

ABSTRACT
Hepatitis C virus (HCV) is a major cause of chronic liver disease and is highly dependent on cellular proteins for virus propagation. To identify the cellular factors involved in HCV propagation, we recently performed protein microarray assays using the HCV nonstructural 5A (NS5A) protein as a probe. Of 90 cellular protein candidates, we selected the soluble resistance-related calcium-binding protein (sorcin) for further characterization. Sorcin is a calcium-binding protein and is highly expressed in certain cancer cells. We verified that NS5A interacted with sorcin through domain I of NS5A, and phosphorylation of the threonine residue 155 of sorcin played a crucial role in protein interaction. Small interfering RNA (siRNA)-mediated knockdown of sorcin impaired HCV propagation. Silencing of sorcin expression resulted in a decrease of HCV assembly without affecting HCV RNA and protein levels. We further demonstrated that polo-like kinase 1 (PLK1)-mediated phosphorylation of sorcin was increased by NS5A. We showed that both phosphorylation and calcium-binding activity of sorcin were required for HCV propagation. These data indicate that HCV modulates sorcin activity via NS5A protein for its own propagation.
IMPORTANCE Sorcin is a calcium-binding protein and regulates intracellular calcium homeostasis. HCV NS5A interacts with sorcin, and phosphorylation of sorcin is required for protein interaction. Gene silencing of sorcin impaired HCV propagation at the assembly step of the HCV life cycle. Sorcin is phosphorylated by PLK1 via protein interaction. We showed that sorcin interacted with both NS5A and PLK1, and PLK1-mediated phosphorylation of sorcin was increased by NS5A. Moreover, calcium-binding activity of sorcin played a crucial role in HCV propagation. These data provide evidence that HCV regulates host calcium metabolism for virus propagation, and thus manipulation of sorcin activity may represent a novel therapeutic target for HCV.
INTRODUCTION
Hepatitis C virus (HCV) is a major causative agent of non-A, non-B hepatitis. HCV infection often leads to chronic hepatitis, liver cirrhosis, and ultimately hepatocellular carcinoma (HCC) (1). HCV belongs to the member of the Hepacivirus genus within the Flaviviridae family. HCV is an enveloped virus with a positive-sense, single-stranded RNA genome of ∼9.6 kb. Its genome encodes a single polyprotein precursor of more than 3,000 amino acids, which is cleaved by both host and viral proteases at the endoplasmic reticulum (ER), yielding structural (core, E1, and E2) and nonstructural (p7 and NS2 to NS5B) proteins (2). The nonstructural 5A (NS5A) protein is a phosphoprotein consisting of 447 amino acid residues. NS5A exists in two forms of polypeptide, p56 and p58, which are phosphorylated at serine residues by cellular kinase (3), and phosphorylation regulates the HCV life cycle (4). NS5A protein forms a part of the HCV RNA replication complex (5) and is involved in liver pathogenesis (6). NS5A is a multifunctional protein that regulates RNA replication, interferon (IFN) resistance, and a variety of cellular signaling pathways (7–10). Furthermore, NS5A is involved in the assembly and maturation of infectious viral particles. Nevertheless, how NS5A participates in virus production has not yet been fully demonstrated.
Soluble resistance-related calcium-binding protein (sorcin) is a 21.6-kDa calcium-binding protein that belongs to the penta-EF-hand family (11). Sorcin maintains a high level of calcium in the ER through calcium channels and exchangers located at the plasma membrane and at the ER/sarcoplasmic reticulum (SR). Sorcin regulates calcium levels by interacting with the ryanodine receptor and SR calcium transport ATPase (SERCA) located in the ER (12). Sorcin activates calcium-ATPase-mediated Ca2+ uptake and restores SR Ca2+ content, plays a crucial role in Ca2+ homeostasis, and prevents ER stress (13–15). Sorcin undergoes Ca2+-dependent conformational changes and translocation from the cytosol to membranes, where it binds to many target proteins including serine-threonine kinase. Sorcin is phosphorylated by the polo-like kinase (PLK1) (16), cyclic AMP (cAMP)-dependent protein kinase (PKA), and calcium-calmodulin dependent kinase II (CaMKII) (17). Phosphorylation of sorcin by PKA inhibits ryanodine receptor activity. Sorcin is differentially expressed in cancer cells and plays an important role in multidrug resistance (MDR) (18, 19). Interestingly, foot-and-mouth disease virus (FMDV) VP1 interacts with sorcin and suppresses tumor necrosis factor alpha (TNF-α) or the Sendai virus (SeV)-induced type 1 interferon response to escape host immune surveillance (20). In addition, sorcin is identified as an antiviral factor involved in a late step of the replication cycle of HCV (21). However, the functional role of sorcin in HCV propagation has not been studied yet.
We recently performed protein microarray analysis to identify host factors interacting with HCV NS5A (22). In the present study, we selected sorcin for further characterization. Protein binding between NS5A and sorcin was verified by both in vitro and coimmunoprecipitation assays. Silencing of sorcin expression resulted in a decrease of HCV infectivity but not of HCV RNA and protein levels. We further showed that sorcin was involved in the assembly step of the HCV life cycle. These data suggest that sorcin is a novel host factor involved in HCV propagation.
MATERIALS AND METHODS
Plasmid constructions and DNA transfection.
Total RNAs were isolated from Huh7.5 cells by using RiboEx (GeneAll), and full-length sorcin was amplified by a primer set (sense, 5′-AAG GTA CCA TGG CGT ACC CGG GGC AT-3′; antisense, 5′-CCG CGG CCG CGA ACA CTC ATG ACA CAT-3′) from cDNA synthesized by using a cDNA synthesis kit (Toyobo) according to the manufacturer's instructions. PCR products were inserted into the NotI and KpnI sites of plasmid pEF6/V5-HisB (Invitrogen). A pEF6B V5-tagged HCV small interfering RNA (siRNA)-resistant sorcin mutant (pEF6/V5-sorcin-Res) was constructed by introducing three mutations at the siRNA binding site using a site-directed mutagenesis kit (Enzynomics). The phosphorylation-defective T155A sorcin mutant, phosphomimetic T155D sorcin mutant, and calcium-binding-defective F112L sorcin mutant were constructed by PCR-based mutagenesis, and each construct was inserted into the corresponding enzyme sites of the plasmid pEF6/V5-HisB. All DNA transfections were performed by using polyethyleneimine (Sigma-Aldrich) as we described previously (23).
Antibodies and chemical.
Antibodies were purchased from the following sources: sorcin antibodies were from Santa Cruz and Abcam, c-Myc antibodies were from Santa Cruz, β-actin antibodies were from Sigma-Aldrich, and V5 antibodies were from Invitrogen; HCV core, NS3, and NS5A antibodies have been described elsewhere (22). BI2536, a PLK1 inhibitor, was purchased from Selleck Chem.
