

ABSTRACT
Gap junctions (GJs) form intercellular channels which directly connect the cytoplasm between neighboring cells to facilitate the transfer of ions and small molecules. GJs play a major role in the pathogenesis of infection-associated inflammation. Mutations of gap junction proteins, connexins (Cxs), cause dysmyelination and leukoencephalopathy. In multiple sclerosis (MS) patients and its animal model experimental autoimmune encephalitis (EAE), Cx43 was shown to be modulated in the central nervous system (CNS). The mechanism behind Cx43 alteration and its role in MS remains unexplored. Mouse hepatitis virus (MHV) infection-induced demyelination is one of the best-studied experimental animal models for MS. Our studies demonstrated that MHV infection downregulated Cx43 expression at protein and mRNA levels in vitro in primary astrocytes obtained from neonatal mouse brains. After infection, a significant amount of Cx43 was retained in endoplasmic reticulum/endoplasmic reticulum Golgi intermediate complex (ER/ERGIC) and GJ plaque formation was impaired at the cell surface, as evidenced by a reduction of the Triton X-100 insoluble fraction of Cx43. Altered trafficking and impairment of GJ plaque formation may cause the loss of functional channel formation in MHV-infected primary astrocytes, as demonstrated by a reduced number of dye-coupled cells after a scrape-loading Lucifer yellow dye transfer assay. Upon MHV infection, a significant downregulation of Cx43 was observed in the virus-infected mouse brain. This study demonstrates that astrocytic Cx43 expression and function can be modulated due to virus stress and can be an appropriate model to understand the basis of cellular mechanisms involved in the alteration of gap junction intercellular communication (GJIC) in CNS neuroinflammation.
IMPORTANCE We found that MHV infection leads to the downregulation of Cx43 in vivo in the CNS. In addition, results show that MHV infection impairs Cx43 expression in addition to gap junction communication in primary astrocytes. After infection, Cx43 did not traffic normally to the membrane to form gap junction plaques, and that could be the basis of reduced functional gap junction coupling between astrocytes. This is an important first step toward understanding how viruses affect Cx43 expression and trafficking at the cellular level. This may provide a basis for understanding how structural alterations of astrocytic gap junctions can disrupt gap junction communication between other CNS cells in altered CNS environments due to infection and inflammation. More specifically, alteration of Cx43 may be the basis of the destabilization of Cx47 in oligodendrocytes seen in and around inflammatory demyelinating plaques in MS patients.
INTRODUCTION
Gap junction communication (GJC) is one of the pivotal requirements in all vertebrates for maintaining cellular homeostasis (1). Gap junctions span the plasma membranes of closely opposed cells to align end to end, forming intercellular channels which allow the exchange of small molecules (less than 1 kDa). They are composed of two hemichannels contributed by two opposing cells. These hemichannels or connexons are formed by six protein subunits, called connexins (Cx). Connexin proteins consist of an intracellular N terminus, four transmembrane domains, two extracellular loops, one cytoplasmic loop, and an intracellular C terminus (2). In the central nervous system (CNS), the major cell type which is coupled by gap junctions is the astrocyte. Astrocytes also form gap junction channels between other neuronal cells to form panglial networks to provide metabolic support and maintain homeostasis in the CNS.
Astrocytes, being the most abundant cells in this panglial system, perform essential metabolic functions by maintaining the balance of fluid, ions, pH, and some neurotransmitters in the CNS (3). Enormous amounts of evidence have proven astrocytes to be the cell population providing metabolic support and trophic support to neurons (4) and oligodendrocytes (5, 6). Astrocytes are well connected through GJCs, forming a glial network which bears an essential role in maintaining metabolic homeostasis and osmotic balance. Astrocytes mainly express the GJ proteins Cx43 and Cx30 (7), of which Cx43 is the more prevalent astrocytic connexin both in vivo and in vitro (8). Cx43 is the most abundant gap junction protein in the brain, where it is found primarily between astrocytes (9), but it also forms a substantial amount of GJCs between oligodendrocytes in combination with Cx47, playing an important role in oligodendrocyte K+ buffering and small metabolite exchange (10).
GJCs allow the passage of small molecules, e.g., glucose and its derivatives (11), and second messengers, such as inositol 1,4,5-trisphosphate, cyclic AMP (cAMP), and Ca2+ (12–15). Apart from exchanging small molecules, connexins interact with a myriad of proteins, including cytoskeletal elements, enzymes, and signaling molecules, through their cytoplasmic domain (16). One report has shown the involvement of astrocytic hemichannels in intercellular Ca2+ signaling via ATP release (17). Although a lot is known about Cx43 coupling in CNS cells, its implication in neurological diseases was not studied very well until the early 1990s. Recent studies demonstrated that functional coupling between astrocytes, as well as in panglial connections, is altered in pathological conditions, more specifically in neurodegenerative disorders. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson's disease results in an immediate yet transient increase in Cx43 in mice but induces no change in functional dye coupling between astrocytes (18). Also, Cx43 is increased in amyloid plaques of Alzheimer's disease patients (19). The infiltration of these amyloid plaques by astrocytic processes might be a cause of elevated local concentrations of Cx43. In contrast, the expression of Cx43 is known to be downregulated in high-grade (grade III and grade IV) human gliomas, which might be related to tumor development (20). Cx43 mutation causes severe pathological conditions, including a dysmyelinating phenotype observed in human oculodentodigital dysplasia (ODDD) (21).
Astrocytes obtained from whole brains of late-gestation litters from heterozygous mice containing a Cx43 null mutation exhibit a reduction in gap junction coupling (8). Interestingly, under such conditions, Cx43 knockout astrocytes can express other types of connexins, like Cx30, Cx40, and Cx45 (22); however, these connexins cannot compensate for Cx43 reduction in astrocytes. Astrocyte-targeted deletion of Cx43 and Cx30 leads to a dysmyelinating phenotype in mice (10). Changes in astrocytic structure and morphology are one of the characteristic features of human MS. In autopsy brains of MS patients, numerous glial fibrillary acidic protein-positive (GFAP+) large reactive astrocytes are observed to be preserved in perivascular areas and around demyelinating plaques, whereas astrocytes are found to be significantly reduced inside necrotic lesions (23). Cx43 is downregulated in the spinal cord white matter of EAE mice (24); however, the precise mechanism of downregulation is poorly understood and demands further investigation.
Recent studies found that in addition to neurodegenerative conditions, neuroinfections also can lead to the alteration of Cx43 expression in vivo. For example, persistent Borna disease virus (BDV) infection led to a global reduction of Cx43 mRNA and protein in infected rats. Although Cx43 reduction was observed in forebrain and hippocampal CA3 regions, Cx43 levels increased in areas associated with the dentate gyrus (25). Another report showed that the E8 protein of bovine papillomavirus type 4 induces the loss of GJIC between primary fibroblasts, but the involvement of specific connexins was not shown (26). Rous sarcoma virus (RSV)-induced transformation of mammalian fibroblasts also disrupts Cx43-mediated gap junction communication (27), and prenatal human influenza virus infection in mice induces the downregulation of Cx43 in neocortex and cerebellum (28). HIV infection of astrocytes induces the opening of Cx43 hemichannels, which results in the dysregulated secretion of dickkopf-1 protein, a soluble inhibitor of Wnt signaling (29). Thus far, these types of studies were restricted mainly to describing either the alteration of expression levels in vivo or alteration of the function of gap junction channels in vitro.
In this context, our study was designed to understand the cellular mechanism of mouse hepatitis virus (MHV)-induced alteration of Cx43 expression and functional channel formation both in vitro in primary astrocytes and in vivo in the CNS of MHV-infected mice.
MHV-A59, a neurotropic strain of coronavirus, causes meningoencephalitis, demyelination, and concurrent axonal loss (30, 31) which mimics certain pathologies of the human CNS demyelinating disease multiple sclerosis (MS). MHV-induced acute neuroinflammation and chronic demyelination in mice is an excellent animal model to study the regulation of Cx43 in astrocytes, as astrocytes are known to get infected by this virus both in vitro and in vivo (32, 33).
In vitro and in vivo studies demonstrated that there was a downregulation of Cx43 expression both at RNA and protein levels. In addition, MHV-A59 infection induced the retention of Cx43 in ER/ERGIC and affected Cx43-mediated GJIC, which might be due to altered Cx43 trafficking. To our knowledge, this is the first report of MHV-induced remodeling of Cx43 expression and induced alteration of GJIC. Thus, the alteration of expression of Cx43 in astrocytes might play a role in MHV-induced neuroinflammation.
MATERIALS AND METHODS
Preparation of mixed glia cultures.
Primary cultures of mixed glia from newborn mice (day 0 to 1) were prepared as described previously (34), with minor modifications. Briefly, after the removal of meninges, brain tissues were minced and incubated in a rocking water bath at 37°C for 30 min in Hanks' balanced salt solution (HBSS; Gibco) in the presence of 300 μg/ml DNase I (Sigma) and 0.25% trypsin (Sigma). Enzyme-dissociated cells were triturated with 0.25% of fetal bovine serum (FBS), followed by a wash and centrifugation (300 × g for 10 min). The pellet was resuspended in HBSS and passed through a 70-μm nylon mesh, followed by a second wash and centrifugation (300 × g for 10 min), and then diluted with astrocyte-specific medium (Dulbecco's modified eagle medium containing 1% penicillin-streptomycin, 1% l-glutamine, and 10% FBS). Cells were plated and allowed to adhere for 1 day in a humidified CO2 incubator at 37°C. After 24 h, all nonadherent cells were removed and fresh astrocyte-specific medium was fed. Adherent cells were maintained in astrocyte-specific medium until confluence with a medium change every 3 to 4 days.
Isolation of primary astrocytes from mixed glia.
