry of Kasumi-3 cells.
FIG 7.
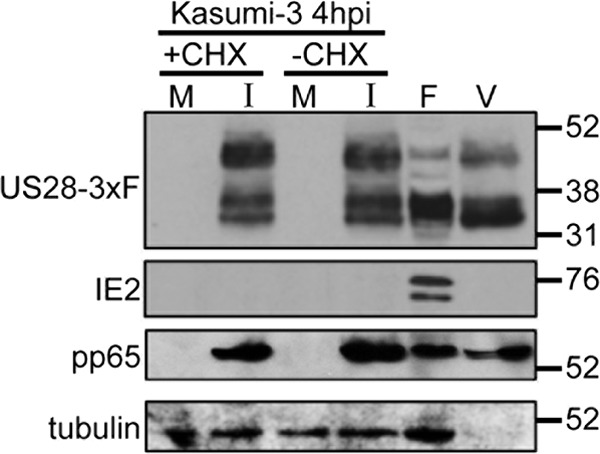
US28 is present in extracellular virions and is brought into newly infected Kasumi-3 cells. (A) Kasumi-3 cells were left untreated (−CHX) or treated with cycloheximide (+CHX), mock infected (M) or infected with US28-3×FLAG (I), and harvested at 4 hpi. Results for cell-free, purified HCMV virions (V) and extracts from lytically infected fibroblasts (F) at 96 hpi are also depicted. US28 was detected by probing immunoblots for the FLAG epitope tag. The blot was reprobed with antibodies directed to IE2 (clone 3A9) (58), pp65 (clone 8A8) (59), and cellular tubulin as a controls. Values at the right of the blot are in kilodaltons.
US28 provided in trans fails to rescue the US28Δ latency phenotype.
To begin to understand how US28 contributes to a successful latent infection, we asked if the presence of US28 in the virion at the time of infection was sufficient. To this end, we UV inactivated either WT or US28Δ virus (designated WT-UV or US28Δ-UV, respectively). We then coinfected Kasumi-3 cells with the following combinations of viral inocula: WT and WT-UV, WT and US28Δ-UV, US28Δ and WT-UV, and US28Δ and US28Δ-UV. We assessed both viral transcripts and the ability of the infected Kasumi-3 cells to transmit virus to naive fibroblasts by coculture. We hypothesized that if virion-associated US28 aids in establishing a latent infection, then coinfection of US28Δ with WT-UV virus would phenotypically resemble a WT (i.e., latent) infection. While infection with WT virus, regardless of the UV-treated virus used for coinfection, resulted in a latent infection (Fig. 8A), we found that virion-associated US28 provided by the WT-UV inoculum was not sufficient to promote a latent infection with US28Δ but displayed a phenotype similar to that of the combination of US28Δ and US28Δ-UV infection (Fig. 8B). UL123 expression levels for WT-UV and US28Δ-UV coinfections with the WT (Fig. 8A, bottom) or US28Δ (Fig. 8B, bottom) did not differ. While the WT coinfections resulted in barely detectable infection of naive fibroblasts (Fig. 8A, top), the US28Δ-coinfected Kasumi-3 cells cocultured with naive fibroblasts resulted in the formation of viral plaques (Fig. 8B, top). As controls to ensure that the UV inactivation of each viral inoculum was successful, we compared the input titers to the non-UV-treated input both qualitatively (Fig. 8C) and quantitatively (Fig. 8D). As expected, inoculum from the WT and US28Δ strains resulted in detectable IE1-positive nuclei (Fig. 8C) and measurable titers by TCID50 assay (Fig. 8D), while inoculum from the WT-UV and US28Δ-UV strains was restricted for growth in each assay. Together, these results suggest that while US28 is necessary for a successful latent infection in both Kasumi-3 cells and CD34+ HPCs, the presence of US28 in the viral particle is not sufficient for HCMV to establish a latent infection in Kasumi-3 cells.
