

ABSTRACT
The core, conserved function of the herpesvirus gH/gL is to promote gB-mediated membrane fusion during entry, although the mechanism is poorly understood. The human cytomegalovirus (HCMV) gH/gL can exist as either the gH/gL/gO trimer or the gH/gL/UL128/UL130/UL131 (gH/gL/UL128-131) pentamer. One model suggests that gH/gL/gO provides the core fusion role during entry into all cells within the broad tropism of HCMV, whereas gH/gL/UL128-131 acts at an earlier stage, by a distinct receptor-binding mechanism to enhance infection of select cell types, such as epithelial cells, endothelial cells, and monocytes/macrophages. To further study the distinct functions of these complexes, mutants with individual charged cluster-to-alanine (CCTA) mutations of gH and gL were combined to generate a library of 80 mutant gH/gL heterodimers. The majority of the mutant gH/gL complexes were unable to facilitate gB-mediated membrane fusion in transient-expression cell-cell fusion experiments. In contrast, these mutants supported the formation of gH/gL/UL128-131 complexes that could block HCMV infection in receptor interference experiments. These results suggest that receptor interactions with gH/gL/UL128-131 involve surfaces contained on the UL128-131 proteins but not on gH/gL. gH/gL/UL128-131 receptor interference could be blocked with anti-gH antibodies, suggesting that interference is a cell surface phenomenon and that anti-gH antibodies can block gH/gL/UL128-131 in a manner that is distinct from that for gH/gL/gO.
IMPORTANCE Interest in the gH/gL complexes of HCMV (especially gH/gL/UL128-131) as vaccine targets has far outpaced our understanding of the mechanism by which they facilitate entry and contribute to broad cellular tropism. For Epstein-Barr virus (EBV), gH/gL and gH/gL/gp42 are both capable of promoting gB fusion for entry into epithelial cells and B cells, respectively. In contrast, HCMV gH/gL/gO appears to be the sole fusion cofactor that promotes gB fusion activity, whereas gH/gL/UL128-131 expands cell tropism through a distinct yet unknown mechanism. This study suggests that the surfaces of HCMV gH/gL are critical for promoting gB fusion but are dispensable for gH/gL/UL128-131 receptor interaction. This underscores the importance of gH/gL/gO in HCMV entry into all cell types and reaffirms the complex as a candidate target for vaccine development. The two functionally distinct forms of gH/gL present in HCMV make for a useful model with which to study the fundamental mechanisms by which herpesvirus gH/gL regulates gB fusion.
INTRODUCTION
Human cytomegalovirus (HCMV), an exemplar of the betaherpesvirus subfamily, is endemic in human populations and causes lifelong persistent infections (1–3). Primary infection of healthy individuals is usually subclinical and asymptomatic; however, in immunocompromised hosts, such as those infected with HIV or transplant recipients on antirejection therapies, primary infection or reactivation can have serious complications. Furthermore, maternal transmission of HCMV to the developing fetus across the placenta can lead to severe congenital birth defects. The diverse nature of HCMV-associated disease is likely related to the ability of the virus to infect many different cell types in the body, including epithelial and endothelial cells, fibroblasts, neurons, dendritic cells, hepatocytes, macrophages, and leukocytes (4–6). To understand the complex fusion machinery of HCMV, ample research has aimed to reconcile HCMV entry mechanisms for infection of various cell types (reviewed in reference 7). The bulk of these studies have revealed at least two distinct entry mechanisms between fibroblasts and epithelial/endothelial cells. It is likely that the mechanisms of entry into other cell types, some of which are difficult to culture in the laboratory, are identical or similar to the mechanisms of entry into either fibroblasts or epithelial/endothelial cells that have been described.
For all herpesviruses, glycoproteins gB and gH/gL make up the core fusion machinery necessary for entry. Herpesvirus genomes also encode accessory or auxiliary proteins that regulate tropism and interact stably or transiently with gH/gL (reviewed in reference 8). HCMV gH/gL exists in extracellular virions bound to either gO or the UL128, UL130, and UL131 (UL128-131) proteins. UL128-131-null mutants replicate well in fibroblast cultures but poorly enter epithelial, endothelial, or dendritic cells (9–12). In contrast, gO-null mutants display impaired entry into all cell types (13–16). Zhou et al. demonstrated that the amount of these two complexes in the virion envelope varies dramatically among different strains of HCMV (17). This may be partly explained by the findings of Murrell et al. indicating that nucleotide polymorphisms within the UL128-131 locus affect mRNA splicing and, thus, the amounts of the proteins available for assembly of the gH/gL/UL128-131 complex (18). Another factor may be the UL148 protein, which was recently characterized by Li et al. as an endoplasmic reticulum (ER) resident protein that influences the assembly of gH/gL/gO and gH/gL/UL128-131 (19). The amounts of gH/gL/gO and gH/gL/UL128-131 in the virion envelope affect the efficiency of entry of cell-free HCMV into different cell types. More recently, Zhou et al. showed that the efficiency of the fusion stage of entry into either fibroblasts or epithelial cells was dependent on abundant gH/gL/gO (20). In contrast, while infection of epithelial cells was dependent on the presence of gH/gL/UL128-131, it was not especially sensitive to the amount of the complex in the virion envelope (20). Together, these observations support a model in which gH/gL/gO is required to promote gB-mediated membrane fusion for infection of all cell types within the tropism range of HCMV, but entry into some cell types, such as epithelial/endothelial cells and some leukocytes, has an additional requirement for gH/gL/UL128-131, which likely acts through an earlier, distinct mechanism. The mechanism of gH/gL/UL128-131 may involve signaling through engagement of cell surface receptors (21–23). This model differs from that for Epstein-Barr virus (EBV), in which both gH/gL and gH/gL/gp42 promote gB-mediated fusion but do so on different cell types (8).
The specific role of the herpesvirus gH/gL in gB-mediated membrane fusion remains unclear. Crystal structures of gH/gL from herpes simplex virus 2 (HSV-2), EBV, and varicella-zoster virus (VZV) have revealed a similar core structure between viruses but do not offer clear pictures of mechanisms (24–26). Recent electron microscopic analysis of HCMV gH/gL/gO and gH/gL/UL128-131 has revealed a global structure of gH/gL similar to that of HSV-2, despite less than 25% amino acid sequence identity (27). Detailed structural and functional information for HCMV gH/gL is notably lacking, and little regarding the surfaces of HCMV gH/gL involved in their roles in either gH/gL/gO or gH/gL/UL128-131 is known. In order to identify the regions of HCMV gH/gL complexes critical for these distinct functions, we generated a library of gH/gL mutants and assessed their ability to promote gB-mediated membrane fusion. Additionally, we characterized their assembly, recognition by monoclonal antibodies, surface expression, and ability to form gH/gL/UL128-131 complexes capable of interfering with HCMV infection.
MATERIALS AND METHODS
Cell lines.
U373 human glioblastoma cells (American Type Culture Collection, Manassas, VA, USA) were grown in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% heat-inactivated bovine growth serum (BGS; Rocky Mountain Biologicals, Inc., Missoula, MT, USA). Retinal pigment epithelial cell line ARPE-19 (American Type Culture Collection, Manassas, VA, USA) was grown in a 1:1 dilution mix of DMEM and Ham's F-12 medium (DMEM–F-12; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Rocky Mountain Biologicals, Inc., Missoula, MT, USA.). Primary human foreskin fibroblasts (HFFs; Life Technologies) were grown in DMEM supplemented with 6% heat-inactivated FBS and 6% BGS. 293IQ cells (Microbix, Toronto, Ontario, Canada) were grown in minimum essential medium (MEM; Life Technologies) supplemented with 10% FBS.