After obtaining confluent monolayers of mixed glial cells as detailed above, the addition of new medium was stopped for 10 days to allow differential adhesion of astrocytes and microglia. To remove the microglia from the astrocyte monolayer, the culture flask was thoroughly agitated in an orbital incubator shaker (180 rpm for 45 min at 37°C), followed by an immediate shake off. Cells suspended in the culture medium then were removed, and the remaining adherent monolayers of astrocytes were used as enriched glial cultures for further experimentation. These enriched primary cultures were quantified for GFAP+ astrocytes with the help of immunofluorescence, cell cytometry, and Western blotting and showed that the cells present in the primary culture were 86.6% ± 1.5% to 90.3% ± 1.5% GFAP+ astrocytes (Fig. 1). Double immunolabeling of these astrocytes with Cx43 and GFAP showed the cells expressed high levels of Cx43, which was present as distinct puncta on the cell surface of two adjacent GFAP+ astrocytes (Fig. 2).
FIG 1.

Enrichment and characterization of astrocytes enriched from mixed glial culture. One confluent monolayer of mixed glial cells (A) and an astrocyte-enriched primary culture (B) isolated from neonatal mouse brain were labeled with anti-GFAP antibody (astrocytic marker; green) and CD11b (microglia marker; red). Cells were counterstained with DAPI (nuclear stain; blue). (C) Visual counting of immunostained cells demonstrated that mixed glial cultures consist of GFAP+ astrocytes (70% ± 3.0%) and CD11b+ microglia (9% ± 2%), whereas astrocyte-enriched cultures demonstrated a significantly higher percentage (90.3% ± 1.5%) of cells that were positive for GFAP (data were mean values ± SEM from three experimental sets having 10 replicates each; ****, P < 0.0001). (D) Five micrograms of total protein from mixed glial cultures as well as astrocyte-enriched cultures was immunoblotted for GFAP and an internal control, γ-actin. (E) Astrocyte-enriched cultures demonstrated 26.3% ± 3.4% higher GFAP expression (normalized to internal control actin) than mixed glial cultures (data were mean values ± SEM from three experiment sets; ***, P < 0.001). (F) For quantification, primary astrocytes were immunostained with FITC-labeled anti-GFAP antibody and subjected to flow cytometric analysis. A total of 86.6% of cells were positive for GFAP (one representative plot of three experiments is shown).
FIG 2.
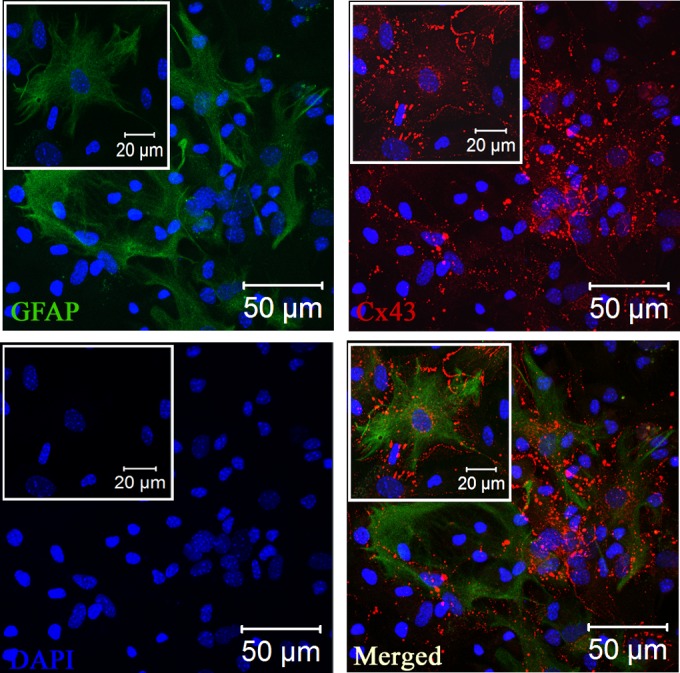
Expression of Cx43 at the cell surface of GFAP+ primary astrocytes. Primary astrocytes were double immune labeled with mouse anti-GFAP (astrocyte marker) and rabbit anti-Cx43 antisera. Cells subsequently were labeled with FITC goat anti-mouse IgG and Texas Red goat anti-rabbit IgG, respectively. Immunostained cells were counterstained with DAPI (blue). Merged images show that GFAP-positive astrocytes express high levels of punctate Cx43 staining at the cell surface. Insets show magnifications of the expression of Cx43 at the cell surface of GFAP+ astrocytes.
Infection of primary astrocytes with MHV-A59.
A neurotropic demyelinating strain of MHV-A59 was used for studying the effect of virus infection on astrocyte gap junction communication (35). Primary astrocytes were infected with inoculation medium (DMEM containing 1% penicillin-streptomycin and 1% glutamine with 2% FBS) containing MHV-A59 at multiplicities of infection (MOI) of 2 and 5 and allowed to adhere for 1 h at 37°C in a humidified CO2 incubator. After 1 h of incubation, infected cells were maintained in astrocyte-specific medium containing 10% normal serum. Cultures were subjected to different cell biological and biochemical assays at different time points postinfection (p.i.) as described for each experiment.
Immunofluorescence.
Immunofluorescence studies were done according to a protocol described previously (36), with minor modifications. For standard immunofluorescence, primary astrocytes were plated on etched glass coverslips and fixed with 4% paraformaldehyde (PFA). Permeabilization was done with phosphate-buffered saline (PBS) containing 0.5% Triton X-100 and blocked with PBS containing 0.5% Triton X-100 and 2.5% heat-inactivated goat serum (PBS-GS). The cells were incubated with primary antisera diluted in blocking solution for 1 h, washed, and then labeled with secondary antisera diluted in blocking solution. Cells then were washed with PBS, mounted with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; VectaShield, Vector Laboratories), and visualized using a Zeiss confocal microscope (LSM710). Images were acquired and processed with Zen2010 software (Carl Zeiss).
Flow cytometry.
Primary astrocytes were trypsinized and washed thrice in flow buffer (PBS containing Ca2+/Mg2+, 2% FBS, 0.01% sodium azide) and centrifuged at 500 × g for 5 min at 4°C. Approximately 1 × 106 cells were added to 5 ml polystyrene round-bottom tubes, washed with 3 ml of flow buffer, and centrifuged at 300 × g for 5 min. Cells were fixed by adding 100 μl BD Perm/Fix buffer (BD Biosciences, San Jose, CA) for 15 min. Following a wash with flow buffer, cells were incubated with mouse anti-GFAP antibody (1:40; Sigma) diluted in BD Perm/Wash buffer (BD Biosciences, San Jose, CA) for 30 min at room temperature (RT). Cells were washed, centrifuged, labeled with goat anti-mouse fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:40), and diluted in BD Perm/Wash buffer for 30 min at RT. After a final wash and centrifugation, cells were resuspended in 200 μl of flow buffer and subjected to flow cytometry using a BD FACSverse flow cytometer and analyzed using BD FACSuite software.
Total protein extraction from astrocyte cultures.
Total proteins from primary astrocytes were extracted as described previously (37), with minor modifications. Primary astrocytes were washed and harvested with PBS containing protease inhibitors. Following centrifugation, cell pellets were resuspended in ice-cold protein extraction buffer containing 25 mM Tris, pH 7.6, 1 mM MgCl2, 1% Triton X-100, 0.5% SDS with 1× EDTA-free complete protease inhibitor (Roche, Mannheim, Germany), and phosphatase inhibitors (1 mM NaVO4 and 10 mM NaF) for 30 min with a mild vortex at regular time intervals. The suspension was centrifuged at 10,000 × g for 15 min at 4°C using an Eppendorf 5415 R centrifuge. The supernatant was taken as whole-cell lysate and protein content was measured using the Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL). The whole-cell lysates were resuspended in Laemmli's sample buffer (38) and subjected to Western blot analysis.
Membrane protein preparation and analysis.
Primary astrocytes were washed, harvested in PBS containing protease inhibitor cocktail (complete protease inhibitor; Roche, Mannheim, Germany) and phosphatase inhibitors (1 mM NaVO4 and 10 mM NaF), and then passed through a Dounce homogenizer 100 times (39). The homogenate was centrifuged at 500 × g for 5 min using an Eppendorf 5415 R centrifuge, and the resulting supernatant was centrifuged at 100,000 × g for 30 min using a Beckman Optima Max ultracentrifuge to obtain a membrane-enriched pellet. To analyze total membrane connexin expression, this pellet was resuspended in Laemmli's buffer and subjected to immunoblot analysis for Cx43.
For detergent solubilization studies, the membrane-enriched pellet was resuspended in PBS with protease and phosphatase inhibitors at 4°C containing 1% Triton X-100 and then incubated for 30 min at 4°C. Care was taken to ensure that the samples were not warmed to RT during the extraction procedure. The sample then was centrifuged at 100,000 × g for 30 min and separated into Triton X-100 soluble supernatant and insoluble pellet fractions (40). The soluble fraction then was diluted into Laemmli's sample buffer, while the insoluble fraction initially was resuspended in PBS with 1% Triton X-100 prior to dilution. Equal volumes of soluble and insoluble fractions were probed for Cx43 by Western blotting.
LY dye transfer assay.
The gap junction functional coupling between astrocytes was determined using scrape loading of Lucifer yellow (LY) in confluent monolayers of primary astrocytes as described previously (41), with minor modifications. Primary astrocytes were scrape loaded with PBS containing 4 mg/ml Lucifer yellow CH (Sigma, Saint Louis, MO). After a 1-min incubation, LY solution was removed from the culture, the culture was washed thoroughly with PBS, and astrocyte-specific medium was added. The distance of LY spread from the scrape-loading point to neighboring cells was imaged using an Olympus IX-81 microscope system with a Hammatzu Orca-1 charge-couple device (CCD) camera, and the distance spread was measured using ImageJ software.