DISCUSSION
Our work suggests that US28 expression during latency in HPCs has biological functions. We have confirmed US28 transcription in latently infected Kasumi-3 cells and primary CD34+ HPCs. The absence of US28 expression in latently infected Kasumi-3 cells results in a significant increase in extracellular virion production. In infected ex vivo-cultured cord blood-derived CD34+ HPCs, US28 expression was critical to suppressing MIEP-driven transcription under latent culture conditions. Furthermore, the absence of US28 expression during latent infection of either Kasumi-3 cells or primary CD34+ HPCs resulted in the transfer of infectious virus to naive fibroblasts. Importantly, the phenotype we observed in each cell type is US28 dependent as infection with US28wt virus displayed results similar to those with WT virus, and, thus, the other three virus-encoded GPCRs do not contribute significantly to this phenotype. Finally, we found that US28 is present in mature virions and is expressed upon entry into Kasumi-3 cells. Together, our findings suggest that US28 functions to aid in the establishment or maintenance of successful latent infections in culture.
US28 is a multifunctional protein, and its functions during lytic infection are well studied (61, 66, 67). This viral GPCR functions as a potent signaling molecule. US28 binds a variety of ligands, including CCL5 (RANTES), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL1, and the only known CXCL1 chemokine, fractalkine (reviewed in reference 48). Interaction of US28 with a ligand induces cellular signaling that results in altering the host cell milieu. US28-induced signaling is cell type dependent (54), suggesting that this viral GPCR functions differently depending on the type of cell in which the protein is expressed. HCMV infects a variety of cell types in vivo, including fibroblasts, endothelial cells, epithelial cells, hematopoietic cells (i.e., monocytes and macrophages), and dendritic cells. At least two other HCMV-encoded GPCRs, US27 (53) and UL78 (55), display cell-type-specific functions. Additionally, deletion of the US28 ORF from the viral genome yields reduced viral growth in epithelial cells, where viral spread is restricted to the cell-to-cell route (61). Together, these data argue that the HCMV-encoded GPCRs have specific functions in cells of various tissue origins and that US28 is no exception in this regard. It is thus not surprising that US28 functions differently in HPCs, which support HCMV latency, than it does in highly permissive cells, such as fibroblasts, in which HCMV undergoes lytic replication. US28Δ infection of fibroblasts does not result in a significant increase of viral growth (Fig. 2) (54), and thus during lytic infection of these cells, it is unlikely that US28 ensures transcriptional suppression of the MIEP as it does in HPCs. It is attractive to speculate that the cellular environment influences the biological functions of US28.
Our finding that US28 copurifies with extracellular virions is not surprising as several groups of investigators have suggested that this protein is indeed incorporated into mature viral particles. Pleskoff et al. speculated that US28 could be associated with the lipid envelope of HCMV, given i expression kinetics and topology of US28 (68). Similarly, Kledal and colleagues also discussed the possibility of US28's inclusion in the viral envelope as they proposed a mechanism by which US28 binds host proteins expressed on the cell surface to aid in fusion, cell-to-cell spread, and dissemination (69). Interestingly, Varnum et al. performed proteomic analyses of HCMV viral particles and dense bodies, and although these investigators detected US28 peptides in their virion preparations, the peptides they identified did not meet their predetermined criteria for inclusion within the list of virion proteins (70). This may be due to the preparatory methods, including the solutions and temperature used to denature the virion proteins, or to the mass spectrometry method itself, which detects the most abundant proteins in a mixture. That recent studies have failed to detect US28 in either fibroblast cell-associated (71) or virion (71, 72) preparations could be due to the strict stringency of the criteria set for these investigations, thereby favoring the more abundant proteins. For example, Buscher et al. characterized the 40 most abundant proteins in virions produced from several HCMV strains. While this group of investigators did identify two other HCMV GPCRs, US27 and UL33, both of these proteins were in the less abundant half of the proteins identified (72). In our analysis, we have probed for US28 via its triple FLAG C-terminal epitope tag, thus increasing the sensitivity for this protein. It is also not surprising to find that US28 copurifies with mature virions, as US27 (53, 70, 73), UL33 (74), and UL78 (55) have been identified as components of the viral particle.
