

ABSTRACT
Following influenza A virus (IAV) infection, development of a robust IAV-specific CD8 T cell response is required for clearance of primary infection and enhances memory protection. Following IAV infection, plasmacytoid dendritic cells (pDC) or CD8α+ DC regulate pulmonary effector CD8 T cell responses within the lung. Without this DC-T cell interaction, insufficient effector CD8 T cells are maintained in the lungs, leading to enhanced morbidity and mortality. Previous studies have demonstrated that pDC are capable of classical presentation or cross-presentation of IAV antigens and could potentially regulate IAV-specific CD8 T cell responses through either mechanism. Our results demonstrate that pDC from the lungs of donor mice infected with an IAV that is not able to replicate in hematopoietic cells (142t-IAV), unlike donor pDC isolated from the lungs of control infected mice, are not able to rescue the host IAV-specific CD8 T cell response from apoptosis. This indicates that pDC must utilize the direct presentation pathway for this rescue. This inability of pDC from 142t-IAV donors to rescue the IAV-specific CD8 T cell response is not due to differences in the overall ability of 142t-IAV to replicate within the lungs or generate defective viral genomes or to differences in levels of costimulatory molecules required for this interaction. We further demonstrate that bypassing the antigen presentation pathway by coating the 142t-IAV pDC with IAV peptide epitopes restores their ability to rescue the IAV-specific CD8 T cell response.
IMPORTANCE IAV continues to be a global health burden, infecting 5 to 20% of the global population annually. Continued investigation into the mechanisms that mediate protective immune responses against IAV is important to improving current vaccination and antiviral strategies antagonistic toward IAV. Our findings presented herein demonstrate a key requirement for pDC promotion of effector CD8 T cell survival: that rather than utilizing cross-presentation, pDC must be infected and utilize the endogenous pathway for presentation of antigens to CD8 T cells during in vivo IAV infections. This suggests that targeting presentation via the endogenous pathway in pDC could be important for the development of unique antiviral cellular therapies.
INTRODUCTION
The development of a cytotoxic CD8 T cell response is key in clearance of influenza A virus (IAV) infection. Following activation of naive CD8 T cells in the lung-draining lymph nodes (dLNs), our studies have demonstrated that effector CD8 T cells must interact with either plasmacytoid DC (pDC) or CD8α+ DC within the lungs in order to avoid apoptosis, generate a full-magnitude CD8 T cell response, and lead to clearance of virus from the host (1, 2). The rescue of the T cells from death by DC during this pulmonary DC-CD8 T cell interaction requires presentation of viral antigen in the context of major histocompatibility complex class I (MHC-I), trans-presentation of interleukin 15 (IL-15), and CD70 (references 2 and 3 and unpublished observations).
During in vivo infection, IAV replicates in both lung epithelial cells and antigen-presenting cells (APCs) (4–7). DC are positioned in the airways and interstitium of the lungs and acquire viral antigens from surrounding dying epithelial cells or through direct infection. DC then process this antigen and express viral peptide-containing MHC-I molecules, which are utilized in the initial activation of naive CD8 T cells in the dLN and for maintaining the CD8 T cells within the lung. The viral peptides or proteins acquired from IAV-infected, dying epithelial cells and during direct infection of the DC are processed and presented via 2 pathways: cross-presentation and the endogenous pathway, respectively (reviewed in reference 8). While CD8α+ DC are well documented to be effective cross-presenters of antigens, pDC are typically not regarded as efficient cross-presenters (9, 10). Interestingly, however, pDC have been demonstrated to cross-present antigens following Toll-like receptor 7 (TLR7) stimulation (11) and produce type I interferon (IFN) following TLR7 activation during IAV infection (12). Therefore, it is possible that pDC could utilize the cross-presentation pathway to present viral antigens to CD8 T cells in the lungs during IAV infection. At this time, whether pDC can cross-present antigens during an in vivo IAV infection remains controversial (6, 13). While a pDC cell line has been demonstrated to cross-present viral antigens in vitro after exposure to IAV (13), exposure of human primary pDC to IAV has been reported to impair their ability to cross-present antigens to CD8 T cells (6). As antigen presentation by either pDC or CD8α+ DC to effector CD8 T cells is required within the lungs during IAV infection, the possibility that these DC subsets could utilize distinct pathways of antigen processing or presentation in order to maintain the pulmonary IAV-specific CD8 T cell response is intriguing. The utilization of disparate pathways by pDC and CD8α+ DC subsets to process and present antigen during their normal rescue of the IAV-specific CD8 T cell response from apoptosis in the lungs during IAV infection (2) would be the first distinction between these two DC subsets in their requirements for acquiring the ability to protect CD8 T cells from apoptosis and drive robust T cell immunity in the lungs. Knowledge of such potential distinctions would aid in further delineating the pathways that would need to be involved in the future design of therapeutic strategies to boost pulmonary CD8 T cell responses during infection. Therefore, we sought to determine whether these DC subsets utilize the endogenous or cross-presentation pathways to present antigens via MHC-I within the lungs during IAV infection.
