

ABSTRACT
Sulfolobus turreted icosahedral virus (STIV), an archaeal virus that infects the hyperthermoacidophile Sulfolobus solfataricus, is one of the most well-studied viruses of the domain Archaea. STIV shares structural, morphological, and sequence similarities with viruses from other domains of life, all of which are thought to belong to the same viral lineage. Several of these common features include a conserved coat protein fold, an internal lipid membrane, and a DNA-packaging ATPase. B204 is the ATPase encoded by STIV and is thought to drive packaging of viral DNA during the replication process. Here, we report the crystal structure of B204 along with the biochemical analysis of B204 mutants chosen based on structural information and sequence conservation patterns observed among members of the same viral lineage and the larger FtsK/HerA superfamily to which B204 belongs. Both in vitro ATPase activity assays and transfection assays with mutant forms of B204 confirmed the essentiality of conserved and nonconserved positions. We also have identified two distinct particle morphologies during an STIV infection that differ in the presence or absence of the B204 protein. The biochemical and structural data presented here are not only informative for the STIV replication process but also can be useful in deciphering DNA-packaging mechanisms for other viruses belonging to this lineage.
IMPORTANCE STIV is a virus that infects a host from the domain Archaea that replicates in high-temperature, acidic environments. While STIV has many unique features, there exist several striking similarities between this virus and others that replicate in different environments and infect a broad range of hosts from Bacteria and Eukarya. Aside from structural features shared by viruses from this lineage, there exists a significant level of sequence similarity between the ATPase genes carried by these different viruses; this gene encodes an enzyme thought to provide energy that drives DNA packaging into the virion during infection. The experiments described here highlight the elements of this enzyme that are essential for proper function and also provide supporting evidence that B204 is present in the mature STIV virion.
INTRODUCTION
In Sulfolobus turreted icosahedral virus (STIV), the open reading frame B204 (previously described as B164) (1, 2) encodes a putative ATPase that is predicted to function as the molecular motor driving viral DNA packaging during infection (3). Interestingly, it has been demonstrated that viruses harboring ATPases homologous to B204 share other common features, such as a spherical morphology, an internal lipid membrane, and a structurally homologous coat protein fold (termed the double β-barrel or double jelly roll fold) (4). The structural and functional assessment of viruses belonging to this putative viral lineage can provide insights on viral infection strategies across the three domains of life. In the case of B204, understanding its function has implications for mechanisms by which an ancient molecular motor may have operated prior to separation of the three domains of life.
Over the past decade, STIV has become one of the most characterized archaeal viruses and is viewed as a model virus for the domain Archaea. Studies on STIV have revealed the identification of a novel viral lysis mechanism (5), unusual features of the STIV virion (6), and a coat protein fold that links STIV to viruses infecting other domains of life (4, 7). Of the predicted 38 open reading frames encoded by STIV (including the more recently identified B129 [8]), 11 have been experimentally investigated through genetic knockout experiments (9), biochemical analysis (2, 10, 11), or structural characterization (5–7, 12–14). While certain steps of the STIV infection cycle are under investigation (11, 15), much remains to be understood, including the processes of viral attachment and viral DNA packaging.
STIV B204 belongs to the FtsK/HerA superfamily (3), whose members share several conserved amino acid sequence motifs. In addition to the conserved Walker A and B motifs which are common to all P-loop ATPases (16, 17), the superfamily contains a third conserved motif that includes an arginine finger (which interacts with the neighboring protomer) (18, 19) and a polar residue (usually Gln, which senses the ATP triphosphate moiety [3]). Within the FtsK/HerA superfamily, B204 belongs to the A32-like DNA-packaging clade, which includes ATPases belonging to viruses from all three domains of life (3, 20, 21). To date, few DNA-packaging ATPases homologous to B204 have been studied in detail. Several reports suggest that members of this clade package their double-stranded DNA (dsDNA) genomes after (and not during) procapsid assembly (22). For example, whole-cell cryotomography of STIV-infected cells suggests that the major capsid protein and internal membrane assemble into procapsids prior to the formation of the mature, genome-packaged particles (22). In bacteriophage PRD1, mutants that lack a functional P9 ATPase result in the formation of empty procapsids, which can be rescued by addition of active P9 ATPase and PRD1 genomic DNA in vitro to yield packaged particles (20). Additional knockout experiments in PRD1 indicate that this virus packages DNA at a unique vertex that extends through to the internal lipid membrane and is comprised of two membrane proteins (P20 and P22), the coat protein P6, and the P9 ATPase (23). A similar model has been proposed recently for STIV2, involving recognition of STIV-DNA with the protein STIV2 B72, assembly of a unique portal complex (which includes STIV2 B204), and translocation of DNA through this complex and subsequently through the lipid membrane (21). Unfortunately, the latest mass spectrometric analysis of the mature STIV2 virion did not show evidence that B204 is present in the particle (21). Similarly, STIV B204 does not appear to be present in the high-resolution cryoelectron microscopy reconstruction of the virion (6). These results do not necessarily correlate with the proposed model unless the portal complex is somehow disassembled prior to cell lysis.
Upon consideration of sequence conservation patterns within the family (3) in addition to structural information from both STIV2 B204 (21) and STIV B204 (current work), we have chosen a series of positions to mutate within the STIV B204 gene. We have biochemically tested these mutants for their ability to (i) hydrolyze ATP in an in vitro ATPase assay and (ii) sustain viral infection in the susceptible host, S. solfataricus P23. Our results provide new biochemical insight into the effect of mutation at both conserved and nonconserved positions within the B204 sequence in the context of the DNA-packaging clade and the larger family. In addition, we demonstrate that highly purified STIV virions contain B204, thereby providing support for the proposed DNA packaging model.
MATERIALS AND METHODS
Virus and host strains.
An infectious clone of STIV was constructed through insertion of the STIV genome into a TOPO vector as previously described (9). The susceptible host Sulfolobus solfataricus 2-2-12 (P23) was evolved from S. solfataricus P2 as previously described (2).
Cloning B204 and construction of B204 mutants in pDEST14 and STIV-TOPO.
Wild-type B204 was PCR amplified from the infectious clone STIV-TOPO (9) and into the Gateway pDEST14 vector (Life Technologies, La Jolla, CA) using the Gateway cloning strategy (Life Technologies) with universal primers and gene-specific amplification primers containing attB sites and a C-terminal 6× histidine tag. A series of site-directed mutants (both conservative and nonconservative changes) targeting conserved and nonconserved residues within the FtsK/HerA superfamily of ATPases were generated. Briefly, B204 mutants in pDEST14 were constructed using the QuikChange mutagenesis strategy (Stratagene, La Jolla, CA) with Phusion high-fidelity DNA polymerase (New England BioLabs [NEB], Ipswich, MA) and degenerate primers. B204 mutations in STIV-TOPO first were constructed using QuikChange mutagenesis in a TOPO subclone containing not only the B204 gene but also flanking regions of the STIV genome whose ends contain the unique restriction enzyme sites BspEI and BstZI7I (which are native to the STIV genome) (9). Once verified by DNA sequencing, these mutant subclones were amplified with primers containing BspEI and BstZI7I sites, digested with these restriction enzymes, and ligated back into the BspEI/BstZI7I-digested STIV vector using T4 DNA ligase (NEB).
