

ABSTRACT
The Asian citrus psyllid, Diaphorina citri, is the natural vector of the causal agent of Huanglongbing (HLB), or citrus greening disease. Together; HLB and D. citri represent a major threat to world citrus production. As there is no cure for HLB, insect vector management is considered one strategy to help control the disease, and D. citri viruses might be useful. In this study, we used a metagenomic approach to analyze viral sequences associated with the global population of D. citri. By sequencing small RNAs and the transcriptome coupled with bioinformatics analysis, we showed that the virus-like sequences of D. citri are diverse. We identified novel viral sequences belonging to the picornavirus superfamily, the Reoviridae, Parvoviridae, and Bunyaviridae families, and an unclassified positive-sense single-stranded RNA virus. Moreover, a Wolbachia prophage-related sequence was identified. This is the first comprehensive survey to assess the viral community from worldwide populations of an agricultural insect pest. Our results provide valuable information on new putative viruses, some of which may have the potential to be used as biocontrol agents.
IMPORTANCE Insects have the most species of all animals, and are hosts to, and vectors of, a great variety of known and unknown viruses. Some of these most likely have the potential to be important fundamental and/or practical resources. In this study, we used high-throughput next-generation sequencing (NGS) technology and bioinformatics analysis to identify putative viruses associated with Diaphorina citri, the Asian citrus psyllid. D. citri is the vector of the bacterium causing Huanglongbing (HLB), currently the most serious threat to citrus worldwide. Here, we report several novel viral sequences associated with D. citri.
INTRODUCTION
Viruses are the most abundant microbes on our planet (1) and are found in all types of organisms. Insects are the largest and most diverse taxonomic class among animals, representing perhaps half of known animals, with more than one million species recognized worldwide (2). Insects are known to be hosts to viruses belonging to various viral taxa, including the Baculoviridae, Parvoviridae, Flaviviridae, Ascoviridae, Togaviridae, Bunyaviridae, and Rhabdoviridae (3). However, the number of currently described viral species infecting insects is relatively low compared to the number of viruses that have been discovered among prokaryotes, plants, and vertebrates. Furthermore, most of the insect viruses described to date have been discovered because of their pathogenic effects on their insect hosts or because they are pathogens of humans, other vertebrates, or economically important plants. Traditional viral detection methods that require prior knowledge of genome sequences may not be suitable for the discovery of new viruses and, in particular, viruses with a high level of genetic diversity. However, in the past decade, the development of high-throughput next-generation sequencing (NGS) technologies and bioinformatics applications has provided new opportunities for discovering viruses in many organisms, including humans (4–7), arthropods (8–29), and plants (30–45). In addition to transcriptome sequencing (RNA-seq) (15, 18, 28), deep sequencing of small RNAs (sRNAs) and the subsequent assembly of the sRNAs have been proven to be promising approaches for the discovery of both RNA and DNA viruses in plant and insect hosts (9, 41, 42, 46–50).
The Asian citrus psyllid, Diaphorina citri Kuwayama, is currently the most important insect associated with worldwide citrus production (51). D. citri is the vector of “Candidatus Liberibacter asiaticus,” the causal agent of Huanglongbing (HLB). HLB, also known as citrus greening disease, is the most devastating disease of citrus trees and currently is a threat to world citrus production (52). HLB is native to Asia (53), and recently the American form, which is believed to have originated in China, was discovered and reported in South America, Mexico, and North America (United States) (54, 55). As there is no cure for HLB, disease control relies on a combination of approaches including insect vector management through chemical and biological control strategies.
In an attempt to discover putative viruses which may be associated with D. citri, we conducted a metagenomic study of populations of D. citri from various locations around the world. We used a multistep analysis pipeline consisting of high-throughput NGS of small RNAs and transcriptomes, de novo sequence assembly, in silico searches for sequence similarities to reference viruses, and confirmation of putative viral sequences via reverse transcription-PCR (RT-PCR) and PCR followed by Sanger sequencing. Here we describe the success of these methods to assess the diversity of viral sequences found in D. citri. Our findings demonstrate the presence of viral sequences from several distinct taxa, including the families Reoviridae, Parvoviridae, Bunyaviridae, the picornavirus superfamily, an unclassified positive-sense single-stranded RNA (ssRNA) virus, and the Wolbachia prophage WO.
MATERIALS AND METHODS
Sample collection and total RNA preparation.
In 2013, RNA samples from D. citri collected from Florida were shipped to our laboratory at the University of California-Davis (UCD). We also received RNA samples from D. citri collected in China and Taiwan, the native geographic regions of D. citri, and from Brazil, where D. citri and HLB are listed as newly emerging. In 2014, we received RNA samples from other citrus-growing regions, including Hawaii, California, Texas, and Pakistan. RNA was extracted by homogenizing 50 to 60 whole wild-caught psyllids in TRIzol reagent according to the manufacturer's instructions (Life Science Research, Carlsbad, CA). D. citri RNAs were qualitatively and quantitatively evaluated on an Experion RNA analysis system using the Experion RNA SdtSens analysis kit (Bio-Rad, Hercules, CA) and a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE), respectively. High-quality RNAs from the samples were selected for construction of small RNA and RNA-seq cDNA libraries for high-throughput NGS.
Small RNA and RNA-seq library construction and sequencing.
Four sRNA libraries were generated from 2.0 μg of total RNA from Brazil, China, Taiwan, and Florida with RNA quality indicator (RQI) values of ≥7 using Illumina's TruSeq small RNA sample preparation kit according to the sample preparation instructions (Illumina, San Diego, CA). Briefly, 5′ and 3′ adapters designed based on the natural structure of small RNAs included in the kit were ligated to each end of the RNA molecules. cDNA was synthesized by reverse transcription (RT) followed by PCR amplification with common and indexed primers. The sRNA libraries were then gel size selected and purified, and the final cDNA libraries were validated on an Experion system using the Experion DNA 1K analysis kit (Bio-Rad). Final concentrations were adjusted to 10 nM, and 10 μl of each sample was shipped to the Beijing Genomics Institute (BGI), Hong Kong, for 50-bp single-read sequencing using the Illumina HiSeq 2000 platform in four lanes.
Since assembly of sRNA is difficult, and obtaining full-genome coverage is challenging due to the short lengths of the generated contiguous sequences (contigs), we also sequenced D. citri total RNA (RNA-seq) using an Illumina HiSeq 2000 platform. rRNA-depleted RNAs from the China, Brazil, Hawaii, and Florida populations were used to construct RNA-seq libraries with the ScriptSeq v2 RNA-Seq library preparation kit by following the manufacturer's instructions (epicenter/Illumina, San Diego, CA). Briefly, following DNase I digestion, rRNA was removed from total RNA using the epicenter/Illumina Ribo-Zero magnetic gold kit. The RNA was then fragmented and further processed according to the manufacturer's protocol. The libraries were multiplexed via PCR with ScriptSeq index PCR primers. Final indexed libraries were validated on the Experion system using the Experion DNA 1K analysis kit (Bio-Rad) and sequenced on the HiSeq 2000 platform (100-bp pair-end sequencing) at the Vincent J. Coates Genomics Sequencing Laboratory at University of California-Berkeley.
Small RNA and RNA-seq analysis and virus genome identification.
Bioinformatics analysis of sRNA and RNA-seq data was performed using the CLC Genomic Workbench software package (CLC Bio-Qiagen, Boston, MA). Briefly, low-quality reads (<0.05) and adapter sequences were first removed from the raw sRNA data set. Trimmed sRNA sequences shorter than 15 nucleotides (nt) were discarded, and the remaining reads were mapped to the available recently assembled D. citri genome (GCF_000475195.1) to remove the host-related reads. Reads were then de novo assembled using two assemblers: the CLC Assembly Cell and Velvet (56), with word size/k-mer values ranging between 15 and 19. We used two assemblers because the length of contigs can vary based on the assembly programs and the parameter setting used for each program are specific, and using two assemblers also can give support of newly identified virus sequences (57). The resulting contigs were compared against the nonredundant viral protein database available in NCBI using BLASTx and tBLASTx at an E value of <10−3 (58). BLAST results were then inspected manually to screen for potential viral sequences.
For the reads derived from RNA-seq, trimming and mapping were performed under the same conditions as described for sRNA. Reads were then assembled with word size/k-mer values ranging from 45 to 65. BLAST searches were conducted using contigs of >200 nt against the nonredundant viral protein database using BLASTx and tBLASTx at an E value of <10−3 (58).
Viral genome sequence validation.
To confirm the presence of the viral sequences identified in different populations of D. citri, RT-PCR and PCR assays were developed using specific primers designed based on de novo-assembled contigs with similarities to viral sequences. The (RT-)PCR products were sequenced by Sanger sequencing. We used RNA extracted from our D. citri colony maintained in the Contained Research Facility (CRF) at UC Davis, which was negative for identified viral sequences here based on (RT-)PCR results as the negative control in all (RT-)PCRs.
Phylogenetic analysis.