HCMV.
HCMV strain TR was derived from the bacterial artificial chromosome (BAC) clone provided by Jay Nelson (Oregon Health and Sciences University, Portland, OR, USA) (28). Infectious HCMV was recovered by electroporation of BAC DNA into MRC-5 fibroblasts as described by Wille et al. (16). Cell-free HCMV was produced by infecting HFFs at 2 PFU per cell. At 8 to 10 days postinfection (when cells were still visually intact), culture supernatants were harvested and cellular contaminants were removed by centrifugation at 1,000 × g for 10 min and again at 6,000 × g for 10 min. Stocks were judged to be cell free by the lack of calnexin and actin in immunoblot analyses and stored at −80°C. Freeze-thaw cycles were avoided.
Ad vectors.
Replication-defective (E1-negative) adenovirus (Ad) vectors that express HCMV TR gH, gL, UL128, UL130, UL131, and gB were previously described (17, 29, 30). Charged cluster-to-alanine (CCTA) mutagenesis was performed by GeneArt (Life Technologies). Ad vector stocks were generated by infecting 293IQ cells at 0.1 PFU/cell for 6 to 10 days. The cells were then pelleted by centrifugation at 600 × g for 15 min, resuspended in DMEM containing 2% FBS, sonicated to release cell-associated virus, and then centrifuged at 3,000 × g for 5 min to clear the cellular debris. Ad stock titers were determined by plaque assay on 293IQ cells. Multiplicities of infection (MOIs) for Ad vectors were determined empirically for each experiment and ranged from 3 to 30 PFU per cell. Because protein expression can vary between stocks of Ad vectors, experiments were performed with Ad vectors at different MOIs to account for the possible effects of under- or overexpression of proteins.
Antibodies.
Monoclonal antibodies (MAbs) specific for HCMV gH (MAbs AP86 and 14-4b) and IE1/IE2 (MAb P63-27) were provided by Bill Britt (University of Alabama, Birmingham, AL) (31–33). Rabbit polyclonal antipeptide antibody against HCMV gL was provided by David Johnson (Oregon Health and Sciences University, Portland, OR) (29).
Immunoprecipitation.
Cell proteins were extracted with Tris-buffered saline extraction buffer (20 mM Tris, pH 6.8, 100 mM NaCl, 1% Triton X-100) at a density of 106 cells/ml and clarified by centrifugation at 16,000 × g for 30 min at 4°C. Immunoprecipitation involved the addition of an MAb hybridoma supernatant overnight, followed by incubation with protein A/G magnetic beads (Pierce/Thermo) for an additional 2 h at 4°C. Captured proteins were eluted from the beads in denaturing gel loading buffer containing 2% SDS and 25 mM dithiothreitol.
Immunoblotting.
Protein samples were separated by SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene difluoride membranes (Bio-Rad) in a buffer containing 25 mM Tris, 192 mM glycine, and 10% methanol. Transferred proteins were detected by immunoblotting with mouse MAbs or rabbit antipeptide antibodies as previously described (17).
Immunofluorescence.
Cells were fixed with 2.5% formaldehyde and permeabilized using phosphate-buffered saline (PBS) containing 0.5% Triton X-100, 0.5% sodium deoxycholate, 1% bovine serum albumin (BSA), and 0.05% sodium azide. Cells were then stained with mouse monoclonal anti-HCMV IE1/IE2 antibody P63-27, followed by Alexa Fluor 488-conjugated anti-mouse immunoglobulin antibody. Cell nuclei were stained with 0.4 μM 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) as described previously (12).
Confocal microscopy.
Cells were seeded in Lab-Tek II 8-well chamber slides (Nunc) and stained for 15 min at 24°C with 10 μg/ml of wheat germ agglutinin conjugated to Alexa Fluor 488 and 2.5 μg/ml Hoechst 33342 (Invitrogen) in HEPES-buffered saline (HBS). After a wash with HBS, the cells were fixed for 15 min at 24°C in 2% formaldehyde in PBS, pH 7.3, followed by several HBS washes. The slides were analyzed with an Olympus FV1000 confocal laser scanning microscope under a 60× oil immersion objective. Digital images were acquired with FluoView software (Olympic Life Science).
Cell-cell fusion.
Cell-cell fusion experiments were performed using ARPE-19 epithelial cells as previously described (30). Briefly, ARPE-19 cells were seeded in 96-well plates and allowed to grow to confluence, and then the cells were transfected with Ad vectors expressing the HCMV gB, gH, and gL proteins as well as enhanced green fluorescent protein for fluorescent visualization of syncytia. Fusion was scored as positive or negative on the basis of observation of syncytia at 48 h postinfection. To control for differences in expression levels between mutant gH/gL combinations, positive or negative fusion for each mutant gH/gL was determined on the basis of at least three independent experiments, each of which involved the titration of gH/gL mutant expression (Ad vector multiplicities).
Cell surface expression of mutant and wild-type gH/gL.
ARPE-19 epithelial cells were seeded in 96-well cell-based enzyme-linked immunosorbent assay (CELISA) culture plates (white wall, clear bottom) and transduced with Ad vectors expressing wild-type or mutant gH/gL combinations. Specific mutants were also tested with Ad vectors expressing the UL128-131 proteins. To measure cell surface gH, cells were incubated for 1 h with AP86, fixed for 30 min, and then incubated for 45 min with secondary antibody. Cells were washed between steps with PBS supplemented with 1% BSA and 5% FBS. Two minutes prior to data collection, wells were incubated with SuperSignal enzyme-linked immunosorbent assay (ELISA) Femto substrate (Thermo), and then chemiluminescence was measured on a BioTek plate reader.
Receptor interference.
Interference assays were performed using a modification of a previously described method (21). Briefly, ARPE-19 epithelial cells were seeded into 96-well culture plates and transduced with Ad vectors expressing a combination of wild-type or mutant gH/gL and the UL128-131 proteins. After 24 h of incubation with or without HCMV-neutralizing antibodies, HCMV strain TR was added onto cells at a dose sufficient to infect approximately 50% of control cells (i.e., cells not transduced with Ad). Forty-eight hours later, HCMV infection was analyzed by immunofluorescence detection of IE1/IE2.
Homology modeling of HCMV gH structure.
A structural model for residues 199 to 724 of HCMV gH (strain TR) was generated using the program MODELLER (34). The published crystal structures of EBV gH (26) and HSV-2 gH (PDB accession numbers 3PHF and 3M1C, respectively) were used as homolog templates, with EBV gH yielding a more ordered and convincing HCMV gH structure model. Model representations were rendered using the UCSF Chimera package. Chimera was developed and is maintained by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
RESULTS
CCTA scanning mutagenesis of HCMV gH/gL.