Western blotting.
For immunoblotting, samples were resolved by SDS-PAGE using a 12% polyacrylamide gel (42), transferred to polyvinylidene difluoride (PVDF) membranes using transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol), and blocked for 1 h at RT using blocking solution (5%, wt/vol, powdered milk dissolved in TBST or Tris-buffered saline containing 0.1%, vol/vol, Tween 20). The samples then were incubated overnight at 4°C in primary antisera diluted in blocking solution, followed by washes with TBST and then a 1-h incubation of horseradish peroxidase (HRP)-conjugated secondary IgG in blocking solution. The immunoblots were washed in TBST, and then immunoreactive bands were visualized using Super Signal WestPico chemiluminescent substrate (Thermo Scientific, Rockford, IL). Densitometric analysis of nonsaturated films was performed using Bio-Rad Quantity One analysis software (Hercules, CA) or using a Syngene G:Box ChemiDoc system and GENESys software.
RNA extraction both in vitro and in vivo.
To extract total RNA from mouse tissues, animals were transcardially perfused with diethyl pyrocarbonate (DEPC)-treated PBS, after which the brain and liver tissues were immediately harvested in RNAlater solution (Ambion) and stored at −20°C for total RNA extraction. Total RNA from astrocytes and brain tissues was isolated by a Qiagen RNeasy minikit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol. RNA was quantified by absorption at 260 nm and stored at −80°C before use. The first-strand cDNA was synthesized from 1 μg of total RNA with random primers and MultiScribe reverse transcriptase from a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). The quality of cDNA was evaluated by amplification of a housekeeping gene encoding glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Upon obtaining the amplicons specific for GAPDH, the 20-μl cDNA samples were treated with 0.5 μl RNase H (Escherichia coli RNase H; 2U/μl stock; Invitrogen) for 1 h at 37°C. The cDNA samples from liver were checked for the presence of virus nucleocapsid amplicon using two specific primers, IZJ5 (5′-GCTCCAACAGTTGGTGCC-3′) and IZJ6 (5′-ACGTAGGACCTTGCTAACTTC-3′), and the resulting amplified fragment of 601 bp was analyzed by 1.2% agarose gel electrophoresis (35).
Real-time quantitative PCR (qPCR) analysis.
The cDNA samples from astrocytes and brain tissues were subjected to real-time PCR using an ABI Fast 7500 real-time PCR system (Applied Biosystems, Foster City, CA) with a 10-μl reaction volume. Each reaction mix was prepared using 2× TaqMan universal PCR master mix (Applied Biosystems), probe/primer mix (Solaris; Thermo Scientific), and 50 ng cDNA template.
Real-time qPCR was done in parallel wells using probe/primer mix specific for the target Cx43 (GJA1) gene and a gene for an internal control, β-actin (ACTB), with TaqMan universal PCR master mix assays performed in replicate. A nontemplate control was included in all experiments. The results of relatively quantitative real-time PCR were analyzed by the comparative threshold cycle (ΔΔCT) method and normalized by β-actin as an internal control. Finally, products amplified by real-time PCR were subjected to 4% agarose gel electrophoresis to confirm the presence of specific amplicons.
Inoculation of mice.
Four-week-old, MHV-free, C57BL/6 mice were intracranially inoculated with 50% of the 50% lethal dose (LD50) of MHV-A59 (2,000 PFU) and were monitored daily for signs and symptoms of disease (30, 35). Mice mock infected with PBS-bovine serum albumin (BSA) were maintained in parallel. The mice were sacrificed at the peak of inflammation (day 5 p.i.). Mice were transcardially perfused with PBS-DEPC (Sigma) and treated with PBS for RNA extraction. Liver and brain tissues were harvested for experimentation.
Isolation of total protein from brain.
To analyze Cx43 expression in vivo, total protein was extracted from brain as described previously (43), with minor modifications. Brains were flash frozen in liquid N2 and immediately dissolved in PBS containing 2% SDS, 1× EDTA-free complete protease inhibitor (Roche), and phosphatase inhibitors (1 mM NaVO4 and 10 mM NaF). The lysates were sonicated thrice on ice at a 30% amplitude of 30 kHz for 0.5 s using a Sartorius Labsonic M sonicator. The samples then were centrifuged for 20 min at 10,000 × g at 4°C in an Eppendorf 5415 R centrifuge. The supernatants were taken and total protein content was estimated with a Pierce BCA protein assay kit (Thermo Scientific). Twenty micrograms of total protein was loaded for each sample, and they were probed for Cx43 as well as for an internal control, γ-actin (Biobharati Lifescience Pvt. Ltd., Kolkata, India), by Western blotting.
Tissue processing and double-label immunofluorescence of frozen sections.
Mock- and MHV-A59-infected mice (day 5 p.i.) were perfused transcardially with PBS, followed by cold PBS containing 4% PFA. Brains were harvested in 4% PFA for 6 h and then placed at 4°C for 4 h in 10% sucrose, followed by 30% sucrose overnight. Tissues were embedded with OCT medium (Tissue Tek, Hatfield, PA), sectioned sagitally with the help of Cryotome (Thermo Scientific) to 10-μm thickness, and mounted on charged glass slides. Immunostaining was done as described previously (32). Frozen tissue sections were washed with PBS at RT to remove cryomatrix. Tissues then were incubated for 1 h at RT with 1 M glycine in PBS to reduce nonspecific cross-linking, followed by a 10-min incubation at RT with 1 mg/ml NaBH4 in PBS to reduce autofluorescence. Slides were washed with PBS and incubated with blocking serum containing PBS with 0.5% Triton X-100 and 2% goat serum (GS). The sections were incubated overnight at 4°C with a primary antiserum diluted in blocking serum, washed, and subsequently incubated with secondary antiserum diluted in PBS with GS for 2 h at RT. All incubations were carried out in a humidified chamber. After PBS washing, sections were mounted with DAPI containing mounting medium and were imaging using an Olympus IX-81 epifluorescence microscope or Zeiss confocal microscope (LSM710), as specified. The images were processed with the help of ImageJ software.
Colchicine treatment of primary astrocytes.
Primary astrocytes were treated with 100 μM colchicine (44) for 24 h (45) to depolymerize the microtubule network, as this concentration of colchicine was found to be effective at disrupting the microtubule network in primary astrocytes in earlier studies. After 24 h of colchicine treatment, primary astrocytes were processed for Cx43 staining as described above.
Statistical analyses.
All values shown are mean values ± SEM (standard errors of the means). Student's unpaired t test was used to identify significant differences in two-group comparisons. The tests were two-tailed. One-way analysis of variance (ANOVA) was used to make statistical comparisons between three experimental groups, after which pairwise comparisons were made using the post hoc Tukey method for multiple testing. For all experiments, statistical significance was set at P < 0.05.
RESULTS
Retention of Cx43 in the perinuclear compartment of MHV-A59-infected astrocytes.
Primary astrocytes were infected with MHV-A59 at MOIs of 1 (Fig. 3) and 2 (Fig. 4). At different times postinfection, cells were fixed and subjected to immunofluorescence. For double labeling, astrocytes were stained for both Cx43 (Fig. 3 and 4, red) and viral antigen/nucleocapsid protein (Fig. 3 and 4, green). Cells were counterstained with DAPI (Fig. 3 and 4, blue).
FIG 3.
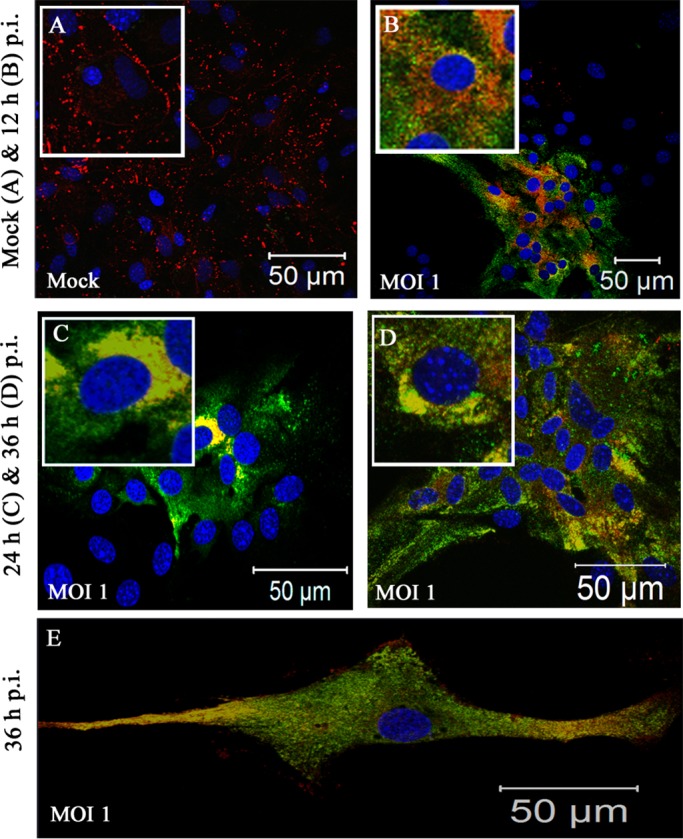
Intracellular localization of Cx43 in MHV-A59-infected primary astrocytes at an MOI of 1. Primary astrocytes were mock infected (A) or infected with MHV-A59 at an MOI of 1 (B to E) and subjected to double-label immunofluorescence with anti-Cx43 antisera (red) and antiviral nucleocapsid (N) antisera (green). Astrocytes demonstrated heterogeneous morphology. At this low viral dose, syncytia were not found to be present, even at 36 h p.i. Infected cells showed characteristic intracellular retention of Cx43 upon viral infection. Astrocytes which were part of a confluent monolayer (B to D), as well as astrocytes which grew as isolated large single cells (E), showed similar retention of Cx43 in an intracellular compartment after infection.
