

ABSTRACT
The herpes simplex virus type 1 (HSV-1) envelope protein pUS9 plays an important role in virus anterograde axonal transport and spread from neuronal axons. In this study, we used both confocal microscopy and transmission electron microscopy (TEM) to examine the role of pUS9 in the anterograde transport and assembly of HSV-1 in the distal axon of human and rat dorsal root ganglion (DRG) neurons using US9 deletion (US9−), repair (US9R), and wild-type (strain F, 17, and KOS) viruses. Using confocal microscopy and single and trichamber culture systems, we observed a reduction but not complete block in the anterograde axonal transport of capsids to distal axons as well as a marked (∼90%) reduction in virus spread from axons to Vero cells with the US9 deletion viruses. Axonal transport of glycoproteins (gC, gD, and gE) was unaffected. Using TEM, there was a marked reduction or absence of enveloped capsids, in varicosities and growth cones, in KOS strain and US9 deletion viruses, respectively. Capsids (40 to 75%) in varicosities and growth cones infected with strain 17, F, and US9 repair viruses were fully enveloped compared to less than 5% of capsids found in distal axons infected with the KOS strain virus (which also lacks pUS9) and still lower (<2%) with the US9 deletion viruses. Hence, there was a secondary defect in virus assembly in distal axons in the absence of pUS9 despite the presence of key envelope proteins. Overall, our study supports a dual role for pUS9, first in anterograde axonal transport and second in virus assembly in growth cones in distal axons.
IMPORTANCE HSV-1 has evolved mechanisms for its efficient transport along sensory axons and subsequent spread from axons to epithelial cells after reactivation. In this study, we show that deletion of the envelope protein pUS9 leads to defects in virus transport along axons (partial defect) and in virus assembly and egress from growth cones (marked defect). Virus assembly and exit in the neuronal cell body are not impaired in the absence of pUS9. Thus, our findings indicate that pUS9 contributes to the overall HSV-1 anterograde axonal transport, including a major role in virus assembly at the axon terminus, which is not essential in the neuronal cell body. Overall, our data suggest that the process of virus assembly at the growth cones differs from that in the neuronal cell body and that HSV-1 has evolved different mechanisms for virus assembly and exit from different cellular compartments.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a neurotropic and neuroinvasive virus that has the ability to infect and remain dormant or latent in neurons of the peripheral nervous system of their human host. After the initial infection of the skin, the virus gains access to the neurons of the dorsal root ganglia via the nerve endings in the innervating skin. Virus reactivation from latency is frequent and results in either asymptomatic virus shedding or recurrent herpes disease (1). Two essential events following reactivation of HSV from latently infected sensory neurons are the transport of the virus down nerves to the nerve terminals (anterograde direction) and subsequent spread of the virus from the nerve terminals across into the skin (2, 3).
Three highly conserved viral envelope proteins, pUS9, gE, and gI, of HSV-1 and pseudorabies virus (PRV), a swine herpesvirus, are important for virus anterograde axonal transport and spread of infection both in vivo and in vitro (4–12). pUS9 is the only one of these three envelope proteins essential for PRV anterograde transport and spread. PRV lacking pUS9 shows a complete defect in axonal sorting of PRV viral particles and structural proteins into axons and a complete block in anterograde transneuronal spread (10, 11). pUS9 has also been shown to be required for the anterograde transport of bovine herpesvirus types 1 and 5 in vivo using animal models (13, 14).
HSV-1 pUS9 has been shown to be required for virus anterograde spread from the trigeminal ganglia to the cornea after infection and spread in the mouse corneal epithelium (15) and also from the retina into the optic nerve after infection of the mouse retinal ganglion cell bodies (6). pUS9, together with gE/gI, has been shown to promote HSV-1 anterograde axonal transport of viral capsids and glycoproteins (mainly gB) and virus spread from distal axons of rat superior cervical ganglia to adjacent nonneuronal cells in vitro (8). Despite extensive studies, it remains unclear how pUS9 mediates anterograde viral transport along axons and its role during virus exit and spread from axons to adjacent cells. In this study, we have used both confocal microscopy and transmission electron microscopy (TEM) in order to examine the role of pUS9 in HSV-1 anterograde axonal transport and virus assembly in the distal axon of human and rat neurons of the dorsal root ganglia (DRG) using US9 deletion (US9−), repair (US9R), and wild-type viruses (strains F, 17, and KOS). In the absence of pUS9, there was a significant decrease in HSV-1 transport along axons but also a marked impairment in virus assembly in distal axons despite the presence of envelope proteins. Our findings support a dual role for pUS9 in HSV-1 anterograde axonal transport and in virus assembly in growth cones in distal axons.
MATERIALS AND METHODS
Cells and viruses.
Virus stocks were passaged in Vero cells, which were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 9% FBS. US9 deletion (US9− [R6607]), and repair (US9R [R6608]) viruses and the F strain were kindly provided by Jennifer LaVail (University of California, San Francisco, CA, USA) (6). F-US9/GFP and F-US9/GFP-R viruses were kindly provided by David Johnson (Oregon Health & Science University, Portland, OR, USA) (8). The KOS strain was provided by Thomas Mettenleiter (16). Strain 17 was provided by Frazer Rixon.
Antibodies.
Rabbit antibody against purified nuclear C capsids (PTNC) was kindly provided by Frazer Rixon, MRC Virology Unit, Institute of Virology, United Kingdom (17). Mouse monoclonal anti-ICP27, mouse monoclonal anti-gE, and rabbit polyclonal anti-gD antibodies were obtained from Abcam, United Kingdom. Mouse monoclonal anti-gC was purchased from Syva Co, USA. Mouse monoclonal anti-gC was purchased from Chemicon International, USA. Alexa Fluor-labeled secondary antibodies were obtained from Life Technologies, USA.
Preparation of human fetal DRG explants.