Protein overexpression and purification.
B204 and mutants were transformed into Rosetta(DE3)pLysS competent cells (EMD Millipore, Billerica, MA). One colony for each mutant was grown in LB media at 37°C overnight, which was subsequently inoculated into 1 liter of LB media and induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density at 650 nm (OD650) of 1.0. The cells were grown overnight at 37°C and harvested 18 h postinduction. The cells subsequently were lysed with buffer A (50 mM HEPES, pH 7.5, 500 mM NaCl, 5% Tween 20, 20 mM imidazole, 10% glycerol) supplemented with lysozyme (1 mg/ml), sonicated, and centrifuged at 30,000 × g. The supernatant was passed through a gravity flow column containing nickel-nitrilotriacetic acid (Ni-NTA; Qiagen, Germantown, MD), washed with 20 column volumes of buffer A and 20 column volumes of buffer B (buffer A without Tween 20), and eluted in buffer C (buffer B supplemented with 250 mM imidazole). The eluate was dialyzed overnight in dialysis buffer (50 mM HEPES, pH 7.5, 500 mM NaCl), concentrated, and stored at −80°C for later use. Proteins were greater than 90% pure (data not shown). Protein that was used for crystallization was further purified on a HiLoad 16/600 Superdex S200 column (GE Healthcare Biosciences, Pittsburgh, PA). The fractions containing the B204 protein were pooled, dialyzed overnight into a buffer containing 10 mM citrate, pH 6.0, 100 mM NaCl, concentrated to 10 mg/ml, and stored at −80°C for later use.
Protein crystallization.
Crystals of B204 were obtained using hanging-drop vapor diffusion with 1 μl of protein and 1 μl of reservoir solution (24). B204 was crystallized using a screen that covered a broad variety of conditions, some of which included magnesium and acidic buffers. The protein crystallized as short rods that were grown for several weeks under the following reservoir conditions: 1.5 M ammonium phosphate dibasic, 0.1 M Tris, pH 8.5. Crystals were obtained without ligands and in the presence of 5 mM adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP) and were optimized over a grid of conditions ranging from 1.2 to 1.7 M ammonium phosphate dibasic in either 0.1 M HEPES buffer (pH 7 or 7.5) or 0.1 M Tris buffer (pH 8.0 or 8.5). Crystals were transferred into a reservoir solution containing 20 to 30% polyethylene glycol 400 and soaked overnight prior to freezing in liquid nitrogen. Diffraction data were collected remotely through beamlines 12-2 and 14-1 at the Stanford Synchrotron Radiation Lightsource (SSRL). Data were processed using XDS (25) and MOSFLM (26) software and scaled using the program Scala from the CCP4 software package (27). The structures were solved by molecular replacement using the program phaser-MR as part of the PHENIX software package (28). A model of STIV B204 was constructed in SwissModel (29, 30) based on the template PDB code 4KFU, the crystal structure of STIV2 B204 complexed with β,γ-methyleneadenosine 5′-triphosphate (AMP-PCP) (21). The structures were built using the program COOT (31), and refinement cycles were performed using Refmac from the CCP4 software package (27) and the Phenix refinement software as part of the Phenix package (28).
ATPase assays.
All ATPase assays were performed with the high-throughput colorimetric ATPase assay kit (Innova Biosciences, Babraham, Cambridge, United Kingdom) that measures the accumulation of inorganic phosphate as a by-product of ATP hydrolysis. In general, 18 μl of enzyme mix containing 0.5 mM ATP and 1.3 μg B204 was added to 42 μl of preheated (80°C) sample mix containing 140 ng plasmid DNA (8,434 bp in length), 2.5 mM MgCl2, 150 mM NaCl, and 10 mM citrate buffer, pH 4.5. Experiments were incubated at 80°C for 3 min and then quenched by the addition of an acidic dye solution called pi-color lock gold (Innova Biosciences). The absorbance values at 650 nm were quantified based on a standard curve constructed from phosphate standard solutions that were prepared using a method similar to that described above. In experiments where the concentration of NaCl was varied, the assays were carried out with NaCl concentrations ranging from 0 to 500 mM. All experiments were performed in triplicate, and each experiment included controls to measure background ATP hydrolysis. In experiments performed with purified STIV particles, the assays were carried out through the substitution of virus (showing quantitative PCR [qPCR] values of 1 × 1012 genomes/ml) in place of the B204 enzyme. All experiments were performed in duplicate. The controls included uninfected cultures of S. solfataricus P23 that were purified and treated similarly to the infected samples (described below).
Transfection of mutated STIV DNA.
Mutated STIV DNA was prepared as previously described (9). Briefly, STIV DNA inserted into the Escherichia coli TOPO plasmid does not replicate in the susceptible host without prior removal of the TOPO plasmid. Plasmid removal was accomplished by digestion with BamHI (which flanks the TOPO sequence) and religation of the STIV genome by T4 DNA ligase (NEB). Sulfolobus solfataricus P23 competent cells were prepared as previously described (9). After transfection, samples were collected every 24 h and STIV replication was measured by qPCR. Standard qPCR methods were followed using SsoFast EvaGreen supermixes (Bio-Rad Laboratories, Hercules, CA) with primers that amplify a 280-bp portion of the major coat protein. B204 mutants were considered successful in transfection experiments if the qPCR values for amplification of the coat protein were 4 to 5 orders of magnitude above background levels. In successful transfection experiments, the presence of the B204 mutation within the STIV genome was confirmed by sequencing of the PCR-amplified B204 gene product using virus particles from the transfection experiment as the template.
Purification of STIV.
STIV was purified as previously described (4). Briefly, concentrated STIV was added to a solution of 38% (wt/vol) cesium sulfate in 25 mM citrate buffer, pH 3.5, and centrifuged at 247,600 × g for 20 h. Two blue bands were apparent near the top of the gradient, termed the upper and lower bands. Each virus band was separately dialyzed into 25 mM citrate buffer, pH 3.5. Uninfected controls containing S. solfataricus P23 were treated similarly and processed through identical cesium sulfate gradients; while no bands were visible for this sample, fractions were collected in the positions of the gradient corresponding to the upper band observed in the STIV cesium sulfate gradients. The upper and lower bands were loaded onto an SDS-PAGE gel (data not shown). Purified virus and control samples were used directly in the ATPase assays. Western blot analysis was performed on samples from the upper and lower bands as well as various concentrations (0.001 to 50 μg) of E. coli-purified B204, serving as a copy number control. Rabbit polyclonal antibodies raised against STIV B204 or A223 proteins were used as the primary antibody, and the Immun-Blot Opti-4CN assay kit (Bio-Rad, Hercules, CA) was used for the detection of the horseradish peroxidase (HRP)-conjugated secondary antibody, goat anti-rabbit IgG (H+L)-HRP conjugate (Bio-Rad). The cesium gradients were repeated multiple times to collect a sufficient quantity of virions for mass spectrometric analysis. Briefly, mass spectrometry was performed on STIV samples prepared by in-solution trypsin digestion of the upper and lower bands. The in-solution digests and peptide matching were performed as previously described (1). Briefly, the digests were run on a Biflex III matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrophotometer (Bruker Daltonics), and experimental peptides were matched with predicted peptide sequences derived from a theoretical trypsin digest of the B204 open reading frame using the Mascot search engine (Matrix Science, Boston, MA). Purified STIV particles also were viewed by a transmission electron microscope (TEM; Leo 912 AB [Zeiss]).