FIG 4.
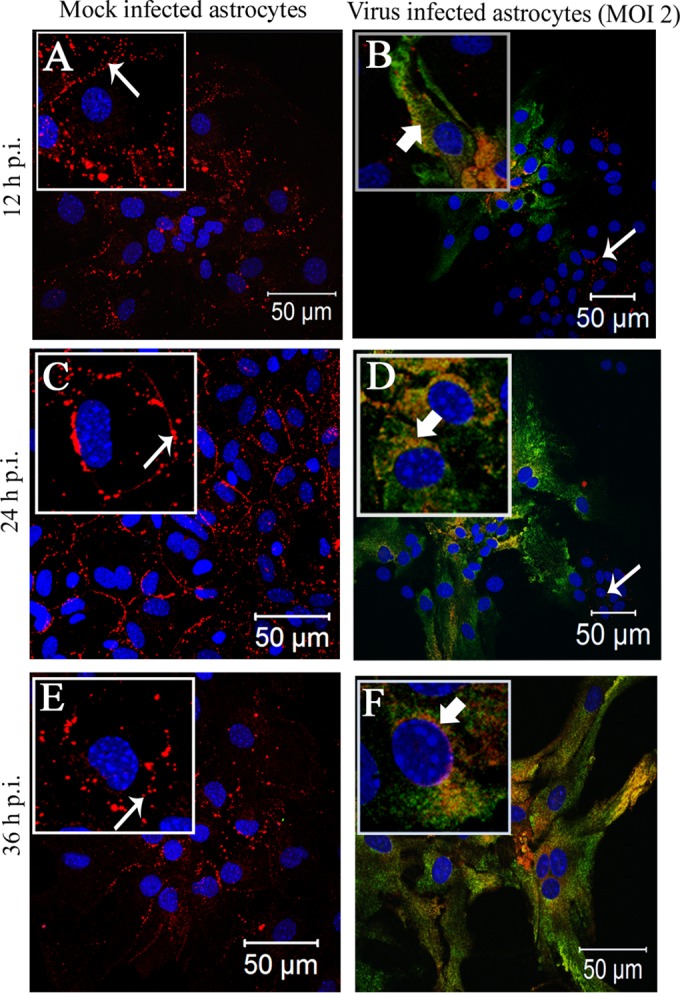
Intracellular localization of Cx43 in MHV-A59-infected primary astrocytes at an MOI of 2. Primary astrocytes were mock infected (A, C, and E) or infected with MHV-A59 at an MOI of 2 (B, D, and F). Cells were subjected to double-label immunofluorescence at 12 h (A and B), 24 h (C and D), and 36 h (E and F) p.i. with anti-Cx43 antisera (red) and anti-N antisera (green). Cells were visualized at 40× on a laser-scanning microscope. (A, C, and E) For the mock-infected cells, Cx43 was localized at the cell surface (thin arrow) with very minimal distribution in the intracellular compartment. In contrast, in MHV-A59-infected cells Cx43 was localized primarily in the intracellular compartment with very minimal distribution at the cell surface and was mostly colocalized with anti-N antisera (B, D, and E, thick arrow). Interestingly, intracellular retention of Cx43 was restricted to infected cells only. Cells negative for viral-N (B and D, thin arrow) did not show retention of Cx43 in an intracellular compartment. At each time point p.i., mock-infected cells (A, C, and E) expressed Cx43 at the cell surface (thin arrow), whereas Cx43 was localized mainly in the perinuclear compartment (B, D, and F, thick arrow) and partially colocalized with viral N protein in infected cells. To better illustrate these observed localization patterns, a single cell from the confluent astrocyte monolayer is shown at higher magnification in an inset.
In culture, a heterogenous population of astrocytes remains visible as aggregations of cells, as well as large individual cells of a fibrous nature. In the lower-dose viral inoculum at an MOI of 1 (Fig. 3B to E), the cells did not form syncytia even at 36 h p.i. Specifically, infected astrocytes, positive for viral nucleocapsid (N) protein, showed Cx43 retention within the intracellular compartment. Mock-infected cultures showed typical Cx43 staining at the cell surface (Fig. 4A, C, and E). Upon MHV-A59 infection at an MOI of 2 (which was the best-optimized experimental condition), Cx43 was localized in the intracellular compartment and mostly colocalized with viral N protein. At 12 h p.i., Cx43 was distributed both in the intracellular compartment (Fig. 4B, thick arrow) as well as at the cell surface (thin arrow). At 24 h p.i., the majority of cells were infected and showed intracellular localization of Cx43 (Fig. 4D, thick arrow) with very little distribution at the cell surface (Fig. 4D, thin arrow). This altered localization of Cx43 was independent of visible cytopathic effect, alteration of cellular morphology, or cell-to-cell fusion. Indeed, prominent visible syncytium formation was not observed, consistent with prior studies on primary astrocytes infected with MHV-A59 (46). Interestingly, both at 12 and 24 h p.i., the alteration of Cx43 expression was restricted to virus-infected cells only. In the MHV-A59-infected culture, the cells which were anti-N negative showed the presence of Cx43 at its cell surface (Fig. 4B and D, thin arrow). At 36 h p.i., similar retention of Cx43 was observed in virus-infected cells only (Fig. 4F). Localization of Cx43 at the single-cell level is shown in insets in each figure. The intracellular compartment retained Cx43, mostly colocalized with the ER marker calnexin (Fig. 5B, thick arrow) and endoplasmic reticulum Golgi intermediate complex (ERGIC) marker β-Cop (Fig. 5D, thick arrow). In contrast, Cx43 was observed predominantly at the cell surface with characteristic punctate staining in mock-infected astrocyte cultures without colocalization with calnexin or β-cop (Fig. 5A and B, thin arrow).
FIG 5.
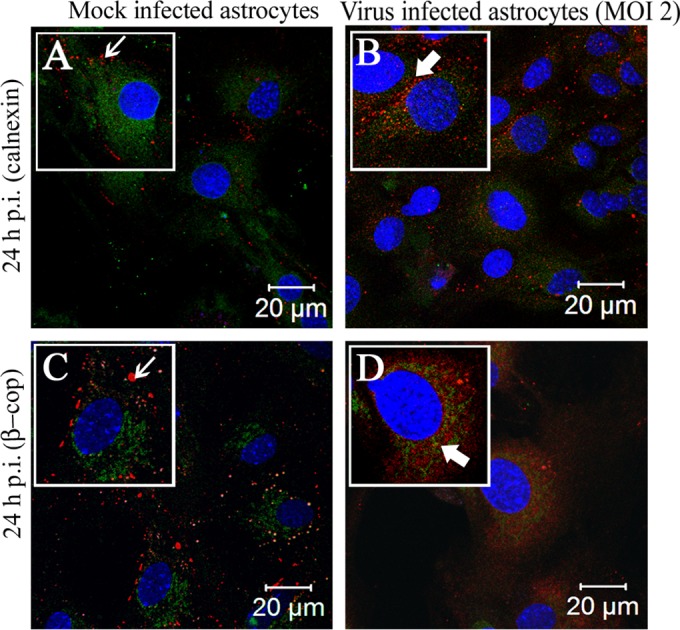
Localization of Cx43 predominantly in the ER/ERGIC of virus-infected cells. Primary astrocytes were mock infected or infected with MHV-A59 at an MOI of 2 and were subjected to double-label immunofluorescence at 24 h p.i. with anti-Cx43 antisera (red) and anti-calnexin (green) or anti-β-cop antisera(green). The images show prominent punctate staining of Cx43 at the cell surface (A and C, thin arrow), forming gap junction plaques, in mock-infected cells. Cx43 in the virus-infected cells, which was retained in the intracellular compartments, mostly colocalized with the ER marker calnexin and/or ERGIC marker β-cop (B and D, thick arrow).
Downregulation of Cx43 protein and RNA due to MHV-A59 infection.
It has been reported previously that systemic human influenza virus infection can cause the downregulation of Cx43 expression in the CNS. Similarly, our study demonstrated that neurotropic MHV infection can cause the downregulation of Cx43.
To determine MHV-A59-induced alteration of Cx43 expression at the protein level, immunoblotting was performed on proteins isolated from mock-infected as well as from MHV-A59-infected astrocytes. Primary astrocytes were infected with MHV-A59 at MOIs of 2 and 5. At 24 h p.i., total protein was extracted and the concentration was measured, and 5 μg of total protein was loaded for immunoblotting, probed with either rabbit anti-Cx43 antibody (Sigma), rabbit anti-GFAP antibody (Sigma), or internal control rabbit anti-γ-actin antibody (Biobharati Lifescience Pvt. Ltd.), and detected with HRP goat anti-rabbit secondary antibody. This experiment demonstrated that due to MHV-A59 infection, Cx43 expression was downregulated compared to the level for the control (Fig. 6A). Virus-mediated altered expression of Cx43 was specific, as GFAP expression was found not to be altered upon virus infection (Fig. 6A).
FIG 6.

Reduction of Cx43 protein expression due to MHV-A59 infection. Primary astrocytes were either mock infected or infected with MHV-A59, and total protein was extracted at 24 h p.i. Protein (5 μg) was resolved in SDS-PAGE, transferred to a PVDF membrane, and immunoprobed. (A) Upon infection with MHV-A59, whole-cell Cx43 protein levels were decreased compared to those of mock-infected cells. The internal loading control γ-actin showed similar signal intensity for all experiments. (B) Densitometric analysis showed that there was a 36.3% ± 3.3% (at an MOI of 2) and 40.8% ± 6.8% (at an MOI of 5) reduction in total Cx43 for MHV-infected cells compared to the level for mock-infected control cells. The total proteins isolated from primary astrocytes also were probed for GFAP, but expression was found to be similar in all experimental groups. The mean ± SEM incidences of six experimental replicates from three different experiments are shown (****, P < 0.0001).