DRG were prepared as previously described (18–21) from human fetal tissue obtained at therapeutic termination following the informed consent of the mother. Protocols were approved by the Western Sydney Local Health District and The University of Sydney Human Research Ethics Committees. The DRG were dissected, cleansed of connective tissue, placed onto Matrigel (BD Biosciences, USA)-coated plastic coverslips, and cultured at 37°C with 5% CO2 in Neurobasal medium (Life Technologies, USA) supplemented with 4 mM l-glutamine (Invitrogen, USA), 2% B-27 supplement (Invitrogen, USA), and 7S nerve growth factor (100 ng/ml) (Sigma, USA) for 5 to 7 days for axon outgrowth prior to HSV-1 infection.
Preparation of dissociated rat neuronal cultures.
DRG neurons were prepared from 4-day-old Wistar rat neonates as previously described (21, 22). Briefly, DRG were dissociated in Hanks calcium- and magnesium-free solution (Invitrogen, USA) plus 0.25% trypsin (Sigma, USA) and 0.05% collagenase (Worthington Biomedical Co., USA) for 30 min at 37°C, followed by DNase (10 mg/ml) (Sigma) for 5 min at 37°C, washed two times by centrifugation at 80 × g, and passed through 35% Percoll (Sigma, USA). The cell pellet was resuspended in 500 μl of Neurobasal medium, plated onto Matrigel-coated plastic coverslips, and cultured at 37°C with 5% CO2 in Neurobasal medium supplemented with 4 mM l-glutamine, 2% B-27 supplement, and 7S nerve growth factor (100 ng/ml) for 3 days prior to HSV-1 infection. The Western Sydney Local Health District and the University of Sydney Animal Research Ethics Committees approved the use of rat neonates.
HSV-1 infection of human DRG explant and rat dissociated neuronal cultures for electron microscope (TEM) and confocal microscopy studies.
DRG cultures were infected with virus (105 PFU/0.5 ml/ganglion for DRG explants or 1 PFU/cell for dissociated neuronal cultures). After 2 h, the inoculum was removed and the cultures were washed three times with fresh medium. The infected cultures were incubated at 37°C under 5% CO2 for 22 to 24 h. The cultures were washed three times in PBS and processed for either confocal microscopy or TEM as previously described (18).
Neuronal cultures in microfluidic devices.
Standard neuron devices (SND450) obtained from Xona Microfluidics, USA were prepared as per the manufacturer's instructions and used to study virus anterograde axonal transport and spread from axons to Vero cells. Cover glasses (24 mm by 40 mm) were plasma bonded to the neuron devices by exposing the surface of both coverglass and device to glow discharge treatment for 10 s (under 39 Pa) with air using a Pelco easiGlow glow discharge cleaning system (Ted Pella, USA). After plasma bonding, the devices were coated by overnight incubation in 0.5 mg/ml poly-d-lysine (Sigma, USA) in borate buffer (pH 8.5) at 37°C with 5% CO2. The devices were then washed three times with water followed by a second overnight incubation in laminin (10 μg/ml) (Sigma, USA) at 37°C with 5% CO2. The devices were washed three times with Neurobasal medium prior to seeding of neuronal cell suspension. Rat DRG dissociated neurons were prepared as described above, and a 10-μl suspension containing approximately 40,000 neurons was added to the somal compartments of the devices. Neurons were grown in Neurobasal medium supplemented with 4 mM l-glutamine, B-27 supplement, and 7S nerve growth factor (100 ng/ml) for 4 to 5 days at 37°C with 5% CO2 to allow the axons to grown into the axonal compartment of the device. A total of 50,000 Vero cells were then added to the axonal compartment of the devices 24 h prior to infection. Ten microliters of virus suspension (5 PFU/cell) was added in the soma (neuronal cell body) compartment. The virus inoculum was removed at 2 h postinfection (hpi), and the cells were washed with Neurobasal medium. The medium in the axonal compartment was removed at 8 hpi and replaced with medium containing Foscarnet (100 μg/ml) (Sigma) to prevent secondary virus spread in Vero cells. Cultures were fixed at 22 to 24 hpi in 3% formaldehyde and 0.1% Triton X-100 in phosphate-buffered saline (PBS).
For each experiment, to check for virus leakage from one compartment to another in the devices during infection, Vero cells were plated in the axonal compartment 24 h prior to addition of viruses to the somal compartment in the absence of neurons. The devices were fixed and immunostained in situ with antibodies to glycoproteins and C capsids. This control was performed in conjunction with mock-infected controls where neurons in somal compartments were mock infected followed by fixation and immunostaining of cultures in situ.
Immunofluorescence and confocal microscopy.
Dissociated DRG cultures in glass coverslips were fixed in 3% formaldehyde for 30 min at room temperature followed by addition of 0.1% Triton X-100 for 5 min and then processed for immunofluorescence studies as previously described (18).
Neuronal cultures in microfluidic devices were fixed in situ by addition of 3% formaldehyde to both somal and axonal compartments and incubation for 30 min at room temperature followed by addition of 0.1% Triton X-100 for 5 min. The cultures were immunostained in situ by addition of primary antibodies to both somal and axonal compartments and incubation overnight at 4°C followed by incubation of secondary antibodies for 1 h at room temperature. The cultures were washed six times after each antibody incubation step in phosphate-buffered saline (PBS). PBS from the last wash was then removed and replaced with Fluoromount G prior to examination using a Leica SP5 II confocal microscope. For axonal transport studies, axons in close proximity to microgrooves from 10 randomly selected fields of view (1,024 by 1,024 pixels) were manually counted. For studies involving virus spread from axons to Vero cells, the total number of ICP27-positive Vero cells in the axonal compartment was manually counted.
Statistical analysis.
For studies of capsid transport along axons in the axonal compartment of the microfluidic devices in the presence or absence of pUS9, the number of capsid-positive axons and the total number of axons observed were recorded under the deleted and repair conditions in each of 3 separate experiments performed for each of 2 deletion/repair virus pairs. Statistical analysis was performed using a generalized linear mixed-effect (GLME) model with a logit link function to investigate the effects of US9 repair versus US9 deletion status and type of virus on the odds of an axon being positive for capsid. In these models, the grouping variable was experiment within virus type. Experiment and virus type were considered random effects and condition (repair versus deleted) as a fixed effect.