A library of mutants with charged cluster-to-alanine (CCTA) mutations was generated by identifying regions within the gH and gL polypeptide sequences that contain extensive amounts of charged amino acids and replacing those charged residues with alanine. Eight charged clusters were targeted within gH, and another eight were targeted within gL. The location of the first residue of each charged cluster is indicated in Fig. 1, and the full sequences of the regions mutated are shown in Table 1. Note that the linear epitope for the HCMV-neutralizing MAb AP86 (amino acids 34 to 43) (33) was not affected by any of the mutations within the gH polypeptide (Fig. 1A). Also note that Cys144 of the gL polypeptide, which has been reported to make a disulfide bond with either gO or UL128 (27), falls within the mutated region beginning at residue 139, but the cysteine itself is left intact (Fig. 1B and Table 1). Each mutant gH and gL was cloned into an adenovirus (Ad) expression vector and is denoted by the numerical position of the first mutated residue of the cluster. Thus, combinatorial expression of wild-type and mutant gH and gL variants yielded a library of 80 distinct gH/gL mutants, each of which contained a mutation within either the gH subunit or the gL subunit, or both.
FIG 1.

CCTA scanning mutagenesis of HCMV gH/gL. Schematic representation of HCMV gH (A) and gL (B) indicating the starting residue of each mutated charged cluster. The designation of gH domains D-I to D-IV was based on homology with the domain designations of EBV gH described by Matsuura et al. (26). Also indicated are the predicted signal peptides (sp), the transmembrane (tm) region of gH, the linear epitope of the neutralizing anti-gH MAb AP86 (33), and cysteine 144 of gL, which was reported by Ciferri et al. to form a disulfide linkage with either gO or UL128 (27). N-term, N terminus; aa, amino acids.
TABLE 1.
CCTA substitutions of HCMV gH and gL

a Enlarged letters (gray) represent charged residues replaced by alanine.
Mutational disruption of the ability of gH/gL to promote gB-mediated membrane fusion.
Although in extracellular HCMV virions gH/gL is mostly bound to either gO or the UL128-131 proteins, gH/gL alone is sufficient to promote gB-mediated membrane fusion in cell-cell fusion experiments (17, 30). To identify critical functional surfaces of gH/gL, mutants were screened for the ability to promote gB-mediated cell-cell fusion. Briefly, the cells were transduced with Ad vectors to express gB and gH/gL (the wild type or each of the mutant gH/gL combinations), and cell-cell fusion was assessed 48 h later (Fig. 2A.) Consistent with the findings of Vanarsdall et al., gB expression alone did not induce cell-cell fusion, but when wild-type gH/gL was coexpressed, large syncytia were readily apparent (30) (Fig. 2A). Fusion could be blocked by the HCMV-neutralizing, anti-gH MAb 14-4b (Fig. 2A) or AP86 (not shown). Mutant gH/gL proteins were scored as positive or negative for fusion on the basis of the results of at least three experiments (Fig. 2B). There were no obvious intermediate phenotypes in terms of the size of the syncytia. Eight gH/gL mutants supported cell-cell fusion indistinguishable from that of wild-type gH/gL. The remaining 72 mutants were negative. Five of the eight gH mutations (H.111, H.271, H.335, H.481, H.671) impaired cell-cell fusion irrespective of which gL was coexpressed. Likewise, six of eight mutants with gL mutations (the L.46, L.63, L.156, L.201, L.230, and L.256 mutants) were nonfunctional irrespective of which gH was coexpressed. In contrast, mutant L.139 was indistinguishable from wild-type gL, and mutant L.244 was functional only with wild-type gH. Of particular note, three gH mutants (the H.188, H.402, and H.510 mutants) were fusion competent when paired with wild-type gL (or mutant L.139). However, each of these three gH mutants was defective when paired with the otherwise functional mutant L.244 (i.e., the combinations of mutations H.188/L.244, H.402/L.244, and H.510/L.244).
FIG 2.
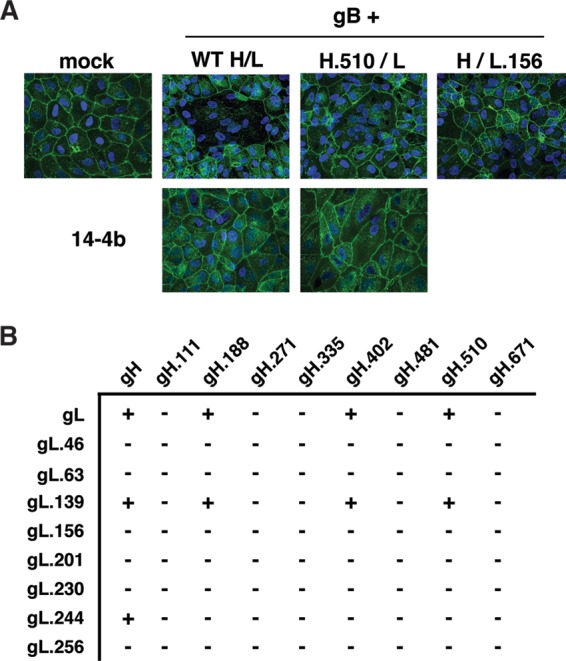
Effect of mutation on cell-cell fusion induced by gB and gH/gL. (A) ARPE-19 cells were transduced with gB and wild-type (WT) or mutant gH/gL in either the presence or absence of MAb 14-4b. Syncytium formation was analyzed by confocal fluorescence microscopy to detect Hoechst 33342-stained nuclei (blue) and wheat germ agglutinin-stained plasma membranes (green). (B) The results of cell-cell fusion assays for all mutant gH and gL combinations. +, gH/gL mutant combinations that resulted in syncytium formation comparable to that for wild-type gH/gL; −, mutant combinations that were impaired for fusion in at least three independent experiments.
Since sequences rich in charged amino acids tend to represent the exposed surfaces of folded proteins rather than the internal core structures, CCTA mutagenesis was expected to disrupt surface-exposed regions of gH/gL while leaving the core heterodimerization intact. To assess weather CCTA mutagenesis grossly affected protein folding and gH/gL dimerization, wild-type and mutant gH and gL were expressed in each of the 80 combinations and coimmunoprecipitated using either of two HCMV-neutralizing anti-gH antibodies. The MAb AP86 recognizes a short, continuous sequence of gH just downstream of the signal peptide (Fig. 1A) (33), while the MAb 14-4b recognizes a previously uncharacterized conformation-dependent epitope on gH (31, 32). Both AP86 and 14-4b captured wild-type gH/gL or wild-type gH monomers with similar efficiencies (Fig. 3A). All 80 of the gH/gL mutants were efficiently pulled down with AP86, suggesting that mutagenesis did not grossly misfold gH or gL enough to affect heterodimerization. All mutant heterodimers containing the H.111, H.188, H.402, and H.510 mutations were also efficiently immunoprecipitated with 14-4b, suggesting that the conformation of the 14-4b epitope was not affected in these cases (Fig. 3B; results for representative mutant H.402/L.244 are shown). However, gH/gL mutants containing either the H.271 or H.335 mutation were less efficiently immunoprecipitated with 14-4b (Fig. 3C; results for representative mutant H.335/L are shown), and mutants containing either the H.481 or H.671 mutation were completely unrecognized by 14-4b (Fig. 3D; results for representative mutant H.671/L.156 are shown). These results were not affected by the gL subunit expressed, consistent with the notion that the 14-4b epitope is entirely contained on the gH polypeptide and is not dependent on gL for its conformation.
FIG 3.