Recently, Langlois et al. described an IAV containing four perfect targeting sequences for a hematopoietic cell-specific microRNA (miR-142) at the 3′ end of the nucleocapsid protein (NP) mRNA of A/PR/8/34 (142t-IAV) (14). As miR-142 is exclusively expressed in cells of hematopoietic origin (15), the NP mRNA is targeted for degradation, efficiently restricting replication of this virus within hematopoietic cells (14). While early RIG-I-mediated type I IFN production was reduced following infection with 142t-IAV, similar viral titers and clearance rates, weight loss, and accumulation of IAV-specific CD8 T cells were observed following infection with 142t-IAV compared to a control A/PR/8/34 (ctrl-IAV) containing random nucleotides instead of the miR142 targeting sequence (14). Further, even though the frequency of CD45+ CD11c+ that are hemagglutinin positive (HA+; i.e., infected) is significantly reduced in 142t-IAV infected mice compared to ctrl-IAV-infected mice, this virus does not appear to be attenuated. Therefore, the use of donor DC subsets from animals infected with 142t-IAV allows us to address the contribution of direct infection of the DC to pulmonary T cell responses.
Herein, we demonstrate that adoptively transferred CD8α+ DC are able to rescue the IAV-specific CD8 T cell response of pulmonary APC-depleted host mice regardless of whether they are purified from the lungs of mice infected with ctrl-IAV or 142t-IAV, suggesting that cross-presentation of viral antigens by CD8α+ DC is sufficient to prevent IAV-specific CD8 T cell apoptosis within the lungs. In contrast, while donor pDC from ctrl-IAV-infected lungs permit development of full magnitude IAV-specific CD8 T cell responses within the lungs of APC-depleted hosts, pDC from the lungs of 142t-IAV-infected donors do not rescue the IAV-specific CD8 T cell response, indicating that direct presentation by pDC is required.
MATERIALS AND METHODS
Mice.
Six- to 8-week-old wild-type (WT) BALB/c mice were obtained from the National Cancer Institute (Frederick, MD). All mice were housed and maintained in the animal care facility at the University of Iowa (Iowa City, IA). All experiments were performed in accordance with regulatory standards and guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Iowa.
Influenza virus infection.
Previously described A/PR/8/34 viruses containing a miR-142 targeting sequence (142t-IAV) or a control, nonsense sequence (ctrl-IAV) were grown in eggs (14). Host mice were anesthetized with isoflurane and infected intranasally (i.n.) with 474 tissue culture infectious units of ctrl-IAV (0.1 50% lethal dose [LD50 dose]) in 50 μl Iscove's modified Dulbecco's medium (IMDM). DC donor mice were infected with 47.4 tissue culture infectious units of ctrl-PR8 or 142t-IAV (0.01 LD50). Infection was quantified by performing plaque assays on lung samples as previously described (14).
Clodronate-liposome treatment.
Pulmonary DC and macrophage depletion was performed as previously described by treatment with liposomes containing dichloromethylene bisphosphonate (clodronate) (2). At 48 h postinfection (p.i.), mice were anesthetized by isoflurane inhalation and administered 75 μl of clodronate-liposomes or phosphate-buffered saline (PBS)-liposomes intranasally.
Peptides.
Influenza virus peptides HA533-541 (IYATVAGSL) and NP147-155 (TYQRTRALV) were synthesized by BioSynthesis Incorporated.
Preparation of cells.
Lungs were pressed through wire mesh to obtain a single cell suspension, which was then enumerated by trypan blue exclusion. For DC preparations, lungs were digested for 25 min at 25°C in media containing 1 mg/ml of collagenase (Sigma) and 0.02 mg/ml of DNase (Sigma) before single-cell preparation. The DC utilized for reconstitution experiments were purified from the lungs of day 6 ctrl-IAV- or 142t-IAV-infected BALB/c donors to 80 to 85% purity using magnetic activated cell sorting (MACS) technology (Miltenyi Biotech) according to the manufacturer's instructions, as previously described (2). Briefly, cells were stained with anti-CD3ε, anti-CD4, anti-CD19, anti-CD49b, anti-SiglecF, and anti-F4/80 monoclonal antibodies (MAbs) conjugated to phycoerythrin (PE) followed by anti-PE microbeads according to the manufacturer's instructions (Miltenyi Biotech). Labeled cells were isolated using an auto-MACS. The negative fraction was labeled with anti-CD8α microbeads according to the manufacturer's instructions (Miltenyi Biotech). Labeled cells were isolated using MACS LS columns, and microbeads were removed from cells using MACS Multisort release reagent from the MACS anti-PE Multisort kit according to the manufacturer's instructions. Labeled cells were then stained with anti-CD45R-PE followed by anti-PE microbeads according to the manufacturer's instructions. Labeled (pDC) and unlabeled (CD8α+ DC) cells were isolated using MACS LS columns, and microbeads were removed as described above. Cells were then diluted in trypan blue and counted using a hemacytometer to enumerate live pDC and CD8α+ DC prior to adoptive transfer. For peptide coating, purified pDC were incubated with 1 μM HA533 or NP147 peptide for 1 h in complete medium at 37°C, washed, and mixed at a 1:1 ratio prior to transfer. Purified cells were resuspended in IMDM, and 2.5 × 104 cells in 50 μl were adoptively transferred intranasally to host mice on day 3 p.i.
MHC-I tetramers.
MHC class I tetramers HA533-541 [H-2K(d)/IYSTVASSL] and NP147-155 [H-2K(d)/TYQRTRALV] were obtained from the National Institute of Allergy and Infectious Disease MHC Tetramer Core Facility (Atlanta, GA).