Estimates of the ln(odds ratio) and associated 95% confidence intervals (CI) were back-transformed to produce estimated odds ratios of observing capsid-positive axons in US9 repair versus US9 deletion viruses.
For studies of virus spread from axons to nonneuronal Vero cells in the axonal compartment, the total number of ICP27-positive Vero cells in the axonal compartment was considered a Poisson variable and log transformed in order to stabilize the variance prior to analysis. The number of ICP27-positive final cells (from 40,000) never exceeded 1,100 in any experiment under either the US9-deleted or repair condition. Linear mixed-effect (LME) models were used to investigate the effects of US9 repair versus deletion and type of virus. In these models, each experiment and virus (US9−, US9R, F-US9/GFP, and F-US9/GFP-R) was considered a random effect, whereas the US9 repair versus US9-deleted condition was considered a fixed effect. Estimates and their 95% confidence intervals were back transformed to produce estimated fold changes in the number of ICP27-positive Vero cells in US9 repair versus deletion.
The statistical software IBM SPSS version 22 and SPLUS 8.2 were used to analyze the data. Two-tailed tests with a significance level of 5% were used throughout.
Transmission electron microscopy.
Coverslips with DRG cultures in situ were fixed in Karnovsky's fixative and processed with TAAB TLV epoxy resin as previously described (18–20) Following polymerization, the coverslips were separated from the resin, leaving the DRG explant and axons exposed on the surface of the block. Areas in the blocks containing mid- to distal regions of axons were selected. Ultrathin sections (70 nm) were collected and examined using either a Philips CM120 BioTWIN equipped with an SIS Morada digital camera at 100 kV or an FEI Tecnai G2 20 TWIN equipped with a FEI Eagle 2K digital camera at 120 kV.
Statistical analysis for TEM studies.
The statistical package IBM SPSS version 22 was used to analyze the data. Two-tailed tests with a significance level of 5% were used throughout. Odds ratios with 95% confidence intervals (95% CI) were used to quantify the strength of association between US9 status and the presence of enveloped capsids in axonal varicosities and growth cones. Quantitation of viral particles in varicosities and growth cones in distal axons was performed for only one set of US9 deletion and repair viruses: US9− and US9R, respectively.
RESULTS
Capsid and envelope (gC and gD) proteins transport into and along axons.
In this study, we used primary DRG neurons grown in both single-chamber and trichamber culture systems and confocal microscopy to examine the role of pUS9 in HSV-1 anterograde axonal transport and virus assembly in the distal axon using wild-type virus (strains F, 17, and KOS) along with US9 deletion and repair viruses from two independent laboratories.
Initial experiments using dissociated primary cultures grown in 24-well plates infected with wild-type (strains F, 17, and KOS) or US9 deletion (US9−) or repair (US9R) viruses showed that there was no significant decrease in the axonal transport of envelope glycoproteins gC, gD, and gE after 24 hpi (Fig. 1 and Table 1).
FIG 1.

Confocal micrographs of dissociated neurons infected with either US9 deletion (US9—) or US9 repair (US9R) virus showing the distribution of glycoproteins gC, gD, and gE in the cell body periphery and along axons (arrows). Neonatal rat DRG neurons were dissociated, pelleted through a 35% Percoll gradient, and plated on Matrigel-coated coverslips. HSV-1-infected cultures were fixed at 24 hpi, permeabilized, and labeled for gC, gD, or gE. Deletion of pUS9 does not inhibit axonal transport of glycoproteins gC, gD, and gE. Bars, 10 μm.
TABLE 1.
Summary of positive immunofluorescence staining results for glycoproteins gC, gD, and gE along axons of rat dissociated neurons infected with F strain, KOS, or US9 deletion and repair viruses at 24 hpi
| Virus | Presence of glycoproteins gC, gD, and gE alonga: |
|
|---|---|---|
| Proximal axons | Mid- to distal axons | |
| F strain | +++ | ++ |
| KOS | +++ | ++ |
| US9− | +++ | ++ |
| US9R | +++ | ++ |
| F-US9/GFP | +++ | ++ |
| F-US9/GFP-R | +++ | ++ |
The intensity of fluorescence signal along axons was compared to the intensity of fluorescence observed in the cell body. The intensity was classified semiquantitatively using the following criteria: +++, ≥intensity of the membrane of the cell body (this region shows the brightest fluorescence); ++, intensity the same as the cytoplasm of the cell body. Approximately 90% of infected neurons had one or more positive axons for glycoproteins. An average of 200 neurons were manually counted per virus in replicate experiments.
Examination of the effects of US9 deletion on capsid anterograde transport in dissociated neuronal cultures is often difficult given that these cultures form extensive network of axons. Therefore, similar experiments were also performed using primary neurons cultured in microfluidic trichamber devices (Fig. 2 to 5). In these experiments, dissociated neurons were seeded into the somal compartments of the devices and cultured for 4 to 5 days to allow axons to grow through microgrooves into the axonal compartments. The neurons in the somal compartments were then infected with two matched pairs of US9 deletion and repair viruses at 5 PFU/cell, respectively, and the cultures were fixed from 22 to 24 hpi. The cultures in the devices were immunostained in situ by addition of antibodies to purified C capsids to both the somal and axonal compartments. No significant differences in viral capsid distribution in the cell body or in the proportion of infected cell bodies (>90%) were observed in the somal compartments of cultures infected with either US9 deletion or repair viruses (data not shown).
FIG 2.

Confocal micrographs of axons in the axonal compartment of neuronal cultures in microfluidic devices infected with US9 deletion (US9− and F-US9/GFP) and US9 repair (US9R and F-US9/GFP-R) viruses. Neonatal rat DRG neurons were dissociated, pelleted through a 35% Percoll gradient, and plated into the somal compartment of microfluidic devices. Cultures were incubated for 5 to 6 days to allow axons to grow into the axonal compartment. Vero cells were added to the axonal compartment 24 h prior to virus addition to the somal compartment. Foscarnet (100 μg/ml) was added to the axonal compartment 8 hpi to prevent secondary virus spread in Vero cells. The cultures were fixed at 22 hpi followed by immunostaining for C capsids (PTNC) and examined using a Leica SP5 confocal microscope. In the absence of pUS9, there was a reduction but not complete block in the anterograde axonal transport of capsids (arrows) to distal axons. Bars, 25 μm.