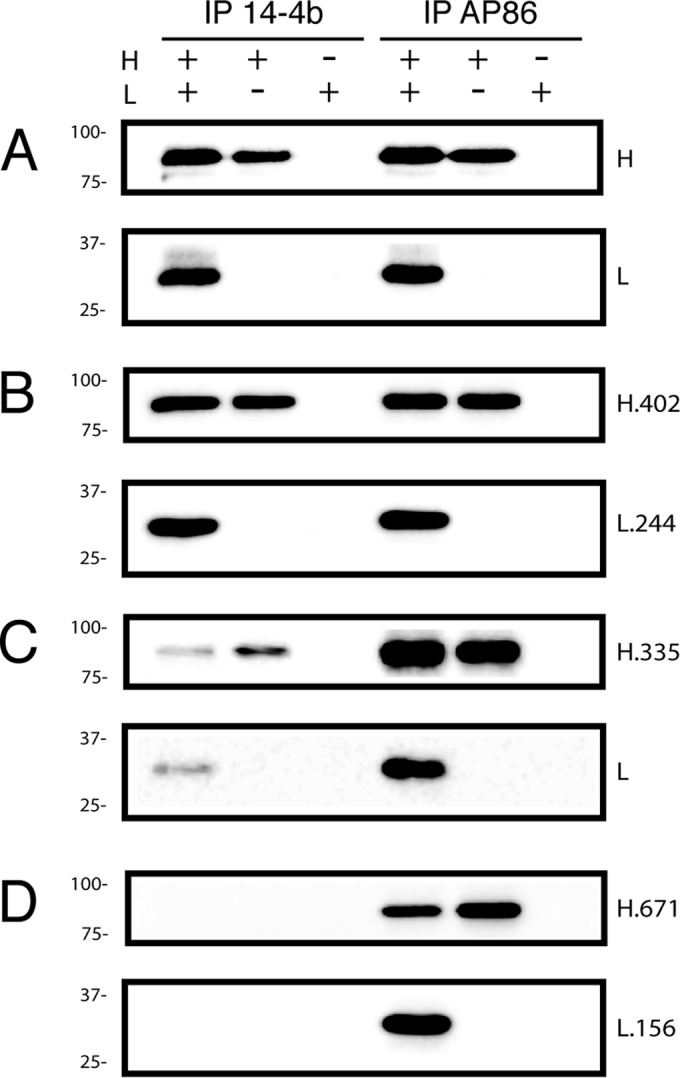
Heterodimerization of gH and gL mutants. Human glioblastoma (U373) cells were transduced with the indicated combination of Ad vectors expressing wild-type (A) or mutant (B to D) gH and gL. Cell extracts were immunoprecipitated (IP) with MAb 14-4b or AP86, separated by reducing SDS-PAGE, and identified by immunoblotting with anti-gH or anti-gL antibodies. Mass standards (in kilodaltons) are indicated to the left of each panel.
Failure of the gH/gL mutants to function in the cell-cell fusion assays might be explained by a lack of surface expression. Although wild-type gH/gL of HCMV is inefficiently exported from the ER unless it is bound by UL128-131 or gO (29, 35), clearly, enough can accumulate on cell surfaces during Ad vector expression to promote gB-mediated cell-cell fusion. Cell surface expression of wild-type or mutant gH/gL dimers was compared by cell-based ELISA (CELISA) (Fig. 4). Approximately 8-fold more gH was found on the cell surface when both wild-type gH and gL were expressed than when gH was expressed without gL. As expected, all of the gH/gL mutants that were positive for fusion were detected at the cell surface (black columns in Fig. 4A to C). The gH mutants that were negative in the fusion assay were expressed poorly at the surface (white columns in Fig. 4A). In contrast, several of the fusion-negative gL mutants were detected on the cell surface at levels comparable to those for wild-type gH/gL (Fig. 4B, L.46, L.63, and L.156 mutants). Other fusion-negative gL mutants were detected on cell surfaces at lower levels (Fig. 4B, L.201, L.230, and L.256 mutants), as was the fusion-positive mutant L.244 (Fig. 4B). The fact that the L.244 mutant was fusion positive, despite reduced surface levels, might indicate that the L.244 mutation enhances fusion. However, given the binary readout of the cell-cell fusion assay, it seems more likely that this reflects a low threshold requirement for surface gH/gL in cell-cell fusion. Interestingly, while the H.510 and L.244 single mutants were each fusion competent, the H.510/L.244 double mutant was fusion negative (Fig. 2). This was unlikely due to a lack of surface expression, since CELISA indicated higher surface expression for the H.510/L.244 double mutant than for the fusion-positive wild-type H/L.244 single mutant (approximately 9,000 and 6,000 relative light units, respectively; Fig. 4B and C).
FIG 4.

Cell surface expression of wild-type and mutant gH/gL complexes. ARPE-19 epithelial cells expressing wild-type or mutant gH/gL complexes were analyzed for surface expression by CELISA using anti-gH MAb AP86. Shown are the numbers of relative light units (RLU) for individual gH mutants (A), individual gL mutants (B), and representative combinations (C). Black and white bars, mutants that tested positive and negative for cell-cell fusion, respectively; horizontal dashed lines, the threshold for surface expression, as defined by the surface detection of gH expressed without gL. Experiments were performed in triplicate, and error bars represent standard errors.
gH/gL mutants support gH/gL/UL128-131 receptor interference.
Transient expression of gH/gL/UL128-131 renders epithelial cells resistant to HCMV infection. This phenomenon, known as receptor interference, is likely due to the sequestration or saturation of gH/gL/UL128-131 receptors by gH/gL/UL128-131 expressed within the cell. Because expression of all of the gH, gL, and UL128-131 proteins is required for this effect, interference is an indicator for the assembly of gH/gL/UL128-131 complexes in a biological environment (i.e., within a cell, as opposed to a detergent extract) (21). To analyze the effects of gH/gL mutations on gH/gL/UL28-131, epithelial cells were transduced with Ad vectors to express a gH/gL mutant either alone or together with the UL128-131 proteins, and subsequent HCMV infection was determined by immunofluorescence detection of immediate early gene expression. Consistent with the findings described in a previous report (21), neither wild-type gH/gL alone nor the UL128-131 proteins alone interfered with subsequent HCMV infection, but when they were expressed together, cells were highly resistant to HCMV infection (Fig. 5A). Mutants that displayed efficient surface expression as gH/gL dimers (Fig. 4), including those that failed to promote gB-mediated cell-cell fusion, reproducibly supported gH/gL/UL128-131 interference (Fig. 5B). Mutants containing L.139 showed no apparent deficiency in supporting gH/gL/UL128-131 interference (Fig. 5B). Note that Cys144 of gL, which was shown to make a disulfide bond either with gO or with UL128 (27), was within the region of L.139 mutations, but Cys144 itself was left intact.
FIG 5.

Effect of gH/gL mutation on assembly of gH/gL/UL128-131. (A) ARPE-19 epithelial cells expressing wild-type or mutant gH/gL heterodimers alone or together with UL128-131 proteins were inoculated with HCMV, and infection was assessed by immunofluorescence detection of immediate early gene expression. (B) Relative HCMV infection was calculated as the percentage of HCMV-infected cells when cells expressed wild-type or mutant gH/gL plus UL128-131 divided by the percentage of HCMV-infected cells when cells expressed only the wild-type or mutant gH/gL dimer. Black bars, gH/gL mutants that were positive for promoting gB-mediated cell-cell fusion; white bars, mutants that were negative for fusion. Representative mutant gH/gL/UL128-131 complexes were tested in triplicate, and error bars represent standard errors.