Reference amino acid sequences of the respective viral RNA-dependent RNA polymerase (RdRp) proteins, and nonstructural (NS) proteins in the case of DNA viruses, were downloaded from GenBank. Multiple amino acid sequence alignments were performed with MUSCLE in MEGA version 6 with the default settings (59). Phylogenetic trees were constructed using the neighbor-joining (NJ) and maximum-likelihood (ML) methods in MEGA6 using the appropriate models for each group of viral sequences with 1,000 bootstraps. GenBank accession numbers of the reference sequences used in the phylogenetic analysis are shown in Table S3 in the supplemental material.
Accession numbers.
All raw reads produced and used in this study were submitted to the National Center for Biotechnology Information (NCBI)'s Sequence Read Archive (SRA) under bioproject accession PRJNA293863. Sequences described in this paper were deposited in GenBank under the accession numbers KT698823 (DcBV L segment), KT698824 (DcBV M segment), KT698825 (DcBV S segment), KT698826 (DcACV RNA1), KT698827 (DcACV RNA2), KT698828 (DcDNV NS2), KT698829 (DcDNV VP1), KT698830 (DcRV segment 1), KT698831 (DcRV segment 8), KT698832 (DcRV segment 2), KT698833 (DcRV segment 10), KT698834 (DcRV segment 3), KT698835 (DcRV segment 4), KT698836 (DcRV segment 7), and KT698837 (DcPLV).
RESULTS
Small RNA profiles and RNA-seq analysis of D. citri.
Because insects use RNA interference (RNAi), which results in the generation of abundant 21- to 30-nt virus-derived sRNAs, as a primary antiviral defense, NGS sequencing of sRNA libraries is a valid approach to identify insect-infecting viruses (9, 30, 60, 61). High-throughput NGS of the sRNA libraries generated 150 million to 200 million usable reads per library, with a length range of 15 to 30 nt. Both CLC Assembly Cell and Velvet assemblers generated more and longer contigs with a word window/k-mer value of 19 after subtracting the host reads. However, as we expected, the majority of the assembled contigs (70%) were ≤400 bp in length. Moreover, the number of reads that mapped to some detected virus-like sequences was low (see Table S2 in the supplemental material), but the mapping nevertheless suggested the presence of viral sequences in our libraries. Therefore, in order to generate longer contigs, four RNA-seq libraries were constructed and sequenced in multiplex. Approximately 50 million to 100 million paired-end raw reads of length 100 were produced from each library. Raw reads were cleaned and assembled de novo as described for the sRNA libraries.
BLASTx searches with all the generated contigs from both sRNA and RNA-seq libraries suggested the presence of viral sequences from several distinct taxa. Viral sequences similar to those within the Reoviridae family and the picornavirus superfamily comprised the majority of viral sequences identified in D. citri (see Table S1). A smaller number of reads and contigs were observed to have similarity with viruses of the families Bunyaviridae and Parvoviridae and with Chronic bee paralysis virus, an unclassified positive-sense single-stranded RNA virus (see Table S1). Furthermore, a bacteriophage-like contig was identified in all D. citri populations. Most of the identified viral sequences shared less than 50% amino acid identity to known viral sequences, suggesting that they represent novel viral sequences. Table S2 in the supplemental material provides a list of all viruses in the viral database that showed significant BLASTx hits to contigs produced in this study.
Contigs related to picorna-like viruses.
Contigs that showed similarity to picorna-like viruses (Iflaviruses) were assembled from sRNA and RNA-seq libraries from the D. citri populations from Brazil, China, and Taiwan (Table 1; see also Table S2). Picorna-like viruses, which belong to the picornavirus superfamily, a major division of eukaryotic positive-strand RNA viruses (62), are a large group of positive-sense, single-stranded RNA viruses which includes important pathogens of humans, plants, and insects. The genomes of viruses in the picornavirus superfamily are characterized by an RdRp, a chymotrypsin-like 3C protease, a putative helicase, and a genome-linked protein (VPg) (62–65). The picornavirus superfamily currently has 14 divergent families of viruses and several unclassified genera and species (62). Five of these families, Picornaviridae, Iflaviridae, Dicistroviridae, Marnaviridae, and Secoviridae, are further classified in the order Picornavirales, which includes extremely diverse viruses and virus-like elements (64). Iflaviruses are members of a relatively newly recognized family called Iflaviridae, members of which all belong to the genus Iflavirus and possess a monopartite, single-stranded, positive-sense RNA genome ranging from 8.5 to 10 kb in length (66). The genome encodes a single polyprotein of ∼3,000 amino acids that is processed to produce a helicase, a protease, an RdRp, and four structural proteins (VP1 to VP4). All known iflaviruses are insect-infecting viruses with a wide range of hosts belonging to the orders Lepidoptera, Hemiptera and Hymenoptera and also bee parasitic mites (11, 12, 14–16, 20, 21, 26, 29, 67). However, a plant-infecting iflavirus-like virus, Tomato matilda virus, was recently reported from tomato (Solanum lycopersicum) (68). There are currently seven definitive groups within the genus Iflavirus recognized in the Ninth Report of the International Committee on Taxonomy of Viruses (69). However, several tentative viruses have been identified that show sequence similarity to the members of the genus Iflavirus yet are classified as unassigned viruses at this time (12, 16, 21, 70, 71). Previous phylogenetic analysis suggested that iflaviruses have evolved from different origins since the viruses infecting insects from the same order do not form a single clade (3, 17).
TABLE 1.
Viral sequences identified in D. citria
| Virus category | Tentative name for putative virus | Closest relative virus (BLASTx) | Tentative virus family or superfamily | Tentative virus genus | Largest sequence length (bp) obtained | Source of representative D. citri populations |
|---|---|---|---|---|---|---|
| dsRNA | DcRV | Nilaparvata lugens reovirus | Reoviridae | Fijivirus | Seg. 1: 4,454 | CH, TW, FL, HW, TX |
| Seg. 2: 3,530 | ||||||
| Seg. 3: 3,814 | ||||||
| Seg. 4: 3,445 | ||||||
| Seg. 7: 2,129 | ||||||
| Seg. 8: 1,737 | ||||||
| Seg. 10: 1,216 | ||||||
| ssRNA | DcPLV | Deformed wing virus | Picorna-like virus | Unclassified | 9,580 | BR, CH, TW |
| ssDNA | DcDNV | Cherax quadricarinatus densovirus | Parvoviridae | Unclassified | NS1: 1,299 | CH, TW, Pak |
| NS2: 1,344 | ||||||
| VP1: 1,767 | ||||||
| VP2: 618 | ||||||
| ssRNA | DcBV | Wuchang cockroach virus 1 | Bunyaviridae | Unclassified | L-seg.: 1,911 | CH, TW |
| M-seg.: 1,852 | ||||||
| S-seg.: 438 | ||||||
| ssRNA | DcACV | Pea enation mosaic virus-2 | Unclassified | Umbravirus | RNA1: 1,111 | CH, CA, TX, FL |
| Chronic bee paralysis virus | Unclassified | Unclassified | RNA2: 1,764 | |||
| dsDNA | WO Prophage | WO prophage | Unclassified phages | Unclassified | 8,615 | All |
DcPLV, Diaphorina citri picorna-like virus; DcRV, Diaphorina citri reovirus; DcDNV, Diaphorina citri densovirus; DcBV, Diaphorina citri bunyavirus; DcACV, Diaphorina citri associated C virus; Seg., segment; CH, China; TW, Taiwan; FL, Florida; BR, Brazil; HW, Hawaii; TX, Texas; CA, California; Pak, Pakistan.
Through bioinformatics analysis of both sRNA and RNA-seq data, we were able to assemble more than 80% of the predicted genome sequence of a putative picorna-like virus tentatively named Diaphorina citri picorna-like virus (DcPLV). The presence of this virus in the RNA samples was subsequently confirmed by RT-PCR and Sanger sequencing using specific primers. Using a primer walking strategy to fill in the gaps, 9,580 nucleotides of the genome of DcPLV were determined (Table 1). Bioinformatics analysis predicted one possible open reading frame (ORF) of 8,496 nt. Sequence analysis showed that the DcPLVs found in the D. citri populations from China, Taiwan, and Brazil shared 97 to 99% nucleotide identity (data not shown), suggesting that they are members of the same species. BLASTx and tBLASTx searches showed that the DcPLVs shared low sequence identity (less than 40%) with members of Iflaviridae at the amino acid level (Table 2). However, DcPLV has a distinctly different genome organization from iflaviruses, which have a single large ORF encoding a polyprotein that is cleaved into both structural and nonstructural proteins with the structural proteins (capsid proteins) located N terminal to the nonstructural proteins (Fig. 1A). In contrast, the DcPLV structural proteins are located C terminal to the nonstructural proteins in the predicted DcPLV polyprotein (Fig. 1B). The organization of the DcPLV genome appears to be most like that of Heterosigma akashiwo RNA virus (HaRNAV), which belongs to the family Marnaviridae (72). The family Marnaviridae consists of a single genus, Marnavirus, with HaRNAV as the type species (73). Interestingly, DcPLV and HaRNAV are phylogenetically distant, sharing only limited (20%) sequence identity at the amino acid level (Fig. 2A). Therefore, we believe that DcPLV is not an iflavirus, a marnavirus, or a dicistrovirus (Fig. 1C) but is a new, unclassified picorna-like virus. A phylogenetic tree generated based on the RdRp placed DcPLV close to the Iflaviridae and related to other members of the order Picornavirales (Fig. 2A). To assess the presence of DcPLV in other D. citri populations, we examined RNA from different populations using RT-PCR with specific primers. The results showed the presence of DcPLV in all of the populations from China and Brazil but not in the populations from the United States, Pakistan, or Taiwan, except one (see Table S4 in the supplemental material).