In order to determine the effect of US9 deletion on transport of capsids into and along distal axons in the axonal compartment, the numbers of capsid-positive axons versus the total number of axons in the axonal compartment from 10 randomly selected fields of view, in close proximity to microgrooves, were counted in three separate experiments for both US9 deletion and US9 repair viruses. Transport of capsids into and along distal axons in the axonal compartment in US9 deletion viruses was significantly reduced in comparison to that in repair viruses (Fig. 2 and 3). Approximately 40% of axons in the axonal compartments of cultures infected with US9 deletion viruses showed positive capsid staining, whereas up to 80% of axons in axonal compartments of cultures infected with repair viruses were positively stained for capsid (Fig. 3). This represented an overall reduction of approximately 50% in capsid transport along axons in the absence of pUS9.
FIG 3.

Transport of capsid along axons in neuronal cultures infected with US9 deletion (US9− and F-US9/GFP) and US9 repair (US9R and F-US9/GFP-R) viruses. Neuronal cultures in the somal compartment of microfluidic devices were infected at 5 PFU/cell and fixed at 22 hpi. The cultures were then stained in situ by addition of antibodies to purified C capsids (PTNC) to both somal and axonal compartments. Axons emerging from the middle grooves into the axonal compartment were counted from 10 randomly selected fields of view. The percentages of capsid-positive (capsid +) versus capsid-negative (capsid −) axons are shown for three separate experiments. The numbers on top of each column represent the total number of axons counted per 10 fields per virus in each experiment. A significant reduction in the number of capsid-positive axons was observed in the absence of pUS9.
Estimates of the odds ratios and associated 95% confidence intervals in observing capsid-positive axons in US9 repair versus US9 deletion were determined to be 5.7 (95% CI, 4.2 to 7.8) for US9R and US9− viruses and 9.0 (95% CI, 6.8 to 11.9) for F-US9/GFP-R and F-US9/GFP viruses, with an overall estimated odds ratio of 7.4 (95% CI, 6.0 to 9.0) indicating a significant reduction (P < 0.001) in capsid transport along axons in the absence of pUS9. In addition, there was no statistically significant difference in the proportions of capsid-positive axons between the two independently generated US9 deletion and repair virus pairs.
Virus spread from axons to nonneuronal cells is significantly reduced in the absence of pUS9.
To investigate whether capsids that reach the distal ends of axons of US9 deletion viruses can exit and spread to nonneuronal cells, Vero cells were added to the axonal compartment of primary cultures of neurons grown in microfluidic devices after axons had reached the axonal compartment. Vero cells were seeded 24 h prior to infection of the somal compartments with US9 deletion and repair viruses. Foscarnet (100 μg/ml) was added to the medium in the axonal compartment at 8 hpi to prevent secondary virus spread in Vero cells. Cultures in the microfluidic devices were then fixed at 22 to 24 hpi and immunostained in situ for ICP27 protein. The numbers of ICP27-positive Vero cells in the axonal compartments of both US9 deletion and repair viruses were counted and compared (Fig. 4 and 5). A marked and significant reduction (∼90%; P < 0.001) in virus spread from axons to Vero cells was observed in cultures infected with the US9 deletion viruses compared to those infected with the repair viruses (Fig. 5). Statistical analysis showed that the estimated fold changes (and 95% CI) in the number of ICP27-positive Vero cells for the US9− and US9R viruses were 9.5 (95% CI, 6.0 to 15.2) and 8.7 (95% CI, 7.2 to 10.3) for the F-US9/GFP and F-US9/GFP-R viruses, with an overall fold change of 9.1-fold and 95% CI of 7.9 to 10.5. In addition, there was no statistically significant difference in the effects of US9 deletion between the two independently generated US9 deletion and repair virus pairs.
FIG 4.

Confocal micrographs of Vero cells in the axonal compartment of neuronal cultures in microfluidic devices infected with US9 deletion (US9− and F-US9/GFP) and US9 repair (US9R and F-US9/GFP-R) viruses. Vero cells were added to the axonal compartment 24 h prior to virus addition to the somal compartment. Foscarnet (100 μg/ml) was added to the axonal compartment 8 hpi and remained in the medium until fixation at 22 hpi. The cultures were then immunostained for ICP27 and examined using a Leica SP5 confocal microscope. A marked and significant reduction (∼90%) in virus spread from axons to Vero cells was observed with the US9 deletion viruses compared to repair viruses. Bars, 25 μm.
FIG 5.
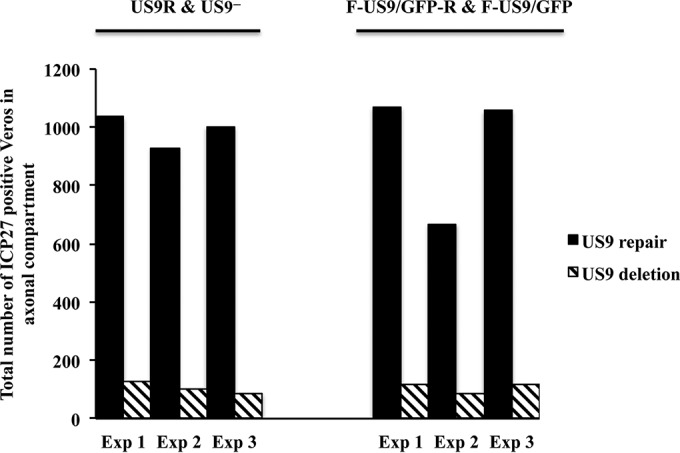
Virus spread from axons to nonneuronal Vero cells in the axonal compartment of neuronal cultures grown in microfluidic devices, after infection with US9 deletion (US9− and F-US9/GFP) and US9 repair (US9R and F-US9/GFP-R) viruses. Foscarnet (100 μg/ml) was added to the axonal compartment 8 hpi, and cultures were fixed at 22 hpi followed by immunostaining for ICP27. The total number of ICP27-positive Vero cells in the axonal compartment was counted for each virus in three independent experiments.