Despite the finding that mutant H.671/L exhibited poor surface expression as a dimer (Fig. 4A), it was capable of supporting gH/gL/UL128-131 interference (Fig. 5B). This suggested the possibility that UL128-131 might rescue surface expression. The UL128-131 proteins were coimmunoprecipitated with mutant H.671/L using the anti-gH MAb AP86 (Fig. 6A). Note that the gL and UL130 associated with mutant H.671 displayed band migration patterns different from those that they displayed when they were associated with wild-type gH. Surface expression for the wild-type and mutant H.671 pentamers was also measured by CELISA (Fig. 6B). As expected, an increase in surface expression was observed for both the wild-type and mutant H.671 pentamers compared to the surface expression observed for the respective wild-type and mutant gH/gL dimers alone (compare Fig. 4A and 6B). However, the surface expression of the mutant H.671 pentamer was lower than that of the wild-type pentamer. Together with the distinct band patterns of gL and UL130 (which likely reflected differences in glycan processing associated with transit from the ER through the Golgi complex), these data are consistent with the notion that pentamer formation leads to an increase in protein maturation and surface expression and that the H.671 mutation reduces the efficiency of this process.
FIG 6.
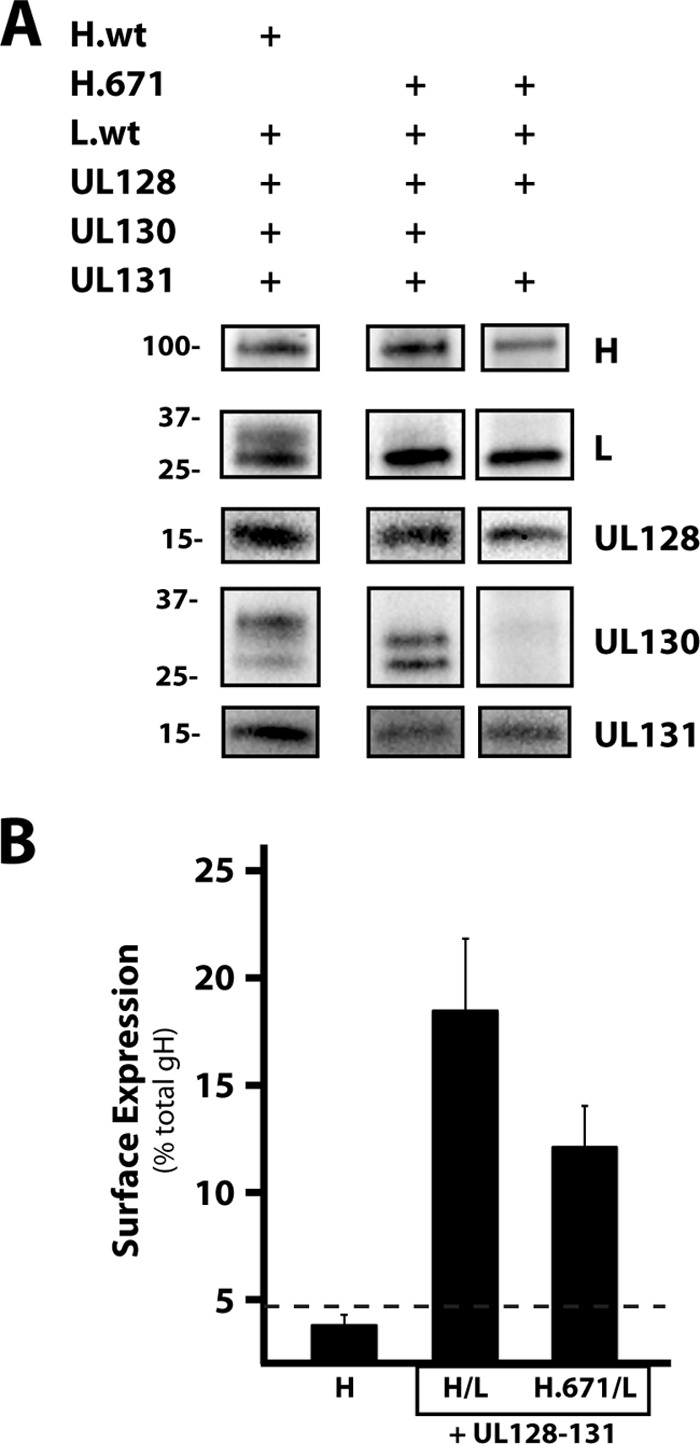
Coimmunoprecipitation and surface expression of H.671/gL/UL128-131. (A) ARPE-19 cells expressing either wild-type gH/gL/UL128-131 or H.671/gL/UL128-131 (or H.671/gL/UL128-131 lacking UL130) were immunoprecipitated with the anti-gH antibody AP86. Samples were separated by SDS-PAGE and analyzed by immunoblotting. Mass markers (in kilodaltons) and protein labels are displayed on the left and right, respectively. H.wt and L.wt, wild-type gH and gL, respectively. (B) Surface expression of wild-type gH/gL and H.671/L when expressed with UL128-131, measured by CELISA. Bars display the amount of surface gH as a percentage of the total amount of gH. Horizontal dashed line, the threshold for surface expression, as defined by the surface detection of gH expressed without gL, Error bars represent standard errors from three experiments.
Anti-gH antibodies can recover the HCMV susceptibility of gH/gL/UL128-131-expressing epithelial cells.
Transient expression of gH/gL/UL128-131 might induce interference by binding a putative receptor(s) in the ER or in the secretory pathway and inhibiting its surface expression. Alternatively, it might be necessary for transiently expressed gH/gL/UL128-131 to first traffic to the cell surface and either bind the receptor(s) there or bind it subsequently within recycling endosomes. To address whether gH/gL/UL128-131–receptor interference involved surface expression, cells expressing gH/gL/UL128-131 complexes were incubated with the MAb 14-4b or AP86 and the level of interference was assessed (Fig. 7). In these experiments, expression of wild-type gH/gL/UL128-131 reduced HCMV infection from 52% to 3% (Fig. 7A and B), and both 14-4b and AP86 were able to recover HCMV infection in cells to 10% and 12%, respectively (Fig. 7H and K). A similar level of interference was observed with mutant H.671/gL/UL128-131 (Fig. 7C), and MAb AP86 recovered HCMV infection to 16% (Fig. 7L), but 14-4b was unable to recover HCMV infection (Fig. 7I). This was consistent with the observations that 14-4b failed to immunoprecipitate any mutants containing the H.671 mutation (Fig. 3D). Together, these results indicate that the anti-gH antibodies 14-4b and AP86 are capable of blocking gH/gL/UL128-131 interference (i.e., recover HCMV infection in gH/gL/UL128-131-expressing epithelial cells), suggesting a cell surface mechanism (see the schematic model in Fig. 8).
FIG 7.