Cell culture.
All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 100 units/ml of penicillin-streptomycin in 5% CO2 at 37°C. Huh7 cells harboring HCV subgenomic replicon derived from genotype 1b and IFN-cured cells were grown as reported previously (23).
GST pulldown assay.
A glutathione S-transferase (GST)-sorcin fusion protein was expressed in Escherichia coli BL21 and purified with glutathione-Sepharose 4B beads (Amersham Biosciences) according to the manufacturer's instructions. HEK293T cells were transfected with the pEF6B Myc-tagged NS5A plasmid. At 36 h after transfection, cells were harvested in lysis buffer. The cell lysates were centrifuged at 13,500 rpm for 15 min, and then the protein concentration was determined by the Bradford assay (Bio-Rad). For an in vitro binding assay, Myc-tagged NS5A was incubated with either GST or a GST-sorcin fusion protein for 2 h at 4°C in cell lysis buffer. The samples were washed five times in lysis buffer, and then bound protein was detected by immunoblot assay.
Luciferase reporter assay.
For a dual-luciferase reporter assay, Huh7.5 cells were cotransfected with a pRL-HL plasmid containing the Renilla luciferase gene under the cytomegalovirus (CMV) promoter and the firefly luciferase gene under the control of the HCV internal ribosome entry site (IRES) and the plasmid indicated in the figures together with the pCH110 reference plasmid. At 48 h after transfection, cells were harvested, and then luciferase assays were performed as we described previously (22).
MTT assay.
Approximately 1.5 × 104 cells seeded on 24-well plates were transfected with the siRNAs indicated in the figures. Cell viability was determined by using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma) assay according to the manufacturer's protocol.
Immunoblot analysis.
Cells were washed twice with phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.5], 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) for 15 min on ice and centrifuged at 12,000 rpm for 10 min at 4°C. The supernatant was collected, and equal amounts of protein were subjected to SDS-PAGE and electrotransferred to a nitrocellulose membrane. The membrane was blocked in Tris-buffered saline (TBS)-Tween (20 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 0.25% Tween 20) containing 5% nonfat dry milk for 1 h and then incubated with the antibodies indicated in the figures. Proteins were detected using an ECL kit (Abfrontier).
Quantification of RNA.
Total RNAs were isolated from cells using RiboEX reagent (GeneAll) according to the manufacturer's instructions. cDNAs were synthesized by using a cDNA synthesis kit (Toyobo) according to the manufacturer's instructions. Quantitative real-time PCR (qRT-PCR) experiments were performed using the following primers: for HCV, 5′-TTA GTA TGA GAG TCG TAC AGC CTC CAG-3′ (sense) and 5′-GGC ATA GAG TGG GTT TAT CCA AGA AAG G-3′ (antisense); for β-actin, 5′-TGA CAG CAG TCG GTT GGA GCG-3′ (sense) and 5′-GAC TTC CTG TAA CAA CGC ATC TCA TA-3′ (antisense); for sorcin, 5′-TCC GCT GTA TGG TTA CTT TC-3′ (sense) and 5′-GGT GAT CTT TCC ATT GGT G-3′ (antisense). qRT-PCR experiments were done using a CFX Connect real-time system (Bio-Rad Laboratories, Hercules, CA) under the following conditions: 15 min at 95°C, followed by 40 cycles of 95°C for 20 s, 55°C for 20 s, and 73°C for 20 s. Seventy-one cycles of 10 s, with 0.5°C temperature increments from 60°C to 95°C, were used for the melting curves.
Immunoprecipitation.
HEK293T cells were cotransfected with V5-tagged sorcin, Myc-tagged NS5A, and Flag-tagged PLK1 plasmids. Total amounts of DNA were adjusted by adding an empty vector. At 48 h after transfection, cells were harvested, and an immunoprecipitation assay was performed as we described previously (24).
RNA interference.
siRNAs targeting sorcin (sorcin siRNA 1, 5′-CUC AGG AUC CGC UGU AUG G-3′ [sense] and 5′-CCA UAC AGC GGA UCC UGA G-3′ [antisense]; sorcin siRNA 2, 5′-AAU GCU GGA UAG AGA UAU GUC-3′ [sense] and 5′-GAC AUA UCU CUA UCC AGC AUU-3′ [antisense]) and the universal negative-control siRNA were purchased from Bioneer (South Korea). An siRNA targeting the 5′ nontranslated region (NTR) of the Jc1 virus (5′-CCUCAAAGAAAAACCAAACUU-3′) was used as a positive control (24). siRNA transfection was performed using Lipofectamine RNAiMax reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol.
Immunofluorescence assay.
Huh7.5 cells grown on cover slides were fixed with 4% paraformaldehyde in PBS for 15 min and then permeabilized with 0.1% Triton X-100 in PBS for 10 min at 37°C. After three washes with PBS, fixed cells were blocked with 1% bovine serum albumin (BSA) in PBS for 1 h at room temperature. The cells were then incubated with an anti-V5 monoclonal antibody and a rabbit anti-NS5A antibody. After three washes with PBS, cells were incubated with either fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG or tetramethylrhodamine isothiocyanate (TRITC)-conjugated donkey anti-rabbit IgG for 1 h at room temperature. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to label nuclei. After three washes with PBS, cells were analyzed using a Zeiss LSM 700 laser confocal microscopy system (Carl Zeiss, Inc., Thornwood, NY).
Protease protection assay.
A protease protection assay was performed as reported previously (25). Briefly, Huh7.5 cells were infected with the Jc1 virus. At 48 h postinfection, cells were transfected with the siRNAs indicated in the figures. At 48 h after transfection, cells were harvested in buffer containing 50 mM Tris-HCl (pH 8.0), 10 mM CaCl2, and 1 mM dithiothreitol (DTT). Cells were then subjected to five cycles of freezing and thawing. Crude cell lysates were either left untreated or treated with 0.5 μg/ml proteinase K (Roche, Mannheim, Germany) for 1 h on ice or treated with 5% Triton X-100 prior to proteinase K treatment. Proteinase K digestion was terminated with the addition of 5 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich) for 10 min on ice. The amount of protease-resistant core was determined by immunoblot assay.
Statistical analysis.
Data are presented as means ± standard deviations (SDs). Student's t test was used for statistical analysis. The asterisks on the figures indicate significant differences, as noted in the legends.
RESULTS
Sorcin is identified as a host factor interacting with NS5A protein.