Capsids in distal axons and in growth cones failed to acquire a viral envelope in the absence of pUS9.
Our confocal data showed that capsids could be detected down to the distal ends of the axon even in the absence of pUS9 (F-US9/GFP virus) (Fig. 6), similar to US9 repair (F-US9/GFP-R) (Fig. 6) and wild-type viruses as previously shown (18). Therefore, in order to visualize capsids in the distal ends of axons and in varicosities and growth cones and to further examine the defect in virus spread from axons in the absence of pUS9, we performed TEM of DRG explants infected with US9 deletion (US9−), US9 repair (US9R), and wild-type (F, 17, and KOS) virus strains. Unenveloped capsids were present in mid-regions of axons infected with US9 deletion and repair viruses as well as in KOS (which also lacks pUS9) (16) and F and 17 strain viruses (Fig. 7 and Table 2). However, most unenveloped capsids were mainly found in axonal varicosities and growth cones of axons infected with US9 deletion (US9−) and KOS (Fig. 8 and Table 2) viruses. Furthermore, there was a marked reduction or absence of enveloped capsids in varicosities and growth cones of axons infected with the US9 deletion and KOS viruses compared to axons infected with wild-type viruses (F and 17) or US9 repair viruses (Fig. 9 and Table 2). Approximately 30 to 70% of capsids in varicosities and growth cones observed in distal axons infected with F and 17 strains and US9 repair (US9R) viruses were fully enveloped compared to less than 6% of capsids found in axons infected with the KOS strain and still lower (<2%) with US9 deletion virus (Table 2). Thus, the odds of observing enveloped capsids in varicosities and growth cones are 45 times higher when pUS9 is present than when deleted (95% CI, 13 to 150; P < 0.001). It is important to note that the rare enveloped capsids observed in growth cones of axons infected with the US9 deletion (US9−) virus and in 4 out of 7 enveloped capsids with KOS virus showed an abnormal tegument that appeared thicker, polarized, and heterogeneous in density, consistent with defective tegument addition to viral capsids (Fig. 9 and Table 2). These rare enveloped capsids with abnormal tegument were not observed in the repair or wild-type viruses examined in this study or in any of our previous studies (18, 19).
FIG 6.

Confocal micrographs of capsid localization in distal axons in the axonal compartment of neuronal cultures in microfluidic devices infected with US9 deletion (F-US9/GFP) and repair (F-US9/GFP-R) viruses at 22 hpi. Concentration of fluorescence for C capsids (arrows) was visible in varicosities and growth cones in distal axons in in both US9 deletion and repair viruses. Bars, 25 μm.
FIG 7.

Electron micrographs of human DRG axons infected with US9 deletion (US9− and F-US9/GFP), US9 repair (US9R and F-US9/GFP-R), KOS, and F strain viruses. Coverslips with explanted human fetal DRG in situ were HSV-1 infected, fixed at 24 hpi, and processed for TEM. Areas containing mid- and distal regions of axons were sectioned and examined using either a Phillips CM120 BioTWIN at 100 kV or an FEI Tecnai G2 20 TWIN at 120 kV. Unenveloped capsids (arrowheads) in mid- to distal axons were observed in all infected axons. Bars, 200 nm.
TABLE 2.
Viral particles in HSV-1-infected human fetal axons, varicosities, and growth conesa
| Virus | Total no. of: |
Count of: |
||||
|---|---|---|---|---|---|---|
| Axons | Var/GC | U/C in mid-axonsb | U/C in Var/GC | E/C in Var/GC | Ab E/C in Var/GC | |
| US9− | 2,445 (517) | 779 (147) | 8 (2) | 52 (27) | 1 (1) | |
| KOS | 2,825 | 1,139 | 26 | 49 | 3 | 4 |
| US9R | 805 | 158 | 10 | 23 | 10 | |
| Strain F | 755 | 100 | 10 | 10 | 5 | |
| Strain 17 | 1,200 | 352 | 6 | 15 | 49 | |
Var/GC, varicosities/growth cones; U/C, unenveloped capsids; E/C, enveloped capsids; Ab, aberrant enveloped capsids. Counts using rat DRG axons are given in parentheses.
No enveloped capsids were observed in mid-axons.
FIG 8.

Electron micrographs of unenveloped capsids (arrowheads) in growth cones and varicosities of human DRG axons infected with US9 deletion (US9− and F-US9/GFP), US9 repair (US9R and FUS9/GFP-R), KOS, and F strain viruses. Coverslips with explanted human fetal DRG in situ were HSV-1 infected, fixed at 24 hpi, and processed for TEM. The arrow in the F-US9/GFP-R micrograph indicates an extracellular virus. Bars, 200 nm.
FIG 9.

Electron micrographs of human DRG axons infected with US9 deletion (US9− and F-US9/GFP), US9 repair (US9R and F-US9/GFP-R), KOS, and F strain viruses. Coverslips with explanted human fetal DRG in situ were HSV-1 infected, fixed at 24 hpi, and processed for TEM. Enveloped capsids (arrows) were present in varicosities and growth cones of axons infected with US9 repair (US9R and FUS9/GFP-R), KOS, and F strain viruses. In axons infected with US9 deletion virus, only rare aberrant enveloped capsids (*) with significantly polarized expansions of the tegument coat were present. These aberrant enveloped capsids (*) were also occasionally present in KOS-infected axons. Bars, 200 nm.