Comparative densitometric analysis revealed a 36.3% ± 3.3% reduction of Cx43 at an MOI of 2 and 40.8% ± 6.8% reduction at an MOI of 5 compared to Cx43 expression in mock-infected astrocytes (Fig. 6B). Compared to mock-infected astrocytes, astrocytes infected at MOIs of both 2 and 5 showed statistically significant alteration of Cx43 (****, P < 0.0001), but the differences in expression levels between groups infected at MOIs of 2 and 5 were not statistically significant. The reduced expression of Cx43 protein implies increased degradation and/or reduced synthesis. Thus, it was further determined whether MHV-A59 infection affects relative Cx43 mRNA expression. Due to the limited availability of primary astrocytes from neonatal mouse brain, further experiments were performed at an MOI of 2 at 24 h p.i., representing the best-optimized experimental condition.
RNA extraction and cDNA synthesis was done from both mock-infected and MHV-A59-infected (at an MOI of 2) cells. Fifty nanograms of cDNA template was used for real-time qPCR analysis for both Cx43 (target gene; GJA1) and β-actin (internal control; ACTB) expression using a TaqMan probe method. Real-time PCR showed that Cx43 transcripts expressed by MHV-A59 primary astrocytes were 3.49- ± 0.25-fold reduced compared to levels for mock-infected astrocytes (Fig. 7A). This Cx43 downregulation was statistically significant (***, P < 0.001). The qPCR product was resolved in 4% agarose gel, and amplicon-specific product was visualized (Fig. 7B).
FIG 7.
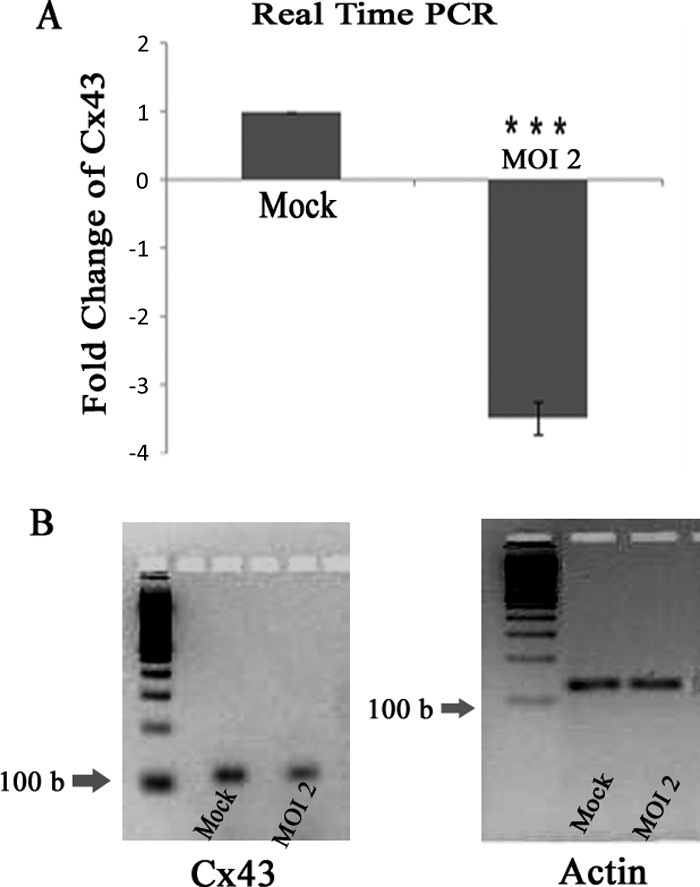
Reduction of Cx43 mRNA abundance in viral infection. Primary astrocytes were infected with MHV-A59 at an MOI of 2, and mock-infected cells were maintained in parallel. RNA was extracted at 24 h p.i., and subsequently cDNA was synthesized. Equal amounts of cDNA template were used for qPCR analysis. The relative expression of Cx43 mRNA was obtained using the ΔΔCT method. (A) Compared to that of mock-infected cells, Cx43 expression was 3.49- ± 0.25-fold downregulated after MHV-A59 infection. The end products of the qPCR of Cx43 and an internal control, β-actin, were resolved in a 4% agarose gel and are shown in panel B. Mean ± SEM incidences from three different experiments are shown (***, P < 0.001).
Alteration of Cx43-mediated gap junction plaque formation due to MHV-A59-induced stress.
Following MHV-A59 infection, the amount of Cx43 expressed in total membrane fractions was reduced compared to that in mock-infected cells (Fig. 8A). Similarly, immunofluorescence, immunoblotting, and real-time qPCR data revealed a total reduction of Cx43 as well as reduced surface expression of Cx43 due to MHV-A59 infection irrespective of viral dose. The amount of Cx43 reduction at the cell surface (visual observation) is not reflected in the reduction at the total protein or RNA level. As previously shown (Fig. 3 and 4), this reduction of Cx43 expression at the cell surface was due to the retention of Cx43 in the ER/ERGIC.
FIG 8.

Confirmation of reduced gap junction plaque formation due to infection using Triton X-100 solubilization. Primary astrocytes were infected with MHV-A59 at an MOI of 2, and mock-infected cells were maintained in parallel. A membrane-enriched fraction was isolated from homogenized cells from each culture, and then protein was solubilized in the presence of 1% Triton X-100 at 4°C. Subsequently, these fractions were loaded in a gel and probed for Cx43. (A) Compared to levels for mock-infected controls, a reduction in both the total membrane fraction as well as the Triton X-100 insoluble fraction was observed upon MHV-A59 infection. (B) The amount of Cx43 present in the total membrane fraction was reduced 38.4% ± 6.9% in virus-infected cells compared to the level for mock-infected cells (means ± SEM; n = 3; *, P < 0.05). In mock-infected cultures, most of the Cx43 (81.6% ± 3.2%) was pooled in the Triton X-100 insoluble (Ins) fraction, whereas only 18.4% ± 3.3% was pooled in the soluble (Sol) fraction. In contrast, in MHV-A59-infected astrocytes (at an MOI of 2 at 24 h p.i.), large fractions of Cx43 (50.6% ± 2.5%) appeared in both the Triton X-100 soluble fraction and the Triton X-100 insoluble gap junction plaques (49.4% ± 2.5%). The insoluble versus soluble fraction ratio was calculated for mock- and virus-infected astrocytes. For virus-infected astrocytes this ratio (1.01) was significantly lower than that for mock-infected astrocytes (5.05; ***, P < 0.001).
Experiments by Musil and Goodenough (40) showed that Cx43 assembled into gap junction plaques is resistant to 1% Triton X-100 solubilization at 4°C, while remaining Cx43 dissolves in the Triton X-100 soluble fraction. Therefore, Triton X-100 solubilization was performed to determine whether MHV-A59 infection promotes the retention of Cx43 in the intracellular compartment instead of it being trafficked to the cell surface to form gap junction plaques.
The membrane fraction was isolated from cells and was solubilized using 1% Triton X-100 at 4°C, separated into detergent-soluble and -insoluble fractions, and probed for Cx43 (39, 40). Mock-infected astrocytes showed that the majority of Cx43 (81.6% ± 3.2%) was pooled into the Triton X-100 insoluble fraction. In contrast, MHV-A59-infected astrocytes accumulated large amounts of Cx43 in the Triton X-100 soluble fraction (50.6% ± 2.5%) (Fig. 8B). Hence, the insoluble/soluble fraction ratio of Cx43 expression was significantly higher (***, P < 0.001) for mock-infected astrocytes than that for virus-infected astrocytes. This experiment demonstrated that upon infection, a significant amount of Cx43 failed to traffic to the cell membrane to form gap junction plaques.
Inhibition of functional gap junction coupling due to virus infection.
LY is a small molecule (457 Da) that moves freely through gap junctions from loaded cells to neighboring ones. Due to MHV-A59 infection there was a clear alteration of Cx43 mRNA expression, protein expression, and trafficking to the cell surface. To understand whether this change could affect functional gap junction communication between astrocytes, cells were scrape loaded with LY and the distance of dye spreading from the tip of the scrape-loading zone (arrow) was measured for mock- and virus-infected cells (Fig. 9A). Upon infection, LY dye spread was significantly reduced, 46.3% ± 0.6% (for an MOI of 2; **, P < 0.01) and 53.6% ± 5.96% (for an MOI of 5; **, P < 0.01), compared to that of mock-infected cells (Fig. 9B). The alteration between groups infected at MOIs of 2 and 5 was not statistically significant. The depletion of Cx43 gap junction plaque formation due to MHV-A59 infection could be the basis of reduced functional gap junction coupling between astrocytes.
FIG 9.

Loss of functional gap junction communication between astrocytes after virus infection. Confluent astrocyte monolayers were infected with MHV-A59 at MOIs of 2 and 5 for 24 h, and mock-infected control cells were maintained in parallel. (A) The cells were scrape loaded with 4 mg/ml Lucifer yellow (LY), which was allowed to diffuse through gap junctions. Following scrape loading of LY (arrow), the dye was transferred to a greater distance in control astrocytes than in infected astrocytes at an MOI of either 2 or 5. The average distance of LY spread was measured and expressed as a ratio of the distance spread in virus-infected versus mock-infected cells. (B) The average dye spread in infected cells was significantly reduced to 46.3% ± 0.6% (for an MOI of 2; **, P < 0.01) or 53.6% ± 5.96% (for an MOI of 5; **, P < 0.01) of the spread measured in mock-infected cells. The small difference in spread between cells infected at an MOI of 2 and 5 was not statically significant. The mean ± SEM measurements of three different experiments are shown.