How does US28 function to aid in a successful latent infection? There are several possibilities. First, it is plausible that US28 induces signaling events that act in trans to ensure suppression of lytic genes. Alternatively, US28-induced signaling could alter the host cell environment, for example, by aiding in the retention of the progenitor-like phenotype of the latently infected cell. The latter function is intriguing as viruses lacking US28 display phenotypes (e.g., cell adhesion and morphological changes) that resemble lytic replication following infection of differentiated HPCs or lytic replication following reactivation. Thus, our future experiments will be aimed at testing the cellular characteristics (e.g., progenitor cell markers versus differentiated cell markers) to discern the cellular changes that are induced upon infections with viruses lacking the US28 ORF. Our data show a requirement for US28 in latent infection. However, it is not clear if the role of US28 is to establish and/or maintain latency. Our finding that coinfection of UV-inactivated WT (WT-UV) virus with US28Δ failed to rescue the US28Δ phenotype suggests that incoming US28 and its subsequent deposition into the infected cell are not sufficient to establish a latent infection (Fig. 8). Thus, perhaps it is the expression of this gene throughout latency that aids in ensuring a successful latent infection. This is an attractive hypothesis as US28 RNA is detected during latency in both the natural and tissue culture settings (4–6, 32, 44, 45, 49). Pinpointing the exact phase of latency for which US28 is required will prove critical for fully understanding the role of this protein during HCMV latency and reactivation. It is attractive to speculate that US28 may serve as an ideal novel therapeutic. A majority of the drugs approved for use by the FDA target cellular GPCRs. Thus, if we can better understand how US28 functions during latency and reactivation, we may be able to exploit this viral GPCR as a novel drug target, which would represent a significant vertical advancement against a human pathogen that currently lacks a vaccine or a cure.
ACKNOWLEDGMENTS
We thank Jason C. Buehler and Felicia D. Goodrum (University of Arizona) for the irradiated stromal feeder cells and invaluable discussions and advice with regard to ex vivo CD34+ HPC experiments. We also thank Wade Sigurdson of the University at Buffalo, SUNY, Confocal Microscope and Flow Cytometry Facility Core for his assistance with fluorescence microscopy. We thank Eain A. Murphy (FORGE Life Sciences) for critical reading of the manuscript.
Funding Statement
The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institutes of Health. Likewise, the American Heart Association had no role in study design, data collection or interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. [DOI] [PubMed] [Google Scholar]
- 2.Hakki M, Chou S. 2011. The biology of cytomegalovirus drug resistance. Curr Opin Infect Dis 24:605–611. doi: 10.1097/QCO.0b013e32834cfb58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronel R, Takayama S, Juwono T, Hertel L. 2015. Dynamics of human cytomegalovirus infection in CD34+ hematopoietic cells and derived Langerhans-type dendritic cells. J Virol 89:5615–5632. doi: 10.1128/JVI.00305-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687–695. doi: 10.1182/blood-2003-12-4344. [DOI] [PubMed] [Google Scholar]
- 5.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci U S A 99:16255–16260. doi: 10.1073/pnas.252630899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hargett D, Shenk TE. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc Natl Acad Sci U S A 107:20039–20044. doi: 10.1073/pnas.1014509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kondo K, Kaneshima H, Mocarski ES. 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci U S A 91:11879–11883. doi: 10.1073/pnas.91.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondo K, Xu J, Mocarski ES. 1996. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc Natl Acad Sci U S A 93:11137–11142. doi: 10.1073/pnas.93.20.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maciejewski JP, Bruening EE, Donahue RE, Mocarski ES, Young NS, St Jeor SC. 1992. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 80:170–178. [PubMed] [Google Scholar]
- 10.Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77:3099–3102. doi: 10.1099/0022-1317-77-12-3099. [DOI] [PubMed] [Google Scholar]