Our confocal data also showed that there was no obvious impairment or blockage of viral capsids resulting in a localized accumulation in the cytoplasm of the cell body, in the axon hillock or proximal axons (Fig. 10). Furthermore, a similar distribution of enveloped capsids in the cytoplasm of the cell body, visualized by colocalization of capsid and glycoproteins, was observed in both US9− and US9R-infected neurons (Fig. 10). Therefore, we used TEM in order to visualize viral capsids in the cytoplasm of the cell body of neurons infected with US9 deletion and repair viruses. The impairment in capsid envelopment in axonal varicosities and growth cones in the absence of pUS9 was not observed in the cell body of neurons infected with the US9 deletion (US9−) virus (Fig. 11 and Table 3). Viral capsids at various stages of assembly in the nucleus and cytoplasm of the cell body and also extracellular virions surrounding the neuronal cell body were observed by TEM in rat DRG neurons infected with US9− (Fig. 11) and F-US9/GFP viruses in a similar way to with control viruses, i.e., the US9 repair (US9R or F-US9/GFP-R), KOS, F, and 17 strains (22; data not shown). In neurons infected with US9 deletion viruses, numerous naked capsids in the nucleus and in the cytoplasm of the cell body were observed (Fig. 11A and C), while enveloped capsids could be seen in the space between inner and outer nuclear membranes (Fig. 11B) as well in close proximity to vesicular membranes in the cytoplasm of the cell body (Fig. 11B and C). In addition, numerous extracellular virions surrounding the cell body were observed (Fig. 11A). Counts of viral particles (enveloped and unenveloped) in the cytoplasm of the cell body of US9−- and US9R-infected human DRG neurons showed the presence of both enveloped and unenveloped capsids in similar proportions (Table 3). These observations suggest that normal virus assembly and exit occur in the cell body of the neuron in the absence of pUS9.
FIG 10.

Confocal micrographs showing colocalization of C capsids and envelope protein gE in dissociated rat neurons infected with US9− and US9R viruses at 24 hpi. The distributions of C capsids (red) and gE (green) in the cytoplasm of the cell body were similar between US9− and US9R viruses, showing no accumulation of capsids with envelope in the cytoplasm of the cell body, axon hillock, or proximal axons in the absence of pUS9. Bars, 10 μm.
FIG 11.

Electron micrographs of rat neurons infected with HSV-1 US9− virus showing normal virus assembly and exit from the neuronal cell bodies in the absence of pUS9. (A) Lower magnification of neuronal cell body showing extracellular virions (arrows). (B) Capsids (arrowheads) in the nucleus and enveloped capsids (open arrows) in the space between inner and outer nuclear membranes (indicated with an asterisk) and in the cytoplasm close to the nucleus. (C) Enveloped (open arrows) and unenveloped (arrowheads) capsids in the cytoplasm of the cell body and extracellular virions (closed arrows). Bars, 200 nm.
TABLE 3.
Viral particles in enveloped versus unenveloped capsids in the cytoplasm of the cell body of US9−- and US9R-infected human DRG neurons
| Virus | No. of virus particles ina: |
|
|---|---|---|
| Enveloped capsids | Unenveloped capsids | |
| US9− | 215 | 153 |
| US9R | 269 | 224 |
Counts represent total number of viral particles in the cell body from 30 cell profiles in replicate experiments for each virus.
DISCUSSION
HSV-1 has evolved mechanisms for its efficient transport along sensory axons and subsequent spread from axons to epithelial cells after reactivation. These two key events in the virus life cycle are mediated by the interaction of viral proteins with the cellular cytoskeleton and proteins involved in intracellular trafficking. The viral envelope protein pUS9 has been shown to be important for virus anterograde axonal transport and spread of infection both in vivo and in vitro for both HSV and PRV (4–11). However, the exact role of pUS9 in anterograde transport of HSV-1 requires further definition.
In this study, we used two independent US9 deletion and repair viruses to infect neurons in the somal compartment of trichamber microfluidic devices where axons penetrate through microgrooves into a separate compartment (axonal compartment) to which Vero cells were added prior to infection. Using immunofluorescence and confocal microscopy, we observed partial reductions (∼50%) in the anterograde axonal transport of capsids but not of envelope glycoproteins (gC, gD, and gE) to distal axons, which were similar between the two pairs of US9 deletion and repair viruses. However, there was a marked (∼90%) and significant reduction in virus spread from axons to Vero cells in the absence of pUS9. Using TEM, a marked reduction or absence of enveloped capsids was observed in growth cones and varicosities in axons infected in the absence of pUS9.
The absence of enveloped capsids in growth cones and varicosities in axons of US9 deletion virus could be explained in two ways. One is that in the absence of pUS9, there is a block in the transport of enveloped capsids from the cell body into axons. Alternatively, there is a defect in envelopment of capsids that reach the varicosities and growth cones in the absence of pUS9. Our observations strongly support the latter alternative. First, we have not observed enveloped capsids in the mid-axon between varicosities and growth cones. Second, our confocal and TEM observations showed no obvious impairment or blockage of viral capsids (enveloped or unenveloped) leading to their localized accumulation in the cytoplasm of the cell body, and similar proportions of both enveloped and unenveloped capsids were present in the cytoplasm of the cell body in the absence of pUS9. Hence, there was a second temporal impairment of virus assembly in distal axons in the absence of pUS9 despite the presence of other envelope proteins. Overall, our results show that deletion of pUS9 leads to defects in the overall anterograde axonal transport of HSV-1 at two sites—virus transport along axons (partial defect) and virus assembly and egress in growth cones prior to transfer to epithelial cells (a marked defect).
Our results using immunofluorescence are consistent with collective defects in combined HSV-1 transport and spread to epithelial cells in vitro and in vivo after pUS9 deletion previously reported by the Johnson and LaVail labs (6, 8). HSV-1 US9 deletion mutants have been also reported to be severely compromised in virus anterograde spread from the trigeminal ganglia to the cornea after infection and spread in the mouse corneal epithelium (15) and also from the retina into the optic nerve after infection of the mouse retinal ganglion cell bodies (6). Our findings that transport of capsids but not glycoproteins along axons in vitro was partially inhibited in the absence of pUS9 correlate with the studies by LaVail et al. showing that pUS9 was required for long distance anterograde transport of capsids but not glycoproteins to axon terminus and for transneuronal spread of infectious virus in vivo (6). Recently, the axonal transport of PRV gM was also found to be independent of pUS9 (23).