TABLE 2.
Query coverage and maximum amino acid identity (BLASTx) between the proteins encoded by the putative viruses found here and the most closely related species or genusa
| Putative virus | Encoded protein | Query % coverage | Maximum % identity | E value | Closely related species or genus | GenBank accession no. |
|---|---|---|---|---|---|---|
| DcRV | Seg. 1_RdRP | 97 | 36 | 0.0 | Nilaparvata lugens reovirus | NP_619776 |
| Seg. 2_136.6KD | 33 | 28 | 2e−18 | NP_619777 | ||
| Seg .3_major core capsid protein | 87 | 29 | 6e−43 | NP_619778 | ||
| Seg. 4_130KD | 99 | 26 | 5e−81 | NP_619779 | ||
| Seg. 7_73.5KD | 96 | 24 | 3e−17 | NP_619782 | ||
| Seg. 8_major outer capsid protein | 87 | 26 | 5e−37 | NP_619775 | ||
| Seg. 10_polypeptide | 82 | 24 | 1e−18 | NP_619774 | ||
| DcPLV | Polyprotein | 70 | 33 | 2e−112 | Deformed wing virus | ADK55526 |
| DcDNV | NS1 | 37 | 33 | 1e−09 | Uncharacterized protein in Diaphorina citri | XP_008482940 |
| NS2 | 96 | 35 | 5e−69 | Cherax quadricarinatus densovirus | YP_009134732 | |
| VP1 | 37 | 31 | 6e−15 | Densovirus SC1065 | AFH02754 | |
| VP2 | 43 | 42 | 2e−14 | Periplaneta fuliginosa densovirus | NP_051016 | |
| DcBV | RdRp | 85 | 31 | 6e−62 | Wuchang cockroach virus 1 | AJG39258 |
| Glycoprotein precursor | 80 | 32 | 1e−25 | AJG39291 | ||
| Nucleocapsid | 90 | 36 | 2e−20 | AJG39319 | ||
| DcACV | RdRp | 70 | 33 | 8e−25 | Pea enation mosaic virus-2 | AAU20330 |
| Hypothetical protein_s2gp2 | 8 | 39 | 0.001 | Chronic bee paralysis virus | YP_001911140 |
DcPLV, Diaphorina citri Picorna-like virus; DcRV, Diaphorina citri reovirus; DcDNV, Diaphorina citri densovirus; DcBV, Diaphorina citri bunyavirus; DcACV, Diaphorina citri associated C virus.
FIG 1.

Schematic illustration of the predicted genome organization of DcPLV. (A) A typical Iflavirus genome showing a single ORF and the encoded structural proteins in the N-terminal region, while nonstructural proteins are located in the C-terminal region; (B) DcPLV predicted genome showing the unique position of the viral proteins in the polyprotein indicated; (C) a typical bipartite Dicistrovirus genome showing two ORFs. The nonstructural proteins are encoded by ORF1, and structural proteins are encoded by ORF2. L, leader protein; VP, virion protein; Hel, superfamily 3 helicase; Vpg, genome-linked protein; Pro, chymotrypsin-like cysteine protease; RdRp, RNA-dependent RNA polymerase; IRES, internal ribosome entry site.
FIG 2.

Maximum-likelihood trees of RdRp/nonstructural (NS2) protein amino acid sequences from representative viruses from picorna-like virus (A), Reoviridae (B), Parvoviridae (C), Bunyaviridae (D), and unclassified viruses, Tombusviridae, and Nodiviridae (E). Phylogenetic trees were constructed using the MEGA 6.0 program, with the LG+G+I evolutionary model for picorna-like virus and unclassified-Tombusviridae-Nodiviridae trees and the WAG+G+F model for Reoviridae, Parvoviridae, and Bunyaviridae trees with 1,000 bootstrap replications. The topology of the NJ trees was similar to that of the ML trees. Table S3 in the supplemental material shows the accession numbers of the reference sequences. DcPLV, Diaphorina citri picorna-like virus; DcRV, Diaphorina citri reovirus; DcDNV, Diaphorina citri densovirus; DcBV, Diaphorina citri bunyavirus; DcACV, Diaphorina citri-associated C virus. Pluses indicate novel viruses discovered in the current study.
Reovirus-like sequences.
Reovirus-like sequences were identified in the D. citri samples from China, Taiwan, Florida, and Hawaii from both sRNA and RNA-seq libraries. These sequences displayed similarities with viruses belonging to the family Reoviridae, genus Fijivirus (Table 1; see also Table S2). RT-PCR and Sanger sequencing confirmed the presence of the identified viral sequences in the above-mentioned samples and also in one sample from Texas (see Table S4). Reoviridae is a family of double-stranded RNA (dsRNA) viruses with members having 10 to 12 genome segments (69). In general, different reoviruses infect a wide range of different hosts, including mammals, birds, reptiles, fish, arthropods, fungi, protists, and plants. Reoviruses infecting plant-feeding hemipteran insects are classified into three genera: Phytoreovirus (74, 75), Fijivirus (76, 77), and Oryzavirus (78). Fijiviruses have 10 linear dsRNA segments encoding 12 proteins (69). Segment lengths can range from 1.4 to 4.5 kb, and the total genome size is about 27 to 30 kb.
In this study, nearly complete nucleotide sequences of seven putative reovirus segments ranging from 1,216 to 4,454 nt in length were identified in D. citri. RT-PCR further confirmed the presence of these sequences in our D. citri RNA extracts, and these sequences were not amplified by PCR, thus suggesting they were derived from dsRNAs. In 2009, a putative reovirus called Diaphorina citri reovirus (DcRV) was reported as a new species from the genus Fijivirus, family Reoviridae, naturally infecting ca. 55% of wild D. citri in Florida (79). However, only partial nucleotide sequences (a range of 400 to 800 nt) of six individual genome segments (predicted segments 1, 2, 4, 7, 8, and 10) of that virus were determined by Sanger sequencing of the cDNA libraries (79). The reovirus-like sequences obtained in the current study showed the highest amino acid similarity to the previously reported DcRV from Florida. However, due to the limited amount of sequence data available for the previously described DcRV, query coverage for our sequences was very low (<30%) for BLAST hits to the previously described DcRV, but in each case amino acid identity was high (≥93%). The second highest score of amino acid similarity belonged to Nilaparvata lugens reovirus (NLRV) (80) and compared to the case with DcRV from Florida, query coverage was much greater with NLRV, likely because a complete genome sequence is available for this virus (Table 2). Consistent with the previous report, our phylogenetic analysis based on segment 1, which encodes the RdRp, suggests that DcRV is a new putative species in the genus Fijivirus, most closely related to NLRV (Fig. 2B). Interestingly, we were not able to identify three predicted genome segments (segments 5, 6 and 9) through our computational analysis. However, by examining dsRNAs extracted from the Hawaiian D. citri population according to the previously described method (81), we observed at least 10 dsRNAs. No dsRNAs were seen in our Californian D. citri (used as a negative control), and the resulting dsRNA pattern closely resembled that of another insect-infecting reovirus, Homalosisca vitripennis reovirus (81), suggesting that predicted segments 5, 6, and 9 were present but could not be identified by our NGS and bioinformatics analysis (Fig. 3). This can be explained if the missing segments are too divergent from known sequences in databases and were thus not recognized by our BLAST searches.
FIG 3.