Antibody staining for flow cytometry.
Single-cell suspensions of lungs were prepared by pressing the tissues through wire mesh screens and plating 1 × 106 cells/well in a 96-well plate and were blocked with rat serum in fluorescence-activated cell sorter (FACS) buffer for 30 min at 4°C.
Following blocking, cells were incubated with FACS buffer containing rat anti-mouse CD8α (53-6.7; BD), rat anti-mouse CD3ε (145-2C11; BD), and HA533 or NP147 tetramers for 30 min at 4°C to identify IAV-specific CD8 T cells as previously described (1, 2). To identify pDC and CD8α+ DC populations, cells were incubated in FACS buffer containing hamster anti-mouse CD11c (HL3; BD), rat anti-mouse CD8α (53-6.7; BD), rat anti-mouse CD11b (M1/70; eBioscience), rat anti-mouse IA/IE (M5/114.15.2; eBioscience), rat anti-mouse CD45R (RA3-6B2; BioLegend), mouse anti-mouse MHC-I (34-1-2S; eBioscience), rat anti-mouse IL-15Rα (DNT15Ra; eBioscience), and rat anti-mouse CD70 (FR70; BioLegend) for 30 min at 4°C. Cells were then fixed in FACS lysis buffer (BD) per the manufacturer's instructions and resuspended in PBS.
For intracellular staining, cells were incubated in FACS buffer with saponin containing goat serum and 2.4G2 for 30 min on ice. Following blocking, cells were incubated with rabbit anti-HA (PY102) for 1 h on ice, washed 3 times, and subsequently incubated with goat anti-rabbit IgG conjugated to fluorescein isothiocyanate (FITC). Following incubation with secondary antibody, cells were washed 3 times and resuspended in PBS. Data were acquired on a BD FACSCanto II or a BD LSR II and analyzed with FlowJo software (TreeStar, Inc.).
Detection of IAV defective interfering particles.
Mice were anesthetized with isoflurane and infected intranasally with 0.1 LD50 of either ctrl-IAV or 142t-IAV. On days 3 and 5 p.i., lungs were harvested and TRIzol extraction was performed to isolate RNA. The SuperScript III One-Step reverse transcription-PCR (RT-PCR) system with Platinum Taq DNA polymerase (Invitrogen) was used to convert A/Puerto Rico/8/34 viral RNA samples into cDNA and to amplify the cDNA by PCR. The PB2 and NA gene segments were amplified from each of the samples utilizing the following previously described primers: for PB2, 5′-GTAGATGCAGCGAAAGCAGGTCAATTAT and 3′-GTAGCAGCAGTAGAAACAAGGTCGTTTT, and for NA, 5′-GTAGATGCAGCGAAAGCAGGGGTTTAAA and 3′-GTAGCAGCAGTAGAAACAAGGAGTTTTT (16). The samples were then loaded onto a 1% agarose gel with 0.012% ethidium bromide and run in Tris-acetate-EDTA buffer, and PB2 primer 3′-GTAGCAGCAGTAGAAACAAGGTCGTTTT was imaged on a Cell Biosciences gel doc.
Statistical analysis.
Data were compiled in graphical format using Prism software (GraphPad Software, San Diego, CA). Error bars in figures represent standard errors of the means (SEMs). Statistical significance for experiments with 2 groups was determined by using unpaired, two-tailed Student's t tests. For experiments containing 3 or more groups, statistical significance was determined by analysis of variance (ANOVA) followed by Tukey's multiple-comparison test.
RESULTS
142t-IAV replicates efficiently in respiratory epithelial cells but not pulmonary pDC.
Intranasal infection with ctrl-IAV or 142t-IAV induced similar weight losses and viral titers within the lungs, confirming that 142t-IAV is not attenuated (Fig. 1) (14). Although this virus is not attenuated, replication of 142t-IAV has been demonstrated to be restricted within the total CD45+ CD11c+ compartment. Therefore, we next confirmed the lack of 142t-IAV replication within pDC and CD8α+ DC. While we observed no difference in the number of pDC recruited to the infected lungs 4 days p.i., the frequency of pDC positive for intracellular HA (indicative of replicating virus) was similar to background levels in the lungs of mice infected with 142t-IAV compared to ctrl-IAV (Fig. 2A to C). This result is consistent with the published expression of miR-142 in pDC (data not shown) (17). Similar to pDC, replication of 142t-IAV was also limited within CD8α+ DC, but again, infection with 142t-IAV did not alter the total number of CD8α+ DC recruited to the lungs on day 4 p.i. (Fig. 2D). These results indicate that infection of DC with 142t-IAV is not permitted and suggest that any antigen presentation by either pDC or CD8α+ DC that occurs via MHC-I during 142t-IAV infection is due to cross-presentation.
FIG 1.
