

ABSTRACT
Tumor suppressor p53 is activated in response to numerous cellular stresses, including viral infection. However, whether murine gammaherpesvirus 68 (MHV68) provokes p53 during the lytic replication cycle has not been extensively evaluated. Here, we demonstrate that MHV68 lytic infection induces p53 phosphorylation and stabilization in a manner that is dependent on the DNA damage response (DDR) kinase ataxia telangiectasia mutated (ATM). The induction of p53 during MHV68 infection occurred in multiple cell types, including splenocytes of infected mice. ATM and p53 activation required early viral gene expression but occurred independently of viral DNA replication. At early time points during infection, p53-responsive cellular genes were induced, coinciding with p53 stabilization and phosphorylation. However, p53-related gene expression subsided as infection progressed, even though p53 remained stable and phosphorylated. Infected cells also failed to initiate p53-dependent gene expression and undergo apoptosis in response to treatment with exogenous p53 agonists. The inhibition of p53 responses during infection required the expression of the MHV68 homologs of the shutoff and exonuclease protein (muSOX) and latency-associated nuclear antigen (mLANA). Interestingly, mLANA, but not muSOX, was necessary to prevent p53-mediated death in MHV68-infected cells under the conditions tested. This suggests that muSOX and mLANA are differentially required for inhibiting p53 in specific settings. These data reveal that DDR responses triggered by MHV68 infection promote p53 activation. However, MHV68 encodes at least two proteins capable of limiting the potential consequences of p53 function.
IMPORTANCE Gammaherpesviruses are oncogenic herpesviruses that establish lifelong chronic infections. Defining how gammaherpesviruses overcome host responses to infection is important for understanding how these viruses infect and cause disease. Here, we establish that murine gammaherpesvirus 68 induces the activation of tumor suppressor p53. p53 activation was dependent on the DNA damage response kinase ataxia telangiectasia mutated. Although active early after infection, p53 became dominantly inhibited as the infection cycle progressed. Viral inhibition of p53 was mediated by the murine gammaherpesvirus 68 homologs of muSOX and mLANA. The inhibition of the p53 pathway enabled infected cells to evade p53-mediated cell death responses. These data demonstrate that a gammaherpesvirus encodes multiple proteins to limit p53-mediated responses to productive viral infection, which likely benefits acute viral replication and the establishment of chronic infection.
INTRODUCTION
Gammaherpesviruses (GHVs) are DNA tumor viruses that include Epstein-Barr virus (EBV), Kaposi's sarcoma-associated herpesvirus (KSHV), and murine gammaherpesvirus 68 (MHV68), among others (1). Like all herpesviruses, GHVs exhibit two distinct infectious cycle phases, lytic replication and latency (2). The lytic cycle is the productive phase of infection and is characterized by temporally regulated viral gene expression, viral DNA replication, and the production of progeny virus (2). Lytic replication enables GHVs to establish a lifelong chronic infection known as latency (3), which is characterized by restricted viral gene expression and viral genome maintenance within host lymphocytes and other cell types (1). Lifelong colonization by GHVs places the infected host at risk for numerous cancers and lymphoproliferative disorders, especially in settings of immunosuppression (4), although mechanisms by which GHVs drive cellular transformation are incompletely defined.
Several recent studies demonstrate that GHVs engage the DNA damage response (DDR) during lytic replication (reviewed in reference 5). The DDR is an ordered, multistep cellular signaling response to potentially genotoxic stresses, including the expression of viral oncogenes that push cell cycle progression (6–8). Following the recognition of damaged DNA by sensor proteins, the DDR cascade is transduced by ataxia telangiectasia mutated (ATM), ATM and Rad 3-related (ATR), or DNA-dependent protein kinase (DNA-PK), kinases of the phosphatidylinositol 3′ kinase-related kinase (PIKK) family (8). These enzymes then target a number of downstream effectors that block cell cycle progression and help repair damaged DNA or induce cell death or senescence if the damage is irreparable (8).
One critical downstream effector of the DDR signaling cascade is tumor suppressor p53, a protein that is essential for limiting the propagation of mutations following genotoxic stresses (9). The importance of p53 in preventing oncogenesis is underscored by the finding that p53 is nonfunctional in approximately half of all human cancers (10, 11). Under normal cellular growth conditions p53 protein is continuously ubiquitinated by mouse double minute 2 (MDM2) and targeted for proteasomal degradation (12, 13). However, DNA damage-responsive kinases can phosphorylate p53, leading to its stabilization and activation. This enables p53-driven transcription of genes involved in cell cycle arrest, DNA repair, or apoptosis (14). Given the capacity of p53 to inhibit cell cycle progression and promote apoptosis, p53 could impose a host restriction on DNA virus replication. This is emphasized by the fact that essentially all DNA viruses encode proteins that inhibit p53 function (5). Indeed, p53 originally was discovered as a host protein bound to simian virus 40 (SV40) large T antigen (15, 16), and it was later demonstrated that T antigen prevented p53 function (17–19).
Following de novo infection of primary macrophages, MHV68 induces the phosphorylation of ATM and histone variant H2AX (20), a classic ATM target in the DDR (21). Consistent with these findings, a phosphoproteomic analysis of lytic MHV68 infection in fibroblasts also identified phosphorylated H2AX and a number of additional ATM targets (22). EBV also triggers ATM activation and phosphorylation of downstream targets, including H2AX, Chk2, NBS1, and p53, during reactivation from latency (23). Interestingly, for both MHV68 and EBV, DDR induction can be actively initiated by a conserved herpesvirus-encoded protein kinase (CHPK), an event that appears to be necessary for efficient viral replication in some settings (20, 24).
Since the cellular function of the DDR is to prevent aberrant cellular replication (8), it stands to reason that DDR activation would limit the replication of GHVs through the induction of host molecules aimed at inhibiting DNA replication, especially activation of p53, unless viral gene products were capable of inhibiting or subverting aspects of the infection-related DDR. Consistent with this notion, a number of GHV proteins inhibit p53 function when expressed in isolation, including v-IRF1, v-IRF3, v-IRF4, RTA, and LANA, as well as a number of viral structural proteins for KSHV and EBNA1 and EBNA3C for EBV (25–32). However, whether p53 inhibition by these proteins (and other lytic cycle proteins) contributes to efficient viral replication is largely unexplored.
While evaluating roles for the MHV68 homolog of the latency-associated nuclear antigen (mLANA) in lytic MHV68 replication, we previously found that LANA-null MHV68 exhibited a lytic replication defect that correlated with enhanced p53 activation and cell death compared to that of wild-type (WT) MHV68 infection (33). We also demonstrated that viral genes were hyperexpressed in the absence of mLANA and that p53 enhanced this phenotype. While these previous studies were consistent with the hypothesis that mLANA controls p53 responses in lytic MHV68 replication, a number of questions still remain. First, whether mLANA directly inhibits p53 responses during MHV68 infection is not known. Our previous studies also were consistent with p53 activation as a consequence of deregulated viral gene expression due to the absence of mLANA (33). Furthermore, although MHV68 activates ATM during lytic replication (20), downstream consequences, such as p53 induction, during WT MHV68 infection have not been evaluated. Second, although the absence of mLANA correlated with increased p53 induction during MHV68 lytic replication, it is not known if p53 functions remain intact or are dominantly inhibited in the infected cell. Finally, it is not yet clear if mLANA is a master regulator of p53 deactivation or if other viral proteins also impact p53-related events in the MHV68-infected cell.
Here, we demonstrate that p53 is induced during lytic MHV68 replication in an ATM-dependent manner. The induction of p53 occurred in multiple cell types, including primary splenocytes in MHV68-infected mice, and correlated with early viral gene expression. Although p53 was transcriptionally active at early time points during the lytic cycle, p53-dependent transcription became repressed as the infectious cycle progressed. Moreover, infection-associated p53 inhibition was dominant, as infected cells became resistant to treatment with potent DNA damage-inducing and p53-stabilizing drugs. We found that both mLANA and the MHV68 shutoff and exonuclease protein, muSOX, were important for inhibiting p53 responses. However, muSOX was not necessary for preventing death when infected cells were treated with exogenous p53-activating agents while mLANA was, suggesting that the two proteins are differentially required for p53 inhibition in specific settings.
MATERIALS AND METHODS
Ethics statement.
Mouse experiments performed for this study were carried out in accordance with NIH, USDA, and UAMS Division of Laboratory Animal Medicine and IACUC guidelines. The protocol supporting this study was approved by the UAMS Institutional Animal Care and Use Committee (animal use protocol 3270). Mice were anesthetized prior to inoculations and sacrificed to minimize pain and distress.
Cell culture and viruses.
Swiss albino 3T3 murine fibroblasts were purchased from the ATCC (CCL-92). ATM−/− and WT murine embryonic fibroblasts (MEFs) were gifts from Vera Tarakanova (Medical College of Wisconsin). MB114 cells were a gift from Linda van Dyk (University of Colorado School of Medicine). All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin-streptomycin, and 2 mM l-glutamine (cMEM). For serum starvation, cells were cultured in DMEM containing 1% FCS, 100 U/ml penicillin-streptomycin, and 2 mM l-glutamine (ssMEM) for 18 to 24 h prior to experiments. Cells were maintained at 37°C in atmosphere containing 5% CO2 and 100% humidity. All viruses used in this study were previously described and include WT MHV68 (34), H2B-yellow fluorescent protein (YFP)-expressing MHV68 (35), RTA-null MHV68 (50.STOP) (36), host shutoff-null MHV68 (ORF37.ΔHS), host shutoff-null marker rescue MHV68 (ORF37.MR [37]), mLANA-null MHV68 (73.STOP), and mLANA-null marker rescue MHV68 (73.MR) (38). Viral stocks were generated as previously described (33). For infections, viral stocks were diluted in cMEM or ssMEM and adsorbed to monolayers of cells. For high-multiplicity infections, cells were inoculated in 1/10 normal culture volume and rocked every 15 min for 1 h. After the removal of inocula, cells were cultured in a normal volume of ssMEM. The time at which virus was added was considered t = 0 h for all time course experiments. For virus growth assays, two cycles of freeze/thaw lysis were performed to extract progeny virions. Lysates were serially diluted, and titers were measured by plaque assay (39).
Immunoblot analyses.
Cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed in-well with radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 20 mM Tris, 2 mM EDTA, 1% NP-40, 0.25% deoxycholate) supplemented with complete mini-EDTA-free protease inhibitors and phosphatase inhibitors (Thermo-Pierce). Insoluble material was pelleted by centrifugation, and 50,000 cell equivalents were boiled in Laemmli sample buffer at 100°C for 8 min (40). Proteins were resolved by SDS-PAGE in 5%, 10%, or 12% polyacrylamide gels. Resolved proteins were transferred to nitrocellulose membranes and probed with antibodies directed to the indicated proteins. As a positive control for p53 induction, MEFs were exposed to 10 Gy of gamma radiation and incubated for 1 h at 37°C. Antigen-antibody complexes were detected by chemiluminescence using horseradish peroxidase (HRP)-conjugated secondary antibodies and Clarity ECL reagent (Bio-Rad). Signal was detected using a ChemiDoc MP digital imaging system (Bio-Rad).
Drugs and antibodies.
Doxorubicin and phosphonoacetic acid (PAA) were purchased from Sigma-Aldrich and diluted in distilled water (dH2O). Tumor necrosis factor alpha (TNF-α; Sigma-Aldrich) was diluted in PBS. Cycloheximide (CHX; Sigma-Aldrich) was diluted in ethanol. KU-55933 (Tocris Pharmaceuticals), nutlin-3a (Cayman Pharmaceuticals), and pifithrin-α (Enzo Life Sciences) were diluted in dimethyl sulfoxide (DMSO). Primary antibodies used in immunoblot analyses include chicken anti-ORF59 (Gallus Immunotech), rabbit anti-mouse p21 (sc-397; Santa Cruz Biotechnology), rabbit anti-ATM (2873; Cell Signaling Technology [CST]), mouse anti-p53 (2524; CST), rabbit anti-phospho-S18 p53 (9284; CST), mouse anti-β-actin (A-5316; Sigma-Aldrich), mouse anti-phospho-S1981 ATM (200-301-400; Rockland Immunochemicals), mouse anti-Chk2 (05-649; Upstate Biotechnology), rabbit polyclonal mLANA (41), and mouse polyclonal MHV68 immune sera (42). Densitometric analyses of p53 levels relative to those of β-actin were carried out with Image Lab software (Bio-Rad). Antibodies used in flow cytometry or immunofluorescence assays include Alexa Fluor 647-conjugated mouse anti-p53 (2533; Cell Signaling Technology), goat anti-green fluorescent protein (GFP; 600-101-215; Rockland Immunochemicals), Alexa Fluor 488-conjugated donkey anti-goat (A-11055), Alexa Fluor 568-conjugated goat anti-mouse (A-11004), and Alexa Fluor 488-conjugated goat anti-chicken (A-11039; Invitrogen).
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR).
Cells were mock infected or infected with MHV68 at a multiplicity of infection (MOI) of 5 PFU/cell. Cells treated with doxorubicin (5 μM) 4 h prior to RNA isolation served as positive controls for p53-induced transcription. RNA was isolated from infected cells at 4, 8, 12, and 18 h postinfection using TRIzol (Ambion) and converted to cDNA as previously described (33). Quantitative PCR was performed on resulting cDNAs according to the manufacturer's guidelines (Agilent). Gene-specific TaqMan probes (Applied Biosystems) for Mdm2 (Mm01233136_m1), Cdkn1a (Mm00432448_m1), and β-actin (Mm00607939_s1) were utilized for quantification. Reactions were performed in a Stratagene Mx3005P thermal cycler with cycling conditions of 2 min at 50°C and then 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Biological duplicate samples were analyzed in technical triplicate using the comparative threshold cycle (ΔΔCT) method as previously described (33). For p53 signaling pathway RT2 profiler transcript arrays (PAMM-027; Qiagen), total RNA was isolated using an miRNeasy minikit (Qiagen) from Swiss albino 3T3 fibroblasts that were mock infected, WT-MHV68 infected, ORF37.ΔHS infected, or 73.STOP infected 18 h postinfection. For nutlin-3a treatments, mock-infected or infected cells were treated with nutlin-3a (10 μM) 4 h before harvest at 18 h postinfection. cDNA was synthesized from 500 ng total RNA using the Qiagen RT2 first-strand kit. Reactions were performed in an Applied Biosystems StepOnePlus PCR system with cycling conditions of 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Biological duplicate samples were analyzed in technical duplicate using the ΔΔCT method with β-actin as the cellular housekeeping transcript control. Nutlin-3a-induced transcription for mock-infected or infected samples was calculated using untreated mock-infected or infected samples.
Cell viability assays.
Cell viability assays were performed essentially as previously described (33), with the following modifications. Cells were infected with WT MHV68, ORF37.ΔHS, or 73.STOP MHV68 at an MOI of 5 PFU/cell and treated with PAA (200 μg/ml) 1 h after MHV68 adsorption to prevent cell death due to viral replication. Cells were treated with vehicle (dH2O), pifithrin-α (30 μM), doxorubicin (8 μM), or TNF-α (25 nM) and cycloheximide (10 μg/ml) 14 h postinfection. Cell viability was assessed 44 h postinfection using a crystal violet-based cytotoxicity assay or CellTiter-Glo (Promega) as previously described (33).
Mouse infections and flow cytometry.
Six- to 8-week-old female C57BL/6J mice were purchased from Jackson Laboratories. Mice were housed under sterile conditions in the animal facility at the University of Arkansas for Medical Sciences in accordance with all federal and university DLAM guidelines. Spleen cell isolation and flow cytometry were performed as previously described (22). Briefly, mice were intraperitoneally mock infected or infected with 106 PFU of H2B-YFP MHV68 (35). Cells were processed for flow cytometry using a Foxp3 transcription factor staining kit (eBioscience). As a positive control for p53 induction, splenocytes from mock-infected animals were exposed ex vivo to 10 Gy of gamma radiation and incubated for 2 h at 37°C. Processed splenocytes were stained to detect p53 and YFP and analyzed using a BD LSR Fortessa flow cytometer. Data were analyzed using FlowJo software.
Immunofluorescence analyses.
For bromodeoxyuridine (BrdU) labeling, 3T3 fibroblasts were grown on glass coverslips and infected with MHV68 at an MOI of 5 PFU/cell. BrdU was added to culture medium 5 h postinfection at a final concentration of 19.3 μM. BrdU-labeled cells were processed for immunofluorescence 18 h postinfection using a BrdU flow kit (BD Pharmingen). After BrdU labeling, cells were stained with anti-ORF59 to detect viral replication complexes. For correlative p53 analyses, cells were fixed 18 h postinfection and processed for immunofluorescence analyses as previously described (33). Cells were stained with chicken anti-ORF59 and mouse anti-p53 to evaluate p53 intensities and p53 localization relative to that of ORF59. Coverslips were mounted on slides using Prolong Gold anti-fade mounting reagent containing 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies) to visualize DNA. For single-cell immunofluorescence analyses, 3T3 fibroblasts were grown in 6-well plates and serum starved prior to infection with MHV68 at an MOI of 0.5 PFU/cell. Cells were mock treated or treated with doxorubicin (5 μM) 14 h postinfection and fixed and processed for immunofluorescence microscopy 4 h later to detect p53 and ORF59. DNA was detected by incubating cells in DAPI-containing PBS prior to imaging. Cells were imaged using a Nikon Eclipse Ti fluorescence microscope. Relative p53 levels were determined by quantifying p53 fluorescence intensity using NIS Elements software for 32 randomly selected, ORF59-negative, or ORF59-positive cells following background correction. Nuclei were identified for quantification using the autoselect region-of-interest function.
RESULTS
Induction of ATM-p53 signaling during MHV68 lytic replication.
MHV68 induces ATM and H2AX phosphorylation, and infected cells exhibit additional DDR-related phosphorylation events during lytic replication in fibroblasts (20, 22, 43–45). However, the kinetics of the MHV68-associated DDR and whether it is associated with p53 activation are not well defined. To better understand DDR signaling and p53 activation during MHV68 lytic infection, we infected p53-competent 3T3 fibroblasts with MHV68 and performed immunoblot analyses to evaluate the activation of ATM, specific downstream effectors, and p53 over time. As a positive control for induction of an ATM-related DDR, uninfected cells were treated with the potent DNA damage-inducing drug doxorubicin. In contrast to mock-infected controls, cells infected with MHV68 exhibited ATM phosphorylation on S1981, Chk2 mobility shift indicative of hyperphosphorylation, and p53 stabilization and phosphorylation on S18, which is a well-characterized residue targeted by ATM for phosphorylation (46, 47) (Fig. 1A). ATM-related signaling events initially were detectable approximately 4 h postinfection, increased to maximal levels by 12 h postinfection, and remained steady for the duration of the time course. Evaluation of the viral immediate-early (IE) protein mLANA, early protein ORF59 (viral DNA polymerase processivity factor), and late structural proteins by immunoblot analyses suggested that ATM-related signaling was amplified as MHV68 progressed through the lytic gene expression cascade (Fig. 1A). Moreover, MHV68 also induced ATM and p53 phosphorylation following infection of MB114 endothelial cells (Fig. 1B) and primary MEFs (see Fig. 3D) (20).
FIG 1.

ATM-p53 response is activated during MHV68 lytic replication. 3T3 fibroblasts (A) or MB114 endothelial cells (B) were mock infected (M) or infected (I) with MHV68 at an MOI of 5 PFU/cell. Cells were treated with 1 μM doxorubicin (Doxo) for 4 h as a positive control for DDR induction. Cells were harvested at the indicated times postinfection (A) or 18 h postinfection (B), SDS-PAGE was performed, and immunoblot analyses were conducted using antibodies specific to the indicated antigens. Detection of β-actin serves as a loading control. (C and D) C57BL/6 mice were intraperitoneally mock inoculated or inoculated with 106 PFU of H2B-YFP-expressing MHV68. Animals were sacrificed 4 days postinfection, and bulk splenocytes were isolated and processed for flow cytometry to detect H2B-YFP (C) and p53 (D). Data shown in panel D are representative histograms for p53. The range of p53 mean fluorescence intensity (MFI) from two independent experiments is shown. Splenocytes from mock-inoculated animals were exposed to 10 Gy of gamma radiation (γ-IR) as a positive control for p53 induction.