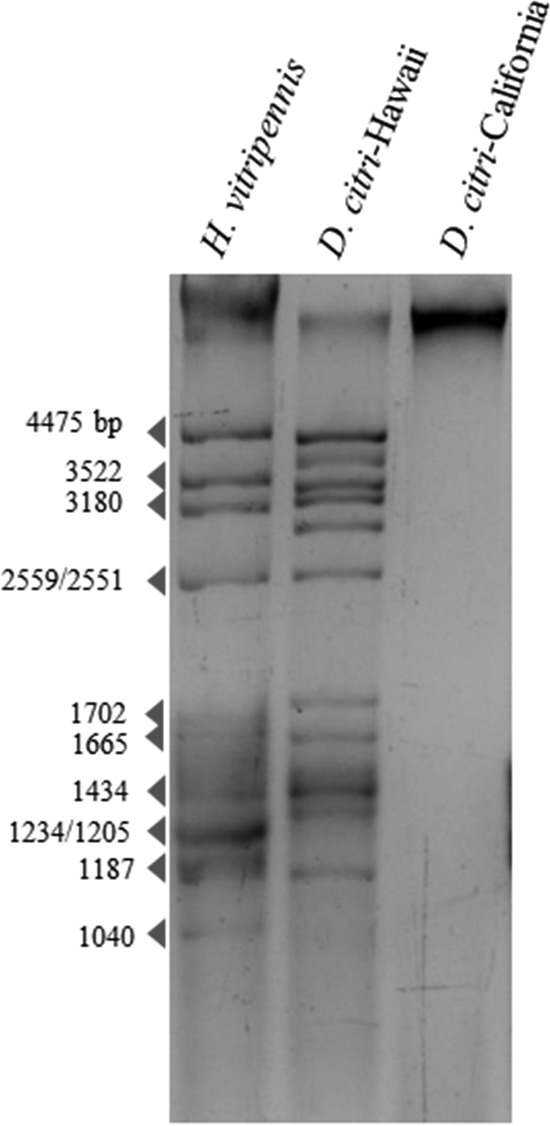
Double-stranded RNAs recovered from the Hawaiian D. citri population. CF11-cellulose-purified dsRNAs were electrophoresed in a 1.5% agarose gel. dsRNAs of Homalodisca vitripennis reovirus (HoVRV; 4,475 to 1040 bp) were used as the size standards. Californian D. citri was used as a negative control.
Densovirus-like sequences.
High-throughput NGS analysis of sRNA and RNA-seq libraries from the China and Taiwan populations revealed densovirus (DNV)-like sequences (Table 1; see also Table S2 in the supplemental material). Similar sequences were also detected in the population from Pakistan and confirmed in the populations from China and Taiwan using PCR (see Table S4). DNVs, which belong to the subfamily Densovirinae in the family Parvoviridae, are characterized by small, nonenveloped virions which contain a linear, single-stranded DNA genome 4 to 6 kb in length (69, 82). DNVs are classified into five genera, Ambidensovirus, Brevidensovirus, Hepandensovirus, Iteradensovirus, and Penstyldensovirus, on the basis of genome characteristics, gene expression strategy, and structure of the terminal hairpins (69, 82, 83). Our analysis generated contigs that displayed significant BLASTx hits against nonstructural (NS) and structural (VP) genomic regions of DNVs. However, amino acid identity between reference sequences and these contigs was low (<40%). In order to obtain more genome coverage, a primer-walking approach was performed using primers based on the contigs generated in our analysis. This approach yielded a product of 4,836 nt, and computational analysis predicted four ORFs, including two overlapping NS genes on the same strand, tentatively called NS1 (1,299 nt) and NS2 (1,344 nt), and two nonoverlapping VP genes on the opposite strand, tentatively called VP1 (1,767 nt) and VP2 (618 nt) (Fig. 4). This putative novel virus is tentatively named Diaphorina citri densovirus (DcDNV). A BLASTx search using the ∼4.8-kb sequence indicated the highest similarity with an ambidensovirus called Cherax quadricarinatus densovirus (GenBank accession no. YP_009134732) (Table 1). Table 2 shows the maximum amino acid identity of each DcDNV-encoded protein to homologous reference proteins. Members of the Ambidensovirus genus have an ambisense genome organization wherein both complementary strands have the capacity to encode functional proteins, a feature that is present among members of the family Parvoviridae (69). All members of Ambidensovirus are pathogenic to their insect hosts, and members of this genus are known to infect insects belonging to at least five orders (Lepidoptera, Diptera, Orthoptera, Odonata, and Hemiptera) (83). A mutualistic association between a novel densovirus, Helicoverpa armigera densovirus-1 (HaDNV-1), and its host, Helicoverpa armigera, has recently been reported (84). However, a phylogenetic tree constructed based on NS2 amino acid sequences placed DcDNV closer to the viruses from the genus Iteradensovirus (Fig. 2C) which possess a monosense genome (69). Interestingly, the NS1 sequence displayed the highest similarity with an uncharacterized insect protein (Table 2).
FIG 4.

Schematic illustration of the predicted genome organization of DcDNV. The 4,836-bp ambisense genome contains four open reading frames (NS1, NS2, VP1, and VP2) which are flanked by inverted terminal repeats (ITR). NS, nonstructural protein; VP, virion protein; NTR, nontranslated region.
Bunyavirus-like sequences.
Sequences similar to the L, M, and S segments of a typical member of the family Bunyaviridae were assembled from the sRNA and RNA-seq libraries from China and Taiwan (Table 1; see also Table S2 in the supplemental material), and the presence of these sequences in D. citri was confirmed by one-step RT-PCR. Bunyaviruses, members of the family Bunyaviridae, have segmented, single-stranded, negative-sense RNA genomes comprised of three RNAs designated L (large), M (medium), and S (small), which together total 11 to 19 kb. The L, M, and S genome segments encode the RdRp, envelope glycoproteins (Gn and Gc), and nucleocapsid protein (N), respectively (69). The family Bunyaviridae includes the genera Orthobunyavirus, Nairovirus, Phlebovirus, Hantavirus, and Tospovirus as well as a recently proposed genus tentatively named Phasmavirus (85, 86). We identified several contigs with amino acid similarity to members of the putative genus Phasmavirus (85). Significant hits included the L, M, and S segments of Wuhan mosquito virus 1 (AJG39267), Wuchang cockraoch virus 1 (AJG39258), and Kigluaik phantom virus (AIA24559) (see Table S2). Table 2 shows the query coverage and the maximum amino acid similarities for individual protein sequences of the putative bunyavirus identified in the current study, which is tentatively named Diaphorina citri bunyavirus (DcBV). DcBV was phylogenetically most closely related to the phasmaviruses based on the analysis of the L segment (Fig. 2D).
Unclassified CBPV-like sequences.
We identified a 1.7-kb contig in the D. citri populations from China and Florida from RNA-seq libraries that displayed 39% amino acid identity to RNA 2 of Chronic bee paralysis virus (CBPV) (Table 2). RT-PCR and Sanger sequencing confirmed and also revealed similar sequences in the populations from California and Texas (see Table S4 in the supplemental material). CBPV is an unclassified single-stranded RNA virus with no close relatives among known sequenced viruses. It infects adult honey bees, resulting in paralysis and death (87). The CBPV genome is comprised of two RNA molecules of 3,674 and 2,305 bases. Both RNA molecules have a 5′ cap structure and are not polyadenylated on the 3′ end (88). A previous phylogenetic study based on the RdRp domains of CBPV suggested an intermediate phylogenetic position for the virus between Nodaviridae and Tombusviridae (87). Although our analysis was unable to identify any sequence related to RNA 1 of CBPV, we detected a contig of 1.1 kb with 33% amino acid sequence identity to the RdRp of tombusviruses (Table 2). Furthermore, the presence of both fragments in the same RNA samples was confirmed by RT-PCR. Therefore, we believe that these two segments belong to a putative novel virus tentatively named Diaphorina citri-associated C virus (DcACV). Phylogenetic analysis based on the RdRp amino acid sequences revealed that DcACV was most closely related to tombusviruses (Fig. 2E).
Bacteriophage-like sequences.
Our approach also generated contigs from both sRNA and RNA-seq libraries prepared from the D. citri samples from China, Taiwan, Brazil, Florida, and Hawaii that displayed 100% identity to nucleotide sequences from Wolbachia phage WO (Table 1; see also Table S2 in the supplemental material). The prokaryotic endosymbiont Wolbachia is present in 66% of all arthropod species (89), and prophage WO is the most widespread bacteriophage, infecting many Wolbachia species (90). To assess the presence of the prophage WO-like sequences in different populations of D. citri, PCR amplifications with primers specific for the minor capsid gene (orf7) (91) were performed, and specific 1-kb amplicons were produced in all populations (data not shown).
DISCUSSION
HLB and its associated natural insect vector, D. citri, are the major threats to the world's citrus industry (52). The goal of the current study was to examine populations of D. citri from both native areas (China and Taiwan) and those where D. citri has more recently emerged (United States and Brazil) through high-throughput NGS of small RNAs and transcriptomes in an attempt to discover putative viruses associated with D. citri. Such metagenomic approaches with similar goals have been successfully implemented to discover highly diverse and novel viruses from field-collected mosquito, bat, and Drosophila fly samples (25, 47, 60, 92–94). A desirable translational outcome of this work would be to identify D. citri-infecting viruses which might have the potential to be used as biological agents to control D. citri and slow the spread of HLB. In this study, we were able to identify and assemble nearly complete genome sequences of several putative novel viruses associated with D. citri, including a picorna-like virus, a reovirus, a densovirus, a bunyavirus, and an unclassified (+) ssRNA virus. We also detected sequences similar to that of Wolbachia phage WO. To trace the identified virus-like sequences in geographically distant D. citri populations, specific primers were designed based on the fragments obtained from bioinformatics analysis and used to screen additional D. citri populations which were not analyzed by NGS, including populations from Brazil, China, Taiwan, Pakistan, and the United States. To the best of our knowledge, this is the first comprehensive high-throughput NGS-based survey of the viral sequences associated with the global population of an agricultural insect pest, and our work demonstrates the success of this approach for field-collected insects.