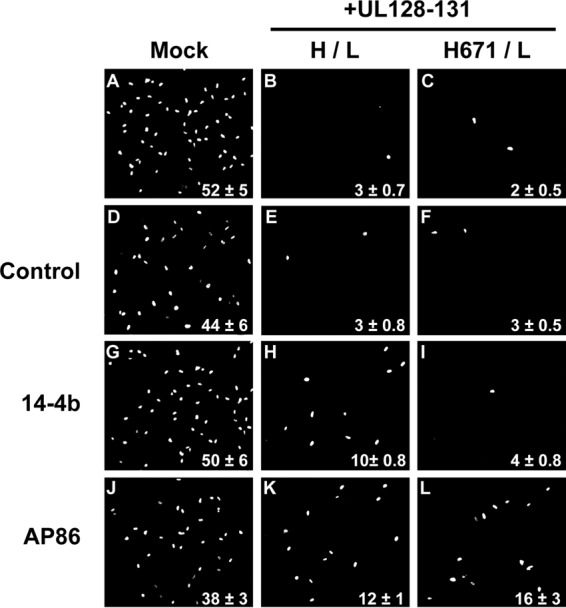
Effect of anti-gH antibodies on receptor interference. (A to C) ARPE-19 cells expressing either the gH/gL/UL128-131 or H.671/gL/UL128-131 complex were inoculated with HCMV, and infection was assessed by immunofluorescence detection of immediate early gene expression. (D to L) Replicate wells were incubated with isotype control IgG (D to F), 14-4b (G to I), or AP86 (J to L) for 24 h prior to HCMV infection. Experiments were performed in triplicate, and the percentage of immediate early gene-positive cells is displayed in the bottom-right corner for each condition.
FIG 8.

Model of gH/gL/UL128-131 receptor interference and blocking by anti-gH antibodies. Step 1, gH/gL/UL128-131 expressed from Ad vectors in epithelial cell is transported to cell surfaces; step 2, interactions with putative receptor molecules render cells resistant to infection by wild-type HCMV; step 3, the addition of anti-gH antibodies to the culture medium prevents interaction between gH/gL/UL128-131 and putative receptors; step 4, cells bearing free putative gH/gL/UL1281-131 receptors are susceptible to HCMV infection.
DISCUSSION
Early characterizations of HSV showing that, without gL, gH was not fully processed or incorporated into progeny virions led to a view of gL as a chaperone for gH (28, 36). In contrast to that view, other analyses of HSV, pseudorabies virus (PRV), and EBV have suggested a more direct role for gL in the fusion function of gH/gL (37–40). The solved crystal structures of EBV and HSV gH/gL provided important insights by showing a very intimate association between gH and gL (25, 26). Thus, another view is that gH and gL are two polypeptide subunits of a single, integrated protein, gH/gL. This makes distinguishing the roles of gH and gL difficult since they are likely mutually required for folding into a functional protein and mutations in one subunit may very well affect the conformation of the other. The mutagenesis strategy reported herein reflects this holistic view of gH/gL by introducing CCTA mutations into the gH and gL subunits of HCMV and then combining the mutant subunits to generate a large set of distinct gH/gL mutants.
Unlike the genomes of alphaherpesviruses, the genomes of beta- and gammaherpesviruses encode proteins that bind the ectodomain of gH/gL, such as gp42 of EBV and gO and the UL128-131 proteins of HCMV. EBV gH/gL can exist in the virion envelope either as unbound gH/gL or as gH/gL/gp42 (reviewed in reference 8). For entry into epithelial cells, EBV gH/gL binds select integrins though a canonical integrin-binding motif (KGD) present on the gH subunit, and conformational changes resulting from this interaction may facilitate gB-mediated membrane fusion on these cells (41, 42). gp42 binding to gH/gL blocks the KDG integrin-binding motif and facilitates interactions with major histocompatibility complex class II on the surfaces of B cells, which promote fusion on these cells (26, 43–45). In both cases, it is likely that gH/gL surfaces are directly involved in the fusion reaction with gB, with gp42 acting as a receptor adaptor that alters specificity and, potentially, the structural rearrangements that occur in gH/gL during the fusion reaction (45). HCMV gH/gL complexes are similar in that they exist in two forms that influence cell tropism, but unlike EBV, only one, gH/gL/gO, appears to function in promoting gB fusion, whereas the other, gH/gL/UL128-131, likely acts through a distinct mechanism to enhance infection of select cell types (20). The specific role of gO is unclear since gH/gL alone is sufficient to promote gB-mediated membrane fusion in cell-cell fusion experiments (30), and this likely involves the binding of cell surface molecules as receptors (30, 46, 47). It has been suggested that HCMV entry involves gH/gL binding to integrins (48). Indeed this parallels the reports of integrin binding by HSV and EBV gH/gL (41, 42, 49), but unlike HSV and EBV gH/gL, HCMV gH/gL lacks a clear RGD or KGD integrin-binding motif.
The goal of our CCTA mutagenesis was to disrupt surface-exposed regions of the gH/gL heterodimer without affecting the folding of the hydrophobic protein core, which would result in globally misfolded, uninformative mutants. The fact that all gH/gL combinations were readily expressed and could be coimmunoprecipitated as dimers argues against the notion of global misfolding because it seems unlikely that grossly misfolded proteins would bind their interaction partners, especially in cases where that interaction partner itself was not mutated. The ability of the conformation-dependent antibody 14-4b to recognize the mutants further supports this notion. Interestingly, two mutations, H.271 and H.335, displayed reduced 14-4b binding in coimmunoprecipitation analyses, suggesting that in these cases mutagenesis indeed perturbed the structure of gH/gL but did not misfold the 14-4b epitope beyond recognition. On the other hand, mutations H.481 and H.671 resulted in a total loss of 14-4b binding. While it is possible that these mutations reflect global misfolding, the fact that the mutants with these mutations formed dimers with gL and, in the case of H.671, formed gH/gL/UL128-131 complexes that were readily detected at the cell surface and induced receptor interference argues otherwise. It seems more likely that these mutations specifically affected regions near the 14-4b epitope.
Atomic resolution structures are available for portions of gH/gL from EBV, HSV-2, PRV, and VZV (24–26, 50). It is likely that HCMV gH/gL shares structural similarities with these homologous proteins. Indeed, recent electron microscopy analyses by Ciferri et al. suggest that HCMV gH/gL adopts a boot-like global structure similar to that of HSV-2 gH/gL (27). This structure is in an orientation governed by the position of the N-terminal domain I of gH relative to the rest of the protein and is likely influenced by interactions with gL (27). We used a computational approach to generate a structure model for a portion of HCMV gH lacking the N-terminal 198 amino acids (Fig. 9A). Existing structures for both EBV gH and HSV-2 gH were used as candidate homolog templates, and EBV yielded a more ordered and convincing structural model corresponding to domains II, III, and IV of EBV gH. The better modeling on EBV gH may reflect the fact that HCMV gH shares 26.6% sequence identity with EBV gH in this region but 21.8% identity with HSV-2 gH. Sequences targeted by CCTA mutations were located on the HCMV gH structural model (Fig. 9B). Interestingly, the mutations H.481 and H.671 are predicted to reside within a large groove between domains III and IV in the homology model. This large groove is homologous to the one described for HSV-2 gH/gL, which contains the 52S epitope, and has been implicated in the direct binding and activation of gB (51). It is possible that this region of HCMV gH/gL is mechanistically analogous, and this might help explain the neutralizing activity of MAb 14-4b. Furthermore, this putative 14-4b epitope is predicted to be very near the epitopes of other HCMV-neutralizing anti-gH antibodies (52, 53).
FIG 9.