FIG 3.

ATM mediates p53 induction, but not viral replication, during MHV68 lytic infection. 3T3 fibroblasts were infected with MHV68 at an MOI of 0.5 PFU/cell (A) or 5 PFU/cell (B). One hour after adsorption, inocula were removed and cells were treated with vehicle (DMSO) or the ATM inhibitor KU-55933 (1 μM). Cells were harvested at the indicated times postinfection and subjected to freeze-thaw lysis, and viral titers were determined by plaque assay. Results are means from triplicate samples. Error bars represent standard deviations. (C) 3T3 fibroblasts were mock infected or infected with MHV68 at an MOI of 5 PFU/cell. One hour after adsorption, inocula were removed and cells were treated with vehicle (DMSO) or ATM inhibitor KU-55933 (1 μM). Cells were harvested in RIPA buffer at the indicated times postinfection, and lysates were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. The detection of β-actin serves as a loading control. (D) ATM+/+ (WT) or ATM−/− (KO) murine embryonic fibroblasts were mock infected or infected with wild-type MHV68 at an MOI of 5 PFU/cell. Mock-infected cells were exposed to 10 Gy of gamma radiation (γ-IR) as a positive control for p53 induction. Samples were harvested at 18 h postinfection and processed for immunoblot analyses as described for panel C.
We next sought to determine whether p53 was stabilized as a consequence of viral replication in vivo. Mice were infected via intraperitoneal inoculation with a recombinant virus that constitutively expresses an H2B-YFP fusion protein from a neutral locus within the MHV68 genome (35). This virus efficiently marks infected cells for detection by flow cytometry. Four days postinoculation, a time point at which productive replication is robust in splenic B cells (48), splenocytes were isolated and stained to detect H2B-YFP and p53 (Fig. 1C and D, respectively). Compared to H2B-YFP− cells, which exhibited p53 levels equivalent to those of mock-infected animals, H2B-YFP+ splenocytes exhibited a ca. twofold increase in p53 staining (Fig. 1D). The increase in p53 detected in H2B-YFP+ cells was equivalent to p53 induction following treatment of naive spleen cells with 10 Gy ionizing radiation, a potent DNA damage and p53-activating stimulus (49). Importantly, the finding that only H2B-YFP+ cells contain increased p53 levels demonstrates that p53 induction is specific to infected cells and is not a simple consequence of inoculation or bystander activation. These data therefore agree with observations of cultured cells and provide in vivo relevance confirming that p53 is induced during lytic MHV68 infection.
Finally, we performed cycloheximide-chase experiments to verify that increased p53 detection was due to the stabilization of the protein during MHV68 infection. Indeed, similar to treatment with doxorubicin, p53 remained stable following cycloheximide-mediated inhibition of new protein synthesis, while p53 was rapidly lost from mock-infected cells (Fig. 2A and B). This observation is consistent with the disruption of MDM2-mediated p53 ubiquitination and degradation due to ATM-mediated p53 phosphorylation during MHV68 infection. Together, these data demonstrate that ATM-related DDR signaling and p53 induction occur as consequences of MHV68 lytic infection of multiple cell types in tissue culture and in vivo.
FIG 2.

MHV68 infection leads to p53 stabilization. (A) 3T3 fibroblasts were mock infected, infected with MHV68 at an MOI of 5 PFU/cell, or treated with 1 μM doxorubicin (Doxo). Cells were treated with cycloheximide (CHX; 100 μg/ml) or vehicle 14 h postinfection or 4 h after doxorubicin treatment. Cells were harvested in RIPA buffer either immediately after CHX or vehicle treatment or every hour for the following 3 h. Proteins were resolved by SDS-PAGE, and immunoblot analyses were performed to detect p53 and β-actin as a loading control. (B) Densitometric analyses were performed on resulting immunoblots to compare levels of p53 between CHX-treated or vehicle-treated samples relative to that for the loading control β-actin, which is not affected by CHX treatment. The graph depicts the percentage of p53 remaining over time in CHX-treated cells relative to that of vehicle-treated controls at equivalent time points.
ATM mediates p53 induction during MHV68 replication.
Given the role of ATM in p53 activation following genotoxic stress (14), we next tested whether ATM was necessary for p53 induction during MHV68 lytic replication. Because ATM is required for MHV68 lytic replication in some settings (20), we first evaluated whether inhibition of ATM with the pharmacologic inhibitor KU-55933 (50) reduced MHV68 replication in 3T3 fibroblasts. We performed titration experiments to define the maximal concentration of KU-55933 that was not overtly cytotoxic to 3T3 fibroblasts (data not shown). We then analyzed MHV68 replication over time at both high and low multiplicities in the presence or absence of KU-55933. In these experiments, KU-55933 treatment did not have any impact on MHV68 replication (Fig. 3A and B). Having established that viral replication was normal in treated cells, we next evaluated whether p53 induction during MHV68 infection was dependent on ATM. Consistent with viral growth assays, immunoblot analyses demonstrated that KU-55933 did not overtly influence viral gene expression. However, both p53 phosphorylation and stabilization were potently inhibited in MHV68-infected cells following KU-55933 treatment (Fig. 3C). As an independent and complementary approach to KU-55933 experiments, we also evaluated p53 induction following MHV68 infection of ATM−/− and ATM+/+ MEFs. Although p53 phosphorylation and stabilization readily occurred in ATM+/+ MEFs following either MHV68 infection or treatment with ionizing radiation as a positive control, p53 phosphorylation and stabilization did not occur in either case for ATM−/− MEFs (Fig. 3D). Therefore, these data implicate ATM as the primary mediator of p53 induction during MHV68 lytic replication and demonstrate that ATM inhibition does not reduce MHV68 gene expression and productive replication in 3T3 fibroblasts.
Defining steps in the infectious cycle at which ATM-p53 induction occur.
Like other herpesviruses, the MHV68 lytic gene expression cascade occurs in three temporally distinct phases: IE, early, and late (51–54). To more precisely identify specific phases of the viral replication cycle that correlate with p53 activation, we evaluated ATM and p53 activation following infection of 3T3 fibroblasts with WT MHV68, RTA-null (50.STOP) MHV68 (36), and WT MHV68 in the presence of the viral DNA polymerase inhibitor PAA. Infection by RTA-null MHV68, which lacks the IE replication and transactivator protein RTA and thus cannot initiate early gene synthesis (36), did not trigger ATM-p53 signaling (Fig. 4). This suggests that neither host cell detection of the viral particle nor expression of other IE proteins, such as mLANA, induce an ATM-p53 response. In contrast, while PAA treatment potently inhibited the expression of late viral antigens, which are dependent on viral DNA replication, PAA did not block ATM-p53 activation by MHV68, as levels of ATM and p53 phosphorylation and p53 stabilization were equivalent to those observed for WT MHV68 infection in the absence of inhibitor. PAA treatment alone did not induce ATM or p53. Thus, these data demonstrate that ATM-p53 activation occurs prior to or coinciding with viral DNA replication through RTA and/or early viral gene expression.
FIG 4.
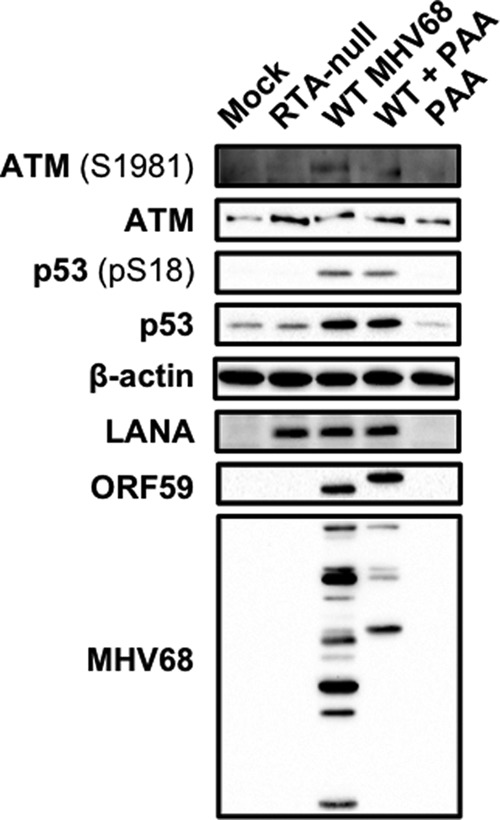
ATM-p53 activation requires viral gene expression downstream of RTA. 3T3 fibroblasts either were mock infected or infected at an MOI of 5 PFU/cell with RTA-null MHV68 (50.STOP) or WT MHV68, or they were mock infected or infected with WT MHV68 in the presence of PAA (200 μg/ml) to inhibit viral DNA replication. Cells were harvested in RIPA buffer 18 h postinfection, and proteins were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. The detection of β-actin serves as a loading control.
p53-responsive genes are differentially regulated during lytic MHV68 infection.
Having established that MHV68 elicits an ATM-p53 response during lytic infection, we next wanted to define the functional consequences of p53 activation on the host cell. To determine if p53 induced during MHV68 infection was functional and capable of driving downstream host gene expression, we performed quantitative reverse-transcription PCR (qRT-PCR) analyses to compare transcription of p53-responsive genes mdm2 and cdkn1a following MHV68 infection. Cells were treated with doxorubicin as a positive control for the induction of p53-mediated transcription. Interestingly, both mdm2 and cdkn1a transcripts were elevated 4 and 8 h postinfection, approximating levels observed in cells treated with doxorubicin (Fig. 5A). However, transcripts decreased to levels detected in mock-infected controls by 12 h and remained low at 18 h postinfection, suggesting that p53-related transcription became inhibited as the infectious cycle progressed. As a broader test of this hypothesis, we utilized p53 transcript arrays to compare MHV68 infection to treatment of cells with nutlin-3a, a highly specific chemotherapeutic p53 agonist that functions by preventing MDM2-mediated proteasomal degradation of p53 (55). Remarkably, compared to the 12 transcripts most potently induced by nutlin-3a treatment, including mdm2, rprm, pmaip1, and cdkn1a, among others, these same transcripts were relatively unchanged at 18 h postinfection with MHV68 (Fig. 5B), further supporting the notion that p53-related transcription is impaired at late time points during MHV68 lytic infection. Together, these data demonstrate that p53 is transcriptionally active at early time points during MHV68 infection but becomes inactive or repressed as the lytic cycle progresses despite the prolonged presence of phosphorylated and stabilized p53 (Fig. 1 and 2).