Downregulation of Cx43 expression in vivo in MHV-A59-infected mice.
Astrocytes, the most abundant glial cells in the CNS, maintain cerebral homeostasis and provide functional support (3). These cells mainly express Cx43 that takes part in the formation of functional syncytia among themselves as well as other CNS cells (47).
To understand whether alteration of Cx43 in vitro could be reflected in vivo, mice were intracranially inoculated with MHV-A59 at 50% of the LD50 or with PBS-BSA for mock-inoculated mice. Tissues were processed at day 5 p.i. (peak of inflammation) from virus-infected as well as mock-inoculated mice to extract RNA along with total protein. As hepatitis and the presence of virus in hepatocytes is the index of viral infection in the acute phase of infection, total RNA was extracted from liver, cDNA was synthesized, and the viral N gene was detected by PCR. Amplification of the viral N gene resulted in a specific PCR product which was obtained from virus-infected mice, but no amplicon was observed for mock-infected mice (Fig. 10A). Upon confirmation of infection, RNA was extracted from brains and cDNA was prepared. These samples were subjected to qPCR and normalized by β-actin as an internal control. With real-time qPCR quantification, the infected mice showed a 3.13- ± 0.06-fold downregulation of Cx43 compared to that of mock (n = 3)-infected mice (Fig. 10B). This difference was statistically significant (****, P < 0.0001). To examine changes in Cx43 expression at the protein level, total protein was extracted from mouse brains and probed for Cx43 as well as γ-actin. Total protein also was probed for the astrocyte marker GFAP. Upon MHV-A59 infection, there was a reduction in total Cx43 protein expression. Noticeably, the reduction of the Cx43 protein expression in brain was more prominent in mice with a higher viral load, as quantified by viral N gene-specific amplicon. A major reduction in phosphorylated isoforms, which are located mainly at the cell membrane and are involved in intracellular signaling (48), was observed upon MHV infection (Fig. 10C). Viral nucleocapsid proteins were detected from the CNS of infected mice, confirming that the reduction of Cx43 expression correlated with MHV-A59 infection (data not shown). Similar to the in vitro data, the change of Cx43 was found to be specifically regulated by MHV infection. The nonphosphorylated form of Cx43 (42 kDa) expression was reduced 20.1% after MHV-A59 infection, whereas a 43.2% reduction was observed in phosphorylated forms of Cx43 (44 kDa and 46 kDa) after viral infection (Fig. 10D). This reduction was statistically significant (*, P < 0.05). GFAP expression was not altered in MHV-infected brain tissues. The reduced Cx43 expression of MHV-A59 in the brain of infected mice at day 5 p.i. resolved later in the disease, as levels became comparable to normal levels in mock-infected mice at day 30 p.i. (Fig. 11).
FIG 10.

Alteration of Cx43 in mouse brain due to MHV-A59 infection. Mice were infected intracranially with MHV-A59 at 50% of the LD50 or with PBS-BSA for mock-infected mice. Mice were sacrificed at day 5 p.i., after which their liver and brain tissues were processed for RNA extraction. cDNA was synthesized from RNA of the brains and livers. To confirm infection, cDNAs from liver were amplified for virus-specific antinucleocapsid primers (IZJ5 and IZJ6). (A) Intracranial injection of mice with the virus showed the presence of nucleocapsid-specific amplicons (Infected lanes 1, 2, and 3) in liver. As expected, no such amplification was noted from mock-inoculated mice (Mock lanes 1 and 2). (B) Real-time qPCR analysis of the RNA samples from brain showed a significant 3.13- ± 0.06-fold reduction in relative abundance of Cx43 mRNA after MHV-A59 infection. The mean ± SEM incidences from three different mice are shown. (****, P < 0.0001; n = 3). Total protein was extracted from brain, and 20 μg of protein was resolved in SDS-PAGE, transferred to a PVDF membrane, and probed for Cx43 or the internal control, γ-actin. (C) Following infection with MHV-A59, the total Cx43 expression level was reduced, with a significant reduction observed for both 44- and 46-kDa isoforms of Cx43. This alteration was specific for Cx43, as GFAP expression was not changed significantly upon virus infection. Infection in mouse brains was confirmed by the presence of viral nucleocapsid protein (data not shown). (D) The nonphosphorylated form of Cx43 (42 kDa) expression was reduced 20.1% after MHV-A59 infection, whereas a 43.2% reduction was observed in phosphorylated forms of Cx43 (44 kDa and 46 kDa) after viral infection. The mean ± SEM incidences from three different animals is shown (*, P < 0.05).
FIG 11.
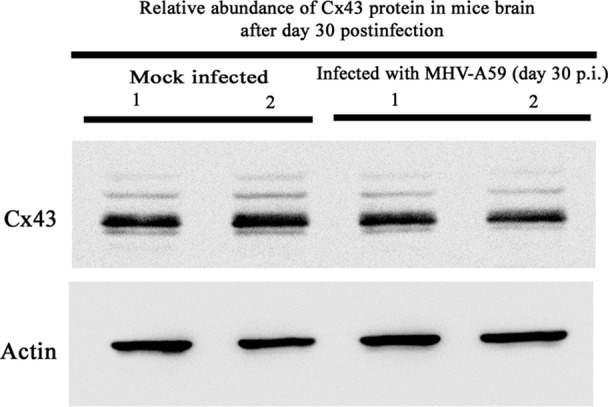
MHV-A59 infection during the chronic phase did not alter Cx43 protein expression in vivo. Mice were infected intracranially with MHV-A59 at 50% of the LD50 or mock infected with PBS-BSA. Mock-infected and virus-infected mice were sacrificed at day 30 p.i., and total protein was extracted from brain tissue. Twenty micrograms of protein was resolved in SDS-PAGE, followed by transfer to a PVDF membrane and probing for Cx43 or the internal control, γ-actin. No observable alteration was observed. Data from two representative infected mice are shown.
Alteration of Cx43 in astrocytes present in mouse brain following MHV-A59 infection.
To examine alterations of Cx43 expression specifically in and around areas of infection, frozen sections were immunostained with either GFAP (red) and viral N (green) or Cx43 (red) and viral N (green) protein, followed by counterstaining of the nucleus with DAPI (blue). GFAP+ astrocytes were found to be infected (Fig. 12D to F, thick arrow) in vivo, although the cell morphology of the infected astrocytes was found be unaltered from that of the mock-infected brain sections (Fig. 12A and C, thin arrow). Cx43 was observed to be expressed abundantly as prominent puncta in mock-infected mouse brains (Fig. 12G and I, thin arrow). In contrast, Cx43 expression was reduced in virus-infected mouse brains (Fig. 12J to L, thick arrow).
FIG 12.

In situ immunofluorescence data on infected brain tissue demonstrated alteration of Cx43 in GFAP-positive astrocytes. Cryosections of brain tissue from MHV-A59-infected and mock-infected mice were double immunolabeled for either GFAP (red) and viral N (green) protein (A to F) or Cx43 (red) and viral N (green) protein (G to L). Cells were counterstained with DAPI. Mock-infected (A to C) and virus-infected (D to F) astrocytes appeared to be morphologically normal (thin arrow in panels A and C and thick arrow in panels D and F). Abundant punctate Cx43 staining was observed (thin arrow in panels G and I) in mock-infected brain tissue. In contrast, significant loss of Cx43 staining was observed in MHV-A59-infected brain tissue (thick arrow in panels J and L).
Alteration of Cx43 expression in colchicine-treated primary astrocytes.
To understand whether the retention of Cx43 in an intracellular compartment could be due to a trafficking defect of Cx43 along microtubules due to virus-specific utilization of the microtubule network, primary astrocytes were treated with colchicine in the absence of virus. Confluent monolayers of primary astrocytes were treated with 100 μM colchicine, a known microtubule-depolymerizing agent. Untreated cells were maintained in parallel as a control. After 24 h, cells were subjected to Cx43 (red) immunolabeling and counterstained with DAPI (blue). Cx43 was present as prominent characteristic puncta in untreated cells (Fig. 13B and C, thin arrow), but colchicine-treated cells showed Cx43 was distributed in an intracellular compartment in a pattern similar to that observed in virus-infected cells (Fig. 13E and F, thick arrow). Hence, virus-induced disruption of tubulin-Cx43 interaction could be a major cause of retention of Cx43 in an intracellular compartment.
FIG 13.

Retention of Cx43 in an intracellular compartment in colchicine-treated primary astrocytes. Primary astrocytes were treated with 100 μM colchicine, a known microtubule-depolymerizing agent, and untreated astrocytes were maintained in parallel as a control. After 24 h, the cells were subjected to immunofluorescence for Cx43 (red), and DAPI (blue) was used to counterstain the nuclei. Untreated astrocytes showed characteristic punctate staining of Cx43 at the cell surface (B and C, thin arrow). In colchicine-treated astrocytes, Cx43 was localized mainly in the intracellular compartment (E and F, thick arrow) and was depleted from the cell surface.
DISCUSSION
In our study, it was shown that MHV-A59 can directly cause infection in primary astrocytes. MHV-A59 infection induced the downregulation of Cx43 at both mRNA and total protein levels. Beyond the alteration of Cx43 synthesis, it was observed that the majority of Cx43 was localized in a perinuclear region of virus-infected cells, mostly colocalizing with viral nucleocapsid (N) protein. This Cx43 pool was found to be unable to reach the cell surface to form Triton X-100 insoluble gap junction plaques. As a result of these alterations, functional gap junction communication was reduced between astrocytes in primary culture, as demonstrated by LY dye transfer assay. This phenomenon was reflected in vivo by the reduction of Cx43 protein and mRNA during MHV-A59-induced acute neuroinflammation in C57BL/6 mice.