We recently performed a protein microarray assay using the NS5A protein as a probe to identify cellular proteins interacting with NS5A (22). Statistically significant proteins were determined as NS5A interactors. Sorcin was identified as one of 90 candidates (Fig. 1A). Sorcin is a protein that participates in the regulation of calcium homeostasis in cells. Since sorcin interacts with FMDV VP1 and modulates the cellular type 1 interferon response (20), we selected sorcin and explored its possible involvement in HCV propagation. To verify the protein array data, we first performed an in vitro binding assay using GST-tagged sorcin purified from E. coli cells and cell lysates expressing Myc-tagged NS5A. The results shown in Fig. 1B indicated that NS5A specifically interacted with sorcin. Coimmunoprecipitation data further demonstrated that NS5A selectively interacted with sorcin (Fig. 1C). Next, we investigated whether NS5A protein interacted with endogenous sorcin. Cell lysates harvested at 4 days after HCV RNA electroporation were immunoprecipitated with either control rabbit serum or an anti-NS5A antibody, and bound proteins were analyzed by immunoblotting with an anti-sorcin antibody. As shown in Fig. 1D, HCV NS5A interacted with endogenous sorcin. These data suggested that sorcin might colocalize with NS5A in HCV-infected cells. To investigate this possibility, Huh7.5 cells were either mock infected or infected with Jc1, followed by transfection with V5-tagged sorcin, and an immunofluorescence assay was performed. Results shown in Fig. 1E demonstrated that sorcin was dispersedly expressed in both the nucleus and the cytoplasm in mock-infected cells (upper panel), whereas sorcin and HCV NS5A were colocalized in the perinuclear region in Jc1-infected cells, as indicated by the yellow fluorescence. Furthermore, we verified that endogenous sorcin was colocalized with NS5A in Jc1-infected cells (Fig. 1F). Colocalization of NS5A and sorcin was further verified by determining both Pearson's and Mander's coefficients. Collectively, these data suggest that NS5A specifically interacts with sorcin both in vitro and in vivo.
FIG 1.

(A) Identification of sorcin in microarray. Ab, antibody. (B) Sorcin interacts with NS5A protein. HEK293T cells were transiently transfected with a Myc-tagged NS5A plasmid. Total cell lysates were harvested and incubated with either purified GST or GST-tagged sorcin. GST-bound protein was detected by immunoblot analysis using an anti-Myc monoclonal antibody. (C) HEK293T cells were cotransfected with Myc-tagged NS5A and V5-tagged sorcin expression plasmids. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-Myc monoclonal antibody, and then bound proteins were detected by immunoblot assay using an anti-V5 monoclonal antibody. Protein expression of Myc-tagged NS5A and V5-tagged sorcin was verified by immunoblotting with the indicated antibody. (D) NS5A protein interacts with the endogenous sorcin in HCV replicating cells. Huh7.5 cells were electroporated with 10 μg of Jc1 RNA. Cells lysates harvested at 4 days after electroporation were immunoprecipitated with either control serum or an anti-NS5A antibody. Bound protein was immunoblotted with an anti-sorcin antibody. Immunoprecipitation efficiency was verified by immunoblot analysis using an anti-NS5A antibody (lower panel). (E) Huh7.5 cells were either mock infected or infected with Jc1 for 4 h. At 48 h postinfection, cells were transfected with V5-tagged sorcin. At 24 h after transfection, cells were fixed in 4% paraformaldehyde, and immunofluorescence staining was performed by using an anti-V5 monoclonal antibody and fluorescein isothiocyanate-conjugated goat anti-mouse IgG to detect V5-tagged sorcin (green); a rabbit anti-NS5A antibody and TRITC-conjugated donkey anti-rabbit IgG were used to detect NS5A (red). Dual staining showed colocalization of sorcin and NS5A as yellow fluorescence in the merged image. Cells were counterstained with DAPI to label nuclei (blue). (F) Huh7.5 cells were either mock infected or infected with Jc1 for 4 h. At 48 h postinfection, cells were fixed in 4% paraformaldehyde, and immunofluorescence staining was performed by using an anti-sorcin monoclonal antibody and TRITC-conjugated donkey anti-mouse IgG to detect endogenous sorcin (red); a rabbit anti-NS5A antibody and fluorescein isothiocyanate-conjugated goat anti-rabbit IgG were used to detect NS5A (green). Dual staining showed colocalization of endogenous sorcin and NS5A as yellow fluorescence in the “Crop” image. IP, immunoprecipitation; IB, immunoblot; M1(R/G), Mander's colocalization coefficient (ratio of red and green fluorophore colocalization).
HCV NS5A interacts with sorcin through domain I of NS5A, and phosphorylation of the threonine residue 155 of sorcin is required for protein interaction with NS5A.
To determine the region in NS5A responsible for sorcin binding, the interaction between sorcin and various deletion mutants of NS5A (Fig. 2A) was determined by a transfection-based coimmunoprecipitation assay. As show in Fig. 2B, sorcin interacted with domain I and with domains I and II but not with domains II and III. This result indicated that domain I was responsible for binding with sorcin. It has been previously reported that sorcin is phosphorylated by PLK1 and that phosphorylation at threonine residue 155 of sorcin is important for sorcin activity (16). To examine whether phosphorylation at threonine residue 155 of sorcin was involved in protein interplay with NS5A, we constructed both phosphorylation-defective T155A and phosphomimetic T155D mutants (Fig. 2C). Using both wild-type and sorcin mutants, we determined the binding capabilities of these mutants with NS5A. As shown in Fig. 2D, the phosphomimetic T155D mutant was capable of interacting with NS5A, whereas the T155A sorcin mutant no longer interacted with NS5A, indicating that the phosphorylation of threonine residue 155 in sorcin was required for the binding activity of sorcin with NS5A.
FIG 2.

HCV NS5A interacts with sorcin through domain I of NS5A, and phosphorylation of the threonine residue 155 of sorcin is involved in protein interaction. (A) Schematic illustration of both the wild type (WT) and mutants of HCV NS5A. (B) HEK293T cells were cotransfected with the indicated combinations of expression plasmids. At 48 h after transfection, cell lysates were immunoprecipitated (IP) with an anti-V5 monoclonal antibody, and bound protein was detected by immunoblot (IB) analysis with an anti-Myc antibody. (C) Schematic illustration of both the wild type and sorcin mutants. (D) HEK293T cells were cotransfected with Myc-tagged NS5A and either the wild type or mutants of V5-tagged sorcin expression plasmids. Cell lysates harvested at 48 h after transfection were immunoprecipitated with an anti-Myc monoclonal antibody, and bound protein was detected by immunoblot analysis using an anti-V5 antibody. aa, amino acids.
PLK1-mediated phosphorylation of sorcin is increased by NS5A.