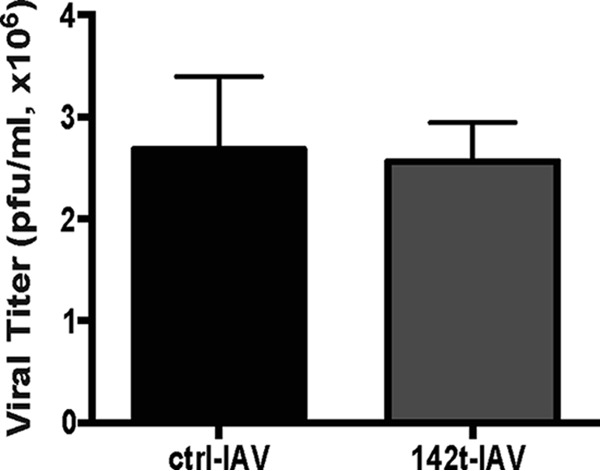
Replication of IAV expressing a miR-142 targeting sequence is not inhibited in the lungs relative to ctrl-IAV. Mice were infected with 0.1 LD50 of ctrl- or 142t-IAV. On day 4 p.i., lungs were harvested and viral titers were determined. Viral titer data represent one experiment performed in duplicate (n = 5 mice/group).
FIG 2.

IAV expressing a miR-142 targeting sequence does not replicate efficiently within pulmonary pDC or CD8α+ DC. Mice were infected with 0.1 LD50 of ctrl- or 142t-IAV. On day 4 p.i., lungs were harvested and HA+ pDC and CD8α+ DC were identified by staining with anti-HA, anti-CD11c, anti-MHC-II, anti-CD8α, and anti-B220. Representative gatings for these populations are shown in panels A and B. In panels C and D, the total number and the frequency of HA+ among total pDC (C) or CD8α+ DC (D) were determined. Data are pooled results from two independent experiments (n = 5 to 8 mice/group). Bars correspond to means ± SEMs. **, P < 0.01. Statistical significance determined by ANOVA followed by Tukey's multiple-comparison test. n.s., not significant.
Replication within pDC but not CD8a+ DC is required for the rescue of the pulmonary IAV-specific CD8 T cell response.
We have previously demonstrated that pDC or CD8α+ DC are sufficient to rescue the pulmonary IAV-specific CD8 T cell response from apoptosis during IAV infection (1, 2). To determine whether pDC and CD8α+ DC need to be directly infected or can utilize cross-presentation to present antigen to activated CD8 T cells within the lungs, we used a methodology similar to what we have previously described (2). We transferred donor pDC or CD8α+ DC purified from the lungs of mice infected with either ctrl- or 142t-IAV into host mice that had been infected with ctrl-IAV and administered clodronate liposomes intranasally 48 h p.i. to deplete phagocytic cells within the lungs. Donor pDC and CD8α+ DC were transferred 24 h following i.n. APC depletion of host mice. As previously described (2), IAV infection generates a robust IAV-specific CD8 T cell response detectable in the lungs on 7 days p.i. that is blunted when phagocytic cells in the airways are depleted 48 h postinfection (Fig. 3). Reconstitution of APC-depleted, ctrl-IAV-infected mice with donor pDC from the lungs of ctrl-IAV-infected mice completely restores both the NP147 and HA533 IAV-specific CD8 T cell responses (Fig. 3). In contrast, donor pDC from the lungs of 142t-IAV-infected mice were not able to rescue either the NP147 or HA533 IAV-specific CD8 T cell response in APC-depleted, ctrl-IAV-infected hosts (Fig. 3). Given that DC-T cell interactions within the lungs are required to rescue the IAV-specific CD8 T cell response from apoptosis (2), these results indicate that cross-presentation alone by pDC is not sufficient for generating a full-magnitude IAV-specific CD8 T cell response. Further, these data suggest that pDC must be directly infected and process and present antigens to effector CD8 T cells in the lungs via the endogenous pathway. Like with pDC, administration of donor CD8α+ DC from the lungs of ctrl-IAV-infected mice to APC-depleted, ctrl-IAV-infected hosts was able to rescue the pulmonary CD8 T cell response against IAV (Fig. 4). However, in contrast to pDC, donor CD8α+ DC from 142t-IAV-infected lungs are able to rescue the IAV-specific CD8 T cell response within the lungs of APC-depleted, ctrl-IAV-infected hosts, indicating that CD8α+ DC are able to efficiently utilize cross-presentation to display IAV peptide–MHC-I complexes to effector CD8 T cells within the lungs (Fig. 3).
FIG 3.