To the best of our knowledge, this is the first report showing that MHV-A59-induced cellular stress may affect Cx43 trafficking to the cell surface. Cx43 was localized in the ER and ERGIC of infected cells instead of forming gap junction plaques. The reduced synthesis and accumulation of Cx43 inside the intracellular compartment successively altered the functional coupling between astrocytes, which might play a major role in altered CNS homeostasis during acute infection. In vivo, a noticeable reduction in 44- and 46-kDa isoforms of Cx43 was observed. These isoforms are known to be located primarily in cell membrane and to form gap junction plaques. Histopathological analyses also showed astrocytes were infected by MHV-A59 in vivo at day 5 p.i. Upon infection, the expression of Cx43 was downregulated and Cx43 localization was found to be altered in and around infected regions of brain. By the chronic stage (day 30 p.i.), Cx43 levels returned to normal.
The current findings indicate that Cx43 mRNA synthesis was reduced in vivo and in vitro. It has been reported that MHV hijacks the host translational machinery to produce its own proteins which may arrest the synthesis of a large number of host cellular mRNAs (49), and virus-induced host translational shutoff can directly downregulate mRNAs that have a short half-life. As a consequence, Cx43 mRNA, with a short half-life (50, 51), may have been downregulated directly due to MHV infection. In addition, current studies have shown that unfolded protein responses (UPR) induced by ER stress result in the fast downregulation of Cx43 expression at both protein and mRNA levels (52). Thus, it is also possible that viral infection-mediated ER stress leads to the accumulation of Cx43 in the perinuclear region, subsequently inducing the downregulation of Cx43 mRNA and protein.
Although it was observed that virus infection in astrocytes induced the retention of Cx43 in the intracellular compartment, the underlying mechanism was not known. Cx43 directly binds to microtubules (53) and gets transported to the cell membrane along the microtubules (54). A recent in vitro study showed a demyelinating strain of MHV is preferentially transported by microtubules in neurons (45). MHV-A59 proteins are also targeted to the Golgi apparatus (GA), inducing fragmentation of the GA and rearrangement of the microtubule network (55). Thus, the virus-specific utilization of the microtubule network may hinder Cx43 transport to the cell surface. Indeed, results here show colchicine-treated astrocytes have similar retention of Cx43 in the intracellular compartment, as seen in virus-infected astrocytes. This experiment showed that in vitro, in the absence of virus, depolymerization of the microtubule network affects Cx43 trafficking in a manner similar to that of viral infection. Results suggest Cx43 and microtubule association during infection needs to be further investigated as a mechanism mediating effects of MHV-A59-induced demyelinating disease.
Results of the current studies provide important insights into the virus-induced alteration of GJIC between astrocytes. Cx43 not only is involved in maintaining homeostasis among astrocytes but also is equally important in forming heterotypic gap junction channels with the oligodendrocytic GJ protein Cx47 present in glial networks in the CNS. Astrocyte/oligodendrocyte heterocellular coupling is important for maintaining the integrity of oligodendrocyte function as well as normal myelin formation. The underlying mechanism predicts that these GJCs play a crucial role in oligodendrocyte K+ ion buffering and metabolite homeostasis (47). In addition, Cx43 also mediates different signaling pathways that might be linked to myelination and maintenance of myelin in vivo. For example, a recent study showed that ATP released from axons mediates the release of leukemia inhibitory factor (LIF) from astrocytes, which in turn induces myelin oligodendrocyte glycoprotein (MOG) production and myelination by mature oligodendrocytes (56). Perhaps astrocyte-to-oligodendrocyte GJCs propagate one or more of these signals (47).
Moreover, Cx43 has been shown to control Cx47 phosphorylation and stability in gap junctions, and the loss of Cx43 may result in the secondary loss of Cx47 at astrocyte/oligodendrocyte GJCs (57). Previous studies have shown that the deletion of astrocytes Cx43 and Cx30 (10) or oligodendrocytes Cx47 and Cx32 (58) can cause pathological conditions featuring demyelination and oligodendrocyte cell death. In a human channelopathy, ODDD, Cx43 mutation can cause the loss of visual function and demyelination, which is similar to effects of MS (59). Furthermore, Cx43/Cx47 is reported to be critical for human myelination under disease conditions (60, 61). The loss of Cx47 in a mouse model causes severe myelin loss and microglial activation (62), which are major pathological hallmarks of MHV-induced demyelination (63) and of human MS. Therefore, the loss of Cx43 in astrocytes initiated during acute neuroinflammation, as observed here, may have a significant impact on oligodendroglial pathology during chronic inflammation which subsequently can contribute to myelin degeneration and expansion of demyelinating lesions (64).
Furthermore, it is evident that astrocyte/oligodendrocyte gap junction communication via Cx43/Cx47 is severely reduced not only in and around chronic MS lesions but also in normal-appearing white matter (NAWM) in MS and therefore may be a contributing factor to disease progression.
Early stages of neuroinflammation in a model of MS provided evidence of marked reduction of Cx43 linked with disruption of GJ communication between astrocytes and oligodendrocytes, but data identifying mechanisms of initial Cx43 alteration were lacking. The current study in a virus-induced neuroinflammatory model suggests a cause-effect relationship between viral infection, trafficking and expression of Cx43, and potentially subsequent demyelination. In summary, our study showed that Cx43 synthesis and its membrane docking machinery were significantly modified in astrocytes due to MHV-A59 infection, which in turn led to the failure of intercellular gap junction communication. The lack of intercellular communication between glial cells may be one of the major cellular determinants of chronic progressive demyelination.
ACKNOWLEDGMENTS
This work was supported by research grants (BT/PR14260/MED/30/437/2010 and BT/PR4530/MED/30/715/2012) from the Department of Biotechnology, India, and the Indian Institute of Science Education and Research Kolkata (IISER-K), India start-up fund, to J.D.S. We thank the Council of Scientific and Industrial Research (CSIR), India, for providing research support to R.B. and the IISER-K Integrated Ph.D. Program for supporting A.B. and K.B.
We thank the IISER-K confocal facility and Ritabrata Ghosh for his confocal assistance. We also thank Kenneth S. Shindler and Michael H. Koval for critically reading and editing the manuscript.
Funding Statement
N/A
REFERENCES
- 1.Panchin YV. 2005. Evolution of gap junction proteins–the pannexin alternative. J Exp Biol 208:1415–1419. doi: 10.1242/jeb.01547. [DOI] [PubMed] [Google Scholar]
- 2.Solan JL, Lampe PD. 2009. Connexin43 phosphorylation: structural changes and biological effects. Biochem J 419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sofroniew MV, Vinters HV. 2010. Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volterra A, Meldolesi J. 2005. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- 5.Wasseff SK, Scherer SS. 2011. Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol Dis 42:506–513. doi: 10.1016/j.nbd.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nair A, Frederick TJ, Miller SD. 2008. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci 65:2702–2720. doi: 10.1007/s00018-008-8059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MV, Spray DC, Willecke K. 1989. Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci U S A 86:10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naus CC, Bechberger JF, Zhang Y, Venance L, Yamasaki H, Juneja SC, Kidder GM, Giaume C. 1997. Altered gap junctional communication, intercellular signaling, and growth in cultured astrocytes deficient in connexin43. J Neurosci Res 49:528–540. [DOI] [PubMed] [Google Scholar]
- 9.Iacobas DA, Urban-Maldonado M, Iacobas S, Scemes E, Spray DC. 2003. Array analysis of gene expression in connexin-43 null astrocytes. Physiol Genomics 15:177–190. doi: 10.1152/physiolgenomics.00062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. 2009. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci 29:7743–7752. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabernero A, Giaume C, Medina JM. 1996. Endothelin-1 regulates glucose utilization in cultured astrocytes by controlling intercellular communication through gap junctions. Glia 16:187–195. doi:. [DOI] [PubMed] [Google Scholar]
- 12.Saez JC, Connor JA, Spray DC, Bennett MV. 1989. Hepatocyte gap junctions are permeable to the second messenger, inositol 1,4,5-trisphosphate, and to calcium ions. Proc Natl Acad Sci U S A 86:2708–2712. doi: 10.1073/pnas.86.8.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charles AC, Merrill JE, Dirksen ER, Sanderson MJ. 1991. Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6:983–992. doi: 10.1016/0896-6273(91)90238-U. [DOI] [PubMed] [Google Scholar]
- 14.Charles AC, Naus CC, Zhu D, Kidder GM, Dirksen ER, Sanderson MJ. 1992. Intercellular calcium signaling via gap junctions in glioma cells. J Cell Biol 118:195–201. doi: 10.1083/jcb.118.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finkbeiner S. 1992. Calcium waves in astrocytes-filling in the gaps. Neuron 8:1101–1108. doi: 10.1016/0896-6273(92)90131-V. [DOI] [PubMed] [Google Scholar]
- 16.Herve JC, Bourmeyster N, Sarrouilhe D, Duffy HS. 2007. Gap junctional complexes: from partners to functions. Prog Biophys Mol Biol 94:29–65. doi: 10.1016/j.pbiomolbio.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Stout CE, Costantin JL, Naus CC, Charles AC. 2002. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem 277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 18.Rufer M, Wirth SB, Hofer A, Dermietzel R, Pastor A, Kettenmann H, Unsicker K. 1996. Regulation of connexin-43, GFAP, and FGF-2 is not accompanied by changes in astroglial coupling in MPTP-lesioned, FGF-2-treated parkinsonian mice. J Neurosci Res 46:606–617. [DOI] [PubMed] [Google Scholar]