We next investigated whether phosphorylation of sorcin was also regulated by NS5A protein. For this purpose, HEK293T cells were cotransfected with Myc-tagged NS5A and either a V5-tagged wild-type sorcin or mutants of sorcin. Results shown in Fig. 3A indicate that phosphorylation levels of both the wild-type protein and the phosphomimetic T155D were highly increased by NS5A, whereas the phosphorylation level of the binding-defective sorcin mutant was slightly increased, indicating that phosphorylation of sorcin was also affected by NS5A. Since NS5A also interacts with PLK1 (26), we further examined the involvement of NS5A and PLK1 in the phosphorylation of sorcin. The experiment shown in Fig. 3B demonstrated that the phosphorylation level of sorcin was increased by NS5A (lane 2). However, the phosphorylation level of sorcin was unaffected by NS5A if PLK1 expression was knocked down (Fig. 3B, lane 3). It was noteworthy that the interaction between NS5A and sorcin was also reduced by knockdown of PLK1. To investigate whether the phosphorylation level of sorcin was upregulated in the context of HCV replication, Huh7.5 cells were either mock infected or infected with Jc1 and then transfected with V5-tagged sorcin. As shown in Fig. 3C, the phosphorylation level of sorcin was elevated in Jc1-infected cells compared to that in mock-infected cells. To investigate whether endogenous sorcin was activated by phosphorylation during HCV infection, we performed a time course experiment using HCV-infected cells to analyze the phosphorylation status of sorcin. Although the phosphorylation level of endogenous sorcin was not profound compared with the level of ectopic expression of sorcin, endogenous sorcin was activated by phosphorylation during HCV infection, and the phosphorylation level of sorcin increased in a time-dependent manner (Fig. 3D). Since sorcin is also phosphorylated by PLK1 (16), we investigated whether an interaction between sorcin and NS5A was affected by a PLK1 kinase inhibitor. Huh7.5 cells cotransfected with V5-tagged sorcin and Myc-tagged NS5A were either left untreated or treated with BI2536. As shown in Fig. 3E, the interaction between sorcin and NS5A was interrupted in PLK1 inhibitor-treated cells, further confirming that PLK1-mediated phosphorylation of sorcin was important for the interaction with NS5A. We further verified that the PLK1 inhibitor suppressed the phosphorylation of sorcin (Fig. 3E, bottom). It has been previously reported that sorcin interacts with PLK1 (16) and that NS5A is coimmunoprecipitated with PLK1 (26). By performing a coimmunoprecipitation assay using HEK293T cells cotransfected with V5-tagged sorcin, Flag-tagged PLK1, and Myc-tagged NS5A, we further demonstrated that both NS5A and PLK1 could be coimmunoprecipitated with sorcin, indicating that sorcin might have dual binding sites for both NS5A and PLK1 proteins (Fig. 3F). These data suggest that NS5A may regulate sorcin activity by tethering sorcin in close proximity to PLK1 in HCV-infected cells.
FIG 3.

Sorcin is phosphorylated at threonine residue 155 by PLK1 in the presence of NS5A. (A) HEK293T cells were cotransfected with Myc-tagged NS5A and V5-tagged wild-type and mutant constructs of sorcin. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-V5 monoclonal antibody and then immunoblotted with an anti-phospho-threonine (p-Thr) monoclonal antibody. Bound proteins were also immunoblotted with an anti-Myc monoclonal antibody. (B) Huh7.5 cells were transfected with either 20 nM negative siRNA or a PLK1-specific siRNA. At 24 h after transfection, cells were further transfected with V5-tagged sorcin in the absence or presence of Myc-tagged NS5A plasmid. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-V5 monoclonal antibody, and bound proteins were immunoblotted with an anti-Myc monoclonal antibody. Knockdown efficiency of PLK1 was verified by immunoblot assay using an anti-PLK1 monoclonal antibody. Phosphorylation of sorcin was analyzed by immunoblotting with an anti-phospho-threonine monoclonal antibody using the same immunoprecipitates. (C) Huh7.5 cells were either mock infected or infected with Jc1 for 4 h and then further transfected with V5-tagged sorcin. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-V5 monoclonal antibody, and bound proteins were immunoblotted with the indicated antibodies. Phosphorylation of sorcin was analyzed by immunoblotting with an anti-phospho-threonine monoclonal antibody using the same immunoprecipitates. (D) Huh 7.5 cells were infected with Jc1 for 4 h and then harvested at the indicated time points. Cell lysates were immunoprecipitated with an anti-sorcin antibody, and bound protein was immunoblotted with an anti-NS5A antibody. Phosphorylation of sorcin was analyzed by immunoblotting with an anti-phospho-threonine monoclonal antibody. p.i., postinfection. (E) Huh7.5 cells were cotransfected with V5-tagged sorcin in the absence or presence of Myc-tagged NS5A plasmid. Cells were either left untreated or treated with 1 μM BI2536 PLK1 inhibitor. At 24 h after treatment, cell lysates were immunoprecipitated with an anti-V5 monoclonal antibody, and bound proteins were immunoblotted with an anti-Myc monoclonal antibody. Expression of sorcin was verified by immunoblotting with an anti-V5 antibody. Phosphorylation of sorcin was analyzed by immunoblotting with an anti-phospho-threonine monoclonal antibody. (F) HEK293T cells were cotransfected with V5-tagged sorcin, Flag-tagged PLK1, and Myc-tagged NS5A. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-V5 antibody, and bound proteins were immunoblotted with either an anti-Flag or an anti-Myc antibody. Protein expression was verified by immunoblotting with the indicated antibodies.
Sorcin is required for HCV propagation.
To investigate the functional involvement of sorcin in HCV propagation, Huh7.5 cells transfected with two different siRNAs targeting sorcin were infected with Jc1, and then both RNA and protein levels were determined. Knockdown of sorcin significantly suppressed the HCV RNA level (Fig. 4A, left panel). Consistently, HCV protein expression levels were also impaired in sorcin knockdown cells (Fig. 4A, right panel). To further investigate the role of sorcin in HCV propagation, naive Huh7.5 cells were infected with Jc1 harvested from the supernatant of the first infection (Fig. 4A), and then both RNA and protein levels of HCV were analyzed. As shown in Fig. 4B, viral infectivity in the second infection was significantly decreased by knockdown of sorcin. To verify this result, we determined the 50% tissue culture infective dose (TCID50) of HCV in sorcin knockdown cells. The data in Fig. 4C show that HCV infectivity was significantly decreased in sorcin knockdown cells. We further demonstrated that treatment of the same concentration of siRNAs displayed no cytotoxicity in Jc1-infected cells, indicating that the silencing effect was specific to sorcin (Fig. 4D). To rule out the off-target effect of a sorcin siRNA, we generated an siRNA-resistant sorcin mutant. As shown in Fig. 4E, exogenous expression of the siRNA-resistant sorcin mutant, but not of wild-type sorcin, restored the HCV protein expression level (lane 4). Collectively, these data suggest that sorcin is required for HCV propagation.
FIG 4.