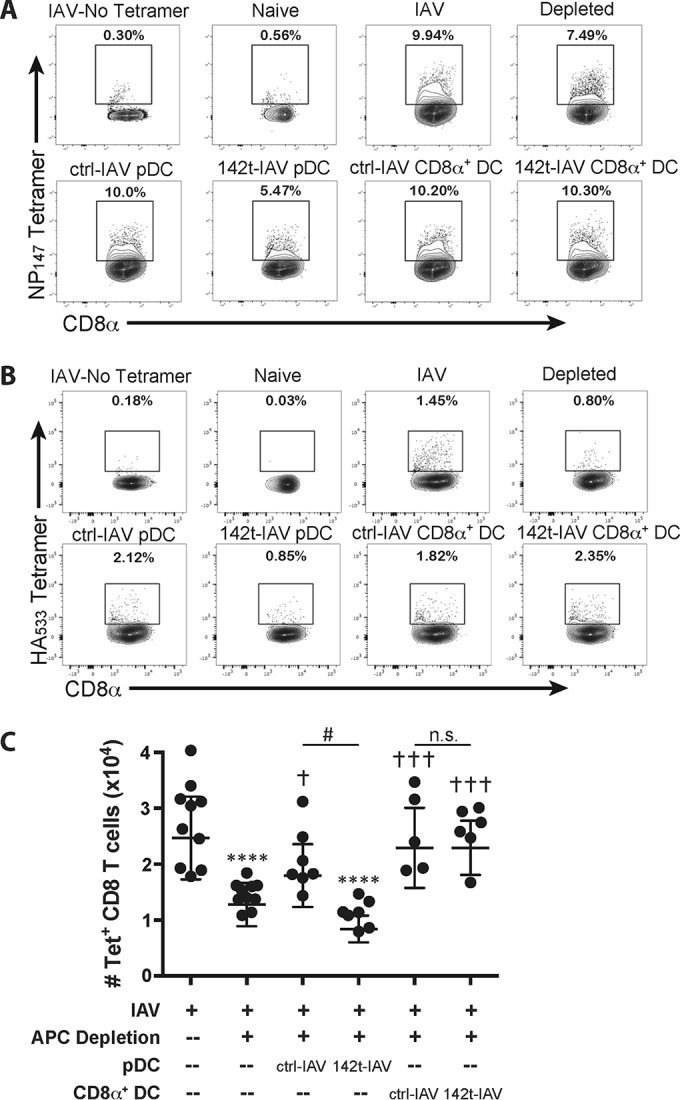
Donor CD8α+ DC, but not pDC, from 142t-IAV-infected lungs are able to rescue the pulmonary IAV-specific CD8 T cell response. Host mice were infected with 0.1 LD50 of ctrl-IAV and administered clodronate liposomes 48 h later. Twenty-four hours following treatment, groups of host mice were administered 2.5 × 104 pDC or CD8α+ DC isolated from the lungs of donor ctrl- or 142t-IAV-infected mice. Four days later, on day 7 p.i., the lungs were harvested and the frequency and number of IAV-specific (HA533- and NP147-positive) CD8 T cells were determined by flow cytometry. Representative flow plots for NP147 (A) and HA533 (B) staining are shown. (C) Total numbers of combined IAV-specific (HA533- and NP147-positive) CD8 T cells were calculated. Pooled results from two independent experiments are depicted (n = 5 to 10 mice/group). Each point corresponds to one mouse, and bars correspond to the means ± SEMs. ****, P < 0.0001 compared to the value for the IAV-infected group; †, P < 0.05 compared to the value for the IAV-infected, APC-depleted group; †††, P < 0.001 compared to the value for the IAV-infected, APC-depleted group; #, P < 0.05 compared to the value obtained with transfer of pDC from ctrl-IAV-infected donors. Statistical significance was determined by ANOVA followed by Tukey's multiple-comparison test.
FIG 4.

pDC from ctrl- and 142t-IAV-infected lungs express similar levels of molecules known to be required for pulmonary rescue of the IAV-specific CD8 T cell response. Mice were infected as described for Fig. 1. On day 6 p.i., lungs were harvested and the expression of MHC-I, IL-15-Ra, and CD70 on the surface of pDC (CD11c+ CD11b− MHC-II+ B220+ CD8a+) from ctrl-IAV-infected (black dotted line) and 142t-IAV-infected (gray line) lungs compared to the isotype control (shaded histogram) was determined. Bar graphs depict MHC-I, IL-15Ra, and CD70 expression as measured by MFI on pDC from ctrl-IAV-infected (black bars) and 142t-IAV-infected (gray bars) lungs. Each bar corresponds to the mean ± SEM from two pooled, independent experiments (n = 10 mice/group).
To date, we have defined several molecules critical during the pulmonary DC-T cell interaction. Presentation of viral peptide in the context of MHC-I, trans-presentation of IL-15, and CD70 on pDC are all required in order to prevent apoptosis of effector IAV-specific CD8 T cells as they enter the lung during IAV infection (data not shown) (2, 3). Therefore, we wanted to verify that the inability of pDC from 142t-IAV-infected lungs to maintain sufficient numbers of IAV-specific CD8 T cells was due to an inability of pDC to cross-present this viral antigen and not due to differential expression of MHC-I, IL-15Rα, or CD70 on pDC during infection with 142t-IAV. Our results show that expression of these molecules on pDC is the same on day 6 p.i. (i.e., the day donor DC are harvested for transfer in Fig. 3) regardless of infection with ctrl- or 142t-IAV (Fig. 4). Further, we also observed no difference in the expression of these molecules on CD8α+ DC during infection with either ctrl- or 142t-IAV (data not shown). These results indicate that the inability of pDC from 142t-IAV-infected mice to rescue the CD8 T cell response is not due to a lack of molecules known to be required on the surface of pDC for proper maintenance of IAV-specific CD8 T cells in the lungs.
Defective interfering genomes have been demonstrated to be produced following IAV infection and can stimulate innate immune responses and enhance DC maturation, which could modulate induction of antiviral CD8 T cell responses (16, 18, 19). Therefore, differences in the amounts of defective genomes produced in the lungs of DC donors infected with ctrl- or 142t-IAV could potentially lead to alterations in antigen presentation capabilities of DC subsets. Our results demonstrate that at both days 3 and 5 p.i., there is no difference in the amounts of defective genomes in the lungs of mice infected with ctrl- or 142t-IAV (Fig. 5). These results reveal that the inability of pDC from the lungs of 142t-IAV-infected mice to rescue the CD8 T cell response is not due to differential exposure to defective IAV.