- 19.Nagy JI, Li W, Hertzberg EL, Marotta CA. 1996. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer's disease. Brain Res 717:173–178. doi: 10.1016/0006-8993(95)01526-4. [DOI] [PubMed] [Google Scholar]
- 20.Huang RP, Hossain MZ, Sehgal A, Boynton AL. 1999. Reduced connexin43 expression in high-grade human brain glioma cells. J Surg Oncol 70:21–24. [DOI] [PubMed] [Google Scholar]
- 21.Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, Jabs EW. 2003. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet 72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, Bennett MV, Spray DC. 2000. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res Brain Res Rev 32:45–56. doi: 10.1016/S0165-0173(99)00067-3. [DOI] [PubMed] [Google Scholar]
- 23.Masaki K, Suzuki SO, Matsushita T, Matsuoka T, Imamura S, Yamasaki R, Suzuki M, Suenaga T, Iwaki T, Kira J. 2013. Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS One 8:e72919. doi: 10.1371/journal.pone.0072919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brand-Schieber E, Werner P, Iacobas DA, Iacobas S, Beelitz M, Lowery SL, Spray DC, Scemes E. 2005. Connexin43, the major gap junction protein of astrocytes, is down-regulated in inflamed white matter in an animal model of multiple sclerosis. J Neurosci Res 80:798–808. doi: 10.1002/jnr.20474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koster-Patzlaff C, Hosseini SM, Reuss B. 2007. Persistent Borna disease virus infection changes expression and function of astroglial gap junctions in vivo and in vitro. Brain Res 1184:316–332. doi: 10.1016/j.brainres.2007.09.062. [DOI] [PubMed] [Google Scholar]
- 26.Faccini AM, Cairney M, Ashrafi GH, Finbow ME, Campo MS, Pitts JD. 1996. The bovine papillomavirus type 4 E8 protein binds to ductin and causes loss of gap junctional intercellular communication in primary fibroblasts. J Virol 70:9041–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crow DS, Beyer EC, Paul DL, Kobe SS, Lau AF. 1990. Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol Cell Biol 10:1754–1763. doi: 10.1128/MCB.10.4.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fatemi SH, Folsom TD, Reutiman TJ, Sidwell RW. 2008. Viral regulation of aquaporin 4, connexin 43, microcephalin and nucleolin. Schizophrenia Res 98:163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orellana JA, Saez JC, Bennett MV, Berman JW, Morgello S, Eugenin EA. 2014. HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J Neurochem 128:752–763. doi: 10.1111/jnc.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR. 1984. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34:597–603. doi: 10.1212/WNL.34.5.597. [DOI] [PubMed] [Google Scholar]
- 31.Das Sarma J. 2010. A mechanism of virus-induced demyelination. Interdiscip Perspect Infect Dis 2010:109239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das Sarma J, Iacono K, Gard L, Marek R, Kenyon LC, Koval M, Weiss SR. 2008. Demyelinating and nondemyelinating strains of mouse hepatitis virus differ in their neural cell tropism. J Virol 82:5519–5526. doi: 10.1128/JVI.01488-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu D, Zhang X. 2006. Differential induction of proinflammatory cytokines in primary mouse astrocytes and microglia by coronavirus infection. Adv Exp Med Biol 581:407–410. doi: 10.1007/978-0-387-33012-9_73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marek R, Caruso M, Rostami A, Grinspan JB, Das Sarma J. 2008. Magnetic cell sorting: a fast and effective method of concurrent isolation of high purity viable astrocytes and microglia from neonatal mouse brain tissue. J Neurosci Methods 175:108–118. doi: 10.1016/j.jneumeth.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Das Sarma J, Fu L, Tsai JC, Weiss SR, Lavi E. 2000. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J Virol 74:9206–9213. doi: 10.1128/JVI.74.19.9206-9213.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das Sarma J, Meyer RA, Wang F, Abraham V, Lo CW, Koval M. 2001. Multimeric connexin interactions prior to the trans-Golgi network. J Cell Sci 114:4013–4024. [DOI] [PubMed] [Google Scholar]
- 37.Laing JG, Manley-Markowski RN, Koval M, Civitelli R, Steinberg TH. 2001. Connexin45 interacts with zonula occludens-1 and connexin43 in osteoblastic cells. J Biol Chem 276:23051–23055. doi: 10.1074/jbc.M100303200. [DOI] [PubMed] [Google Scholar]
- 38.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 39.Koval M, Harley JE, Hick E, Steinberg TH. 1997. Connexin46 is retained as monomers in a trans-Golgi compartment of osteoblastic cells. J Cell Biol 137:847–857. doi: 10.1083/jcb.137.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musil LS, Goodenough DA. 1991. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol 115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li W, Hertzberg EL, Spray DC. 2005. Regulation of connexin43-protein binding in astrocytes in response to chemical ischemia/hypoxia. J Biol Chem 280:7941–7948. doi: 10.1074/jbc.M410548200. [DOI] [PubMed] [Google Scholar]
- 42.Lampe PD, Kurata WE, Warn-Cramer BJ, Lau AF. 1998. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci 111(Part 6):833–841. [DOI] [PubMed] [Google Scholar]
- 43.Ezan P, Andre P, Cisternino S, Saubamea B, Boulay AC, Doutremer S, Thomas MA, Quenech'du N, Giaume C, Cohen-Salmon M. 2012. Deletion of astroglial connexins weakens the blood-brain barrier. J Cerebral Blood Flow Metab 32:1457–1467. doi: 10.1038/jcbfm.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ou JW, Kumar Y, Alioua A, Sailer C, Stefani E, Toro L. 2009. Ca2+- and thromboxane-dependent distribution of MaxiK channels in cultured astrocytes: from microtubules to the plasma membrane. Glia 57:1280–1295. doi: 10.1002/glia.20847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biswas K, Das Sarma J. 2014. Effect of microtubule disruption on neuronal spread and replication of demyelinating and nondemyelinating strains of mouse hepatitis virus in vitro. J Virol 88:3043–3047. doi: 10.1128/JVI.02545-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lavi E, Suzumura A, Hirayama M, Highkin MK, Dambach DM, Silberberg DH, Weiss SR. 1987. Coronavirus mouse hepatitis virus (MHV)-A59 causes a persistent, productive infection in primary glial cell cultures. Microb Pathog 3:79–86. doi: 10.1016/0882-4010(87)90066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orthmann-Murphy JL, Abrams CK, Scherer SS. 2008. Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci 35:101–116. doi: 10.1007/s12031-007-9027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Solan JL, Lampe PD. 2007. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membrane Biol 217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raaben M, Groot Koerkamp MJ, Rottier PJ, de Haan CA. 2007. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol 9:2218–2229. doi: 10.1111/j.1462-5822.2007.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin PC, Shen CC, Liao CK, Jow GM, Chiu CT, Chung TH, Wu JC. 2013. HYS-32, a novel analogue of combretastatin A-4, enhances connexin43 expression and gap junction intercellular communication in rat astrocytes. Neurochem Int 62:881–892. doi: 10.1016/j.neuint.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 51.Fong JT, Kells RM, Falk MM. 2013. Two tyrosine-based sorting signals in the Cx43 C-terminus cooperate to mediate gap junction endocytosis. Mol Biol Cell 24:2834–2848. doi: 10.1091/mbc.E13-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang T, Wan Y, Zhu Y, Fang X, Hiramatsu N, Hayakawa K, Paton AW, Paton JC, Kitamura M, Yao J. 2009. Downregulation of gap junction expression and function by endoplasmic reticulum stress. J Cell Biochem 107:973–983. doi: 10.1002/jcb.22202. [DOI] [PubMed] [Google Scholar]
- 53.Giepmans BN, Verlaan I, Hengeveld T, Janssen H, Calafat J, Falk MM, Moolenaar WH. 2001. Gap junction protein connexin-43 interacts directly with microtubules. Curr Biol 11:1364–1368. doi: 10.1016/S0960-9822(01)00424-9. [DOI] [PubMed] [Google Scholar]
- 54.Francis R, Xu X, Park H, Wei CJ, Chang S, Chatterjee B, Lo C. 2011. Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS One 6:e26379. doi: 10.1371/journal.pone.0026379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lavi E, Wang Q, Weiss SR, Gonatas NK. 1996. Syncytia formation induced by coronavirus infection is associated with fragmentation and rearrangement of the Golgi apparatus. Virology 221:325–334. doi: 10.1006/viro.1996.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. 2006. Astrocytes promote myelination in response to electrical impulses. Neuron 49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.May D, Tress O, Seifert G, Willecke K. 2013. Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of Connexin43 protein in astrocytes. J Neurosci 33:7985–7996. doi: 10.1523/JNEUROSCI.5874-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. 2003. Connexins are critical for normal myelination in the CNS. J Neurosci 23:5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Bock M, Kerrebrouck M, Wang N, Leybaert L. 2013. Neurological manifestations of oculodentodigital dysplasia: a Cx43 channelopathy of the central nervous system? Front Pharmacol 4:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloglu H, Nurnberg P, Hubner C, Weschke B, Gartner J. 2004. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet 75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bugiani M, Al Shahwan S, Lamantea E, Bizzi A, Bakhsh E, Moroni I, Balestrini MR, Uziel G, Zeviani M. 2006. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology 67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- 62.Tress O, Maglione M, Zlomuzica A, May D, Dicke N, Degen J, Dere E, Kettenmann H, Hartmann D, Willecke K. 2011. Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans. PLoS Genet 7:e1002146. doi: 10.1371/journal.pgen.1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chatterjee D, Biswas K, Nag S, Ramachandra SG, Das Sarma J. 2013. Microglia play a major role in direct viral-induced demyelination. Clin Dev Immunol 2013:510396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Markoullis K, Sargiannidou I, Schiza N, Hadjisavvas A, Roncaroli F, Reynolds R, Kleopa KA. 2012. Gap junction pathology in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol 123:873–886. doi: 10.1007/s00401-012-0978-4. [DOI] [PubMed] [Google Scholar]