Sorcin is required for HCV propagation. (A) Huh7.5 cells were transfected with a 20 nM concentration of the indicated siRNAs. At 2 days after siRNA transfection, cells were infected with Jc1. At 48 h postinfection both RNA and protein levels were analyzed by qRT-PCR and immunoblot assay, respectively. (B) Naive Huh7.5 cells were infected with Jc1 harvested from cultured supernatants of the experiment shown in panel A. At 48 h postinfection, both HCV RNA and protein levels were determined. Results are presented as a percentage of negative siRNA (means ± SDs; n = 3). The asterisks indicate significant differences (**, P < 0.01; ***, P < 0.001) from the value for the negative control. (C) Huh7.5 cells were transfected with a 20 nM concentration of the indicated siRNAs. At 2 days after transfection, cells were infected with green fluorescent protein-tagged Jc1. At 48 h postinfection, HCV infectivity was determined by limiting dilution assays. Infected cells were assessed by fluorescence microscope. TCID50, 50% tissue culture infectious dose. (D) Huh7.5 cells were transfected with a 20 nM concentration of the indicated siRNAs, and cell viability was assessed by MTT assay. (E) Huh7.5 cells were transfected with the indicated siRNAs. At 24 h after siRNA transfection, cells were further transfected with either a wild-type or siRNA-resistant sorcin mutant plasmid, followed by Jc1 infection. At 48 h postinfection, cell lysates were immunoblotted using the indicated antibodies. Positive (or Pos), HCV-specific siRNA targeting the 5′ nontranslated region (NTR) of Jc1; negative (or Neg), the universal negative-control siRNA; V5-sorcin WT, V5-tagged sorcin wild type; V5-sorcin-Res, V5-tagged siRNA-resistant sorcin mutant.
Sorcin is not involved in the replication and translation steps of the HCV life cycle.
To examine which steps of the HCV life cycle were required for sorcin, Huh7.5 cells were transiently transfected with increasing amounts of sorcin expression plasmid together with pRL-HL and a β-galactosidase plasmid as we reported previously (23), and then luciferase activity was determined. As shown in Fig. 5A, overexpression of sorcin had no effects on HCV IRES-dependent translation. To further explore if sorcin was involved in HCV replication, sorcin expression in Huh7.5 cells harboring HCV subgenomic replicon was impaired by knockdown of siRNAs, and both RNA and protein levels were analyzed. As shown in Fig. 5B, silencing of sorcin exerted no effect on intracellular HCV RNA and protein levels in subgenomic replicon cells derived from genotype 1b. We further verified the consistent results in subgenomic replicon cells derived from genotype 2a (Fig. 5C). These data suggested that sorcin might be involved in other steps of the HCV life cycle.
FIG 5.

Sorcin is not involved in the replication and translation steps of the HCV life cycle. (A) A schematic diagram of the pRL-HL plasmid is shown at top. Huh7.5 cells were transiently transfected with increasing amounts of V5-tagged sorcin expression plasmid together with pRL-HL dual luciferase and a pCH110 β-galactosidase plasmid. At 48 h after transfection, relative luciferase activity was determined (bottom). BGH-pA denotes bovine growth hormone polyadenylation signal sequence. (B) Huh7.5 cells harboring an HCV subgenomic replicon derived from genotype 1b were transfected with the indicated siRNAs. At 72 h after siRNA transfection, both RNA (left) and protein (right) levels were analyzed by qRT-PCR and immunoblot assay, respectively. (C) Huh6 cells harboring an HCV subgenomic replicon derived from genotype 2a were treated as described for panel B, and both RNA (left) and protein (right) levels were determined. Positive, HCV-specific siRNA targeting the 5′ nontranslated region (NTR) of Jc1; negative (or Neg), universal negative-control siRNA.
Sorcin is required for the assembly or release stage of the HCV life.
To investigate if sorcin was required for a later stage of the HCV life cycle, Huh7.5 cells were infected with the Jc1 virus for 4 h. At 48 h postinfection, cells were further transfected with either a negative siRNA or a sorcin-specific siRNA or with an apolipoprotein E (ApoE) siRNA as a control. Although the mRNA level of sorcin was significantly suppressed by siRNA treatment, the intracellular HCV RNA level was unaffected (Fig. 6A). Consistently, the extracellular HCV RNA level was not influenced by knockdown of sorcin (Fig. 6B). Interestingly, when naive Huh7.5 cells were infected with Jc1 harvested from the culture supernatant of the experiment shown in Fig. 6A, intracellular HCV RNA levels in the second infection were decreased by 60% in sorcin knockdown cells (Fig. 6C), indicating that sorcin might be involved in virion assembly or release. To verify these results, intracellular HCV infectivity was determined. Huh7.5 cells infected with Jc1 were transfected with the siRNAs indicated in Fig. 6D. Either at 24 h or 48 h postinfection, cells were disrupted by repeated cycles of freezing and thawing. By infection of naive Huh7.5 cells with cellular supernatant, relative intracellular HCV infectivity was determined by qRT-PCR. As shown in Fig. 6D, intracellular HCV infectivity was significantly decreased in sorcin knockdown cells. We further verified that the HCV protein level was similarly reduced by a second infection in sorcin knockdown cells (Fig. 6E). These data suggested that sorcin might be involved in either the assembly or release stage of the HCV life cycle.
FIG 6.

Sorcin is required for a late step of the HCV life cycle. (A and B) Huh7.5 cells were infected with Jc1 for 4 h. At 48 h postinfection, cells were transfected with a 20 nM concentration of the indicated siRNAs. At 24 h after siRNA transfection, medium was replaced with fresh DMEM containing antibiotics. At the indicated time points, intracellular RNA levels (A) and extracellular HCV RNA levels (B) were determined by qRT-PCR. (C) Naive Huh7.5 cells were infected with Jc1 harvested from culture supernatants of the experiment shown in panel A, and viral infectivity was determined by measuring intracellular HCV RNA levels by qRT-PCR. (D) Huh7.5 cells treated as described in panel A were lysed with three cycles of freezing and thawing and centrifuged at 15,000 × g for 15 min in a 4°C microcentrifuge. The supernatant was collected to determine intracellular HCV infectivity. Naive Huh7.5 cells were infected with Jc1 harvested from the intracellular supernatant for 4 h. At 48 h postinfection, relative intracellular HCV infectivity was determined by qRT-PCR. (E) Huh7.5 cells were infected with Jc1 for 4 h (upper panel). At 48 h postinfection, cells were transfected with a 20 nM concentration of the indicated siRNAs. At the indicated time points, protein levels were determined by immunoblot assays using the indicated antibodies. For the second infection (lower panel), naive Huh7.5 cells were infected with Jc1 harvested from culture supernatants of the experiment described for the upper panel. At 48 h postinfection, protein levels were determined by immunoblot assays using the indicated antibodies. Negative (or Neg), universal negative-control siRNA.
Sorcin is involved in the capsid assembly step of the HCV life cycle.