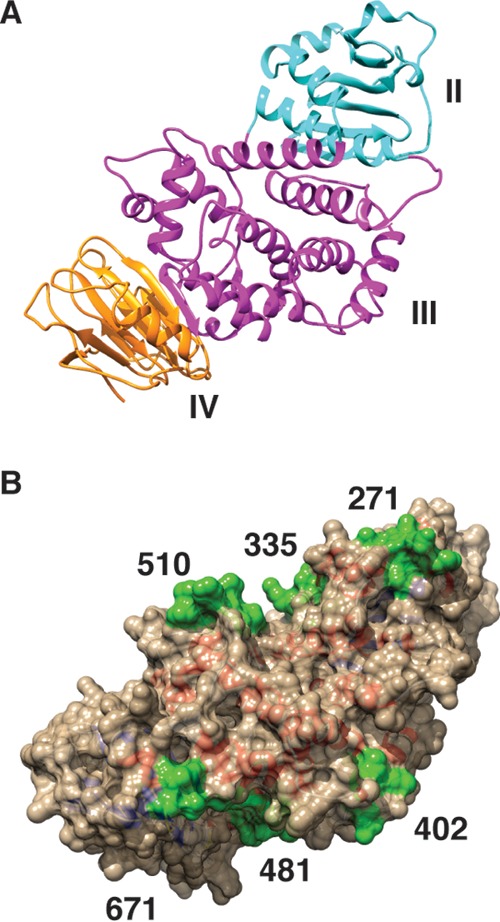
Homology model of HCMV gH. HCMV gH (residues 199 to 724; strain TR) was computationally analyzed using the solved structure of EBV gH as a homolog template in MODELLER. (A) Ribbon representation of the predicted HCMV gH structure, with domain assignments corresponding to those of EBV gH. Cyan, domain II; magenta, domain III; orange, domain IV. (B) Space-filling representation of the HCMV gH structure model with the locations of CCTA mutagenesis highlighted in green.
The failure of some mutants to promote fusion was explained by reduced cell surface expression, but several fusion-impaired mutants were detected on cell surfaces at levels similar to the level of wild-type gH/gL. All mutants with mutations in gH that resulted in a lack of fusion displayed greatly reduced levels of surface expression. In contrast, several mutants with gL mutations were defective for fusion but displayed surface expression levels comparable to the level for wild-type gH/gL. These observations suggest that the surfaces of gH may be more critical for proper maturation and surface expression, while regions near the gH-gL interface may directly participate in fusion either by binding to receptors or by interacting with gB. The H.510 and L.244 mutants were functional in fusion experiments when paired with their wild-type counterparts but were defective when paired with one another (H.510/L.244). One interpretation is that this combination of gH and gL mutations disrupted the conformation of critical surfaces distinct from the exact mutation sites, highlighting the interdependence of gH and gL for proper folding and function. On the other hand, many mutations were sufficient to result in a fusion defect regardless of their counterpart subunit. For example, mutants with the L.156 and L.201 mutations each failed to promote gB-mediated membrane fusion regardless of which gH was coexpressed. This may indicate that gL surfaces are directly involved in the mechanism of fusion. However, since gH and gL are likely mutually dependent on one another for proper folding, it is also possible that these gL mutations distorted critical surfaces on gH. The library of gH/gL mutants will next be tested for their ability to load gO and function in the context of HCMV replication.
Many of the mutants supported the assembly of gH/gL/UL128-131 complexes, as judged by receptor interference analyses. This was a surprising result and stands in stark contrast to the reports that similar CCTA mutagenesis of the UL128-131 proteins greatly disrupted gH/gL/UL128-131 (54–56). Given that gH/gL/UL128-131 likely binds cell type-specific receptors to facilitate infection (21, 22), these observations suggest that the receptor-binding surfaces lie on UL128-131 themselves and gH/gL may serve as a scaffold or platform for presentation of the UL128-131 proteins. A recent analysis by Ciferri et al. indicated that binding of UL128-131 to gH/gL involves a disulfide bond between gL-Cys144 and UL128-Cys162 (27). Even though Cys144 falls within the alanine cluster of the L.139 mutant, no substantial loss of HCMV interference was observed when this mutant was expressed as a gH/gL/UL128-131 complex. It could be that the gL-UL128 interaction does not require charged residues and may be governed by hydrophobic surfaces to preserve the core structure.
The observation that receptor interference could be at least partly blocked (i.e., HCMV infection could be recovered) by neutralizing, anti-gH antibodies may have wider implications for the mechanisms of neutralization on specific cell types. Previous studies have reported that anti-gH antibodies block HCMV infection more effectively on epithelial cells than on fibroblasts (57, 58). The analysis reported herein demonstrates that anti-gH antibodies can block gB-mediated cell-cell fusion independently of receptor-based HCMV interference, suggesting that these antibodies may be capable of neutralizing the distinct mechanisms of both the trimer and pentamer.
In conclusion, HCMV gH/gL likely shares many structural and mechanistic characteristics with gH/gL of other herpesviruses, but there are clearly some important differences. This study reports the generation of a large library of HCMV gH/gL mutants and their functional characterization. Further analysis of this mutant library will help evaluate HCMV gH/gL complexes as vaccine targets, as well as shed light on their mechanisms of action.
ACKNOWLEDGMENTS
We are grateful to Bill Britt for providing antibodies, Scott Wetzel and Lou Herritt of the University of Montana Center for Environmental Health Sciences Confocal Microscopy Core Facility for technical assistance, and Momei Zhou and Qin Yu for insightful discussions and critical reviews of the manuscript. We also thank Tiffani Howard and David Johnson for allowing us to adapt schematic cartoon images.
B.J.R. is supported by grants from the National Institutes of Health (5-R01-AI097274-02 and PG20GM103546).