NGS and bioinformatics analyses alone are very useful for virus identification but yield only viral sequences, some of which may not be representative of viruses actually infecting the host sampled. Therefore, we took several approaches in attempts to assess if the viral sequences identified in this study represented viruses of D. citri. We intentionally chose sRNA NGS because this has proven to be a powerful means to identify active RNA and DNA viruses of both plants and insects (42, 49, 95). Both insects and plants use RNAi as a primary defense against virus infections. RNAi activity results in distinct populations of overlapping sRNAs derived from the RNAi-targeted viral genomes, and in insects, 21-nt sRNAs represent the primary size class (9, 60, 61, 96–98). In our study, the size distribution of sRNAs mapping to the putative viruses had a prominent peak at 21 nt (data not shown), suggesting that these putative viruses may originate from viral infections of D. citri and may be processed by the antiviral RNAi machinery. In a recent study, the presence of virus-derived 21-nt small RNAs was used to support the majority of identified putative viruses in Drosophila melanogaster as bona fide Drosophila infections (60). As a second approach, we used RT-PCR, PCR, and Sanger sequencing to further confirm the origin of our sequences and their identities. When we performed PCR with DNA extracted from D. citri for all of the RNA virus-like sequences identified in this study, no products were amplified. In contrast, RT-PCR did give the expected products, confirming that the sequences represented nonintegrated RNA virus sequences. We did amplify products for DcDNV by both RT-PCR and PCR, but Southern blot hybridization analysis using DNA extracted from a Taiwanese D. citri population and a probe based on our DcDNV sequence showed a single DNA molecule of ∼5 kb, suggesting that the DcDNV sequences were not integrated into the genome of D. citri (data not shown). Finally, our phylogenetic analyses also strongly support the idea that all of the putative viruses reported here are closely related to known insect-specific viruses. Taken together, our cumulative data indicate that all of the putative viruses identified in this study are in fact episomal viruses and do not represent genomic integration events or contamination. Moreover, the fact that similar viral sequences were found in geographically distant D. citri populations collected at different times and from different plants suggests that the viral sequences identified in this study are likely not derived from environmental factors, such as ingested plant material. Finally, the goal of the present study was to utilize metagenomic approaches to identify novel putative viruses associated with D. citri, and in that we were successful. While it is very likely that many viruses of D. citri remain to be discovered, here we have identified four putative RNA viruses and one putative DNA virus that are good candidates for further study.
All of the virus sequences identified here represent new putative viruses. We detected DcPLV in RNA samples from China, Taiwan, and Brazil. The genome organization of DcPLV suggests that it is a new unclassified picorna-like virus which is phylogenetically close to Iflaviridae, a relatively newly recognized insect virus family. The reo-like virus DcRV has been previously described to occur in a subset of the natural D. citri population in Florida, and here we provide additional genome sequence information and also evidence for its incidence in D. citri from Hawaii, China, and Taiwan.
Identification of a densovirus from RNA libraries is not surprising. Detection of DNA viruses via small-RNA and total-RNA sequencing has been previously reported (42, 47, 99). In the case of transcriptome sequencing, viral mRNA transcribed from DNA genomes and even small amounts of DNA have been implicated as explanations for the detection of a DNA virus via RNA sequencing (99). The underlying mechanism for detection of DNA viruses by sequencing of small RNA libraries has been provided by evidence that antisense transcripts with the potential to form dsRNA by base pairing with sense transcripts are produced in different families of DNA viruses, and these could be targeted by RNAi activity (95, 100–102). It is worth mentioning that densoviruses have some features that make them attractive for use as biological control agents (103). In fact, developing genetically modified densoviruses to express genes of interest and their successful application in vitro has been discussed as an alternative approach to control mosquito populations (103–105).
The putative bunyalike virus identified in this study displayed the highest amino acid similarities with newly discovered bunyavirids (18, 106). In 2014, Ballinger et al. discovered the most divergent group of bunyavirids, informally referred to as phasmaviruses, including Kigluaik phantom virus (KIGV) and Nome phantom virus (NOMV) in phantom midges (106). The phasmaviruses shared only 30% amino acid identity with the RdRps of other bunyaviruses (106). One year later, another group (18) sequenced RNAs extracted from 70 arthropod species in China, and they discovered sequences in the mosquito (Wuhan mosquito virus 1), cockroach (Wuchang cockraoch virus 1), and water strider (Gerridae) (Sanxia water strider virus 2) that displayed high amino acid similarities to phasmaviruses. During preparation of the manuscript, two other novel bunyavirids isolated from mosquito cell lines, Jonchet virus (JONV) and Ferak virus (FERV), were described (85). JONV and FERV phylogenetically branch from an old common ancestor in a similar manner to unclassified Phasmaviruses (85). The discovery of this new group of bunyaviruses in distantly related arthropods highlights the importance of these viruses as potential emerging agents.
Two CBPV-related viruses, Anopheline-associated C virus (AACV) and Dansoman virus, have recently been reported from mosquito field populations and Drosophila melanogaster, respectively (25, 60). These two viruses displayed protein sequence identities of <30% to the reference CBPV RNA 2, which is consistent with our observation in this study for DcACV. Thus, these recent discoveries show that the incidence of CBPV-related viruses is not restricted to only honeybees and that this group of viruses probably has a wide distribution.
Metagenomic approaches for virus discovery have been used for a range of organisms, mostly including those of potential medical importance. Compared with the viral communities found among mosquitoes and bats by approaches similar to those used in this study (13, 18, 25, 107, 108), the diversity of viruses found in this agricultural pest insect was much lower. Obviously, host range of the target sampled can play an important role in the diversity of viral sequences detected. Mosquitoes are blood-feeding arthropods and are known to feed on a wide range of sources, including humans, nonhuman primates, other mammals, birds, and even plant nectar (13, 25). As the second most diverse group of mammals, bats are natural reservoirs of many emerging viruses and feed on a diverse array of biota (107, 109, 110). Thus, for bats and mosquitoes, the viral sequences detected through metagenomics likely reflect viruses infecting the host sampled as well as biota on which these hosts feed. It is only recently that metagenomic approaches have been applied to viral discovery in insect pests of plants. For example, a vector-enabled metagenomics study of Bemisia tabaci from a single site in Florida resulted in the detection of mostly plant viruses and some insect virus-like sequences (111). Unlike the animal-feeding targets mentioned above (bats and mosquitoes), D. citri feeds almost exclusively on citrus. Thus, our analyses supporting the idea that the putative viruses described here are active insect viruses suggests that D. citri, rather some other unknown insect, is their likely host.
In summary, by pairing Illumina high-throughput NGS technology with a dedicated bioinformatics workflow, we obtained a snapshot of the viral sequences associated with D. citri populations from various world regions. Additionally, this initial characterization sheds light on the diversity of putative viruses in non-blood-feeding insects and will aid further studies in identifying and finding biotechnological applications for insect viruses. However, the data presented here likely do not depict the total diversity of viral sequences in D. citri. Sample collection was a limitation in this study. Samples were collected in different geographic locations and at different times and probably reflect different ages of D. citri. We also do not know enough about the genetic diversity among the D. citri populations studied here, and it has been demonstrated that the viromes of mosquitoes and bats vary by species, age, space, time, and sample type (13, 107).
Supplementary Material
ACKNOWLEDGMENTS
We thank our colleagues who provided us D. citri RNA: W. O. Dawson, H. Wuriyanghan, H.-Y Yeh, Y. Cen, A. M. Khan, M. A. Machado, T. da Silva, D. M. Galdeano, D. Jenkins, C. Higashi, A. Chow, D. Morgan, K. Pelz-Stelinski, and K. Godfrey. We thank Raj Nandety for helping with the Velvet program. We also thank T. Nguyen, D. J. Coyle, and M. Wohfeil for lab assistance. We thank Massimo Turina for critical review of the manuscript. We thank the reviewers of the manuscript for their constructive and helpful comments.
This project was supported by the awards 13-002NU-781 and 2015-70016-23011 from the U.S. Department of Agriculture (USDA).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02793-15.