FIG 5.

Analysis of p53-responsive gene expression during MHV68 lytic replication. (A) 3T3 fibroblasts were either mock treated or treated with 5 μM doxorubicin (Doxo) or were mock infected or infected with MHV68 at an MOI of 5 PFU/cell. RNA was isolated from cells 4 h after doxorubicin treatment or at the indicated times postinfection, and quantitative RT-PCR analyses were performed to detect murine mdm2 or cdkn1a (p21) transcripts. Data represent the change (n-fold) in transcript levels normalized to the levels for β-actin, as determined using the ΔΔCT method for treated or infected samples relative to mock-treated or mock-infected samples. Data represent mean values from two independent experiments analyzed in triplicate. Error bars represent the range of data. (B) 3T3 fibroblasts were either mock treated or treated with 5 μM nutlin-3a or were mock-infected or infected with MHV68 at an MOI of 5 PFU/cell. RNA was isolated from cells 4 h after nutlin-3a treatment or 18 h postinfection, and quantitative RT-PCR analyses were performed using RT2 profiler p53 signaling pathway PCR arrays. Data represent the change (n-fold) in transcript levels normalized to the levels for β-actin, as determined using the ΔΔCT method for treated or infected samples relative to mock-treated or mock-infected samples. Data represent mean values from two independent experiments. Error bars represent the range of data.
Dominant inhibition of p53 responses during MHV68 infection.
Since one would expect the level of p53-related transcription to correlate with high levels of stabilized p53 protein observed at 12 h and 18 h postinfection, the time-dependent reduction in p53-responsive gene expression observed during MHV68 infection suggests that MHV68 impairs p53 functions during lytic viral replication. If p53 functions are specifically inhibited by the virus, we hypothesized that infected cells would become resistant to treatment with exogenous p53-activating stimuli as a consequence. To test this hypothesis, we conducted a time course experiment in which mock-infected and MHV68-infected cells were treated with doxorubicin at different times after infection. Cells were harvested 4 h after drug treatment to allow time for activation of ATM-p53 responses. We then performed immunoblot analyses to compare and contrast p53 stabilization, p53 phosphorylation, and induction of p21 protein, a p53-regulated CDK inhibitor encoded by the CDKN1A gene, which we tested in qRT-PCR experiments. Analogous to mock-infected controls, infected cells exhibited p53 activation and increased p21 expression when treated with doxorubicin prior to 8 h postinfection (Fig. 6A). However, p53 stabilization, p53 phosphorylation, and p21 induction were reduced when infected cells were treated with doxorubicin 10 h postinfection. Doxorubicin responsiveness was completely abolished in infected cells by 14 h postinfection. These data strongly suggest that p53 responses are repressed as the MHV68 lytic cycle progresses.
FIG 6.

MHV68-infected cells become nonresponsive to p53 agonists as infection progresses. (A) 3T3 fibroblasts were mock infected or infected with wild-type MHV68 at an MOI of 5 PFU/cell. Cells were treated with vehicle (DMSO) or doxorubicin (5 μM) 4 h prior to harvest at the indicated times postinfection. Cells were harvested in RIPA buffer at the indicated times postinfection, and lysates were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. Detection of β-actin serves as a loading control. (B and C) 3T3 fibroblasts were infected with MHV68 at an MOI of 0.5 PFU/cell. Cells were treated with vehicle or doxorubicin (5 μM) 14 h postinfection. Cells were fixed with 10% formalin 18 h postinfection and processed for immunofluorescence microscopy. Processed cells were stained to detect viral antigen ORF59 and p53. DNA was detected with DAPI. (C) Relative fluorescent intensities of p53 in ORF59-negative or ORF59-positive cells from panel B were quantified in multiple images using Nikon NIS Elements software. Data represent relative p53 fluorescence intensity from 32 randomly selected cells for each condition.
We next sought to determine if lack of p53 responsiveness was intrinsic to the infected cell or a general response to the infected culture. Thus, we performed low-MOI infections followed by quantitative, single-cell immunofluorescence analyses to directly compare p53 inducibility in infected and uninfected cells in the same culture following treatment with doxorubicin. We first evaluated p53 induction in individual cells without doxorubicin treatment. Productively infected cells were identified by the presence of early gene product ORF59. In agreement with immunoblot analyses, cells that stained positive for ORF59 exhibited ca. 2-fold more p53 staining than ORF59-negative cells by 18 h postinfection (Fig. 6B and C). In contrast, treatment with doxorubicin yielded a ca. 8-fold increase in p53 staining for ORF59-negative cells but less than a 2-fold increase for ORF59-positive cells. Therefore, these data indicate that p53 nonresponsiveness is a cell-intrinsic phenotype that correlates with the expression of proteins involved in viral DNA replication.
We next tested whether p53 was similarly unresponsive to direct stimulation by treatment with the p53 agonist nutlin-3a. 3T3 fibroblasts were mock infected or infected with MHV68 at an MOI of 5 PFU/cell, subsequently were treated with vehicle, doxorubicin (5 μM), or nutlin-3a (10 μM) 14 h postinfection, and then were harvested 4 h later (Fig. 7A). Indeed, MHV68-infected cells failed to respond to nutlin-3a treatment by induction of p21 protein, indicating that MHV68 replication compromises the p53 response to multiple exogenous p53 stresses. Finally, we reasoned that if p53 responses were inhibited during MHV68 infection, then p53-related transcription also should be hindered following treatment with exogenous activators of p53. Unfortunately, treatment with doxorubicin resulted in high sample-to-sample variation using qRT-PCR arrays. Therefore, we used nutlin-3a as the p53 agonist for array experiments. Importantly, MHV68-infected cells also were resistant to p53-mediated p21 induction following nutlin-3a treatment (Fig. 7A). We performed qRT-PCR experiments to determine if p53-dependent transcription was hindered in cells infected with MHV68. In agreement with immunoblot and immunofluorescence data, p53-regulated transcripts were less induced in infected cells than in mock-infected controls following nutlin-3a treatment (Fig. 7B). This indicates that infected cells exhibit a reduced or repressed p53 transcriptional response to an external p53 activation stimulus. Taken together, these three complementary experimental approaches demonstrate that p53 responses become inhibited as MHV68 progresses through the lytic replication cycle. Moreover, the viral block to p53 responses apparently is dominant over exogenous p53-activating stimuli.
FIG 7.

MHV68 infection prevents p21 induction following p53 stimulation. (A) 3T3 fibroblasts were mock infected or infected with MHV68 at an MOI of 5 PFU/cell. Twelve hours postinfection, cells were treated with DMSO, doxorubicin (Doxo; 5 μM), or nutlin-3a (10 μM) for 4 h prior to harvest. Cells were harvested in RIPA buffer 16 h postinfection, and proteins were resolved by SDS-PAGE. Immunoblot analyses were performed using antibodies to detect the indicated proteins. Detection of β-actin serves as a loading control. (B) 3T3 fibroblasts were infected with MHV68 at an MOI of 5 PFU/cell and either mock treated or treated with 5 μM nutlin-3a 14 h postinfection. RNA was isolated from cells 18 h postinfection, and quantitative RT-PCR analyses were performed using RT2 profiler p53 signaling pathway PCR arrays. Data represent the change (n-fold) in transcript levels normalized to the levels for β-actin, as determined using the ΔΔCT method for vehicle-treated versus drug-treated samples. For comparison purposes, data for uninfected cells are reproduced from Fig. 5. Data represent mean values from two independent experiments. Error bars represent the range of data.
Viral DNA replication is not required for p53 inhibition.
We next sought to define mechanisms whereby MHV68 might inhibit p53. In immunofluorescence analyses of p53 in infected cells, we noted that p53 appeared to colocalize with ORF59 in viral replication complexes (Fig. 8A). In agreement with this observation, ORF59 staining corresponded to areas of active DNA replication, which were detected by BrdU incorporation, in nuclei of infected cells. Since infected cells also lost p53 responsiveness relatively late in the infectious cycle, we were curious if newly replicated viral DNA and/or late viral proteins might sequester p53 as a means to prevent its functions during lytic MHV68 infection. This idea was proposed previously for p53 inactivation during human cytomegalovirus (HCMV) replication (56). To test this hypothesis, we evaluated induction of p53 and p21 by doxorubicin in mock-infected and infected cells that also were treated with inhibitors of viral DNA synthesis, PAA, cidofovir, or acyclovir. While mock-infected cells responded to doxorubicin with p53 activation and p21 induction in the presence of each of these drugs, infected cells were not responsive to doxorubicin treatment in either the absence or presence of viral DNA replication inhibitors (Fig. 8B). The reduction in late viral antigens detected with MHV68 antiserum demonstrates drug effectiveness (Fig. 8B), which we also confirmed in control plaque assays that were performed in parallel (data not shown). While these data do not completely rule out the possibility that viral genomes can sequester p53 during MHV68 infection, they do indicate that p53 sequestration by newly replicated viral DNA is not necessary for p53 inactivation. Since late viral gene expression is dependent on viral DNA replication (51, 57), these results also demonstrate that late proteins are not essential for p53 inhibition. These findings therefore highlight potential roles for IE and early viral gene products in restricting p53.
FIG 8.

p53 localizes to MHV68 replication compartments, yet viral DNA replication is not required for p53 inactivation. (A) 3T3 fibroblasts were mock infected (top) or infected with MHV68 at an MOI of 5 PFU/cell (bottom). Cells in the upper and lower panels were incubated in the presence of 20 μM BrdU for 12 h prior to fixation at 18 h postinfection. Cells were stained for indirect immunofluorescence microscopy using antibodies to the indicated proteins. (B) 3T3 fibroblasts were mock infected or infected with MHV68 at an MOI of 5 PFU/cell. Inocula were removed 1 h after adsorption, and cells were treated with cidofovir, PAA, or acyclovir as indicated. Cells were treated with vehicle (DMSO) or doxorubicin (5 μM) 4 h prior to harvest 18 h postinfection. Cells were harvested in RIPA buffer at the indicated times postinfection, and lysates were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. Detection of β-actin serves as a loading control.