To precisely investigate whether sorcin was involved in HCV assembly, a protease protection assay was performed as reported previously (25). Huh7.5 cells infected with Jc1 were transfected with the siRNAs indicated in Fig. 7A. At 48 h after transfection, cells were either left untreated, treated with proteinase K, or treated with Triton X-100 and proteinase K. The protease-resistant core protein level was then determined by immunoblot assay. In the negative-siRNA-treated cells, core protein was relatively resistant to protease digestion (Fig. 7A, lane 2), suggesting that the core protein might acquire a membrane coat in Jc1-infected cells. Meanwhile, knockdown of sorcin displayed remarkable reduction of protected core protein (Fig. 7A, lane 5), indicating that the core was highly sensitive to protease in the absence of sorcin. Since our siRNA-resistant sorcin mutant rescued the effect of sorcin knockdown in HCV propagation (Fig. 4E), we asked whether protease sensitivity in sorcin knockdown cells could be reversed by expression of an siRNA-resistant sorcin mutant. For this purpose, Huh7.5 cells infected with Jc1 were transfected with the siRNAs indicated in Fig. 7B. At 24 h after siRNA transfection, cells were further transfected with an empty vector, an siRNA-resistant sorcin mutant, and a phosphorylation-defective siRNA-resistant sorcin mutant plasmid. As shown in Fig. 7B, the core protein level in cells expressing the siRNA-resistant sorcin mutant (lane 8) was restored compared with that in vector transfected cells (lane 5). Nevertheless, cells expressing the phosphorylation-defective siRNA-resistant sorcin mutant were unable to recover the core protein level (Fig. 7B, lane 11), indicating that phosphorylation of sorcin was involved in capsid formation. These data suggest that sorcin might be involved in the capsid assembly stage of the HCV life cycle.
FIG 7.

Sorcin is involved in the assembly step of the HCV life cycle. (A) Huh7.5 cells were infected with Jc1 for 4 h. At 48 h postinfection, cells were transfected with a 20 nM concentration of either a negative or sorcin-specific siRNA. At 48 h after transfection, cells were either left untreated (lanes 1, 2, 4, and 5), treated with 0.5 μg/ml proteinase K (lanes 2 and 5) for 1 h on ice, or treated with 5% Triton X-100 prior to proteinase K treatment (lanes 3 and 6). The amounts of protease-resistant core protein were determined by immunoblot assay. (B) Huh7.5 cells were infected with Jc1 and transfected with a 20 nM concentration of either negative or sorcin-specific siRNA as described for panel A. At 24 h after siRNA transfection, cells were transfected with vector, an siRNA-resistant sorcin mutant (sorcin-Res), and a phosphorylation-defective siRNA-resistant sorcin mutant (sorcin-Res+T155A) expression plasmid. At 48 h after transfection, samples were immunoblotted with the indicated antibodies. Negative, universal negative-control siRNA.
Calcium-binding activity of sorcin is required for HCV propagation.
Sorcin is a calcium-binding protein, and the calcium-binding domain contains eight α-helices (A to H) organized in five calcium-binding motifs (EF1-EF5). EF2 and EF3 are linked by helix D and have a high affinity with metal (27). It has been previously reported that an F112L mutant of sorcin displays a marked decreased in Ca2+ affinity (28). We therefore asked if calcium-binding activity was required for HCV propagation. To explore this possibility, we first constructed an F112L sorcin mutant and examined its binding ability with NS5A by a coimmunoprecipitation assay. As shown in Fig. 8A, the calcium-binding-defective F112L sorcin mutant demonstrated an impaired interaction with NS5A compared to that of the wild-type sorcin (Fig. 8A, lane 6 versus lane 5). To further investigate the effect of the F112L mutation on HCV propagation, we mutated the phenylalanine residue at amino acid 112 to leucine using an siRNA-resistant sorcin mutant as a template and named it sorcin-Res+F112L. As demonstrated in Fig. 8B, knockdown of sorcin impaired HCV protein expression (lane 3), and transient expression of the siRNA-resistant sorcin mutant restored the HCV protein expression level (lane 4). However, the calcium-binding-defective F112L in the siRNA-resistant sorcin mutant was unable to restore the HCV protein expression level (Fig. 8B, lane 5), indicating that sorcin with its calcium-binding affinity was required for HCV propagation.
FIG 8.
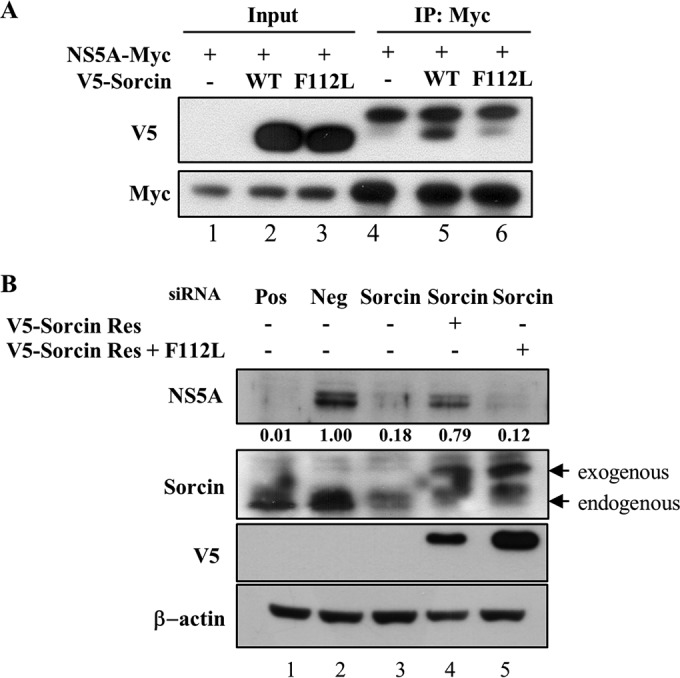
Calcium-binding activity of sorcin is required for HCV propagation. (A) HEK293T cells were cotransfected with a Myc-tagged NS5A and V5-tagged wild-type or mutant sorcin plasmid. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-Myc monoclonal antibody, and bound proteins were immunoblotted with an anti-V5 antibody. Immunoprecipitation efficiency was verified by immunoblotting with an anti-Myc monoclonal antibody using the same lysates. (B) Huh7.5 cells were transfected with the indicated siRNAs. At 24 h after siRNA transfection, cells were transfected with the indicated combinations of expression plasmids and then infected with Jc1 for 4 h. At 48 h postinfection, cell lysates were immunoblotted using the indicated antibodies. Band intensities of HCV NS5A protein were normalized against those of β-actin. Pos, HCV-specific siRNA targeting the 5′ nontranslated region (NTR) of Jc1; Neg, universal negative-control siRNA; V5-sorcin-Res, V5-tagged siRNA-resistant sorcin mutant; V5-sorcin-Res+F112L, V5-tagged siRNA-resistant calcium-binding-defective sorcin mutant.