REFERENCES
- 1.Weitz JS, Wilhelm SW. 2012. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol Rep 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman A. 2009. Numbers of living species in Australia and the world. Australian Biodiversity Information Services, Toowoomba, Australia. [Google Scholar]
- 3.Asgari S, Karyan N. 2010. Insect virology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 4.Chiu CY. 2013. Viral pathogen discovery. Curr Opin Microbiol 16:468–478. doi: 10.1016/j.mib.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipkin WI, Firth C. 2013. Viral surveillance and discovery. Curr Opin Virol 3:199–204. doi: 10.1016/j.coviro.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. 2008. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med 358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- 7.Towner JS, Sealy TK, Khristova ML, Albarino CG, Conlan S, Reeder SA, Quan PL, Lipkin WI, Downing R, Tappero JW, Okware S, Lutwama J, Bakamutumaho B, Kayiwa J, Comer JA, Rollin PE, Ksiazek TG, Nichol ST. 2008. Newly discovered ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog 4:e1000212. doi: 10.1371/journal.ppat.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zirkel F, Kurth A, Quan PL, Briese T, Ellerbrok H, Pauli G, Leendertz FH, Lipkin WI, Ziebuhr J, Drosten C, Junglen S. 2011. An insect nidovirus emerging from a primary tropical rainforest. mBio 2:e00077-11. doi: 10.1128/mBio.00077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li WX, Ding SW. 2010. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci U S A 107:1606–1611. doi: 10.1073/pnas.0911353107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasilakis N, Forrester NL, Palacios G, Nasar F, Savji N, Rossi SL, Guzman H, Wood TG, Popov V, Gorchakov R, Gonzalez AV, Haddow AD, Watts DM, da Rosa AP, Weaver SC, Lipkin WI, Tesh RB. 2013. Negevirus: a proposed new taxon of insect-specific viruses with wide geographic distribution. J Virol 87:2475–2488. doi: 10.1128/JVI.00776-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sparks ME, Gundersen-Rindal DE, Harrison RL. 2013. Complete genome sequence of a novel iflavirus from the transcriptome of Halyomorpha halys, the brown marmorated stink bug. Genome Announc 1(6):e00910-13. doi: 10.1128/genomeA.00910-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith G, Macias-Munoz A, Briscoe AD. 2014. Genome sequence of a novel iflavirus from mRNA sequencing of the butterfly Heliconius erato. Genome Announc 2(3):e00398-14. doi: 10.1128/genomeA.00398-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng TF, Willner DL, Lim YW, Schmieder R, Chau B, Nilsson C, Anthony S, Ruan Y, Rohwer F, Breitbart M. 2011. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS One 6:e20579. doi: 10.1371/journal.pone.0020579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murakami R, Suetsugu Y, Nakashima N. 2014. Complete genome sequences of two iflaviruses from the brown planthopper, Nilaparvata lugens. Arch Virol 159:585–588. doi: 10.1007/s00705-013-1850-0. [DOI] [PubMed] [Google Scholar]
- 15.Murakami R, Suetsugu Y, Kobayashi T, Nakashima N. 2013. The genome sequence and transmission of an iflavirus from the brown planthopper, Nilaparvata lugens. Virus Res 176:179–187. doi: 10.1016/j.virusres.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Millan-Leiva A, Jakubowska AK, Ferre J, Herrero S. 2012. Genome sequence of SeIV-1, a novel virus from the Iflaviridae family infective to Spodoptera exigua. J Invertebr Pathol 109:127–133. doi: 10.1016/j.jip.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Liu S, Vijayendran D, Carrillo-Tripp J, Miller WA, Bonning BC. 2014. Analysis of new aphid lethal paralysis virus (ALPV) isolates suggests evolution of two ALPV species. J Gen Virol 95:2809–2819. doi: 10.1099/vir.0.069765-0. [DOI] [PubMed] [Google Scholar]
- 18.Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, Qin XC, Xu J, Holmes EC, Zhang YZ. 2015. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 4:e05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Junglen S, Drosten C. 2013. Virus discovery and recent insights into virus diversity in arthropods. Curr Opin Microbiol 16:507–513. doi: 10.1016/j.mib.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson H, Dhaygude K, Lindstrom S, Helantera H, Sundstrom L, Trontti K. 2013. A metatranscriptomic approach to the identification of microbiota associated with the ant Formica exsecta. PLoS One 8:e79777. doi: 10.1371/journal.pone.0079777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geng P, Li W, Lin L, de Miranda JR, Emrich S, An L, Terenius O. 2014. Genetic characterization of a novel iflavirus associated with vomiting disease in the Chinese oak silkmoth Antheraea pernyi. PLoS One 9:e92107. doi: 10.1371/journal.pone.0092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delwart EL. 2007. Viral metagenomics. Rev Med Virol 17:115–131. doi: 10.1002/rmv.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dayaram A, Galatowitsch M, Harding JS, Arguello-Astorga GR, Varsani A. 2014. Novel circular DNA viruses identified in Procordulia grayi and Xanthocnemis zealandica larvae using metagenomic approaches. Infect Genet Evol 22:134–141. doi: 10.1016/j.meegid.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 24.Cox-Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA, Quan PL, Briese T, Hornig M, Geiser DM, Martinson V, vanEngelsdorp D, Kalkstein AL, Drysdale A, Hui J, Zhai J, Cui L, Hutchison SK, Simons JF, Egholm M, Pettis JS, Lipkin WI. 2007. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- 25.Cook S, Chung BY, Bass D, Moureau G, Tang S, McAlister E, Culverwell CL, Glucksman E, Wang H, Brown TD, Gould EA, Harbach RE, de Lamballerie X, Firth AE. 2013. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS One 8:e80720. doi: 10.1371/journal.pone.0080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi JY, Kim Y-S, Wang Y, Shin SW, Kim I, Tao XY, Liu Q, Roh JY, Kim JS, Je YH. 2012. Complete genome sequence of a novel picorna-like virus isolated from Spodoptera exigua. J Asia Pac Entomol 15:259–263. doi: 10.1016/j.aspen.2012.01.006. [DOI] [Google Scholar]
- 27.Chen YF, Gao F, Ye XQ, Wei SJ, Shi M, Zheng HJ, Chen XX. 2011. Deep sequencing of Cotesia vestalis bracovirus reveals the complexity of a polydnavirus genome. Virology 414:42–50. doi: 10.1016/j.virol.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Liu S, Bonning BC. 2015. Genome sequence of a novel iflavirus from the leafhopper Graminella nigrifrons. Genome announcements 3(2):e00323-15. doi: 10.1128/genomeA.00323-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrillo-Tripp J, Krueger EN, Harrison RL, Toth AL, Miller WA, Bonning BC. 2014. Lymantria dispar iflavirus 1 (LdIV1), a new model to study iflaviral persistence in lepidopterans. J Gen Virol 95:2285–2296. doi: 10.1099/vir.0.067710-0. [DOI] [PubMed] [Google Scholar]
- 30.Thekke-Veetil T, Sabanadzovic S, Keller KE, Martin RR, Tzanetakis IE. 2012. Genome organization and sequence diversity of a novel blackberry Ampelovirus, abstr 191. In Proc 22nd Int Conf Virus Other Transmissible Dis Fruit Crops, Rome, Italy. [Google Scholar]
- 31.Mumford R, Adams IP, Glover R, Hany U, Boonham N. 2011. From high throughput to high output clondiag microarrays and 454 sequence for viral detection and discovery, abstr 17. In Proc BARD-Sponsored Workshop—Microarrays Next-Generation Sequencing Detect Identification Plant Viruses, Beltsville, MD. [Google Scholar]
- 32.Dombrovsky A, Glanz E, Sapkota R, Lachman O, Bronstein M, schnitzer T, Antignus Y. 2011. Next-generation sequencing a rapid and reliable method to obtain sequence data of the genomes of undescribed plant viruses, p17–19. In Proc BARD-Sponsored Workshop—Microarrays Next-Generation Sequencing Detect Identification Plant Viruses, Beltsville, MD. [Google Scholar]
- 33.Chiumenti M, Roberto R, Bottalico G, Campanale A, de Stradis A, Minafra A, Boscia D, Savino V, Martelli GP. 2012. Virus sanitation and deep sequence analysis of fig, p 284 In Proc 22nd Int Conf Virus Other Transmissible Dis Fruit Crops, Rome, Italy. [Google Scholar]
- 34.Candresse T, Marais A, Faure C, Carriere S, Gentit P. 2012. Use of 454 pyrosequencing for the fast and efficient characterization of known or novel viral agents in Prunus materials, p 282 In Proc 22nd Int Conf Virus Other Transmissible Dis Fruit Crops, Rome, Italy. [Google Scholar]
- 35.Vives MC, Velazquez K, Pina JA, Moreno P, Guerri J, Navarro L. 2013. Identification of a new enamovirus associated with citrus vein enation disease by deep sequencing of small RNAs. Phytopathology 103:1077–1086. doi: 10.1094/PHYTO-03-13-0068-R. [DOI] [PubMed] [Google Scholar]
- 36.Roy A, Choudhary N, Guillermo LM, Shao J, Govindarajulu A, Achor D, Wei G, Picton DD, Levy L, Nakhla MK, Hartung JS, Brlansky RH. 2013. A novel virus of the genus Cilevirus causing symptoms similar to citrus leprosis. Phytopathology 103:488–500. doi: 10.1094/PHYTO-07-12-0177-R. [DOI] [PubMed] [Google Scholar]