MHV68 host shutoff protein muSOX regulates the p53 pathway.
We next sought to identify specific viral proteins that mediate the dominant inhibition of p53 responses during lytic MHV68 infection, and we focused on two particular viral genes for these tests, ORF37 and ORF73. ORF37 encodes the MHV68 conserved herpesvirus alkaline exonuclease homolog muSOX. muSOX performs a host shutoff function by targeting specific cellular transcripts for degradation (58–60). Moreover, orf37 is transcribed with early kinetics (54), which is consistent with the timing of p53 inhibition during MHV68 infection. To test a potential role for muSOX in repressing p53 responses, we utilized ORF37.ΔHS, an MHV68 recombinant virus that harbors a single point mutation (R443I) in muSOX that ablates its host shutoff activity while retaining its exonuclease activity, which is necessary for efficient packaging of viral DNA (37). We performed immunoblot analyses to evaluate p53 stabilization and phosphorylation as well as p21 induction over time following infection of 3T3 fibroblasts with ORF37.ΔHS or its marker rescue control, ORF37.MR. Although both ORF37.ΔHS and ORF37.MR infections led to increased p53 stabilization and phosphorylation, these events appeared to be more potently elicited by MHV68 infection in the absence of host shutoff activity (Fig. 9A). Similarly, rather than decreasing over time, as was observed for WT MHV68 (Fig. 6A) and ORF37.MR, p21 was induced at later time points following ORF37.ΔHS infection (Fig. 9A). These findings suggest that muSOX host shutoff function targets the p53 pathway during lytic MHV68 infection.
FIG 9.

muSOX host shutoff function influences the p53 signaling pathway. 3T3 fibroblasts were mock infected or infected with a host shutoff-null mutant of MHV68 (ORF37.ΔHS) or WT marker rescue control (ORF37.MR) at an MOI of 5 PFU/cell. (A) Cells were harvested at the indicated time postinfection. (B) Cells were treated with doxorubicin (1 μM) or vehicle (DMSO) 14 h postinfection, and lysates were harvested in RIPA buffer 18 h postinfection. Proteins were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies to detect the indicated proteins. Detection of β-actin serves as a loading control.
We next tested whether muSOX host shutoff function contributes to dominant inhibition of p53 during MHV68 replication. As before, cells were mock infected or infected with either ORF37.ΔHS or ORF37.MR for 14 h prior to treatment with doxorubicin. Cells were harvested 4 h later, and p53 activation and p21 induction were evaluated. While mock-infected cells and WT ORF37.MR infections recapitulated previous results, ORF37.ΔHS infections exhibited such high levels of p53 stabilization, phosphorylation, and p21 induction, greater than those of positive controls, that it was difficult to discern whether doxorubicin-mediated induction of p53 signaling was hindered or intact in the absence of host shutoff by ORF37 (Fig. 9B). However, we also evaluated transcriptional responses to p53 agonists in cells infected with ORF37.ΔHS. Unlike cells infected with WT MHV68, ORF37.ΔHS-infected cells remained largely responsive to nutlin-3a treatment, with 7 of the 12 most inducible transcripts still responding to drug treatment (Table 1). Together, these data suggest that muSOX participates in the deactivation of p53 responses during MHV68 infection.
TABLE 1.
Summary of nutlin-3a-inducible p53-responsive gene expression during MHV68 infection
| Gene product | Expression by infection type |
|||||||
|---|---|---|---|---|---|---|---|---|
| Mock |
WT MHV68 |
73.stop MHV68 |
37.ΔHS MHV68 |
|||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Mdm2 | 7.02 | 0.474 | 4.81 | 0.969 | 6.84 | 0.953 | 5.77 | 1.33 |
| Pmaip1 | 4.36 | 0.271 | 2.49 | 0.678 | 5.34 | 0.421 | 6.33 | 2.54 |
| Btg2 | 3.29 | 0.488 | 3.06 | 1.04 | 5.01 | 1.32 | 2.86 | 1 |
| Rb1 | 2.88 | 0.287 | 1.52 | 0.03 | 3.43 | 0.186 | 3.86 | 0.051 |
| Dapk1 | 2.81 | 0.091 | 0.939 | 0.305 | 2.52 | 0.073 | 3.69 | 0.0223 |
| Fas | 2.8 | 0.075 | 1.98 | 0.2 | 2.43 | 0.128 | 6.18 | 0.336 |
| Apaf1 | 2.52 | 0.124 | 1.71 | 0.027 | 2.68 | 0.358 | 3.59 | 0.343 |
| Cdkn1a | 2.29 | 0.118 | 1.79 | 0.406 | 3.11 | 0.064 | 2.4 | 0.508 |
| Egr1 | 2.1 | 0.083 | 1.44 | 0.229 | 1.51 | 0.059 | 0.656 | 0.067 |
| Sesn2 | 2 | 0.221 | 1.5 | 0.288 | 2.38 | 0.034 | 1.68 | 0.001 |
| Bbc3 | 1.92 | 0.291 | 1.82 | 0.681 | 3.48 | 0.61 | 1.48 | 0.023 |
| Ppm1d | 2.1 | 0.134 | 1.83 | 0.035 | 2.44 | 0.05 | 2.67 | 0.142 |
mLANA inhibits p53 during MHV68 infection.
We next tested the role of the MHV68 LANA homolog, mLANA, in p53 deactivation during MHV68 lytic replication. mLANA is encoded by the conserved ORF73 gene, and we previously demonstrated that mLANA-null MHV68 caused greater p53 induction than WT MHV68 at early times postinfection (33). Increased p53 induction also correlated with enhanced cell death, which was partially ameliorated in p53−/− MEFs (33). Although these data suggest that mLANA blocks p53 activation during MHV68 infection, it was not clear from our previous experiments whether mLANA directly hindered p53 responses. To test this, we performed immunoblot analyses to evaluate effects of doxorubicin treatment on p53 activation and p21 induction in the absence of mLANA expression. In contrast to cells infected with WT MHV68, treatment of cells infected with 73.STOP, an mLANA-null MHV68 recombinant (38), resulted in increased p53 stabilization and phosphorylation (Fig. 10). While WT MHV68-infected cells did not produce p21 when doxorubicin treated, p21 was inducible during 73.STOP infection, albeit at a lower level than that observed for mock-infected cells. Since cells infected with ORF37.ΔHS maintained strong p21 expression (Fig. 9A), the general reduction in p21 levels is consistent with a role for muSOX-mediated host shutoff occurring despite the absence of mLANA. Immunofluorescence assays performed in parallel to detect ORF59 confirmed that all cells in these experiments indeed were infected and progressing through the lytic replication cycle (not shown). Thus, p53 inducibility observed during 73.STOP infection was not simply a result of inefficient infection by mLANA-null MHV68. These data suggest that mLANA is directly involved in limiting p53 activation during MHV68 infection.
FIG 10.
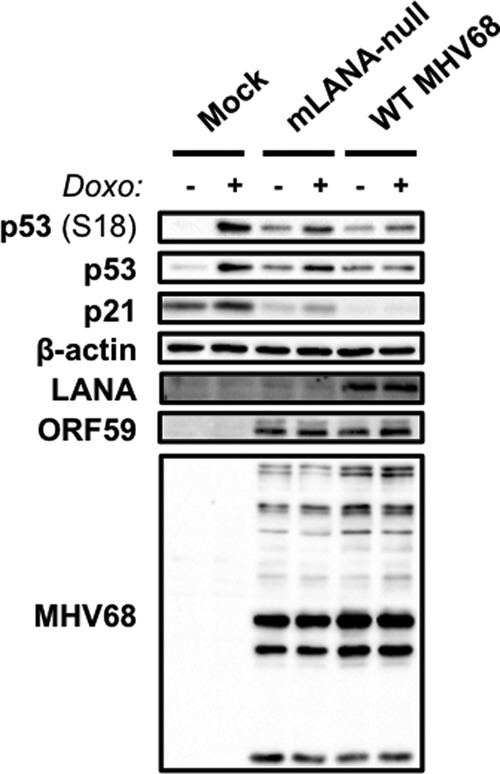
mLANA is necessary for p53 inactivation during MHV68 replication. 3T3 fibroblasts were mock infected or infected with WT MHV68 or mLANA-null MHV68 (73.STOP) at an MOI of 5 PFU/cell. Cells were treated with doxorubicin (1 μM) or vehicle (DMSO) 14 h postinfection, and lysates were harvested in RIPA buffer 18 h postinfection. Proteins were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies to detect the indicated proteins. Detection of β-actin serves as a loading control.
We and others previously demonstrated that mLANA functions as a transcriptional regulator (41, 61). As a complement to immunoblot experiments, we also evaluated whether cells infected with mLANA-null MHV68 were transcriptionally responsive to nutlin-3a treatment. In contrast to the attenuated response observed in cells infected with WT MHV68, cells infected with mLANA-null MHV68 remained largely responsive to nutlin-3a treatment (Table 1). This indicates that mLANA, like muSOX, participates in dampening p53-mediated transcriptional responses to exogenous p53-inducing stimuli. Together, these data demonstrate that mLANA also participates in blocking p53 function during MHV68 lytic replication.
MHV68 infection protects cells from p53-driven cell death.
We hypothesized that if p53 were functionally inactivated during MHV68 infection, then MHV68-infected cells would resist p53-driven cell death induced by exogenous stimuli. We therefore tested whether MHV68-infected cells were sensitive to doxorubicin-induced cell death relative to mock-infected controls. Having established that PAA treatment did not ablate the capacity of MHV68 to repress p53 responses, we used PAA to limit cell death associated with end-stage viral replication in these experiments. Treatment of mock-infected cells with doxorubicin resulted in the death of ca. 50% of cells (Fig. 11A). In contrast, cells that were infected with MHV68 prior to doxorubicin treatment were largely resistant to drug-induced death. To establish that cell death following doxorubicin treatment was mediated primarily by p53, we treated cells with pifithrin-α, a pharmacologic inhibitor of p53 function (62), and found that p53 inhibition with pifithrin-α did indeed prevent cell death following doxorubicin treatment of mock-infected cells. This finding confirms p53 involvement in the cell death response to doxorubicin treatment in 3T3-SA fibroblasts. Finally, as a test to determine if viral inhibition of cell death was specific to the p53 pathway or represented a general nonresponsiveness to exogenous cell death stimuli, we also evaluated cell death responses following treatment with TNF-α and cycloheximide. Unlike treatment with doxorubicin, both mock-infected and infected cells underwent cell death following TNF-α/cycloheximide treatment, demonstrating that MHV68-infected cells remain responsive to certain cell death stimuli. Thus, these data provide strong evidence that p53-directed cell death responses are blocked as a functional consequence of virus-mediated p53 inhibition during lytic MHV68 infection. These data also indicate that viral gene expression and/or host signaling events that occur prior to viral DNA replication and late gene expression are sufficient to prevent the p53-mediated death of the infected cell.