Comparison of STIV B204 to sequences in the environmental metagenome database.
The environmental metagenome includes viral and cellular DNA assembled as contigs from various hot springs within Yellowstone National Park, including Nymph Lake 10 (NL10), Nymph Lake 17 (NL17), Nymph Lake 18 (NL18), and Crater Hills (CH) (32, 33). The B204 amino acid sequence was queried against the metagenome database using tblastn to identify contigs that shared 30% or higher identity to STIV B204. The sequences were aligned using MUSCLE (34).
PDB accession numbers.
The PDB accession numbers for the crystal structures of apo and AMP-PNP-bound STIV B204 are 4R2H and 4R2I, respectively.
RESULTS
The crystal structure of B204.
B204 was crystallized in the presence and absence of the nonhydrolyzable ATP analog adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP), and diffraction data were collected to a resolution of 2.05 Å and 1.96 Å, respectively (Table 1 depicts data collection and refinement statistics). The structure of STIV B204 is represented by a central nine-stranded β-sheet decorated with seven α-helices (Fig. 1a). Strand 8 of the β-sheet, which sits between strands 7 and 9, runs antiparallel to all other strands. B204 from STIV and STIV2 (21) share a high level of amino acid sequence identity (90%) and are structurally homologous; a structural alignment of both enzymes complexed with nonhydrolyzable ATP analogs (PDB codes 4R2I and 4KFU) yields a root-mean-square deviation (RMSD) of 1.21 Å for superposition of 200 C-α atoms. A comparison of the active-site residues between these two complexes reveals that most amino acids are in similar orientations for both structures; however, the γ-phosphate group from each ATP analog are in different positions, causing different amino acid side chains to be in close proximity to this moiety within each structure (Fig. 1b). These differences in γ-phosphate orientation could be attributed to the utilization of different ATP analogs (AMP-PNP for STIV B204 and AMP-PCP for STIV2 B204) in crystallization experiments (35) or to the absence of a divalent cation in crystal structures of STIV B204. Despite the lack of metal ion supplementation in crystallization experiments, a tetrahedrally coordinated zinc ion was observed in both apo and AMP-PNP-bound crystal structures in the same location as that observed for structures of STIV2 B204 (21).
TABLE 1.
Crystallography data collection and refinement statistics
| Parameter | Value(s) for: |
|
|---|---|---|
| B204 apo | B204 AMP-PNP | |
| PDB code | 4R2H | 4R2I |
| Data collection and processing | ||
| Wavelength (Å) | 1.03317 | 0.97950 |
| Resolution range (Å) | 38.65–1.96 (2.029–1.959)a | 44.08–2.05 (2.123–2.05) |
| Space group | P21 21 21 | P21 21 21 |
| Unit cell dimensions (α = β = γ = 90°) | a = 51.9, b = 58.0, c = 67.4 | a = 52.4, b = 58.3, c = 67.3 |
| Total reflections (no.) | 274,398 (26,967) | 243,037 (24,879) |
| Unique reflections (no.) | 15,165 (1,495) | 13,061 (1,327) |
| Multiplicity | 6.3 (6.3) | 3.4 (3.5) |
| Completeness (%) | 100.0 (100.00) | 100.0 (100.0) |
| Mean I/sigma(I) | 7.4 (2.5) | 8.6 (1.7) |
| Rmergeb | 0.061 (0.315) | 0.073 (0.426) |
| Refinement statistics | ||
| Rworkc | 0.1806 | 0.1829 |
| Rfreed | 0.2273 | 0.2361 |
| RMSD | ||
| Bond lengths (Å) | 0.008 | 0.017 |
| Bond angle (°) | 1.1 | 1.7 |
| Average B factor (Å2) | ||
| Protein | 33.6 | 31.1 |
| Ligand | No ligand | 39.0 |
| Solvent | 36.5 | 32.0 |
| No. of atoms | ||
| Protein | 1,684 | 1,722 |
| Ligand | 0 | 32 |
| Solvent | 89 | 59 |
| Ramachandran statistics (%) | ||
| Favored regions | 97.04 | 95.1 |
| Allowed regions | 2.96 | 4.9 |
| Outliers | 0 | 0 |
Statistics for the highest-resolution shell are in parentheses.
Rmerge = ∑hkl ∑i|Ii(hkl) − <I(hkl)>|/∑hkl ∑i|Ii(hkl), where Ii(hkl) is the observed intensity and <I(hkl)> is the mean intensity of reflection hkl over all measurements of Ii(hkl).
Rwork = ∑||Fobs| − |Fcalc||/∑|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
Rfree equals Rwork but is only applied to a test set of reflections (5%) that is not included in the crystallographic refinement.
FIG 1.

(a) Global structure of STIV B204 shaded in rainbow colors from N terminus (blue) to C terminus (red). (b) Differences in orientation and stabilization of the γ-phosphate of the ATP analog (AMP-PNP or AMP-PCP) within crystal structures of STIV B204 (left; PDB code 4R2I) and STIV2 B204 (right; PDB code 4KFU), respectively. The magnesium ion is represented as a green sphere in STIV2 B204. The β-3 strand is represented by a β-bridge between the main-chain O atom of Tyr55 and the main-chain amino hydrogen atom of Val70.
The structures of apo and AMP-PNP-bound STIV B204 align with an RMSD of 0.26 Å for superposition of 204 C-α atoms. Three histidine residues from the C-terminal histidine tag can be seen in the electron density for the AMP-PNP-bound structure, which are stabilized by several residues belonging to neighboring B204 monomers, including Lys 93 from one symmetry mate and Arg 160 and Lys 181 from another. Only one histidine can be seen in the apo crystal structure, which is similarly stabilized by Lys 93 from a neighboring symmetry mate. These contacts most likely aid in the packing and overall stabilization of the crystal lattice for both structures. The crystal structures of B204 contain one molecule per asymmetric unit; these monomers arrange in the crystal lattice such that they form two chained networks comprised of weak monomer-monomer interactions (Fig. 2). Given that B204 and other members of the family form oligomeric ring structures (usually hexamers) (3), the lattice represented here most likely is an artifact of crystallization and is probably not relevant for its biological function. For this reason, we are not able to unambiguously assign how the Arg finger (Arg127 in B204) from one monomer interacts with the neighboring protomer in what is predicted to be the functionally relevant oligomeric complex.
FIG 2.