- 37.Roossinck MJ, Saha P, Wiley GB, Quan J, White JD, Lai H, Chavarria F, Shen G, Roe BA. 2010. Ecogenomics: using massively parallel pyrosequencing to understand virus ecology. Mol Ecol 19(Suppl 1):S81–S88. [DOI] [PubMed] [Google Scholar]
- 38.Quito-Avila DF, Jelkmann W, Tzanetakis IE, Keller K, Martin RR. 2011. Complete sequence and genetic characterization of Raspberry latent virus, a novel member of the family Reoviridae. Virus Res 155:397–405. doi: 10.1016/j.virusres.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Pallett DW, Ho T, Cooper I, Wang H. 2010. Detection of Cereal yellow dwarf virus using small interfering RNAs and enhanced infection rate with Cocksfoot streak virus in wild cocksfoot grass (Dactylis glomerata). J Virol Methods 168:223–227. doi: 10.1016/j.jviromet.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Loconsole G, Saldarelli P, Doddapaneni H, Savino V, Martelli GP, Saponari M. 2012. Identification of a single-stranded DNA virus associated with citrus chlorotic dwarf disease, a new member in the family Geminiviridae. Virology 432:162–172. doi: 10.1016/j.virol.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Li R, Gao S, Hernandez AG, Wechter WP, Fei Z, Ling KS. 2012. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS One 7:e37127. doi: 10.1371/journal.pone.0037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R. 2009. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology 388:1–7. doi: 10.1016/j.virol.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 43.Hany U, Adams IP, Glover R, Bhat AI, Boonham N. 2014. The complete genome sequence of Piper yellow mottle virus (PYMoV). Arch Virol 159:385–388. doi: 10.1007/s00705-013-1824-2. [DOI] [PubMed] [Google Scholar]
- 44.Barba M, Czosnek H, Hadidi A. 2014. Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses 6:106–136. doi: 10.3390/v6010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams IP, Glover RH, Monger WA, Mumford R, Jackeviciene E, Navalinskiene M, Samuitiene M, Boonham N. 2009. Next-generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol Plant Pathol 10:537–545. doi: 10.1111/j.1364-3703.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, Vijayendran D, Bonning BC. 2011. Next generation sequencing technologies for insect virus discovery. Viruses 3:1849–1869. doi: 10.3390/v3101849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma M, Huang Y, Gong Z, Zhuang L, Li C, Yang H, Tong Y, Liu W, Cao W. 2011. Discovery of DNA viruses in wild-caught mosquitoes using small RNA high throughput sequencing. PLoS One 6:e24758. doi: 10.1371/journal.pone.0024758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nandety RS, Fofanov VY, Koshinsky H, Stenger DC, Falk BW. 2013. Small RNA populations for two unrelated viruses exhibit different biases in strand polarity and proximity to terminal sequences in the insect host Homalodisca vitripennis. Virology 442:12–19. doi: 10.1016/j.virol.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Parameswaran P, Sklan E, Wilkins C, Burgon T, Samuel MA, Lu R, Ansel KM, Heissmeyer V, Einav S, Jackson W, Doukas T, Paranjape S, Polacek C, dos Santos FB, Jalili R, Babrzadeh F, Gharizadeh B, Grimm D, Kay M, Koike S, Sarnow P, Ronaghi M, Ding SW, Harris E, Chow M, Diamond MS, Kirkegaard K, Glenn JS, Fire AZ. 2010. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog 6:e1000764. doi: 10.1371/journal.ppat.1000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Munster M, Dullemans AM, Verbeek M, van den Heuvel JF, Reinbold C, Brault V, Clerivet A, van der Wilk F. 2003. Characterization of a new densovirus infecting the green peach aphid Myzus persicae. J Invertebr Pathol 84:6–14. doi: 10.1016/S0022-2011(03)00013-2. [DOI] [PubMed] [Google Scholar]
- 51.Grafton-Cardwell EE, Stelinski LL, Stansly PA. 2013. Biology and management of Asian citrus psyllid, vector of the Huanglongbing pathogens. Annu Rev Entomol 58:413–432. doi: 10.1146/annurev-ento-120811-153542. [DOI] [PubMed] [Google Scholar]
- 52.Da Graca JV. 2013. Etiology, history, worldwide situation of Huanglongbing. 3rd Int Citrus Quarantine Pest Workshop, Manzanillo, Mexico. [Google Scholar]
- 53.OAR. 1919. Disease of economic plants in southern China. Philipp Agric 8:109–135. [Google Scholar]
- 54.Coletta-Filho HD, Targon MLPN, Takita MA, De Negri JD, Pompeu J Jr, Machado MA. 2004. First report of the causal agent of Huanglongbing (“Candidatus Liberibacter asiaticus”) in Brazil. Plant Dis 88:1,382.3. [DOI] [PubMed] [Google Scholar]
- 55.Sutton B, Duan YP, Halbert S, Sun XA, Schubert T, Dixon W. 2005. Detection and identification of citrus huanglongbing (greening) in Florida, USA, workshop H11. Second Int Citrus Canker Huanglongbing Res Workshop, Orlando, FL. [Google Scholar]
- 56.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu S, Chen Y, Bonning BC. 2015. RNA virus discovery in insects. Curr Opin Insect Sci 8:54–61. doi: 10.1016/j.cois.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 58.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 59.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Webster CL, Waldron FM, Robertson S, Crowson D, Ferrari G, Quintana JF, Brouqui JM, Bayne EH, Longdon B, Buck AH, Lazzaro BP, Akorli J, Haddrill PR, Obbard DJ. 2015. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLoS Biol 13:e1002210. doi: 10.1371/journal.pbio.1002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aguiar ER, Olmo RP, Paro S, Ferreira FV, de Faria IJ, Todjro YM, Lobo FP, Kroon EG, Meignin C, Gatherer D, Imler JL, Marques JT. 2015. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res 43:6191–6206. doi: 10.1093/nar/gkv587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koonin EV, Wolf YI, Nagasaki K, Dolja VV. 2008. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat Rev Microbiol 6:925–939. doi: 10.1038/nrmicro2030. [DOI] [PubMed] [Google Scholar]
- 63.Banerjee R, Weidman MK, Echeverri A, Kundu P, Dasgupta A. 2004. Regulation of poliovirus 3C protease by the 2C polypeptide. J Virol 78:9243–9256. doi: 10.1128/JVI.78.17.9243-9256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le Gall O, Christian P, Fauquet CM, King AM, Knowles NJ, Nakashima N, Stanway G, Gorbalenya AE. 2008. Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T = 3 virion architecture. Arch Virol 153:715–727. doi: 10.1007/s00705-008-0041-x. [DOI] [PubMed] [Google Scholar]
- 65.Paul A. 2002. Possible unifying mechanism of picornavirus genome replication, p 227–246. In Semler B, Wimmer E (ed), Molecular biology of picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 66.Berman JJ. 2012. Taxonomic guide to infectious diseases: understanding the biologic classes of pathogenic organisms. Academic Press, Amsterdam, The Netherlands. [Google Scholar]
- 67.Perera OP, Snodgrass GL, Allen KC, Jackson RE, Becnel JJ, O'Leary PF, Luttrell RG. 2012. The complete genome sequence of a single-stranded RNA virus from the tarnished plant bug, Lygus lineolaris (Palisot de Beauvois). J Invertebr Pathol 109:11–19. doi: 10.1016/j.jip.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 68.Saqib M, Wylie SJ, Jones MGK. 2015. Serendipitous identification of a new Iflavirus-like virus infecting tomato and its subsequent characterization. Plant Pathol 64:519–527. doi: 10.1111/ppa.12293. [DOI] [Google Scholar]
- 69.King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed). 2012. Virus taxonomy. Classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 70.Oliveira DC, Hunter WB, Ng J, Desjardins CA, Dang PM, Werren JH. 2010. Data mining cDNAs reveals three new single stranded RNA viruses in Nasonia (Hymenoptera: Pteromalidae). Insect Mol Biol 19(Suppl 1):S99–S107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ryabov EV. 2007. A novel virus isolated from the aphid Brevicoryne brassicae with similarity to Hymenoptera picorna-like viruses. J Gen Virol 88:2590–2595. doi: 10.1099/vir.0.83050-0. [DOI] [PubMed] [Google Scholar]
- 72.Lang AS, Culley AI, Suttle CA. 2004. Genome sequence and characterization of a virus (HaRNAV) related to picorna-like viruses that infects the marine toxic bloom-forming alga Heterosigma akashiwo. Virology 320:206–217. doi: 10.1016/j.virol.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 73.Tai V, Lawrence JE, Lang AS, Chan AM, Culley AI, Suttle CA. 2003. Characterization of HaRNAV, a single-stranded RNA virus causing lysis of Heterosigma akashiwo (Raphidophyceae). J Phycol 39:343–352. doi: 10.1046/j.1529-8817.2003.01162.x. [DOI] [Google Scholar]
- 74.Miura KI, Kimura I, Suzuki N. 1966. Double-stranded ribonucleic acid from rice dwarf virus. Virology 28:571–579. doi: 10.1016/0042-6822(66)90242-X. [DOI] [PubMed] [Google Scholar]