FIG 5.
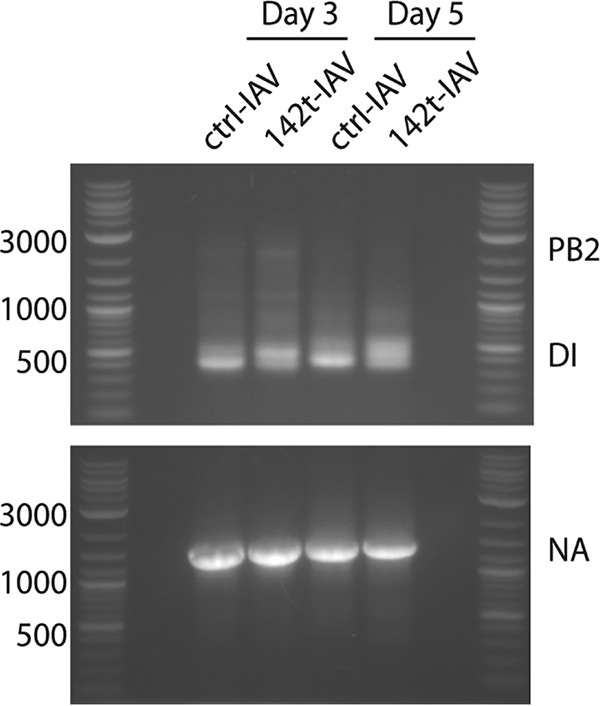
Defective interfering particles (DI) are similar in ctrl- and 142t-IAV-infected lungs. Mice were infected with 0.1 LD50 of ctrl- or 142t-IAV. On days 3 and 5 postinfection, lungs were harvested and processed in TRIzol to collect viral RNA. PCR was performed for the PB2 gene segment to determine the amount of DI in the lungs. As a control, PCR was also performed for the NA gene segment.
Coating pDC from 142t-IAV donors with IAV peptide prior to transfer restores the ability of pDC to rescue the pulmonary IAV-specific CD8 T cell response.
While we observed no differences in the molecules known to be required for the rescue of the IAV-specific CD8 T cell response or defective IAV genome levels within the lungs, it is possible other, currently undefined molecules could be differentially regulated during 142t-IAV infection, potentially impacting the ability of pDC to present viral antigen to effector CD8 T cells within the lungs. If this is the case, providing exogenous viral peptides that correspond to known IAV CD8 T cell epitopes to donor pDC purified from the lungs of 142t-IAV mice should not be able to restore the ability of 142t-IAV pDC to rescue the pulmonary, IAV-specific CD8 T cell response. Therefore, we next incubated pDC purified from the lungs of 142t-IAV-infected donors with exogenous HA533 or NP147 peptide, mixed the peptide-pulsed pDC populations at a 1:1 ratio, and transferred the donor pDC to ctrl-IAV-infected, APC-depleted mice. While donor pDC purified from 142t-IAV-infected lungs could not recue the IAV-specific CD8 T cell response, bypassing antigen processing by coating these cells with IAV peptide prior to transfer restored their ability to rescue the IAV-specific CD8 T cell response in the lungs of APC-depleted mice (Fig. 6). Together, these results demonstrate that the failure of pDC from 142t-IAV-infected lungs to rescue the IAV-specific CD8 T cell response is indeed due to the inability of pDC to cross-present IAV antigen within the lungs and not an inability to present antigen on the cell surface, insufficient expression of required surface molecules (MHC-I, IL-15Rα, and CD70), or differences in their prior exposure to defective genomes. Together, our results suggest that while CD8α+ DC are able to utilize cross-presentation in order to rescue the IAV-specific CD8 T cell response, pDC are not and require direct infection along with the endogenous pathway to present viral antigen in order for this rescue to occur. Importantly, these studies are the first, to our knowledge, to examine the ability of pDC to cross-present antigen within the lungs during IAV infection without first priming or infecting pDC in vitro or analyzing processing and presentation of a model antigen not normally present during an in vivo IAV infection (6, 11–13, 20).
FIG 6.
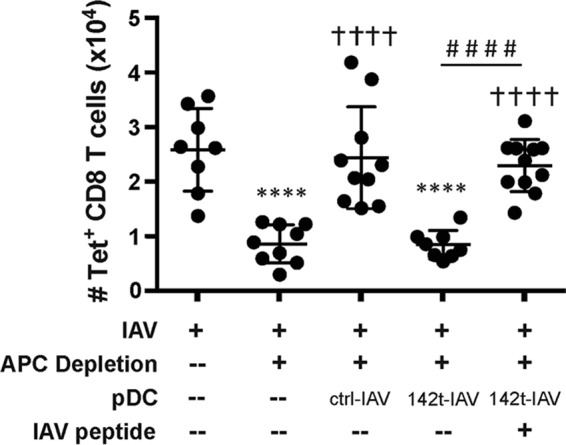
Coating donor 142t-IAV pDC with IAV peptides restores their ability to rescue the pulmonary IAV-specific CD8 T cell response. Host mice were infected with IAV and pulmonary APC were depleted as done for Fig. 4. Twenty-four hours following APC depletion, groups of host mice were administered 2.5 × 104 donor ctrl- or 142t-IAV pDC isolated from the lungs of infected mice or 2.5 × 104 IAV peptide-pulsed pDC from the lungs of 142t-IAV-infected donor mice. Four days later, on day 7 p.i., the lungs were harvested and the total number of IAV-specific (HA533- and NP147-positive) CD8 T cells was determined by flow cytometry. Pooled results from two independent experiments are depicted (n = 8 to 11 mice/group). Each point corresponds to one mouse, and bars correspond to the means ± SEMs. ****, P < 0.0001 compared to the value for the IAV-infected group; †††, P < 0.001 compared to the value for the IAV-infected, APC-depleted group; ††††, P < 0.0001 compared to the value for the IAV-infected, APC-depleted group; ####, P < 0.0001 compared to the value obtained with transfer of pDC from 142t-IAV-infected donors. Statistical significance determined by ANOVA followed by Tukey's multiple-comparison test.