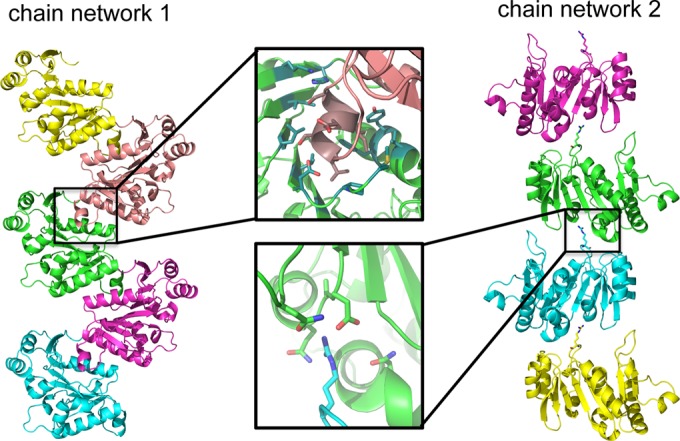
Network of chains present in the apo STIV B204 crystal lattice. Chain network 1 is dominated by interactions generated from the insertion of the α2 helix in a groove in the β-sheet of the neighboring monomer, while chain network 2 interacts only with Arg14 from the β1-α1 loop of one monomer and a pocket of negatively charged amino acids from another monomer, defined by the α4-helix, α5-β7 loop, and the β8-β9 loop. These interactions are also observed for the AMP-PNP-bound structure.
A model of the B204 oligomer was constructed as detailed previously (21) through superposition of B204 monomers onto the hexameric structure of the motor domain of FtsK. The modeled hexamer highlights the proximity of Arg127 to the ATP binding site of the neighboring protomer and also depicts several basic amino acids protruding into the central tunnel, including Lys166 and Lys170 (Fig. 3). It is noteworthy that in this model, the C terminus of each B204 monomer extends along the base of the modeled hexamer with the 6× histidine tag projecting outwards into solvent.
FIG 3.

B204 hexamer model based on the hexameric P. aeruginosa FtsK motor domain (PDB code 2IUU). (a) A global view of the B204 hexamer. The smallest internal diameter of the tunnel (the side-chain-to-side-chain distance) measures approximately 21.5 Å. The black boxes around the central ring and between two protomers of the hexamer are viewed as close-ups in panels b and c, respectively. (b) Two positively charged tunnel residues that protrude into the central ring of the hexamer. (c) The crystallographic orientation of B204 Arg127 places it 12 Å from the γ-phosphate of ATP from the neighboring protomer in the modeled hexamer; however, rotation of the side chain to a more extended conformation positions this amino acid even closer (approximately 6 Å) to the γ-phosphate group.
In vitro ATPase assays with B204.
Purified B204 protein was assayed for ATPase activity in vitro using a malachite green assay, which colorimetrically measures the formation of inorganic phosphate over time. Through trial and error, it was found that the ATPase activity of the enzyme was DNA dependent; therefore, all assays were supplemented with plasmid DNA. The ATPase assay was tested across a pH range of 3 to 6. B204 assays were performed at a temperature of 80°C and a pH of 4.5, as these conditions were determined to be optimal for STIV B204 (data not shown) and STIV2 B204 (21). Additionally, B204 was assayed at a range of sodium chloride concentrations and found to be most active at 150 mM NaCl (Fig. 4). Notably, lower or higher concentrations of NaCl caused sharp decreases in ATPase activity.
FIG 4.
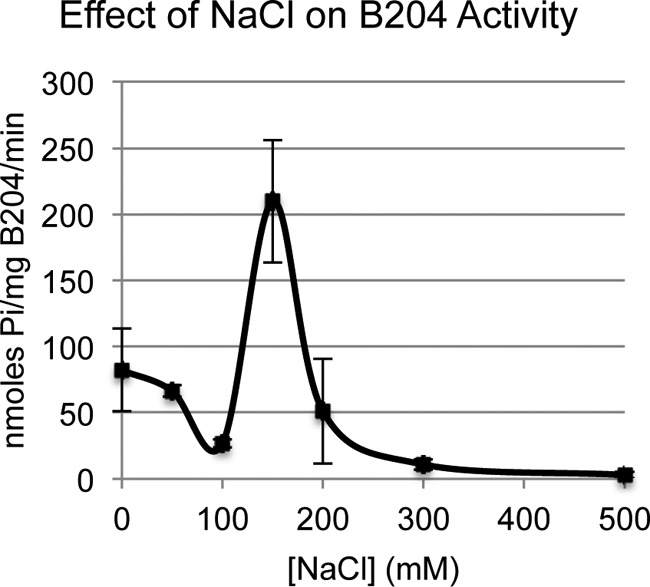
Maximum levels of B204 ATPase activity are apparent for a narrow NaCl concentration range.
Based on both structural information reported above and prior knowledge of enzymes belonging to this superfamily, a total of 20 B204 mutants were constructed; these mutants and their ATPase activities in the presence of dsDNA are reported in Table 2. Collectively, the activity profiles of these mutants span a broad range; mutations at Arg14, Lys17, Glu49, Glu104, and Asp152 and the mutant K13M displayed no detectable ATPase activity, while mutations at Arg127, Gln138, Lys166, and Lys170 and the mutant K13R displayed measurable ATPase activities (Table 2). In general, these results support the essentiality of a number of amino acid residues within B204, most of which are conserved among ATPases belonging to the larger FtsK/HerA superfamily.
TABLE 2.
B204 mutant activities in ATPase assays and transfection experiments
| Mutant | Target motif/predicted function | In vitro ATPase assay (nmol Pi/mg protein/min) | Transfection experiment resulta |
|---|---|---|---|
| WT | 315 ± 30 | + | |
| K13M | Walker A: GRMRSGKS | No detectable activity | Not tested |
| K13R | Walker A: GRRRSGKS | 128 ± 41 | – |
| R14M | Walker A: GRKMSGKS | No detectable activity | Not tested |
| R14K | Walker A: GRKKSGKS | No detectable activity | Not tested |
| K17M | Walker A: GRKRSGMS | No detectable activity | – |
| K17R | Walker A: GRKRSGRS | No detectable activity | – |
| E49K | Putative function in ATP binding/hydrolysis | No detectable activity | – |
| E49L | No detectable activity | – | |
| E49N | No detectable activity | – | |
| E49Q | No detectable activity | Not tested | |
| E104Q | Walker B: VLIIDQ | No detectable activity | – |
| R127E | Arginine finger | 54.4 ± 3.6 | – |
| R127K | 27.8 ± 3.6 | – | |
| R127M | 42.9 ± 12.4 | – | |
| Q138E | Triphosphate-sensing residue | 44.5 ± 9.5 | – |
| Q138N | 88.0 ± 17.5 | + | |
| D152A | Stabilizing residue | No detectable activity | – |
| D152N | No detectable activity | – | |
| K166A | Tunnel residue | 80.1 ± 8.6 | + |
| K170A | Tunnel residue | 675 ± 62 | + |
A dash indicates that the mutant generated qPCR values on par with those of uninfected controls, around 1 × 108 genomes/ml. The plus symbol indicates that the mutant generated qPCR values at levels comparable to that of the wild type, around 1 × 1012 to 1 × 1013 genomes/ml. The mutant particles generated from the first round of infection were capable of reinfecting the host. The boldface letters indicate the position being mutated in the context of the motif of interest.