- 11.Minton EJ, Tysoe C, Sinclair JH, Sissons JG. 1994. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J Virol 68:4017–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14+ monocytes and CD34+ cells. PLoS Pathog 9:e1003366. doi: 10.1371/journal.ppat.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol 84:5594–5604. doi: 10.1128/JVI.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith MS, Bentz GL, Alexander JS, Yurochko AD. 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J Virol 78:4444–4453. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albright ER, Kalejta RF. 2013. Myeloblastic cell lines mimic some but not all aspects of human cytomegalovirus experimental latency defined in primary CD34+ cell populations. J Virol 87:9802–9812. doi: 10.1128/JVI.01436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dosa R, Burian K, Gonczol E. 2005. Human cytomegalovirus latency is associated with the state of differentiation of the host cells: an in vitro model in teratocarcinoma cells. Acta Microbiol Immunol Hung 52:397–406. doi: 10.1556/AMicr.52.2005.3-4.11. [DOI] [PubMed] [Google Scholar]
- 17.Duan YL, Ye HQ, Zavala AG, Yang CQ, Miao LF, Fu BS, Seo KS, Davrinche C, Luo MH, Fortunato EA. 2014. Maintenance of large numbers of virus genomes in human cytomegalovirus-infected T98G glioblastoma cells. J Virol 88:3861–3873. doi: 10.1128/JVI.01166-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128. doi: 10.1016/j.gene.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 19.Keller MJ, Wu AW, Andrews JI, McGonagill PW, Tibesar EE, Meier JL. 2007. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J Virol 81:6669–6681. doi: 10.1128/JVI.01524-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CH, Lee GC, Chan YJ, Chiou CJ, Ahn JH, Hayward GS. 1999. Factors affecting human cytomegalovirus gene expression in human monocyte cell lines. Mol Cells 9:37–44. [PubMed] [Google Scholar]
- 21.Luo MH, Fortunato EA. 2007. Long-term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J Virol 81:10424–10436. doi: 10.1128/JVI.00866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meier JL. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J Virol 75:1581–1593. doi: 10.1128/JVI.75.4.1581-1593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol 86:9854–9865. doi: 10.1128/JVI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Penkert RR, Kalejta RF. 2013. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 4:e00298-13. doi: 10.1128/mBio.00298-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81:9109–9120. doi: 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turtinen LW, Seufzer BJ. 1994. Selective permissiveness of TPA differentiated THP-1 myelomonocytic cells for human cytomegalovirus strains AD169 and Towne. Microb Pathog 16:373–378. doi: 10.1006/mpat.1994.1037. [DOI] [PubMed] [Google Scholar]
- 27.Yee LF, Lin PL, Stinski MF. 2007. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology 363:174–188. doi: 10.1016/j.virol.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 28.Sinclair JH, Reeves MB. 2013. Human cytomegalovirus manipulation of latently infected cells. Viruses 5:2803–2824. doi: 10.3390/v5112803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol 83:5615–5629. doi: 10.1128/JVI.01989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weekes MP, Tan SY, Poole E, Talbot S, Antrobus R, Smith DL, Montag C, Gygi SP, Sinclair JH, Lehner PJ. 2013. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340:199–202. doi: 10.1126/science.1235047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keyes LR, Hargett D, Soland M, Bego MG, Rossetto CC, Almeida-Porada G, St Jeor S. 2012. HCMV protein LUNA is required for viral reactivation from latently infected primary CD14+ cells. PLoS One 7:e52827. doi: 10.1371/journal.pone.0052827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poole E, Walther A, Raven K, Benedict CA, Mason GM, Sinclair J. 2013. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J Virol 87:4261–4271. doi: 10.1128/JVI.03497-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Avdic S, Cao JZ, Cheung AK, Abendroth A, Slobedman B. 2011. Viral interleukin-10 expressed by human cytomegalovirus during the latent phase of infection modulates latently infected myeloid cell differentiation. J Virol 85:7465–7471. doi: 10.1128/JVI.00088-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheung AK, Gottlieb DJ, Plachter B, Pepperl-Klindworth S, Avdic S, Cunningham AL, Abendroth A, Slobedman B. 2009. The role of the human cytomegalovirus UL111A gene in down-regulating CD4+ T-cell recognition of latently infected cells: implications for virus elimination during latency. Blood 114:4128–4137. doi: 10.1182/blood-2008-12-197111. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins C, Garcia W, Godwin MJ, Spencer JV, Stern JL, Abendroth A, Slobedman B. 2008. Immunomodulatory properties of a viral homolog of human interleukin-10 expressed by human cytomegalovirus during the latent phase of infection. J Virol 82:3736–3750. doi: 10.1128/JVI.02173-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poole E, Avdic S, Hodkinson J, Jackson S, Wills M, Slobedman B, Sinclair J. 2014. Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 Secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J Virol 88:13947–13955. doi: 10.1128/JVI.02424-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Connor CM, Vanicek J, Murphy EA. 2014. Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J Virol 88:5524–5532. doi: 10.1128/JVI.00481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poole E, McGregor Dallas SR, Colston J, Joseph RS, Sinclair J. 2011. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34+ progenitors. J Gen Virol 92:1539–1549. doi: 10.1099/vir.0.031377-0. [DOI] [PubMed] [Google Scholar]
- 39.Poole E, Lau JC, Sinclair J. 2015. Latent infection of myeloid progenitors by human cytomegalovirus protects cells from FAS-mediated apoptosis through the cellular IL-10/PEA-15 pathway. J Gen Virol 96:2355–2359. doi: 10.1099/vir.0.000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chee MS, Bankier AT, Beck S, Bohni R, Brown CM, Cerny R, Horsnell T, Hutchison CA III, Kouzarides T, Martignetti JA, Preddie E, Satchwell C, Tomlinson P, Weston KM, Barrell BG. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol 154:125–169. [DOI] [PubMed] [Google Scholar]
- 41.Murphy E, Rigoutsos I, Shibuya T, Shenk TE. 2003. Reevaluation of human cytomegalovirus coding potential. Proc Natl Acad Sci U S A 100:13585–13590. doi: 10.1073/pnas.1735466100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung AK, Abendroth A, Cunningham AL, Slobedman B. 2006. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 108:3691–3699. doi: 10.1182/blood-2005-12-026682. [DOI] [PubMed] [Google Scholar]
- 45.Patterson BK, Landay A, Andersson J, Brown C, Behbahani H, Jiyamapa D, Burki Z, Stanislawski D, Czerniewski MA, Garcia P. 1998. Repertoire of chemokine receptor expression in the female genital tract: implications for human immunodeficiency virus transmission. Am J Pathol 153:481–490. doi: 10.1016/S0002-9440(10)65591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vieira J, Schall TJ, Corey L, Geballe AP. 1998. Functional analysis of the human cytomegalovirus US28 gene by insertion mutagenesis with the green fluorescent protein gene. J Virol 72:8158–8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welch AR, McGregor LM, Gibson W. 1991. Cytomegalovirus homologs of cellular G protein-coupled receptor genes are transcribed. J Virol 65:3915–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Munnik SM, Smit MJ, Leurs R, Vischer HF. 2015. Modulation of cellular signaling by herpesvirus-encoded G protein-coupled receptors. Front Pharmacol 6:40. doi: 10.3389/fphar.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beisser PS, Laurent L, Virelizier JL, Michelson S. 2001. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J Virol 75:5949–5957. doi: 10.1128/JVI.75.13.5949-5957.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asou H, Suzukawa K, Kita K, Nakase K, Ueda H, Morishita K, Kamada N. 1996. Establishment of an undifferentiated leukemia cell line (Kasumi-3) with t(3;7)(q27;q22) and activation of the EVI1 gene. Jpn J Cancer Res 87:269–274. doi: 10.1111/j.1349-7006.1996.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Umashankar M, Goodrum F. 2014. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol Biol 1119:99–112. doi: 10.1007/978-1-62703-788-4_7. [DOI] [PubMed] [Google Scholar]