FIG 11.
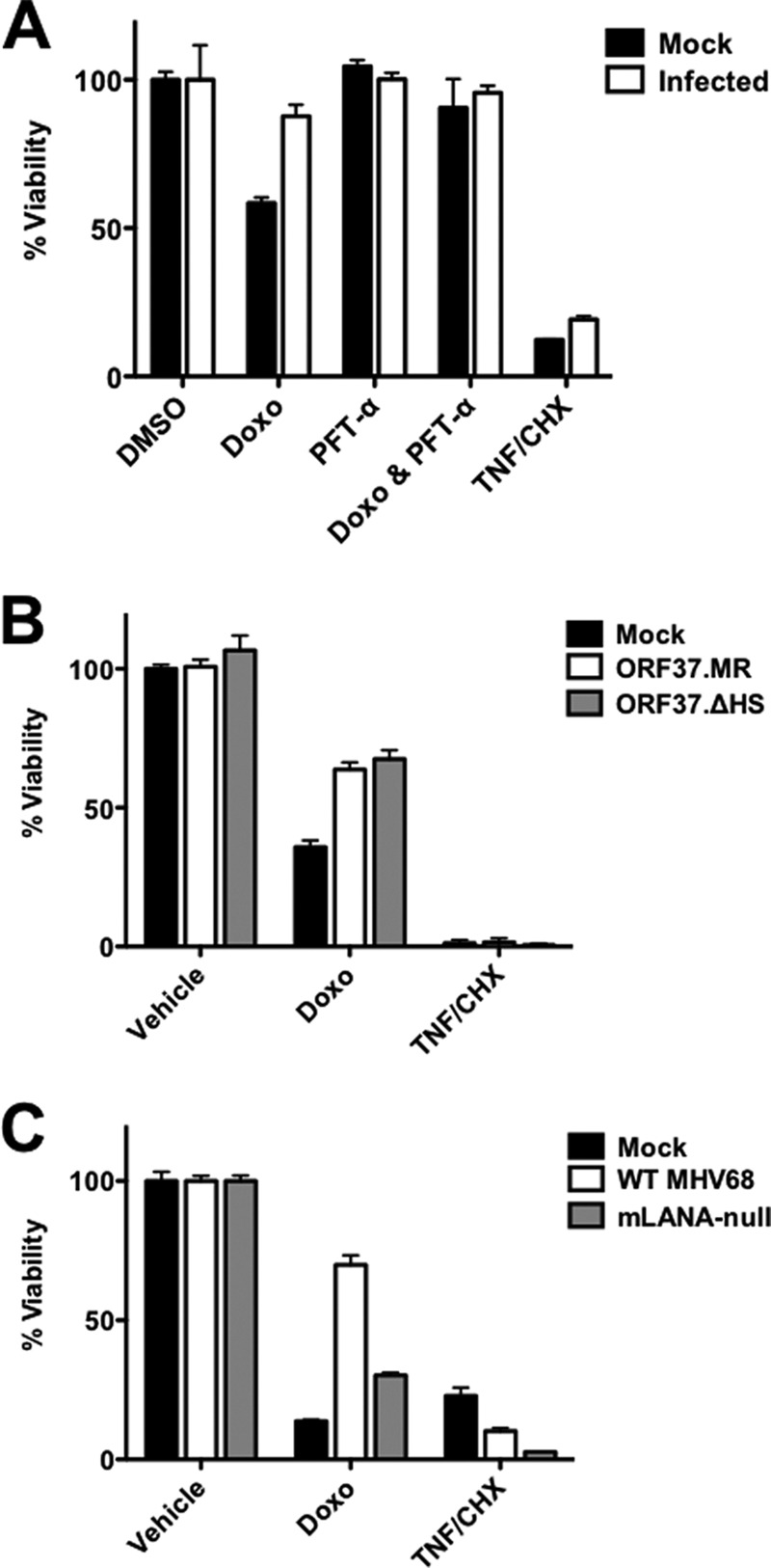
Protection from p53-mediated cell death during MHV68 infection requires mLANA but not muSOX host shutoff function. 3T3 fibroblasts were mock infected or infected with WT MHV68 (A), ORF37.ΔHS MHV68 (B), or mLANA-null MHV68 (C) at an MOI of 5 PFU/cell in the presence of PAA (200 μg/ml). In panel A, the indicated samples were treated with pifithrin-α (PFT-α; 30 μM) at the time of adsorption to inhibit p53. Where indicated, cells were treated with doxorubicin (8 μM) or vehicle (DMSO) 14 h postinfection, and cell viability was determined 44 h postinfection. As a p53-independent cell death stimulus, the indicated samples were treated with 25 nM TNF-α and 10 μg/ml cycloheximide (CHX). Cell viability was determined using a crystal violet retention assay. Data represent percentages of cell death for treated samples relative to that for vehicle-treated controls. Results are means from triplicate samples. Error bars represent standard errors of the means.
Given that muSOX and mLANA were important for preventing p53-related signaling and transcription, we reasoned that these proteins also would be necessary for preventing doxorubicin-induced cell death. We therefore evaluated whether cells infected with either ORF37.ΔHS or mLANA-null MHV68 were resistant to doxorubicin treatment by following the experimental scheme used for WT MHV68. Surprisingly, cells infected with ORF37.ΔHS remained largely resistant to doxorubicin treatment, surviving essentially as well as cells infected with ORF37.MR (Fig. 11B). In contrast, while ORF73.MR infection led to a reduction in cell death following doxorubicin treatment, cells infected with ORF73.STOP MHV68 remained susceptible to drug treatment (Fig. 11C). For all viruses tested, infection did not prevent cell death caused by TNF-α/cycloheximide treatment. These data indicate that although muSOX host shutoff function limits p53 signaling during MHV68 infection, this activity is not essential for inhibiting p53-mediated cell death triggered by an exogenous stimulus in this experimental setting. Conversely, these findings support the conclusion that mLANA is necessary for preventing functional outcomes of p53 responses during lytic MHV68 infection.
DISCUSSION
MHV68 interactions with ATM during lytic replication.
Previous work with MHV68, EBV, and KSHV demonstrated that GHVs activate DDR signaling during productive viral replication. EBV induces a DDR during ZTA-driven reactivation, evidenced by ATM activation and phosphorylation of downstream targets (23). KSHV reactivation following inducible RTA expression similarly promotes enhanced H2AX phosphorylation (45) as well as the activation of a full DDR (63). Tarakanova and colleagues previously demonstrated that MHV68 infection of primary macrophages also induces ATM and H2AX phosphorylation (20), and we likewise observed the phosphorylation of H2AX and other ATM targets in a global phosphoproteomic analysis of MHV68 lytic infection (22). Work presented here reiterates these previous studies and further defines the timing of the DDR induced during MHV68 lytic infection. Data obtained using replication-defective viruses and inhibitors of viral DNA replication strongly suggest that either RTA itself or early genes transcribed downstream of RTA, not viral DNA replication per se or viral late proteins, activate the DDR during MHV68 replication. These findings are analogous to those for DDR induction during herpes simplex virus 1 (HSV1) and HSV2 lytic replication or KSHV reactivation, all of which require viral gene expression but not viral DNA synthesis (23, 63, 64). These findings also agree with DDR induction occurring due to the expression of the viral kinase encoded by ORF36 (20) or expression of the viral cyclin D ortholog encoded by ORF72, which is suggested by the observation that k-cyclin induces a DDR in endothelial cells (65). The finding that a cyclin/CDK phosphorylation signature is prominent in cells productively infected with MHV68 and that MHV68 cyclin is important for phosphorylation changes induced during lytic replication further support this idea (22). Thus, our data establish a specific stage in the MHV68 replication cycle at which ATM signaling is activated during lytic replication in fibroblasts.
We found that infection of fibroblasts and endothelial cells promoted ATM phosphorylation, which correlated with p53 phosphorylation. However, infection-associated p53 phosphorylation and stabilization did not occur when ATM was inhibited with KU-55933 or after infection of ATM-deficient fibroblasts. Although we have previously identified many signaling pathways triggered by MHV68 lytic infection of fibroblasts that also are known to engage p53 (22, 39), the current data demonstrate that ATM is the primary mediator of p53 phosphorylation and stabilization during MHV68 infection of these cells. Whether ATM also is necessary for p53 phosphorylation in B cells remains to be determined but could be tested initially in ATM-deficient mice (66).
It is interesting that ATM clearly is not required for MHV68 replication in fibroblasts or acute replication in vivo (our data and references 20 and 67), although it is required for low-MOI infection of BMDMs (20) and reactivation from latent infection of peritoneal exudate cells (68). ATM inhibition also does not block ORF59 expression during induced KSHV reactivation (63). Given differential requirements for ATM in GHV replication (20, 24, 44, 63, 67–69), it is reasonable to suggest that ATM is situationally required for viral replication, perhaps in terminally differentiated cell types. However, since p53 was stabilized and transcriptionally active early during infection, these findings do indicate that viral induction of a DDR can have consequences on the infected cell, especially activation of downstream effectors such as p53. Hence, it will be of interest to determine if ATM activation is orchestrated by specific viral proteins in cell types that do not require a DDR for replication or if ATM activation simply occurs as a cellular response to infection. This would be interesting, given that Shah and O'Shea recently demonstrated that adenovirus triggers a virus-specific DDR that differs from a typical cellular response to extrinsic mediators of DNA damage (70). Nonetheless, we speculate that the requirement for ATM in MHV68 acute replication in certain settings necessitates that the virus encode proteins that inhibit downstream consequences of ATM activation, such as p53 activity, as a means to enable efficient lytic replication. This notion is supported by our findings that MHV68 encodes at least two factors that attenuate p53 responses during lytic replication.