Transfection of B204 mutant STIV DNA into the host.
Based on the results from the in vitro ATPase assay, several mutants were tested in STIV transfection experiments (Table 2). Briefly, these mutants were constructed within an infectious clone of STIV viral DNA and subsequently transfected into the susceptible host, S. solfataricus P23. It was found that mutants displaying no detectable ATPase activity in vitro did not lead to virus production (assayed by qPCR and electron microscopic visualization). The mutants Q138N, K166A, and K170A, which displayed approximately 28%, 25%, and over 200% of wild-type activity in the in vitro assay, respectively, all were viable in the transfection experiments (Table 2). The mutants K13R and Q138E and the mutations at Arg127, which displayed activities ranging from 9 to 41% of wild-type activity in the in vitro assay, were not viable in transfection experiments (Table 2).
B204 is present in the mature virion.
The question of whether B204 is present in the mature STIV virion remains controversial. For this reason, STIV particles were extensively purified by ultracentrifugation using a cesium sulfate gradient as previously described (4). This process generated two blue bands (termed upper band and lower band) that contained morphologically distinct virus particles; electron micrographs of negatively stained samples revealed that the upper band contained particles with clear icosahedral morphology, including sharply defined edges, while the lower band showed particles with a less clearly defined morphology (Fig. 5a). The upper and lower bands were dialyzed into 25 mM citrate buffer, pH 3.5, and separately analyzed in a variety of experiments, including ATPase assays, Western blot analysis, qPCR analysis, and mass spectrometry analysis (Fig. 5a to d and data not shown). Quantification of packaged virus particles by qPCR (expressed as genomes/milliliter) indicated that only the upper band contained packaged copies of the STIV genome; the qPCR signal observed for the lower band was similar to that of uninfected control reactions (Fig. 5a). In ATPase assays, the upper band demonstrated significantly higher levels of ATPase activity than the lower band or uninfected control reactions (Fig. 5d). Furthermore, in-solution trypsin digestion of the upper and lower bands followed by mass spectrometry analysis revealed that peptides matching the amino acid sequence of B204 were present only in the upper band (data not shown).
FIG 5.

STIV purified via cesium gradient. (a) Depiction of the resulting bands achieved via cesium gradient for an STIV sample, shown as upper and lower blue bands with accompanying electron micrographs of particles observed for each band. (b) Western blot analysis on protein-normalized samples from the upper and lower bands using antibodies raised against B204 and A223 proteins. E. coli-purified B204 protein is labeled “control.” Molecular mass standards are shown in kilodaltons. (c) Western blot analysis of a series of E. coli-purified B204 standards loaded at various concentrations ranging from 0.001 μg to 50 μg. The upper band of the STIV cesium purification is also loaded on this gel. (d) In vitro ATPase activity comparisons between unpurified STIV (prior to Cs2SO4 separation), the upper band, and the lower band of the cesium gradient. The control sample did not contain any particles; details regarding the preparation of the control sample are described in Materials and Methods.
A Western blot analysis using B204 antibodies with protein-normalized loading of the upper and lower bands further confirmed the presence of B204 in the upper band and its absence from the lower band (Fig. 5b). To support evidence for the presence of virus-like particles in both bands, an identical Western blot analysis was performed with antibodies against A223 (a structural protein that forms the penton base at each turret vertex in the STIV particle) and revealed that this protein is present in relatively equal amounts in the upper and lower bands. A third Western blot included the upper band alongside a series of E. coli-purified B204 standards loaded in quantities ranging from 0.001 μg to 50 μg (Fig. 5c). The band intensity of B204 from the upper band falls between the 0.01-μg and 0.1-μg standards, which correspond to 2.51 × 1011 to 2.51 × 1012 molecules of B204. By dividing this range into the number of STIV genomes loaded for the upper band (Fig. 5a, 10 μl of 4.81 × 1012 genomes/ml or 4.81 × 1010 genomes/ml), we estimated that there are around 5 to 52 copies of B204 per STIV virion.
B204-like sequences present in the metagenome.
Sequences showing 30% identity or higher to the STIV B204 gene were aligned and compared to identify conservation patterns present within these closely related proteins (see the supplemental material). Seven of the 10 positions mutated in this study were largely conserved across B204-like sequences, including Lys13, Lys17, Glu49, Glu104, Arg127, Gln138, and Asp152. One of the tunnel residues, Lys170, was conserved as a Lys or Arg in the sequence alignment while the other tunnel position, Lys166, was usually a charged residue (Lys, Asp, or Glu). The most interesting residue was Arg14, which was conserved as either an Arg or a Gly among these aligned sequences.
DISCUSSION
Amino acids serving essential roles in ATP binding and hydrolysis.
STIV B204 is an ATPase that has been suggested to provide energy via ATP hydrolysis to power the packaging of its viral DNA into the STIV virion (20, 21). This enzyme belongs to the A32-like DNA-packaging clade of the larger FtsK/HerA superfamily (3). The Walker A and B motifs are two conserved motifs that are present among P-loop NTPases and generally function in ATP binding and ATP hydrolysis, respectively (17). It was found that both conservative (K17R) and nonconservative (K17M) mutations at this position (which belongs to the Walker A motif) yielded no detectable ATPase activity in both the in vitro assay and transfection experiments, confirming the essentiality of Lys17. Recently, it was proposed that Glu49 in STIV2 B204 (which is only conserved in some members of the DNA-packaging clade within the superfamily) assumes the role of Glu104 (Walker B motif), given that it is in closer proximity to the water nucleophile thought to be responsible for ATP hydrolysis. Our results indicate that all conservative and nonconservative substitutions at these positions (E104Q, E49K, E49L, E49N, and E49Q) are inactive in both in vitro assays and transfection experiments and demonstrates that both Glu49 and Glu104 serve essential roles for B204 that cannot, in this case, be articulated from structural information alone. Given that all enzymes behaved similarly to the wild type during purification with comparable yields and protein purity (data not shown), all enzymes discussed here were not expected to lack activity due to misfolding or instability.
Two highly conserved positions in members of the FtsK/HerA superfamily include the arginine finger (Arg127) located after the α4 helix and a glutamine residue (Gln138) located at the C-terminal end of the β6 strand. In many ATPases, the arginine finger has been reported to be involved in nucleotide binding and/or hydrolysis through interaction with the β- or γ-phosphate of NTP in the active site of the neighboring protomer (18, 19). Given that B204 crystallizes in a lattice that most likely is not functionally relevant, it is not surprising that we are unable to observe the active conformation for Arg127 as described above. We chose several amino acid substitutions to make at this position (R127E, R127K, and R127M) in an attempt to ascertain whether changes in amino acid charge, size, or shape at position 127 would affect activity. In B204, all three R127 mutants displayed detectable levels of ATPase activity in the in vitro assay; however, these activities were lower than those of the wild type, and none of the mutants were viable in transfection experiments. Therefore, it is likely that these mutants cannot sustain sufficient levels of ATPase activity to function normally during the STIV infection cycle.