- 52.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 53.O'Connor CM, Shenk T. 2011. Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J Virol 85:3700–3707. doi: 10.1128/JVI.02442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller WE, Zagorski WA, Brenneman JD, Avery D, Miller JL, O'Connor CM. 2012. US28 is a potent activator of phospholipase C during HCMV infection of clinically relevant target cells. PLoS One 7:e50524. doi: 10.1371/journal.pone.0050524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Connor CM, Shenk T. 2012. Human cytomegalovirus pUL78 G protein-coupled receptor homologue is required for timely cell entry in epithelial cells but not fibroblasts. J Virol 86:11425–11433. doi: 10.1128/JVI.05900-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69:7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang D, Yu QC, Schroer J, Murphy E, Shenk T. 2007. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc Natl Acad Sci U S A 104:20037–20042. doi: 10.1073/pnas.0709704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cuevas-Bennett C, Shenk T. 2008. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J Virol 82:9525–9536. doi: 10.1128/JVI.00946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bechtel JT, Shenk T. 2002. Human cytomegalovirus UL47 tegument protein functions after entry and before immediate-early gene expression. J Virol 76:1043–1050. doi: 10.1128/JVI.76.3.1043-1050.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noriega VM, Gardner TJ, Redmann V, Bongers G, Lira SA, Tortorella D. 2014. Human cytomegalovirus US28 facilitates cell-to-cell viral dissemination. Viruses 6:1202–1218. doi: 10.3390/v6031202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mokros T, Rehm A, Droese J, Oppermann M, Lipp M, Hopken UE. 2002. Surface expression and endocytosis of the human cytomegalovirus-encoded chemokine receptor US28 is regulated by agonist-independent phosphorylation. J Biol Chem 277:45122–45128. doi: 10.1074/jbc.M208214200. [DOI] [PubMed] [Google Scholar]
- 65.Miller WE, Houtz DA, Nelson CD, Kolattukudy PE, Lefkowitz RJ. 2003. G-protein-coupled receptor (GPCR) kinase phosphorylation and beta-arrestin recruitment regulate the constitutive signaling activity of the human cytomegalovirus US28 GPCR. J Biol Chem 278:21663–21671. doi: 10.1074/jbc.M303219200. [DOI] [PubMed] [Google Scholar]
- 66.Stropes MP, Miller WE. 2008. Functional analysis of human cytomegalovirus pUS28 mutants in infected cells. J Gen Virol 89:97–105. doi: 10.1099/vir.0.83226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stropes MP, Schneider OD, Zagorski WA, Miller JL, Miller WE. 2009. The carboxy-terminal tail of human cytomegalovirus (HCMV) US28 regulates both chemokine-independent and chemokine-dependent signaling in HCMV-infected cells. J Virol 83:10016–10027. doi: 10.1128/JVI.00354-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pleskoff O, Treboute C, Alizon M. 1998. The cytomegalovirus-encoded chemokine receptor US28 can enhance cell-cell fusion mediated by different viral proteins. J Virol 72:6389–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kledal TN, Rosenkilde MM, Schwartz TW. 1998. Selective recognition of the membrane-bound CX3C chemokine, fractalkine, by the human cytomegalovirus-encoded broad-spectrum receptor US28. FEBS Lett 441:209–214. doi: 10.1016/S0014-5793(98)01551-8. [DOI] [PubMed] [Google Scholar]
- 70.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reyda S, Buscher N, Tenzer S, Plachter B. 2014. Proteomic analyses of human cytomegalovirus strain AD169 derivatives reveal highly conserved patterns of viral and cellular proteins in infected fibroblasts. Viruses 6:172–188. doi: 10.3390/v6010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buscher N, Paulus C, Nevels M, Tenzer S, Plachter B. 2015. The proteome of human cytomegalovirus virions and dense bodies is conserved across different strains. Med Microbiol Immunol 204:285–293. doi: 10.1007/s00430-015-0397-y. [DOI] [PubMed] [Google Scholar]
- 73.Margulies BJ, Gibson W. 2007. The chemokine receptor homologue encoded by US27 of human cytomegalovirus is heavily glycosylated and is present in infected human foreskin fibroblasts and enveloped virus particles. Virus Res 123:57–71. doi: 10.1016/j.virusres.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Margulies BJ, Browne H, Gibson W. 1996. Identification of the human cytomegalovirus G protein-coupled receptor homologue encoded by UL33 in infected cells and enveloped virus particles. Virology 225:111–125. doi: 10.1006/viro.1996.0579. [DOI] [PMC free article] [PubMed] [Google Scholar]