McGraw et al. reported that pUS9 deletion impaired anterograde spread of HSV-1 in vivo using animal models and in vitro using Campenot chambers, although this was partial and subject to strain variation (24). However, their in vitro findings were based on HSV-1 spread from axons to Vero cells in the absence of any agent to limit the secondary virus spread in Vero cells. Howard et al. (8) and our studies here prevented such spread by using human immunoglobulin and the HSV DNA polymerase inhibitor foscarnet, respectively, in the media added to the axonal compartment where Vero cells were plated to prevent secondary virus spread after infection of the somal compartment. In our experience, in the absence of foscarnet, there was a massive amplification of HSV-1 infection in Vero cells, which masked the true spread defects of US9 deletion mutants (unpublished results). In addition, our findings show consistent results for US9 deletion with different virus strains (F and KOS).
Recently, Johnson and colleagues showed that deletion of either the pUS9, gE, or gE/gI extracellular domain alone led to partial inhibition of capsid transport into axons, whereas deletion of both pUS9 and gE completely blocked capsid transport into axons and virus spread from axons into epithelial cells (8, 12). They proposed two models to explain the roles of pUS9 and gE/gI. The first model is the loading hypothesis, in which in the absence of pUS9 and gE/gI, there is a failure of capsid- and glycoprotein-containing vesicles from the trans-Golgi network (TGN) being loaded onto kinesin motors and being transported into proximal axons. pUS9 and gE/gI may promote capsid transport into axons by interacting with tegument proteins coating unenveloped capsids. Alternatively in the adaptor hypothesis, cytoplasmic domains of pUS9 and gE/gI can interact directly with kinesin motors or kinesin adaptors to mediate transport of vesicles containing HSV glycoproteins (i.e., gB) along axons (8). Recent studies in PRV have shown that gE and gI are required for efficient KIF1A-mediated anterograde transport of PRV by facilitating the interaction between pUS9 and the kinesin-3 motor KIF1A (25, 26).
Our findings are similar to those of Howard et al. (8) in showing that US9 deletion causes a partial inhibition of the anterograde axonal transport of capsids but extend beyond to show that in the absence of pUS9, there is a second impairment in secondary envelopment of capsids that reach the axon terminus or growth cones. The deletion of pUS9 does not completely inhibit anterograde axonal transport of HSV-1 as capsids can be detected in this study along axons up to the distal ends by confocal microscopy and TEM. Similar findings have been recently reported for PRV in which deletion of pUS9 did not impair fast axonal transport of PRV particles that bypass the initial sorting barrier in the proximal axons and are able to reach the axon terminals (27). The unique defect reported in the present study is supported by our findings of aberrant tegument formation in occasional capsids observed in growth cones in the absence of pUS9. Direct comparative studies using TEM were undertaken to quantify the number of unenveloped and enveloped viral particles in varicosities and growth cones of pUS9 deletion virus (US9−) and the KOS strain and, as controls, pUS9 repair (US9R) and strain 17 and F viruses. Marked phenotypic similarities between both the pUS9 deletion strain and KOS strain viruses were found, especially a significant reduction or absence of enveloped capsids in the growth cones. All KOS strains examined to date, including the KOS strain used in this study, have a stop codon resulting in truncation of pUS9 (which removes the transmembrane region and results in loss of pUS9 expression) and also an extension in the envelope protein, pUS8A (16). This was subsequently confirmed by full sequencing of two KOS strains (16; our laboratory, unpublished data). The impairment in secondary envelopment was slightly less severe in the KOS strain, where some capsids (∼6%) in growth cones were fully enveloped, and this finding is consistent with KOS strains being able to cause recurrent herpes in vivo.
The unique impairment in secondary assembly and egress was only observed in axonal varicosities and growth cones and was not seen in the cell body of the neurons. Thus, this finding strongly suggests a major and essential role for pUS9 in HSV-1 assembly in growth cones at the axon terminus but not in the TGN in the cell body of neurons. This suggests a redundant function for pUS9 in the cell body that is absent in the growth cone, where pUS9 is essential. We hypothesize that pUS9 may be substituting for viral or cellular protein (perhaps from the TGN or TGN-derived vesicles) only found in the cell body, or alternatively pUS9 is interacting with a growth cone-specific protein not found in the cell body. The next step is to determine which domains in pUS9 facilitate HSV-1 anterograde axonal transport and egress from neurons (28).
It is now evident from this and previous studies (8, 12) that the anterograde transport and spread of HSV-1 are complex and probably consist of at least 3 phases: (i) loading of capsids and viral tegument and envelope proteins containing vesicles onto microtubules in the cell body, (ii) transport of capsids (unenveloped or enveloped) and vesicles containing tegument and envelope proteins by molecular motors along microtubules in the axon to the axon terminus, and (iii) virus assembly and/or exit in the terminal growth cones and varicosities. It is also clear that more than one HSV-1 protein may be mediating anterograde axonal transport and also that final virus assembly and exit from growth cones differ from those of the cell body in their use of HSV-1 proteins.
ACKNOWLEDGMENTS
We thank Karen Byth, Westmead Hospital, Australia, for extensive assistance in statistical analysis. We thank David Johnson, Oregon Health & Science University, USA, for providing the F-US9/GFP and F-US9/GFP-R viruses and advice on the trichamber systems. We thank Jennifer LaVail, University of California, San Francisco, USA, for providing the F strain, US9−, and US9R viruses. We also thank Thomas Mettenleiter, Friedrich Loeffler Institute, Germany, for providing the KOS virus and Frazer Rixon, University of Glasgow, for providing the strain 17 virus. We also thank Levina Dear, Carol Robinson, and Emma Kettle, Electron Microscope Laboratory, a joint facility of ICPMR, Westmead Hospital, Westmead Research Hub and the University of Sydney, for assistance with electron microscopy. We thank Laurence Cantrill, The University of Sydney, and Hong Yu, The Westmead Institute for Medical Research, for assistance with confocal microscopy. We thank the staff of the Research Holding, ICPMR, Westmead Hospital, and the Biological Services Facility at The Westmead Institute for Medical Research for ongoing assistance with the supply of rat neonates.