The conserved glutamine residue in B204 (Gln138) is analogous to the triphosphate-sensing residue in other ATPases within the AAA+ family and among SFI and SFII helicases (36–38). In B204, the mutants Q138E and Q138N displayed 14% and 28% of the wild-type in vitro ATPase activity, respectively. B204 Q138N was successful in transfection experiments while Q138E was not, suggesting that although side chain length can vary at this position, the amido group is essential for maintaining wild-type levels of ATPase activity.
Essentiality of amino acids involved with interprotomer associations.
The crystal structure of STIV B204 reveals that Asp152 has the potential to participate in at least two salt bridge interactions at different phases of the ATPase reaction: (i) between Asp152 and the Arg127 finger (Fig. 6a) and (ii) between Asp152 from one protomer and a rotated conformation of Lys13 from the neighboring protomer (Fig. 5b) in the context of the modeled hexamer. Position 152 is somewhat conserved for viral ATPases belonging to the DNA-packaging clade of the superfamily; however, larger structural alignments indicate that this conservation does not extrapolate to the entire family (3, 20). Nevertheless, this position is essential in B204 and probably other members of the DNA-packaging clade, as both D152A and D152N do not display activity in either in vitro activity assays or transfection experiments. Based on these results, it appears that Asp152 most likely serves a critical role in stabilization of residues involved with interprotomer interactions.
FIG 6.
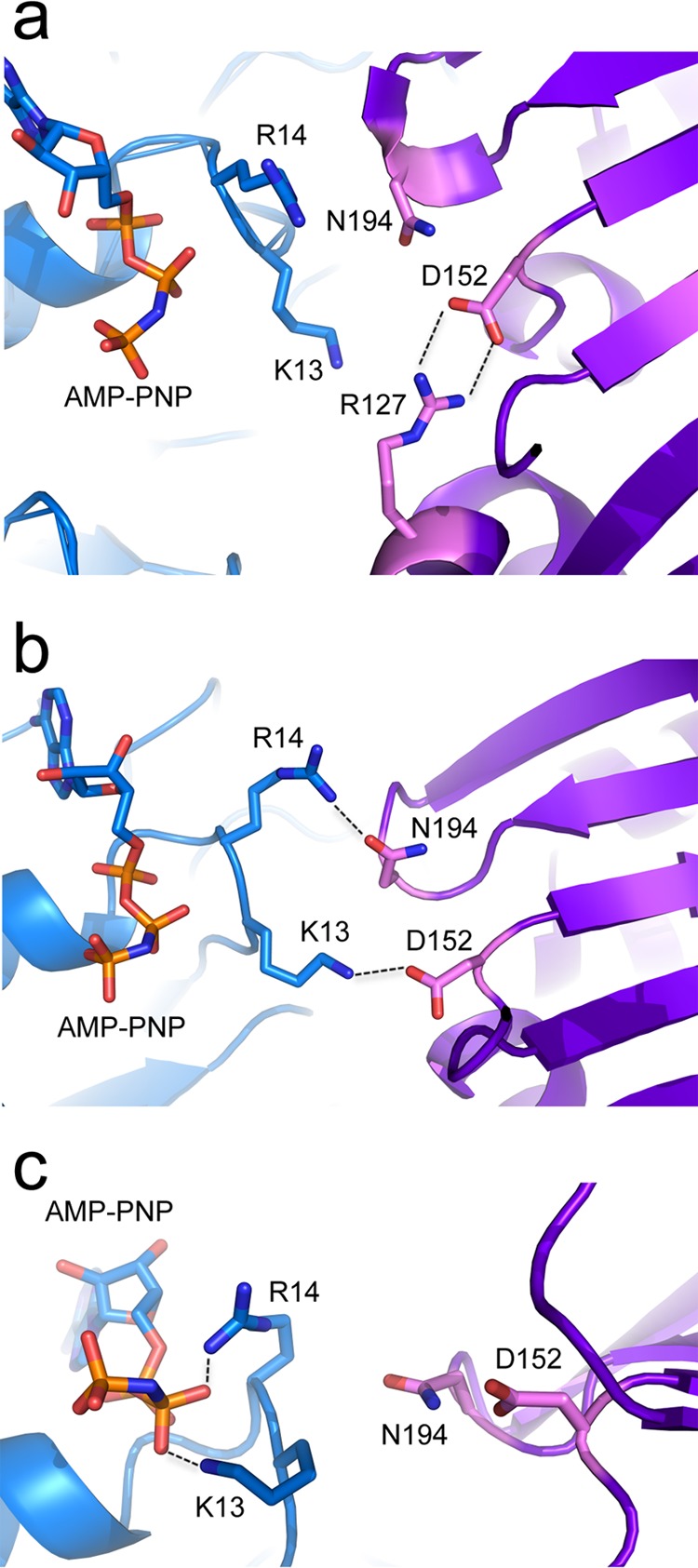
Experimental and modeled interactions of functional residues in B204. All images show two monomers (blue and purple) in the context of the modeled hexamer. (a) Experimentally observed electrostatic interactions between D152 and Arg127 (present in both apo and AMP-PNP-bound crystal structures of STIV B204). (b) Modeled hydrogen bond and electrostatic interactions between Arg14-Asn194 and Lys13-Asp152, respectively. (c) Modeled interactions between the β-phosphate of AMP-PNP and Lys13 or Arg14. It is noteworthy that these two amino acids also can be rotated into positions that would stabilize the γ-phosphate of AMP-PNP.
Putative roles for pseudoconserved residues within the Walker A motif.
In the crystal structure of B204 complexed with AMP-PNP, the side chains of Lys13 and Arg14 are not in close proximity to the nucleotide or any other residues. However, in the context of the modeled B204 hexamer, the side chains from both of these residues can be modeled into orientations that allow them to interact with polar side chains of the neighboring protomer or the β- or γ-phosphate of AMP-PNP (Fig. 6b and c). Both of these conformations may be catalytically relevant and most likely are triggered during different phases of the reaction, serving roles in interprotomer interactions and/or γ-phosphate stabilization during ATP hydrolysis. For Lys13, we found that mutation to arginine but not methionine was sufficient for a detectable level of ATPase activity within the in vitro assay; however, neither mutant was viable in transfection experiments. For Arg14, none of the mutants yielded any detectable levels of activity in either the in vitro assays or transfection experiments. While Lys13 is strictly conserved within the alignment of B204-like sequences from the metagenome, it is not conserved among the larger family and is only slightly conserved within the DNA-packaging clade. Interestingly, at position 14, approximately half of the sequences from the metagenome alignment contain arginine, while the other half contain glycine (see Fig. S1 in the supplemental material), and this position also is usually a glycine within the larger family (3). Because Arg14 demonstrates an essential functional role in B204, we hypothesize that another yet-unidentified amino acid is replacing this role in enzymes that contain a glycine at this position. Curiously, Arg14 corresponds to Lys14 in STIV2 B204; however, our experiments demonstrate that R14K is inactive in both in vitro and transfection assays. This result suggests that interactions between Arg14 and surrounding residues and/or with ATP are highly precise, and that this amino acid has evolved a uniquely essential role in B204 and B204-like sequences. In general, these findings highlight that there exist a larger number of amino acid positions serving essential roles within these molecular motors than what would be predicted from the analysis of conservation patterns.