Inhibition of p53 during MHV68 lytic replication.
Many DNA viruses usurp the DDR to efficiently replicate (5). Since p53 activation by DDR kinases enforces cell cycle arrest and ultimately apoptosis (5, 8), cellular programs that presumably would antagonize viral replication, it is not surprising that p53 inactivation is a common theme for DNA viruses (5). Although active and responsive to external stimuli early during MHV68 infection, p53 became repressed and nonresponsive as the infectious cycle progressed, apparently prior to or coinciding with viral DNA replication. This led us to test the role of muSOX, which is encoded by the early gene ORF37, in repressing p53-related signaling. While the manipulation of host stress responses is a well-documented function for viral host shutoff systems (71–73), descriptions of p53 signaling being affected by host shutoff previously were lacking. Our data demonstrate that muSOX host shutoff function is necessary to limit p53 signaling events during the MHV68 lytic cycle (Fig. 9), a function not previously appreciated for this viral protein. Whether muSOX directly targets the p53 pathway for inhibition or p53 signaling is hyperactivated during ORF37.ΔHS MHV68 infection due to the lack of control of viral gene expression remains to be determined.
ORF37.ΔHS MHV68 exhibits cell type-specific replication defects and altered virion protein composition (74). These phenotypes correlate with attenuation in vivo (74). Although muSOX host shutoff function was not a necessity for protection from cell death induced by exogenous p53-activating treatments, it remains possible that muSOX hindrance of p53 signaling is important in other cell types or settings. Along these lines, it is reasonable to suggest that p53 hyperactivation in the absence of muSOX host shutoff activity contributes to the altered virion protein composition phenotype. This would agree with our previous demonstration that p53 drives viral gene expression (33), perhaps compounding the increased presence of viral transcripts due to the lack of degradation by muSOX (74). Thus, it will be of interest to determine if ORF37.ΔHS viruses produced in p53-deficient cells exhibit ratios of virion proteins that more closely resemble those of WT virus.
We also demonstrate here that the dominant inhibition of p53 requires the MHV68 LANA homolog. We previously observed that mLANA-null MHV68 exhibited more rapid p53 induction and cell death during infection of nontransformed murine fibroblasts (33). We further showed that p53-deficient MEFs were partially protected from death caused by mLANA-null infection and that mLANA-null virus replicated to higher titers in p53-deficient MEFs (33). The current work extends our previous studies and clarifies certain unresolved issues. First, data presented here demonstrate that mLANA indeed is necessary for p53 blockade in a productively infected cell. While the previous data suggested this was the case, we had not directly demonstrated the need for mLANA in preventing p53 activity during infection. Second, here we establish that ATM-p53 signaling is triggered during infection by WT MHV68, even in the presence of mLANA (Fig. 1). This finding indicates that mLANA does not enforce a wholesale blockade to ATM-driven p53 activation as soon as infection occurs. However, as we previously demonstrated that mLANA-null MHV68, but not WT MHV68, replicated to higher titers following infection of p53-deficient fibroblasts than after infection of p53-competent cells (33), the capacity of mLANA to attenuate DDR-p53 signaling clearly impacts viral replication efficiency and further suggests that p53 serves an antiviral function during MHV68 infection.
While we establish here that muSOX and mLANA limit p53 functions during MHV68 lytic replication, the precise mechanisms by which this is accomplished remains to be determined. It is well established that muSOX functions to regulate the levels of both viral and host transcripts during viral replication (58, 74, 75). This includes host factors involved in antiviral processes, signaling, cellular responses to stress, and cell death (74–76). Thus, it is reasonable to suggest that muSOX degrades transcripts involved in p53 responses during infection. mLANA is a DNA binding transcription factor that regulates the expression of genes involved in cell cycle control (61) and can repress a viral promoter contained within the MHV68 terminal repeats (41). Although viral and host genes controlled by mLANA during lytic replication are not known, we speculate that mLANA binds to p53-responsive promoters and represses their activation. With this in mind, it is interesting that viruses containing mutations in the DNA binding domain of mLANA exhibited replication defects and viral gene hyperexpression that recapitulate the phenotype of mLANA-null MHV68 (41). It is possible that mLANA regulates transcription of ORF37 or other viral genes involved in p53 inhibition. Such a possibility could explain the requirement for mLANA, but not muSOX, in protecting infected cells from doxorubicin-induced death. Moreover, it also will be of interest to evaluate viral replication and p53 responses for infections with MHV68 viruses in which both muSOX and mLANA are mutated. Finally, it is worth noting that mLANA is expressed with immediate-early kinetics. This means mLANA likely is expressed within cells at times when p53 was active and still responsive to external stimuli. This leads us to speculate that mLANA is not sufficient in and of itself to prevent p53 function. This seems to agree with findings for KSHV; although LANA is expressed in latently infected primary effusion lymphoma (PEL) cell lines, its presence alone is insufficient to make them refractory to p53 agonists (77, 78). Conversely, reactivating PEL cells are resistant to treatment with p53 agonists (79). Together, these findings strongly suggest that viral and/or host factors in addition to LANA homologs are involved in p53 deactivation during rhadinovirus replication.
With this in mind, while our data demonstrate that muSOX and mLANA are important for deactivating p53 signaling during MHV68 infection, our data do not rule out the possibility that additional factors also suppress p53 functions. A screen of KSHV proteins identified RTA and several other viral proteins capable of repressing p53 transcriptional activity and cell death responses (30). Whether these viral proteins function to restrict p53 during lytic viral replication was not explored, and it is not yet known if MHV68 homologs exhibit similar functions. The observation that p53 localized to sites of viral DNA replication during MHV68 infection (Fig. 8), which also occurs during HSV1, HCMV, and EBV lytic replication (23, 80, 81), suggests that proteins involved in viral DNA replication sequester p53 or that p53 binding sites in viral DNA act as a molecular sponge to inhibit p53. Interestingly, ectopically expressed p53 was recruited to replication complex-like structures following transfection of cells with plasmids encoding components of the HSV1 replication machinery (82), raising the possibility that conserved proteins involved in MHV68 DNA replication also participate in p53 inactivation.
ACKNOWLEDGMENTS
We are grateful to Linda van Dyk, Britt Glaunsinger, and Vera Tarakanova for providing reagents that enabled our studies. We acknowledge Jason Stumhofer and Andrea Harris for help with flow cytometry experiments and analyzing data.
This work was supported by Public Health Service award R01-CA167065 from the National Cancer Institute and P20-GM103625 from the National Institute for General Medical Sciences.
REFERENCES
- 1.Barton E, Mandal P, Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol 29:351–397. doi: 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
- 2.Boehmer PE, Nimonkar AV. 2003. Herpes virus replication. IUBMB Life 55:13–22. doi: 10.1080/1521654031000070645. [DOI] [PubMed] [Google Scholar]
- 3.Moser JM, Farrell ML, Krug LT, Upton JW, Speck SH. 2006. A gammaherpesvirus 68 gene 50 null mutant establishes long-term latency in the lung but fails to vaccinate against a wild-type virus challenge. J Virol 80:1592–1598. doi: 10.1128/JVI.80.3.1592-1598.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cesarman E. 2011. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett 305:163–174. doi: 10.1016/j.canlet.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weitzman MD, Weitzman JB. 2014. What's the damage? The impact of pathogens on pathways that maintain host genome integrity. Cell Host Microbe 15:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lilley CE, Schwartz RA, Weitzman MD. 2007. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol 15:119–126. doi: 10.1016/j.tim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. 2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J 29:943–955. doi: 10.1038/emboj.2009.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine AJ. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323–331. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 10.Joerger AC, Fersht AR. 2007. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- 11.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. 2002. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat 19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 12.Kubbutat MH, Jones SN, Vousden KH. 1997. Regulation of p53 stability by Mdm2. Nature 387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 13.Marine JC, Lozano G. 2010. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ 17:93–102. doi: 10.1038/cdd.2009.68. [DOI] [PubMed] [Google Scholar]
- 14.Lavin MF, Gueven N. 2006. The complexity of p53 stabilization and activation. Cell Death Differ 13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 15.Lane DP, Crawford LV. 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 16.Linzer DI, Levine AJ. 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 17.McCormick F, Clark R, Harlow E, Tjian R. 1981. SV40 T antigen binds specifically to a cellular 53 K protein in vitro. Nature 292:63–65. doi: 10.1038/292063a0. [DOI] [PubMed] [Google Scholar]
- 18.Rushton JJ, Jiang D, Srinivasan A, Pipas JM, Robbins PD. 1997. Simian virus 40 T antigen can regulate p53-mediated transcription independent of binding p53. J Virol 71:5620–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mietz JA, Unger T, Huibregtse JM, Howley PM. 1992. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO J 11:5013–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HW. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 22.Stahl JA, Chavan SS, Sifford JM, MacLeod V, Voth DE, Edmondson RD, Forrest JC. 2013. Phosphoproteomic analyses reveal signaling pathways that facilitate lytic gammaherpesvirus replication. PLoS Pathog 9:e1003583. doi: 10.1371/journal.ppat.1003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y, Isomura H, Ishizaki K, Tsurumi T. 2005. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem 280:30336–30341. doi: 10.1074/jbc.M500976200. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, Desai P, Ambinder RF, Hayward GS, Qian J, Zhu H, Hayward SD. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10:390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seo T, Park J, Lee D, Hwang SG, Choe J. 2001. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J Virol 75:6193–6198. doi: 10.1128/JVI.75.13.6193-6198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin YC, Nakamura H, Liang X, Feng P, Chang H, Kowalik TF, Jung JU. 2006. Inhibition of the ATM/p53 signal transduction pathway by Kaposi's sarcoma-associated herpesvirus interferon regulatory factor 1. J Virol 80:2257–2266. doi: 10.1128/JVI.80.5.2257-2266.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. 2001. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol 75:429–438. doi: 10.1128/JVI.75.1.429-438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HR, Toth Z, Shin YC, Lee JS, Chang H, Gu W, Oh TK, Kim MH, Jung JU. 2009. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J Virol 83:6739–6747. doi: 10.1128/JVI.02353-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friborg J Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894. [DOI] [PubMed] [Google Scholar]