DISCUSSION
Utilizing DC as therapeutics to aid in the development of immune responses against pathogenic infections, autoimmunity, and cancer has great potential (21). Our work has demonstrated that both pDC and CD8α+ DC administered i.n. are able to boost IAV-specific CD8 T cell response in the lungs (2, 3) and could theoretically be utilized as a DC-based therapeutic to aid in the development of robust, local IAV-specific CD8 T cell responses to clear infection. In this context, pDC would likely be the better candidate in development of such a treatment, as pDC are numerous and comprise approximately 1/3 of the DCs within human peripheral blood (20). Therefore, pDC might be able to be purified, matured, and coated with IAV peptides to boost IAV-specific CD8 T cell responses in the lungs after IAV infection is already established and the window of priming within dLN has occurred. Such a therapeutic could be useful in situations of pandemic IAV outbreak where a large portion of the population is suddenly at high risk for potentially damaging IAV infections as their preexisting immunity from former IAV infections and vaccinations is rendered ineffective. Further, lymphopenia is a hallmark of severe IAV infection, and preventing pulmonary apoptosis of effector CD8 T cells might aid in viral clearance and reduction of severe disease outcomes during highly pathogenic IAV infection (22–24). Currently, understanding the requirements and abilities of pDC from IAV-infected hosts is the first step in designing such a therapeutic treatment.
The ability of pDC to cross-present antigen during IAV infection remains controversial. While pDC have been demonstrated to cross-present IAV antigen in vivo and in vitro, such cross-presentation has only been demonstrated when pDC were first stimulated with TLR ligands, such as imiquimod or resiquimod, in vitro (9, 11, 13). Further, direct infection of human pDC with IAV impairs their ability to cross-present exogenous antigens (6). By utilizing an IAV that cannot replicate in hematopoietic cells, we were able to limit any presentation of antigens by pDC from 142t-IAV-infected lungs to cross-presentation. Given this, it is possible that the amount of TLR7 stimulation during in vivo IAV infection may not be sufficient to induce cross-presentation by pulmonary pDC. The addition of TLR7 stimulation in excess in vivo or ex vivo might allow pDC to cross-present antigens to effector IAV-specific CD8 T cells within the lungs, and perhaps even enhance the overall antiviral response within the lungs by increasing type I IFN production by pDC (12). However, any use of TLR7 as a therapy to attempt to induce pDC cross-presentation should proceed with caution, as excessive, systemic TLR7 stimulation can result in damaging, overt inflammation either from pDC or generally within the lung (25).
In agreement with the literature, our results demonstrate CD8α+ DC are potent presenters of antigen and are able to utilize both the direct (endogenous) and cross-presentation (exogenous) pathways to present viral antigen to CD8 T cells via MHC-I (reviewed in reference 8). However, recent studies have demonstrated that not all CD8α+ DC are efficient cross-presenters and that this ability is licensed by exposure to TLR ligands, damage-associated molecular patterns (DAMPs), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-3 (26–28). Whether cytokine and/or pattern- or damage-associated molecular pattern signaling is required to license pDC for cross-presentation, and what exactly those signals are, remains underexplored. The ability of the CD8α+ DC to cross-present suggests that the necessary signals required for licensing of cross-presentation are present in the lungs of IAV-infected mice. This therefore suggests that the signals required for cross-presentation licensing in pDC may be distinct from those utilized by CD8α+ DC (27) or that the pDC are in a location or environment distinct from CD8α+ DC within the lungs.
Cumulatively, these results demonstrate that while CD8α+ DC are able to utilize cross-presentation of antigens to generate a full magnitude, IAV-specific CD8 T cell responses in the lung pDC do not and instead appear to rely solely on direct, endogenous presentation of IAV viral antigen to rescue the IAV-specific CD8 T cell response from apoptosis. These results also more generally demonstrate that pDC do not typically cross-present IAV antigen within the lungs during in vivo IAV infections.
ACKNOWLEDGMENTS
We thank Zeb Zacharias for technical assistance. We also thank Steven Varga for his critical evaluation of the manuscript.
This work was supported by NIH grants AI071085 (to K.L.L.), T32 AI007533 (to E.A.H.), and T32 AI007485 (to E.A.H.).