- 75.Asamizu T, Summers D, Motika MB, Anzola JV, Nuss DL. 1985. Molecular cloning and characterization of the genome of wound tumor virus: a tumor-inducing plant reovirus. Virology 144:398–409. doi: 10.1016/0042-6822(85)90281-8. [DOI] [PubMed] [Google Scholar]
- 76.Isogai M, Uyeda I, Lindsten K. 1998. Taxonomic characteristics of fijiviruses based on nucleotide sequences of the oat sterile dwarf virus genome. J Gen Virol 79(Part 6):1479–1485. [DOI] [PubMed] [Google Scholar]
- 77.Marzachí C, Boccardo G, Nuss DL. 1991. Cloning of the maize rough dwarf virus genome: molecular confirmation of the plant-reovirus classification scheme and identification of two large nonoverlapping coding domains within a single genomic segment. Virology 180:518–526. doi: 10.1016/0042-6822(91)90065-J. [DOI] [PubMed] [Google Scholar]
- 78.Yan J, Kudo H, Uyeda I, Lee SY, Shikata E. 1992. Conserved terminal sequences of rice ragged stunt virus genomic RNA. J Gen Virol 73(Part 4):785–789. [DOI] [PubMed] [Google Scholar]
- 79.Mizuri Marutani-Hert WBH, Catherine Katsar S, Xiomara Sinisterra H, David Hall G, Charles Powell A. 2009. Reovirus-like sequences isolated from adult asian citrus psyllid, (Hemiptera: Psyllidae: Diaphorina citri). Fla Entomol 92:314–320. doi: 10.1653/024.092.0216. [DOI] [Google Scholar]
- 80.Noda H, Ishikawa K, Hibino H, Omura T. 1991. A reovirus in the brown planthopper, Nilaparvata lugens. J Gen Virol 72(Part 10):2425–2430. [DOI] [PubMed] [Google Scholar]
- 81.Spear A, Sisterson MS, Stenger DC. 2012. Reovirus genomes from plant-feeding insects represent a newly discovered lineage within the family Reoviridae. Virus Res 163:503–511. doi: 10.1016/j.virusres.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 82.Bergoin M, Tijssen P. 2000. Molecular biology of Densovirinae. Contrib Microbiol 4:12–32. doi: 10.1159/000060329. [DOI] [PubMed] [Google Scholar]
- 83.Shike H, Dhar AK, Burns JC, Shimizu C, Jousset FX, Klimpel KR, Bergoin M. 2000. Infectious hypodermal and hematopoietic necrosis virus of shrimp is related to mosquito brevidensoviruses. Virology 277:167–177. doi: 10.1006/viro.2000.0589. [DOI] [PubMed] [Google Scholar]
- 84.Xu P, Liu Y, Graham RI, Wilson K, Wu K. 2014. Densovirus is a mutualistic symbiont of a global crop pest (Helicoverpa armigera) and protects against a baculovirus and Bt biopesticide. PLoS Pathog 10:e1004490. doi: 10.1371/journal.ppat.1004490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marklewitz M, Zirkel F, Kurth A, Drosten C, Junglen S. 2015. Evolutionary and phenotypic analysis of live virus isolates suggests arthropod origin of a pathogenic RNA virus family. Proc Natl Acad Sci U S A 112:7536–7541. doi: 10.1073/pnas.1502036112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walter CT, Barr JN. 2011. Recent advances in the molecular and cellular biology of bunyaviruses. J Gen Virol 92:2467–2484. doi: 10.1099/vir.0.035105-0. [DOI] [PubMed] [Google Scholar]
- 87.Ribière M, Olivier V, Blanchard P. 2010. Chronic bee paralysis: a disease and a virus like no other? J Invertebr Pathol 103(Suppl 1):S120–S131. [DOI] [PubMed] [Google Scholar]
- 88.Olivier V, Blanchard P, Chaouch S, Lallemand P, Schurr F, Celle O, Dubois E, Tordo N, Thiery R, Houlgatte R, Ribiere M. 2008. Molecular characterisation and phylogenetic analysis of chronic bee paralysis virus, a honey bee virus. Virus Res 132:59–68. doi: 10.1016/j.virusres.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 89.Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren JH. 2008. How many species are infected with Wolbachia? A statistical analysis of current data. FEMS Microbiol Lett 281:215–220. doi: 10.1111/j.1574-6968.2008.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gavotte L, Henri H, Stouthamer R, Charif D, Charlat S, Bouletreau M, Vavre F. 2007. A survey of the bacteriophage WO in the endosymbiotic bacteria Wolbachia. Mol Biol Evol 24:427–435. [DOI] [PubMed] [Google Scholar]
- 91.Masui S, Kamoda S, Sasaki T, Ishikawa H. 2000. Distribution and evolution of bacteriophage WO in Wolbachia, the endosymbiont causing sexual alterations in arthropods. J Mol Evol 51:491–497. [DOI] [PubMed] [Google Scholar]
- 92.Wang J, Moore NE, Murray ZL, McInnes K, White DJ, Tompkins DM, Hall RJ. 2015. Discovery of novel virus sequences in an isolated and threatened bat species, the New Zealand lesser short-tailed bat (Mystacina tuberculata). J Gen Virol 96:2442–2452. doi: 10.1099/vir.0.000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Donaldson EF, Haskew AN, Gates JE, Huynh J, Moore CJ, Frieman MB. 2010. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J Virol 84:13004–13018. doi: 10.1128/JVI.01255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coffey LL, Page BL, Greninger AL, Herring BL, Russell RC, Doggett SL, Haniotis J, Wang C, Deng X, Delwart EL. 2014. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 448:146–158. doi: 10.1016/j.virol.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bronkhorst AW, van Cleef KW, Vodovar N, Ince IA, Blanc H, Vlak JM, Saleh MC, van Rij RP. 2012. The DNA virus Invertebrate iridescent virus 6 is a target of the Drosophila RNAi machinery. Proc Natl Acad Sci U S A 109:E3604–E3613. doi: 10.1073/pnas.1207213109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mueller S, Gausson V, Vodovar N, Deddouche S, Troxler L, Perot J, Pfeffer S, Hoffmann JA, Saleh MC, Imler JL. 2010. RNAi-mediated immunity provides strong protection against the negative-strand RNA vesicular stomatitis virus in Drosophila. Proc Natl Acad Sci U S A 107:19390–19395. doi: 10.1073/pnas.1014378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marques JT, Wang JP, Wang X, de Oliveira KP, Gao C, Aguiar ER, Jafari N, Carthew RW. 2013. Functional specialization of the small interfering RNA pathway in response to virus infection. PLoS Pathog 9:e1003579. doi: 10.1371/journal.ppat.1003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kemp C, Mueller S, Goto A, Barbier V, Paro S, Bonnay F, Dostert C, Troxler L, Hetru C, Meignin C, Pfeffer S, Hoffmann JA, Imler JL. 2013. Broad RNA interference-mediated antiviral immunity and virus-specific inducible responses in Drosophila. J Immunol 190:650–658. doi: 10.4049/jimmunol.1102486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dacheux L, Cervantes-Gonzalez M, Guigon G, Thiberge JM, Vandenbogaert M, Maufrais C, Caro V, Bourhy H. 2014. A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses. PLoS One 9:e87194. doi: 10.1371/journal.pone.0087194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lin YT, Kincaid RP, Arasappan D, Dowd SE, Hunicke-Smith SP, Sullivan CS. 2010. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA 16:1540–1558. doi: 10.1261/rna.1967910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Akbergenov R, Si-Ammour A, Blevins T, Amin I, Kutter C, Vanderschuren H, Zhang P, Gruissem W, Meins F Jr, Hohn T, Pooggin MM. 2006. Molecular characterization of geminivirus-derived small RNAs in different plant species. Nucleic Acids Res 34:462–471. doi: 10.1093/nar/gkj447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carlson J, Suchman E, Buchatsky L. 2006. Densoviruses for control and genetic manipulation of mosquitoes. Adv Virus Res 68:361–392. doi: 10.1016/S0065-3527(06)68010-X. [DOI] [PubMed] [Google Scholar]
- 104.Gu JB, Dong YQ, Peng HJ, Chen XG. 2010. A recombinant AeDNA containing the insect-specific toxin, BmK IT1, displayed an increasing pathogenicity on Aedes albopictus. Am J Trop Med Hyg 83:614–623. doi: 10.4269/ajtmh.2010.10-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Suzuki Y, Niu G, Hughes GL, Rasgon JL. 2014. A viral over-expression system for the major malaria mosquito Anopheles gambiae. Sci Rep 4:5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ballinger MJ, Bruenn JA, Hay J, Czechowski D, Taylor DJ. 2014. Discovery and evolution of bunyavirids in arctic phantom midges and ancient bunyavirid-like sequences in insect genomes. J Virol 88:8783–8794. doi: 10.1128/JVI.00531-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ge X, Li Y, Yang X, Zhang H, Zhou P, Zhang Y, Shi Z. 2012. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol 86:4620–4630. doi: 10.1128/JVI.06671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, Delwart E. 2010. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol 84:6955–6965. doi: 10.1128/JVI.00501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. 2006. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev 19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shi Z. 2010. Bat and virus. Protein Cell 1:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rosario K, Capobianco H, Ng TF, Breitbart M, Polston JE. 2014. RNA viral metagenome of whiteflies leads to the discovery and characterization of a whitefly-transmitted carlavirus in North America. PLoS One 9:e86748. doi: 10.1371/journal.pone.0086748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.