This work was supported by grants 570849 and AP1069193 from the Australian National Health and Medical Research Council to A. L. Cunningham, D. J. Diefenbach, and M. Miranda-Saksena and bridging grants from the Westmead Millennium Foundation to M. Miranda-Saksena and the Westmead Charitable Trust to D. J. Diefenbach.
REFERENCES
- 1.Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. 2008. Transport and egress of herpes simplex virus in neurons. Rev Med Virol 18:35–51. doi: 10.1002/rmv.560. [DOI] [PubMed] [Google Scholar]
- 2.Penfold ME, Armati P, Cunningham AL. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc Natl Acad Sci U S A 91:6529–6533. doi: 10.1073/pnas.91.14.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mikloska Z, Sanna PP, Cunningham AL. 1999. Neutralizing antibodies inhibit axonal spread of herpes simplex virus type 1 to epidermal cells in vitro. J Virol 73:5934–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brideau AD, Card JP, Enquist LW. 2000. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J Virol 74:834–845. doi: 10.1128/JVI.74.2.834-845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomishima MJ, Enquist LW. 2001. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J Cell Biol 154:741–752. doi: 10.1083/jcb.200011146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LaVail JH, Tauscher AN, Sucher A, Harrabi O, Brandimarti R. 2007. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience 146:974–985. doi: 10.1016/j.neuroscience.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snyder A, Polcicova K, Johnson DC. 2008. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J Virol 82:10613–10624. doi: 10.1128/JVI.01241-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howard PW, Howard TL, Johnson DC. 2013. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J Virol 87:403–414. doi: 10.1128/JVI.02465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draper JM, Huang G, Stephenson GS, Bertke AS, Cortez DA, LaVail JH. 2013. Delivery of herpes simplex virus to retinal ganglion cell axon is dependent on viral protein Us9. Invest Ophthalmol Vis Sci 54:962–967. doi: 10.1167/iovs.12-11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyman MG, Feierbach B, Curanovic D, Bisher M, Enquist LW. 2007. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J Virol 81:11363–11371. doi: 10.1128/JVI.01281-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyman MG, Curanovic D, Enquist LW. 2008. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog 4:e1000065. doi: 10.1371/journal.ppat.1000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howard PW, Wright CC, Howard T, Johnson DC. 2014. Herpes simplex virus gE/gI extracellular domains promote axonal transport and spread from neurons to epithelial cells. J Virol 88:11178–11186. doi: 10.1128/JVI.01627-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butchi NB, Jones C, Perez S, Doster A, Chowdhury SI. 2007. Envelope protein Us9 is required for the anterograde transport of bovine herpesvirus type 1 from trigeminal ganglia to nose and eye upon reactivation. J Neurovirol 13:384–388. doi: 10.1080/13550280701375433. [DOI] [PubMed] [Google Scholar]
- 14.Chowdhury SI, Onderci M, Bhattacharjee PS, Al-Mubarak A, Weiss ML, Zhou Y. 2002. Bovine herpesvirus 5 (BHV-5) Us9 is essential for BHV-5 neuropathogenesis. J Virol 76:3839–3851. doi: 10.1128/JVI.76.8.3839-3851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polcicova K, Biswas PS, Banerjee K, Wisner TW, Rouse BT, Johnson DC. 2005. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc Natl Acad Sci U S A 102:11462–11467. doi: 10.1073/pnas.0503230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Negatsch A, Mettenleiter TC, Fuchs W. 2011. Herpes simplex virus type 1 strain KOS carries a defective US9 and a mutated US8A gene. J Gen Virol 92:167–172. doi: 10.1099/vir.0.026484-0. [DOI] [PubMed] [Google Scholar]
- 17.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J Virol 83:6610–6623. doi: 10.1128/JVI.02655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saksena MM, Wakisaka H, Tijono B, Boadle RA, Rixon F, Takahashi H, Cunningham AL. 2006. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J Virol 80:3592–3606. doi: 10.1128/JVI.80.7.3592-3606.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miranda-Saksena M, Boadle RA, Aggarwal A, Tijono B, Rixon FJ, Diefenbach RJ, Cunningham AL. 2009. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J Virol 83:3187–3199. doi: 10.1128/JVI.01579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miranda-Saksena M, Boadle R, Cunningham AL. 2014. Preparation of herpes simplex virus-infected primary neurons for transmission electron microscopy. Methods Mol Biol 1144:223–234. doi: 10.1007/978-1-4939-0428-0_15. [DOI] [PubMed] [Google Scholar]
- 21.Aggarwal A, Miranda-Saksena M, Boadle RA, Kelly BJ, Diefenbach RJ, Alam W, Cunningham AL. 2012. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J Virol 86:6123–6137. doi: 10.1128/JVI.07016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miranda-Saksena M, Boadle RA, Armati P, Cunningham AL. 2002. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J Virol 76:9934–9951. doi: 10.1128/JVI.76.19.9934-9951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kratchmarov R, Enquist LW, Taylor MP. 2015. Us9-independent axonal sorting and transport of the pseudorabies virus glycoprotein gM. J Virol 89:6511–6514. doi: 10.1128/JVI.00625-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGraw HM, Awasthi S, Wojcechowskyj JA, Friedman HM. 2009. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J Virol 83:8315–8326. doi: 10.1128/JVI.00633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, Enquist LW. 2012. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12:806–814. doi: 10.1016/j.chom.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, Enquist LW. 2013. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J Virol 87:9431–9440. doi: 10.1128/JVI.01317-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daniel GR, Sollars PJ, Pickard GE, Smith GA. 2015. Pseudorabies virus fast-axonal transport occurs by a pUS9-independent mechanism. J Virol 89:8088–8091. doi: 10.1128/JVI.00771-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diefenbach RJ, Davis A, Miranda-Saksena M, Fernandez MA, Kelly BJ, Jones CA, LaVail JH, Xue J, Lai J, Cunningham AL. 2016. The basic domain of herpes simplex virus 1 pUS9 recruits kinesin-1 to facilitate egress from neurons. J Virol 90:2102–2111. doi: 10.1128/JVI.03041-15. [DOI] [PMC free article] [PubMed] [Google Scholar]