DISCUSSION
NS5A is a pleiotropic protein and has been shown to play an essential role in viral replication. To identify host proteins interacting with NS5A, we recently employed protein array screening and identified approximately 90 human candidates as HCV NS5A interactors (22). Since sorcin interacts with the VP1 protein of FMDV and regulates the host immune response (20), we selected sorcin to explore its involvement in HCV propagation. Sorcin regulates intracellular calcium homeostasis (14, 15, 19) and plays a role in the induction of drug resistance in human cancers (18). Sorcin is overexpressed in human cancer as an adaptive mechanism to prevent ER stress and escape apoptosis triggered by chemotherapeutic agents (19). Inhibition of sorcin expression by sorcin-targeting RNA interference results in a decrease in drug resistance (18).
In this study, a protein interaction between sorcin and NS5A was confirmed by both in vitro and coimmunoprecipitation assays. We verified that endogenous sorcin colocalized with NS5A in the cytoplasm of Jc1-infected cells. The interaction between sorcin and NS5A was mediated by domain I of NS5A. We further demonstrated that the phosphorylation status of threonine residue 155 in sorcin regulated binding activity of sorcin with NS5A. It has been reported that sorcin interacts with PLK1 in a calcium-dependent manner and is phosphorylated at threonine residue 155 by PLK1 (16). Sorcin also induces PLK1 autophosphorylation to regulate kinase activity. It has also been reported that PLK1 physically interacts with NS5A and phosphorylates it and thereby indirectly regulates HCV replication (26). Since sorcin is phosphorylated by PLK1 and since phosphorylation of sorcin is important for sorcin activity (16), we explored the possible involvement of the phosphorylation of sorcin in protein-protein interactions. Indeed, we demonstrated that phosphorylation at threonine residue 155 of sorcin played a crucial role in the interaction with the HCV NS5A protein. By using a PLK1 inhibitor, we further verified that PLK1-mediated phosphorylation of sorcin was required for the interaction with NS5A. Interestingly, the PLK1-mediated phosphorylation level of sorcin was increased by NS5A. In fact, we demonstrated that sorcin interacted with both NS5A and PLK1. All these data suggest that NS5A may regulate sorcin activity through PLK1 in HCV-infected cells.
We then explored the functional involvement of sorcin in HCV propagation. HCV infectivity was significantly decreased in sorcin knockdown cells. We demonstrated that sorcin was not involved in HCV replication and IRES-mediated translation. However, intracellular HCV infectivity was significantly reduced in sorcin knockdown cells, indicating that sorcin might be involved in the assembly or release step of the HCV life cycle. By protease protection assay, we indeed demonstrated that the HCV core protein was protected by cellular sorcin and thereby that sorcin played a crucial role in the envelopment of the viral core protein into membranes. We further showed that a phosphorylation-defective sorcin mutant was unable to protect the HCV core protein. Together, these data suggest that sorcin may be involved in the assembly or release step of the HCV life cycle. However, how the interaction between sorcin and NS5A affects HCV assembly needs further investigation. Using a genome-wide siRNA screen, Li et al. reported that sorcin was identified as an antiviral factor involved in a late step of the replication cycle of HCV (21). Interestingly, our data showed that the expression level of ApoE was decreased in sorcin knockdown cells (Fig. 6E) and that transient expression of a sorcin-resistant mutant partially restored the ApoE level in sorcin knockdown cells (data not shown). Since ApoE likely is required for an intracellular maturation step of HCV particles (29), we hypothesized that sorcin might partially regulate HCV assembly and release via ApoE. Nevertheless, how sorcin and ApoE are interconnected in HCV assembly and release is not clear. Moreover, how sorcin is involved in the HCV life cycle remains largely undefined.
Since sorcin is a calcium-binding protein, we then investigated the effect of the calcium-binding activity of sorcin on HCV propagation. We showed that a calcium-binding-defective F112L sorcin mutant displayed impairment in an interaction with NS5A. Most importantly, a calcium-binding-defective sorcin mutant no longer played a positive role in HCV propagation. We further demonstrated that both phosphorylation and the calcium-binding activity of sorcin played a crucial role in protein-protein interactions. We reasoned that both the phosphorylation and calcium-binding sites are so closely located that phosphorylation of sorcin might regulate Ca2+-binding capability thorough conformational rearrangement. It has also been reported that sorcin interacts with the sarcoplasmic reticulum (SR) Ca2+-ATPase and modulates excitation-contraction coupling in the heart by activating Ca2+ uptake (13). Furthermore, sorcin is phosphorylated at serine 178 by PKA, and PKA phosphorylation of sorcin increases Ca2+ sensitivity and translocates sorcin to the SR (13). Since the Ca2+ level in the ER regulates virus maturation and cellular localization of viral proteins in human immunodeficiency virus (30) and rotavirus (31), HCV may regulate the calcium affinity of sorcin to facilitate virus propagation. However, further studies are needed to elucidate the precise role of sorcin in HCV propagation.
ACKNOWLEDGMENTS
We thank Charles Rice (The Rockefeller University) for Huh7.5 cells, Ralf Bartenschlager (University of Heidelberg) for the Jc1 construct, and Hyukjin Cha (Sogang University) and Hyungshin Yim (Hanyang University) for the PLK1 antibody.
This work was supported by the Basic Science Research Program (2015R052697) from the Ministry of Science, ICT and Future Planning, South Korea, and by the Korean Health Technology R&D Project (HI13C1746), Ministry of Health and Welfare, South Korea .