Putative roles for B204 tunnel residues.
The hexameric model of B204 shows a number of amino acids protruding into the central ring, including the long, basic side chains from Lys166 and Lys170 (Fig. 3). It is possible that positively charged residues such as these help regulate the translocation of negatively charged DNA through the central channel and into the virion during the packaging process. The mutants K166A and K170A were active in the in vitro assay (Table 2) and were viable in transfection experiments. Notably, K170A displayed 2-fold higher activity than the wild type in the in vitro assay, and perhaps this increase in activity in the more sensitive ATPase assay occurred at the expense of a loss in charge within the tunnel, which could affect translocation regulation in the context of the virion. These results suggest that tunnel residues are not individually detrimental to the packaging of DNA; however, cumulative mutations at these positions may alter charge balance within the tunnel and could challenge efficient DNA packaging. Future experiments will address the additive effects of tunnel mutations.
The effect of sodium chloride on B204 ATPase activity.
It was determined that the ATPase activity of B204 in the in vitro assays was significantly affected by the concentration of sodium chloride, with maximum activity achieved at a concentration of 150 mM (Fig. 4). There is some precedent for the inhibitory effect of sodium chloride on ATPases belonging to this family; for example, the ATPase TrwK, which is a homolog of VirB4 (the closest structural homolog to STIV and STIV2 B204), is inactive in a buffer containing 200 mM NaCl but also displays minimal activity in buffers containing very low levels of NaCl (39). Additionally, the ATPase homolog of VirB4, named TraB, assembles the active hexamer only in the absence of sodium chloride (40). Another homolog, PRD1 P9 from the DNA-packaging clade of the superfamily, was reported to experience nearly optimal levels of activity in a DNA-packaging assay in the absence of sodium chloride or calcium chloride (20). While it does appear that B204 requires a certain level of sodium chloride to achieve maximum activity, we did not observe higher-order oligomers in gel filtration experiments conditioned with high or low concentrations of sodium chloride (data not shown). These results are consistent with those for STIV2 B204, which also was observed as a monomer in solution and in a crystal lattice. Future experiments will explore whether other salts can be substituted for NaCl to achieve higher levels of ATPase activity and whether these substitutions generate higher-order oligomers in solution. Subsequent crystallization experiments employing conditions that stimulate higher-order oligomers will shed light on the structural features important for this oligomerization process and will build on our current understanding of viral DNA packaging among this family of viruses.
B204 is present within the virion.
The experiments discussed here provide supporting evidence for the existence of an active B204 ATPase present within the virion. Mass spectrometric analysis confirms the presence of the B204 protein and ATPase activity in purified virions (associated with the upper band in the cesium gradients), whereas virions lacking B204 also lack ATPase activity (associated with the lower band of the cesium gradient). Electron micrographs of the upper band clearly depict virus particles with a sharply defined exterior (Fig. 5b) and contain packaged viral DNA. The lower band from the same cesium gradient shows particles with less clearly defined edges and lack packaged DNA. The particles in the lower band are likely to represent empty STIV particles at an earlier phase of the infection cycle. As mentioned previously, structural analysis indicates that STIV B204 does not appear to be present in the virion (6). We speculate this is a result of the low copy number of B204 within the assembled virions and the failure to separate full and empty capsids prior to structure determination. Future studies will be focused on single particle reconstruction of both empty and packaged virus particles.
The current model for STIV DNA packaging proposes that there exists a motor complex at a unique site on the virion (21). This motor complex includes the functional B204 ATPase oligomer, thought to exist as a hexameric or dodecameric ring through which DNA is translocated using energy provided by NTP hydrolysis. Our results correlate well with this model, given that the expected number of B204 copies per virion (6 to 12) falls within the range calculated in our Western blot experiments. The collection of experiments discussed here affirms the long-debated question of the presence of B204 in the mature STIV virion. Future experiments will begin to address the roles that other STIV proteins play in this DNA-packaging process.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NSF-DEB 1342876.
Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The Macromolecular Diffraction Laboratory at Montana State University was supported, in part, by a grant from the Murdock Foundation.
We acknowledge Brian Eilers and Stephen Keable for their assistance with crystallization experiments and X-ray diffraction data collection, Ben Bolduc for the compilation of a list of B204-like sequences from the metagenome, and also the Mass Spectrometry, Proteomics, and Metabolomics Facility at Montana State University for processing all samples analyzed by mass spectrometry. Polyclonal antibodies for B204 and A223 were produced by the Animal Research Center at Montana State University.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02435-15.