The experiments were designed by E.P.S., J.-M.L., and B.J.R. Experiments were performed by E.P.S., J.-M.L., and E.E.E. Data were analyzed by E.P.S., J.-M.L., E.E.E., and B.J.R. The manuscript was prepared by E.P.S., J.-M.L., and B.J.R.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Britt WJ. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. [DOI] [PubMed] [Google Scholar]
- 2.Boppana SB, Fowler KB, Pass RF, Rivera LB, Bradford RD, Lakeman FD, Britt WJ. 2005. Congenital cytomegalovirus infection: association between virus burden in infancy and hearing loss. J Pediatr 146:817–823. doi: 10.1016/j.jpeds.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 3.Streblow DN, Orloff SL, Nelson JA. 2007. Acceleration of allograft failure by cytomegalovirus. Curr Opin Immunol 19:577–582. doi: 10.1016/j.coi.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plachter B, Sinzger C, Jahn G. 1996. Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res 46:195–261. doi: 10.1016/S0065-3527(08)60073-1. [DOI] [PubMed] [Google Scholar]
- 5.Sinzger C, Grefte A, Plachter B, Gouw AS, The TH, Jahn G. 1995. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J Gen Virol 76:741–750. doi: 10.1099/0022-1317-76-4-741. [DOI] [PubMed] [Google Scholar]
- 6.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanarsdall AL, Johnson DC. 2012. Human cytomegalovirus entry into cells. Curr Opin Virol 2:37–42. doi: 10.1016/j.coviro.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87:2451–2460. doi: 10.1099/vir.0.81921-0. [DOI] [PubMed] [Google Scholar]
- 12.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J Virol 74:7720–7729. doi: 10.1128/JVI.74.17.7720-7729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang XJ, Adler B, Sampaio KL, Digel M, Jahn G, Ettischer N, Stierhof YD, Scrivano L, Koszinowski U, Mach M, Sinzger C. 2008. UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J Virol 82:2802–2812. doi: 10.1128/JVI.01550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J Virol 84:2585–2596. doi: 10.1128/JVI.02249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou M, Yu Q, Wechsler A, Ryckman BJ. 2013. Comparative analysis of gO isoforms reveals that strains of human cytomegalovirus differ in the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope. J Virol 87:9680–9690. doi: 10.1128/JVI.01167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murrell I, Tomasec P, Wilkie GS, Dargan DJ, Davison AJ, Stanton RJ. 2013. Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J Virol 87:10489–10500. doi: 10.1128/JVI.01546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li G, Nguyen CC, Ryckman BJ, Britt WJ, Kamil JP. 2015. A viral regulator of glycoprotein complexes contributes to human cytomegalovirus cell tropism. Proc Natl Acad Sci U S A 112:4471–4476. doi: 10.1073/pnas.1419875112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou M, Lanchy JM, Ryckman BJ. 2015. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol 89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci U S A 105:14118–14123. doi: 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loughney JW, Rustandi RR, Wang D, Troutman MC, Dick LWJ, Li G, Liu Z, Li F, Freed DC, Price CE, Hoang VM, Culp TD, DePhillips PA, Fu TM, Ha S. 2015. Soluble human cytomegalovirus gH/gL/pUL128-131 pentameric complex, but not gH/gL, inhibits viral entry to epithelial cells and presents dominant native neutralizing epitopes. J Biol Chem 290:15985–15995. doi: 10.1074/jbc.M115.652230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nogalski MT, Chan GC, Stevenson EV, Collins-McMillen DK, Yurochko AD. 2013. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog 9:e1003463. doi: 10.1371/journal.ppat.1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, Uematsu Y, Lilja AE, Arvin AM, Carfi A. 2015. A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci U S A 112:6056–6061. doi: 10.1073/pnas.1501176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciferri C, Chandramouli S, Donnarumma D, Nikitin PA, Cianfrocco MA, Gerrein R, Feire AL, Barnett SW, Lilja AE, Rappuoli R, Norais N, Settembre EC, Carfi A. 2015. Structural and biochemical studies of HCMV gH/gL/gO and pentamer reveal mutually exclusive cell entry complexes. Proc Natl Acad Sci U S A 112:1767–1772. doi: 10.1073/pnas.1424818112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol 67:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol 82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J Virol 82:11837–11850. doi: 10.1128/JVI.01623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogner E, Reschke M, Reis B, Reis E, Britt W, Radsak K. 1992. Recognition of compartmentalized intracellular analogs of glycoprotein H of human cytomegalovirus. Arch Virol 126:67–80. doi: 10.1007/BF01309685. [DOI] [PubMed] [Google Scholar]
- 32.Britt WJ, Vugler L, Butfiloski EJ, Stephens EB. 1990. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J Virol 64:1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urban M, Britt W, Mach M. 1992. The dominant linear neutralizing antibody-binding site of glycoprotein gp86 of human cytomegalovirus is strain specific. J Virol 66:1303–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2006. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5:Unit 5.6. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryckman BJ, Chase MC, Johnson DC. 2010. Human cytomegalovirus TR strain glycoprotein O acts as a chaperone promoting gH/gL incorporation into virions but is not present in virions. J Virol 84:2597–2609. doi: 10.1128/JVI.02256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W, Chen F, Klyachkin Y, Sham YY, Geraghty RJ. 2014. Mutations in the amino terminus of herpes simplex virus type 1 gL can reduce cell-cell fusion without affecting gH/gL trafficking. J Virol 88:739–744. doi: 10.1128/JVI.02383-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J Virol 71:7687–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plate AE, Smajlovic J, Jardetzky TS, Longnecker R. 2009. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J Virol 83:7678–7689. doi: 10.1128/JVI.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan Q, Lin E, Spear PG. 2009. Insertional mutations in herpes simplex virus type 1 gL identify functional domains for association with gH and for membrane fusion. J Virol 83:11607–11615. doi: 10.1128/JVI.01369-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A 106:20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chesnokova LS, Hutt-Fletcher LM. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J Virol 85:13214–13223. doi: 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirschner AN, Lowrey AS, Longnecker R, Jardetzky TS. 2007. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J Virol 81:9216–9229. doi: 10.1128/JVI.00575-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Rowe CL, Jardetzky TS, Longnecker R. 2012. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio 3:e00290-11. doi: 10.1128/mBio.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Mohl BS, Chen J, Jiang W, Mellins ED, Longnecker R, Zhou ZH, Jardetzky TS. 2014. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog 10:e1004309. doi: 10.1371/journal.ppat.1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vanarsdall AL, Chase MC, Johnson DC. 2011. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J Virol 85:11638–11645. doi: 10.1128/JVI.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wille PT, Wisner TW, Ryckman B, Johnson DC. 2013. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 4:e00332-13. doi: 10.1128/mBio.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med 11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gianni T, Salvioli S, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. 2013. alphavbeta6- and alphavbeta8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog 9:e1003806. doi: 10.1371/journal.ppat.1003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gompels UA, Carss AL, Saxby C, Hancock DC, Forrester A, Minson AC. 1991. Characterization and sequence analyses of antibody-selected antigenic variants of herpes simplex virus show a conformationally complex epitope on glycoprotein H. J Virol 65:2393–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fouts AE, Comps-Agrar L, Stengel KF, Ellerman D, Schoeffler AJ, Warming S, Eaton DL, Feierbach B. 2014. Mechanism for neutralizing activity by the anti-CMV gH/gL monoclonal antibody MSL-109. Proc Natl Acad Sci U S A 111:8209–8214. doi: 10.1073/pnas.1404653111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciferri C, Chandramouli S, Leitner A, Donnarumma D, Cianfrocco MA, Gerrein R, Friedrich K, Aggarwal Y, Palladino G, Aebersold R, Norais N, Settembre EC, Carfi A. 2015. Antigenic characterization of the HCMV gH/gL/gO and pentamer cell entry complexes reveals binding sites for potently neutralizing human antibodies. PLoS Pathog 11:e1005230. doi: 10.1371/journal.ppat.1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schuessler A, Sampaio KL, Sinzger C. 2008. Charge cluster-to-alanine scanning of UL128 for fine tuning of the endothelial cell tropism of human cytomegalovirus. J Virol 82:11239–11246. doi: 10.1128/JVI.01069-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schuessler A, Sampaio KL, Scrivano L, Sinzger C. 2010. Mutational mapping of UL130 of human cytomegalovirus defines peptide motifs within the C-terminal third as essential for endothelial cell infection. J Virol 84:9019–9026. doi: 10.1128/JVI.00572-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schuessler A, Sampaio KL, Straschewski S, Sinzger C. 2012. Mutational mapping of pUL131A of human cytomegalovirus emphasizes its central role for endothelial cell tropism. J Virol 86:504–512. doi: 10.1128/JVI.05354-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Macagno A, Bernasconi NL, Vanzetta F, Dander E, Sarasini A, Revello MG, Gerna G, Sallusto F, Lanzavecchia A. 2010. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol 84:1005–1013. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang XJ, Sampaio KL, Ettischer N, Stierhof YD, Jahn G, Kropff B, Mach M, Sinzger C. 2011. UL74 of human cytomegalovirus reduces the inhibitory effect of gH-specific and gB-specific antibodies. Arch Virol 156:2145–2155. doi: 10.1007/s00705-011-1105-x. [DOI] [PubMed] [Google Scholar]