We declare that we have no conflicts of interest.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Giannini C, Brechot C. 2003. Hepatitis C virus biology. Cell Death Differ 10(Suppl 1):S27–S38. doi: 10.1038/sj.cdd.4401121. [DOI] [PubMed] [Google Scholar]
- 2.Lindenbach BD, Rice CM. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 3.Lemay KL, Treadaway J, Angulo I, Tellinghuisen TL. 2013. A hepatitis C virus NS5A phosphorylation site that regulates RNA replication. J Virol 87:1255–1260. doi: 10.1128/JVI.02154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79:3187–3194. doi: 10.1128/JVI.79.5.3187-3194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 6.Shimakami T, Hijikata M, Luo H, Ma YY, Kaneko S, Shimotohno K, Murakami S. 2004. Effect of interaction between hepatitis C virus NS5A and NS5B on hepatitis C virus RNA replication with the hepatitis C virus replicon. J Virol 78:2738–2748. doi: 10.1128/JVI.78.6.2738-2748.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 8.Pawlotsky JM, Germanidis G, Neumann AU, Pellerin M, Frainais PO, Dhumeaux D. 1998. Interferon resistance of hepatitis C virus genotype 1b: relationship to nonstructural 5A gene quasispecies mutations. J Virol 72:2795–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong G, Waris G, Tanveer R, Siddiqui A. 2001. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-κB. Proc Natl Acad Sci U S A 98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macdonald A, Crowder K, Street A, McCormick C, Saksela K, Harris M. 2003. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J Biol Chem 278:17775–17784. doi: 10.1074/jbc.M210900200. [DOI] [PubMed] [Google Scholar]
- 11.Xie X, Dwyer MD, Swenson L, Parker MH, Botfield MC. 2001. Crystal structure of calcium-free human sorcin: a member of the penta-EF-hand protein family. Protein Sci 10:2419–2425. doi: 10.1110/ps.ps.36701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyers MB, Pickel VM, Sheu SS, Sharma VK, Scotto KW, Fishman GI. 1995. Association of sorcin with the cardiac ryanodine receptor. J Biol Chem 270:26411–26418. doi: 10.1074/jbc.270.44.26411. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto T, Hisamatsu Y, Ohkusa T, Inoue N, Sato T, Suzuki S, Ikeda Y, Matsuzaki M. 2005. Sorcin interacts with sarcoplasmic reticulum Ca2+-ATPase and modulates excitation-contraction coupling in the heart. Basic Res Cardiol 100:250–262. doi: 10.1007/s00395-005-0518-7. [DOI] [PubMed] [Google Scholar]
- 14.Padar S, van Breemen C, Thomas DW, Uchizono JA, Livesey JC, Rahimian R. 2004. Differential regulation of calcium homeostasis in adenocarcinoma cell line A549 and its taxol-resistant subclone. Br J Pharmacol 142:305–316. doi: 10.1038/sj.bjp.0705755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maddalena F, Sisinni L, Lettini G, Condelli V, Matassa DS, Piscazzi A, Amoroso MR, La Torre G, Esposito F, Landriscina M. 2013. Resistance to paclitxel in breast carcinoma cells requires a quality control of mitochondrial antiapoptotic proteins by TRAP1. Mol Oncol 7:895–906. doi: 10.1016/j.molonc.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lalioti VS, Ilari A, O'Connell DJ, Poser E, Sandoval IV, Colotti G. 2014. Sorcin links calcium signaling to vesicle trafficking, regulates Polo-like kinase 1 and is necessary for mitosis. PLoS One 9:e85438. doi: 10.1371/journal.pone.0085438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anthony DF, Beattie J, Paul A, Currie S. 2007. Interaction of calcium/calmodulin-dependent protein kinase II delta C with sorcin indirectly modulates ryanodine receptor function in cardiac myocytes. J Mol Cell Cardiol 43:492–503. doi: 10.1016/j.yjmcc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Colotti G, Poser E, Fiorillo A, Genovese I, Chiarini V, Ilari A. 2014. Sorcin, a calcium binding protein involved in the multidrug resistance mechanisms in cancer cells. Molecules 19:13976–13989. doi: 10.3390/molecules190913976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddalena F, Laudiero G, Piscazzi A, Secondo A, Scorziello A, Lombardi V, Matassa DS, Fersini A, Neri V, Esposito F, Landriscina M. 2011. Sorcin induces a drug-resistant phenotype in human colorectal cancer by modulating Ca2+ homeostasis. Cancer Res 71:7659–7669. doi: 10.1158/0008-5472.CAN-11-2172. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Wang J, Liu J, Li Z, Wang Y, Xue Y, Li X, Cao H, Zheng SJ. 2013. Engagement of soluble resistance-related calcium binding protein (sorcin) with foot-and-mouth disease virus (FMDV) VP1 inhibits type I interferon response in cells. Vet Microbiol 166:35–46. doi: 10.1016/j.vetmic.2013.04.028. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Zhang YY, Chiu S, Hu Z, Lan KH, Cha H, Sodroski C, Zhang F, Hsu CS, Thomas E, Liang TJ. 2014. Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog 10:e1004163. doi: 10.1371/journal.ppat.1004163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park C, Min S, Park EM, Lim YS, Kang S, Suzuki T, Shin EC, Hwang SB. 2015. Pim kinase interacts with nonstructural 5A protein and regulates hepatitis C virus entry. J Virol 89:10073–10086. doi: 10.1128/JVI.01707-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim YS, Tran HT, Park SJ, Yim SA, Hwang SB. 2011. Peptidyl-prolyl isomerase Pin1 is a cellular factor required for hepatitis C virus propagation. J Virol 85:8777–8788. doi: 10.1128/JVI.02533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pham LV, Ngo HT, Lim YS, Hwang SB. 2012. Hepatitis C virus non-structural 5B protein interacts with cyclin A2 and regulates viral propagation. J Hepatol 57:960–966. doi: 10.1016/j.jhep.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Gentzsch J, Brohm C, Steinmann E, Friesland M, Menzel N, Vieyres G, Perin PM, Frentzen A, Kaderali L, Pietschmann T. 2013. Hepatitis C virus p7 is critical for capsid assembly and envelopment. PLoS Pathog 9:e1003355. doi: 10.1371/journal.ppat.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YC, Su WC, Huang JY, Chao TC, Jeng KS, Machida K, Lai MM. 2010. Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J Virol 84:7983–7993. doi: 10.1128/JVI.00068-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zamparelli C, Macquaide N, Colotti G, Verzili D, Seidler T, Smith GL, Chiancone E. 2010. Activation of the cardiac Na+-Ca2+ exchanger by sorcin via the interaction of the respective Ca2+-binding domains. J Mol Cell Cardiol 49:132–141. doi: 10.1016/j.yjmcc.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franceschini S, Ilari A, Verzili D, Zamparelli C, Antaramian A, Rueda A, Valdivia HH, Chiancone E, Colotti G. 2008. Molecular basis for the impaired function of the natural F112L sorcin mutant: X-ray crystal structure, calcium affinity, and interaction with annexin VII and the ryanodine receptor. FASEB J 22:295–306. [DOI] [PubMed] [Google Scholar]
- 29.Lee JY, Acosta EG, Stoeck IK, Long G, Hiet MS, Mueller B, Fackler OT, Kallis S, Bartenschlager R. 2014. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J Virol 88:12422–12437. doi: 10.1128/JVI.01660-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zegarra-Moran O, Rasola A, Rugolo M, Porcelli AM, Rossi B, Galietta LJ. 1999. HIV-1 nef expression inhibits the activity of a Ca2+-dependent K+ channel involved in the control of the resting potential in CEM lymphocytes. J Immunol 162:5359–5366. [PubMed] [Google Scholar]
- 31.Michelangeli F, Liprandi F, Chemello ME, Ciarlet M, Ruiz MC. 1995. Selective depletion of stored calcium by thapsigargin blocks rotavirus maturation but not the cytopathic effect. J Virol 69:3838–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]