- 30.Chudasama P, Konrad A, Jochmann R, Lausen B, Holz P, Naschberger E, Neipel F, Britzen-Laurent N, Sturzl M. 2015. Structural proteins of Kaposi's sarcoma-associated herpesvirus antagonize p53-mediated apoptosis. Oncogene 34:639–649. doi: 10.1038/onc.2013.595. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy G, Komano J, Sugden B. 2003. Epstein-Barr virus provides a survival factor to Burkitt's lymphomas. Proc Natl Acad Sci U S A 100:14269–14274. doi: 10.1073/pnas.2336099100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saha A, Murakami M, Kumar P, Bajaj B, Sims K, Robertson ES. 2009. Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J Virol 83:4652–4669. doi: 10.1128/JVI.02408-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forrest JC, Paden CR, Allen RD III, Collins J, Speck SH. 2007. ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication. J Virol 81:11957–11971. doi: 10.1128/JVI.00111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adler H, Messerle M, Wagner M, Koszinowski UH. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol 74:6964–6974. doi: 10.1128/JVI.74.15.6964-6974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins CM, Speck SH. 2012. Tracking murine gammaherpesvirus 68 infection of germinal center B cells in vivo. PLoS One 7:e33230. doi: 10.1371/journal.pone.0033230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavlova IV, Virgin HW, Speck SH. 2003. Disruption of gammaherpesvirus 68 gene 50 demonstrates that Rta is essential for virus replication. J Virol 77:5731–5739. doi: 10.1128/JVI.77.10.5731-5739.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richner JM, Clyde K, Pezda AC, Cheng BY, Wang T, Kumar GR, Covarrubias S, Coscoy L, Glaunsinger B. 2011. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog 7:e1002150. doi: 10.1371/journal.ppat.1002150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moorman NJ, Willer DO, Speck SH. 2003. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J Virol 77:10295–10303. doi: 10.1128/JVI.77.19.10295-10303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stahl JA, Paden CR, Chavan SS, MacLeod V, Edmondson RD, Speck SH, Forrest JC. 2012. Amplification of JNK signaling is necessary to complete the murine gammaherpesvirus 68 lytic replication cycle. J Virol 86:13253–13262. doi: 10.1128/JVI.01432-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Paden CR, Forrest JC, Tibbetts SA, Speck SH. 2012. Unbiased mutagenesis of MHV68 LANA reveals a DNA-binding domain required for LANA function in vitro and in vivo. PLoS Pathog 8:e1002906. doi: 10.1371/journal.ppat.1002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gargano LM, Forrest JC, Speck SH. 2009. Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J Virol 83:1474–1482. doi: 10.1128/JVI.01717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem 280:8156–8163. doi: 10.1074/jbc.M411405200. [DOI] [PubMed] [Google Scholar]
- 44.Singh VV, Dutta D, Ansari MA, Dutta S, Chandran B. 2014. Kaposi's sarcoma-associated herpesvirus induces the ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J Virol 88:2821–2834. doi: 10.1128/JVI.03126-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jackson BR, Noerenberg M, Whitehouse A. 2014. A novel mechanism inducing genome instability in Kaposi's sarcoma-associated herpesvirus infected cells. PLoS Pathog 10:e1004098. doi: 10.1371/journal.ppat.1004098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakagawa K, Taya Y, Tamai K, Yamaizumi M. 1999. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol Cell Biol 19:2828–2834. doi: 10.1128/MCB.19.4.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lakin ND, Jackson SP. 1999. Regulation of p53 in response to DNA damage. Oncogene 18:7644–7655. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 48.Tibbetts SA, Loh J, Van Berkel V, McClellan JS, Jacoby MA, Kapadia SB, Speck SH, Virgin HW. 2003. Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J Virol 77:7696–7701. doi: 10.1128/JVI.77.13.7696-7701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulikov R, Boehme KA, Blattner C. 2005. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol 25:7170–7180. doi: 10.1128/MCB.25.16.7170-7180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 51.Ahn JW, Powell KL, Kellam P, Alber DG. 2002. Gammaherpesvirus lytic gene expression as characterized by DNA array. J Virol 76:6244–6256. doi: 10.1128/JVI.76.12.6244-6256.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ebrahimi B, Dutia BM, Roberts KL, Garcia-Ramirez JJ, Dickinson P, Stewart JP, Ghazal P, Roy DJ, Nash AA. 2003. Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J Gen Virol 84:99–109. [DOI] [PubMed] [Google Scholar]
- 53.Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, Tong L, Sun R. 2003. Transcription program of murine gammaherpesvirus 68. J Virol 77:10488–10503. doi: 10.1128/JVI.77.19.10488-10503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng BY, Zhi J, Santana A, Khan S, Salinas E, Forrest JC, Zheng Y, Jaggi S, Leatherwood J, Krug LT. 2012. Tiled microarray identification of novel viral transcript structures and distinct transcriptional profiles during two modes of productive murine gammaherpesvirus 68 infection. J Virol 86:4340–4357. doi: 10.1128/JVI.05892-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 56.Rosenke K, Samuel MA, McDowell ET, Toerne MA, Fortunato EA. 2006. An intact sequence-specific DNA-binding domain is required for human cytomegalovirus-mediated sequestration of p53 and may promote in vivo binding to the viral genome during infection. Virology 348:19–34. doi: 10.1016/j.virol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 57.Johnson LS, Willert EK, Virgin HW. 2010. Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe 7:516–526. doi: 10.1016/j.chom.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Covarrubias S, Richner JM, Clyde K, Lee YJ, Glaunsinger BA. 2009. Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J Virol 83:9554–9566. doi: 10.1128/JVI.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glaunsinger BA, Ganem DE. 2006. Messenger RNA turnover and its regulation in herpesviral infection. Adv Virus Res 66:337–394. doi: 10.1016/S0065-3527(06)66007-7. [DOI] [PubMed] [Google Scholar]
- 60.Glaunsinger B, Ganem D. 2004. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell 13:713–723. doi: 10.1016/S1097-2765(04)00091-7. [DOI] [PubMed] [Google Scholar]
- 61.Ottinger M, Pliquet D, Christalla T, Frank R, Stewart JP, Schulz TF. 2009. The interaction of the gammaherpesvirus 68 orf73 protein with cellular BET proteins affects the activation of cell cycle promoters. J Virol 83:4423–4434. doi: 10.1128/JVI.02274-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. 1999. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 63.Hollingworth R, Skalka GL, Stewart GS, Hislop AD, Blackbourn DJ, Grand RJ. 2015. Activation of DNA damage response pathways during lytic replication of KSHV. Viruses 7:2908–2927. doi: 10.3390/v7062752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. 2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A 102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koopal S, Furuhjelm JH, Jarviluoma A, Jaamaa S, Pyakurel P, Pussinen C, Wirzenius M, Biberfeld P, Alitalo K, Laiho M, Ojala PM. 2007. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog 3:1348–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86:159–171. doi: 10.1016/S0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 67.Kulinski JM, Leonardo SM, Mounce BC, Malherbe L, Gauld SB, Tarakanova VL. 2012. Ataxia telangiectasia mutated kinase controls chronic gammaherpesvirus infection. J Virol 86:12826–12837. doi: 10.1128/JVI.00917-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kulinski JM, Darrah EJ, Broniowska KA, Mboko WP, Mounce BC, Malherbe LP, Corbett JA, Gauld SB, Tarakanova VL. 2015. ATM facilitates mouse gammaherpesvirus reactivation from myeloid cells during chronic infection. Virology 483:264–274. doi: 10.1016/j.virol.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hagemeier SR, Barlow EA, Meng Q, Kenney SC. 2012. The cellular ataxia telangiectasia-mutated kinase promotes Epstein-Barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J Virol 86:13360–13370. doi: 10.1128/JVI.01850-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shah GA, O'Shea CC. 2015. Viral and cellular genomes activate distinct DNA damage responses. Cell 162:987–1002. doi: 10.1016/j.cell.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khaperskyy DA, Emara MM, Johnston BP, Anderson P, Hatchette TF, McCormick C. 2014. Influenza a virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog 10:e1004217. doi: 10.1371/journal.ppat.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taddeo B, Esclatine A, Roizman B. 2002. The patterns of accumulation of cellular RNAs in cells infected with a wild-type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. Proc Natl Acad Sci U S A 99:17031–17036. doi: 10.1073/pnas.252588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taddeo B, Esclatine A, Zhang W, Roizman B. 2003. The stress-inducible immediate-early responsive gene IEX-1 is activated in cells infected with herpes simplex virus 1, but several viral mechanisms, including 3′ degradation of its RNA, preclude expression of the gene. J Virol 77:6178–6187. doi: 10.1128/JVI.77.11.6178-6187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abernathy E, Clyde K, Yeasmin R, Krug LT, Burlingame A, Coscoy L, Glaunsinger B. 2014. Gammaherpesviral gene expression and virion composition are broadly controlled by accelerated mRNA degradation. PLoS Pathog 10:e1003882. doi: 10.1371/journal.ppat.1003882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Clyde K, Glaunsinger BA. 2011. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS One 6:e19655. doi: 10.1371/journal.pone.0019655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abernathy E, Glaunsinger B. 2015. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology 479-480:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petre CE, Sin SH, Dittmer DP. 2007. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J Virol 81:1912–1922. doi: 10.1128/JVI.01757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sarek G, Kurki S, Enback J, Iotzova G, Haas J, Laakkonen P, Laiho M, Ojala PM. 2007. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J Clin Investig 117:1019–1028. doi: 10.1172/JCI30945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sarek G, Ma L, Enback J, Jarviluoma A, Moreau P, Haas J, Gessain A, Koskinen PJ, Laakkonen P, Ojala PM. 2013. Kaposi's sarcoma herpesvirus lytic replication compromises apoptotic response to p53 reactivation in virus-induced lymphomas. Oncogene 32:1091–1098. doi: 10.1038/onc.2012.118. [DOI] [PubMed] [Google Scholar]
- 80.Wilcock D, Lane DP. 1991. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 349:429–431. doi: 10.1038/349429a0. [DOI] [PubMed] [Google Scholar]
- 81.Fortunato EA, Spector DH. 1998. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J Virol 72:2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhong L, Hayward GS. 1997. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J Virol 71:3146–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]