REFERENCES
- 1.McGill J, Legge KL. 2009. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J Immunol 183:4177–4181. doi: 10.4049/jimmunol.0901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGill J, Van Rooijen N, Legge KL. 2008. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med 205:1635–1646. doi: 10.1084/jem.20080314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGill J, Van Rooijen N, Legge KL. 2010. IL-15 trans-presentation by pulmonary dendritic cells promotes effector CD8 T cell survival during influenza virus infection. J Exp Med 207:521–534. doi: 10.1084/jem.20091711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hao X, Kim TS, Braciale TJ. 2008. Differential response of respiratory dendritic cell subsets to influenza virus infection. J Virol 82:4908–4919. doi: 10.1128/JVI.02367-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smed-Sörensen A, Chalouni C, Chatterjee B, Cohn L, Blattmann P, Nakamura N, Delamarre L, Mellman I. 2012. Influenza A virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells. PLoS Pathog 8:e1002572. doi: 10.1371/journal.ppat.1002572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.VanoOsten Anderson R, McGill J, Legge KL. 2010. Quantification of the frequency and multiplicity of infection of respiratory- and lymph node-resident dendritic cells during influenza virus infection. PLoS One 5:e12902. doi: 10.1371/journal.pone.0012902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joffre OP, Segura E, Savina A, Amigorena S. 2012. Cross-presentation by dendritic cells. Nat Rev Immunol 12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 9.den Haan JM, Lehar SM, Bevan MJ. 2000. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med 192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pooley JL, Heath WR, Shortman K. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J Immunol 166:5327–5330. doi: 10.4049/jimmunol.166.9.5327. [DOI] [PubMed] [Google Scholar]
- 11.Mouriès J, Moron G, Schlecht G, Escriou N, Dadaglio G, Leclerc C. 2008. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood 112:3713–3722. doi: 10.1182/blood-2008-03-146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 13.Lui G, Manches O, Angel J, Molens JP, Chaperot L, Plumas J. 2009. Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS One 4:e7111. doi: 10.1371/journal.pone.0007111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langlois RA, Varble A, Chua MA, Garcia-Sastre A, Tenoever BR. 2012. Hematopoietic-specific targeting of influenza A virus reveals replication requirements for induction of antiviral immune responses. Proc Natl Acad Sci U S A 109:12117–12122. doi: 10.1073/pnas.1206039109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foa R, Schliwka J, Fuchs U, Novosel A, Muller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frensing T, Heldt FS, Pflugmacher A, Behrendt I, Jordan I, Flockerzi D, Genzel Y, Reichl U. 2013. Continuous influenza virus production in cell culture shows a periodic accumulation of defective interfering particles. PLoS One 8:e72288. doi: 10.1371/journal.pone.0072288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mildner A, Chapnik E, Manor O, Yona S, Kim KW, Aychek T, Varol D, Beck G, Itzhaki ZB, Feldmesser E, Amit I, Hornstein E, Jung S. 2013. Mononuclear phagocyte miRNome analysis identifies miR-142 as critical regulator of murine dendritic cell homeostasis. Blood 121:1016–1027. doi: 10.1182/blood-2012-07-445999. [DOI] [PubMed] [Google Scholar]
- 18.Tapia K, Kim WK, Sun Y, Mercado-Lopez X, Dunay E, Wise M, Adu M, Lopez CB. 2013. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog 9:e1003703. doi: 10.1371/journal.ppat.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yount JS, Kraus TA, Horvath CM, Moran TM, Lopez CB. 2006. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J Immunol 177:4503–4513. doi: 10.4049/jimmunol.177.7.4503. [DOI] [PubMed] [Google Scholar]
- 20.Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, Bhardwaj N. 2003. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood 101:3520–3526. doi: 10.1182/blood-2002-10-3063. [DOI] [PubMed] [Google Scholar]
- 21.Steinman RM, Banchereau J. 2007. Taking dendritic cells into medicine. Nature 449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. 2013. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Criswell BS, Couch RB, Greenberg SB, Kimzey SL. 1979. The lymphocyte response to influenza in humans. Ame Rev Respir Dis 120:700–704. [DOI] [PubMed] [Google Scholar]
- 24.Dolin R, Richman DD, Murphy BR, Fauci AS. 1977. Cell-mediated immune responses in humans after induced infection with influenza A virus. J Infect Dis 135:714–719. doi: 10.1093/infdis/135.5.714. [DOI] [PubMed] [Google Scholar]
- 25.Gunzer M, Riemann H, Basoglu Y, Hillmer A, Weishaupt C, Balkow S, Benninghoff B, Ernst B, Steinert M, Scholzen T, Sunderkotter C, Grabbe S. 2005. Systemic administration of a TLR7 ligand leads to transient immune incompetence due to peripheral-blood leukocyte depletion. Blood 106:2424–2432. doi: 10.1182/blood-2005-01-0342. [DOI] [PubMed] [Google Scholar]
- 26.Dresch C, Leverrier Y, Marvel J, Shortman K. 2012. Development of antigen cross-presentation capacity in dendritic cells. Trends Immunol 33:381–388. doi: 10.1016/j.it.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Nierkens S, Tel J, Janssen E, Adema GJ. 2013. Antigen cross-presentation by dendritic cell subsets: one general or all sergeants? Trends Immunol 34:361–370. doi: 10.1016/j.it.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sathe P, Pooley J, Vremec D, Mintern J, Jin JO, Wu L, Kwak JY, Villadangos JA, Shortman K. 2011. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J Immunol 186:5184–5192. doi: 10.4049/jimmunol.1002683. [DOI] [PubMed] [Google Scholar]