REFERENCES
- 1.Maaty WS, Ortmann AC, Dlakic M, Schulstad K, Hilmer JK, Liepold L, Weidenheft B, Khayat R, Douglas T, Young MJ, Bothner B. 2006. Characterization of the archaeal thermophile Sulfolobus turreted icosahedral virus validates an evolutionary link among double-stranded DNA viruses from all domains of life. J Virol 80:7625–7635. doi: 10.1128/JVI.00522-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortmann AC, Brumfield SK, Walther J, McInnerney K, Brouns SJJ, van de Werken HJG, Bothner B, Douglas T, van der Oost J, Young MJ. 2008. Transcriptome analysis of infection of the archaeon Sulfolobus solfataricus with Sulfolobus turreted icosahedral virus. J Virol 82:6784–6784. doi: 10.1128/JVI.00912-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iyer LM, Makarova KS, Koonin EV, Aravind L. 2004. Comparative genomics of the FtsK-HerA superfamily of pumping ATPases: implications for the origins of chromosome segregation, cell division and viral capsid packaging. Nucleic Acids Res 32:5260–5279. doi: 10.1093/nar/gkh828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice G, Tang L, Stedman K, Roberto F, Spuhler J, Gillitzer E, Johnson JE, Douglas T, Young M. 2004. The structure of a thermophilic archaeal virus shows a double-stranded DNA viral capsid type that spans all domains of life. Proc Natl Acad Sci U S A 101:7716–7720. doi: 10.1073/pnas.0401773101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brumfield SK, Ortmann AC, Ruigrok V, Suci P, Douglas T, Young MJ. 2009. Particle assembly and ultrastructural features associated with replication of the lytic archaeal virus Sulfolobus turreted icosahedral virus. J Virol 83:5964–5970. doi: 10.1128/JVI.02668-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veesler D, Ng TS, Sendamarai AK, Eilers BJ, Lawrence CM, Lok SM, Young MJ, Johnson JE, Fu CY. 2013. Atomic structure of the 75 MDa extremophile Sulfolobus turreted icosahedral virus determined by CryoEM and X-ray crystallography. Proc Natl Acad Sci U S A 110:5504–5509. doi: 10.1073/pnas.1300601110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khayat R, Tang L, Larson ET, Lawrence CM, Young M, Johnson JE. 2005. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc Natl Acad Sci U S A 102:18944–18949. doi: 10.1073/pnas.0506383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maaty WS, Selvig K, Ryder S, Tarlykov P, Hilmer JK, Heinemann J, Steffens J, Snyder JC, Ortmann AC, Movahed N, Spicka K, Chetia L, Grieco PA, Dratz EA, Douglas T, Young MJ, Bothner B. 2012. Proteomic analysis of Sulfolobus solfataricus during Sulfolobus turreted icosahedral virus infection. J Proteome Res 11:1420–1432. doi: 10.1021/pr201087v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wirth JF, Snyder JC, Hochstein RA, Ortmann AC, Willits DA, Douglas T, Young MJ. 2011. Development of a genetic system for the archaeal virus Sulfolobus turreted icosahedral virus. Virology 415:6–11. doi: 10.1016/j.virol.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 10.Snyder JC, Brumfield SK, Peng N, She QX, Young MJ. 2011. Sulfolobus turreted icosahedral virus c92 protein responsible for the formation of pyramid-like cellular lysis structures. J Virol 85:6287–6292. doi: 10.1128/JVI.00379-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snyder JC, Samson RY, Brumfield SK, Bell SD, Young MJ. 2013. Functional interplay between a virus and the ESCRT machinery in Archaea. Proc Natl Acad Sci U S A 110:10783–10787. doi: 10.1073/pnas.1301605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larson ET, Reiter D, Young M, Lawrence CM. 2006. Structure of A197 from Sulfolobus turreted icosahedral virus: a crenarchaeal viral glycosyltransferase exhibiting the GT-A fold. J Virol 80:7636–7644. doi: 10.1128/JVI.00567-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larson ET, Eilers B, Menon S, Reiter D, Ortmann A, Young MJ, Lawrence CM. 2007. A winged-helix protein from sulfoblobus turreted icosahedral virus points toward stabilizing disulfide bonds in the intracellular proteins of a hyperthermophilic virus. Virology 368:249–261. doi: 10.1016/j.virol.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 14.Larson ET, Eilers BJ, Reiter D, Ortmann AC, Young MJ, Lawrence CM. 2007. A new DNA binding protein highly conserved in diverse crenarchaeal viruses. Virology 363:387–396. doi: 10.1016/j.virol.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 15.Snyder JC, Brumfield SK, Kerchner KM, Quax TEF, Prangishvili D, Young MJ. 2013. Insights into a viral lytic pathway from an archaeal virus-host system. J Virol 87:2186–2192. doi: 10.1128/JVI.02956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha-subunits and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leipe DD, Wolf YI, Koonin EV, Aravind L. 2002. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol 317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- 18.Enemark EJ, Joshua-Tor L. 2008. On helicases and other motor proteins. Curr Opin Struct Biol 18:243–257. doi: 10.1016/j.sbi.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singleton MR, Dillingham MS, Wigley DB. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 20.Stromsten NJ, Bamford DH, Bamford JK. 2005. In vitro DNA packaging of PRD1: a common mechanism for internal-membrane viruses. J Mol Biol 348:617–629. doi: 10.1016/j.jmb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Happonen LJ, Oksanen E, Liljeroos L, Goldman A, Kajander T, Butcher SJ. 2013. The structure of the NTPase that powers DNA packaging into Sulfolobus turreted icosahedral virus 2. J Virol 87:8388–8398. doi: 10.1128/JVI.00831-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu CY, Wang K, Gan L, Lanman J, Khayat R, Young MJ, Jensen GJ, Doerschuk PC, Johnson JE. 2010. In vivo assembly of an archaeal virus studied with whole-cell electron cryotomography. Structure 18:1579–1586. doi: 10.1016/j.str.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stromsten NJ, Bamford DH, Bamford JKH. 2003. The unique vertex of bacterial virus PRD1 is connected to the viral internal membrane. J Virol 77:6314–6321. doi: 10.1128/JVI.77.11.6314-6321.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McPherson A. 1982. The preparation and analysis of protein crystals. John Wiley & Sons, New York, NY. [Google Scholar]
- 25.Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26:795–800. doi: 10.1107/S0021889893005588. [DOI] [Google Scholar]
- 26.Leslie AGW, Powell HR. 2007. Processing diffraction data with MOSFLM, p 41–51. In Read RJ, Sussman JL (ed), Evolving methods for macromolecular crystallography, vol 245 Springer, New York, NY. doi: 10.1007/978-1-4020-6316-9_4. [DOI] [Google Scholar]
- 27.Collaborative Computational Project Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 30.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. 2009. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 4:1–13. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Bolduc B, Shaughnessy DP, Wolf YI, Koonin EV, Roberto FF, Young M. 2012. Identification of novel positive-strand RNA viruses by metagenomic analysis of Archaea-dominated Yellowstone hot springs. J Virol 86:5562–5573. doi: 10.1128/JVI.07196-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bolduc B, Wirth JF, Mazurie A, Young MJ. 2015. Viral assemblage composition in Yellowstone acidic hot springs assessed by network analysis. ISME J 9:2162–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:1–19. doi: 10.1186/1471-2105-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flachner B, Kovari Z, Varga A, Gugolya Z, Vonderviszt F, Naray-Szabo G, Vas M. 2004. Role of phosphate chain mobility of MgATP in completing the 3-phosphoglycerate kinase catalytic site: binding, kinetic, and crystallographic studies with ATP and MgATP. Biochemistry 43:3436–3449. doi: 10.1021/bi035022n. [DOI] [PubMed] [Google Scholar]
- 36.Gorbalenya AE, Koonin EV. 1993. Helicases–amino-acid-sequence comparisons and structure-function-relationships. Curr Opin Struct Biol 3:419–429. doi: 10.1016/S0959-440X(05)80116-2. [DOI] [Google Scholar]
- 37.Iyer LM, Leipe DD, Koonin EV, Aravind L. 2004. Evolutionary history and higher order classification of AAA plus ATPases. J Struct Biol 146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Neuwald AF, Aravind L, Spouge JL, Koonin EV. 1999. AAA(+): a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9:27–43. [PubMed] [Google Scholar]
- 39.Arechaga I, Pena A, Zunzunegui S, Fernandez-Alonso MD, Rivas G, de la Cruzi F. 2008. ATPase activity and oligomeric state of TrwK, the VirB4 homologue of the plasmid R388 type IV secretion system. J Bacteriol 190:5472–5479. doi: 10.1128/JB.00321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durand E, Oomen C, Waksman G. 2010. Biochemical dissection of the ATPase TraB, the VirB4 homologue of the Escherichia coli pKM101 conjugation machinery. J Bacteriol 192:2315–2323. doi: 10.1128/JB.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.