

ABSTRACT
The principles underlying membrane binding and assembly of retroviral Gag proteins into a lattice are understood. However, little is known about how these processes are related. Using purified Rous sarcoma virus Gag and Gag truncations, we studied the interrelation of Gag-Gag interaction and Gag-membrane interaction. Both by liposome binding and by surface plasmon resonance on a supported bilayer, Gag bound to membranes much more tightly than did matrix (MA), the isolated membrane binding domain. In principle, this difference could be explained either by protein-protein interactions leading to cooperativity in membrane binding or by the simultaneous interaction of the N-terminal MA and the C-terminal nucleocapsid (NC) of Gag with the bilayer, since both are highly basic. However, we found that NC was not required for strong membrane binding. Instead, the spacer peptide assembly domain (SPA), a putative 24-residue helical sequence comprising the 12-residue SP segment of Gag and overlapping the capsid (CA) C terminus and the NC N terminus, was required. SPA is known to be critical for proper assembly of the immature Gag lattice. A single amino acid mutation in SPA that abrogates assembly in vitro dramatically reduced binding of Gag to liposomes. In vivo, plasma membrane localization was dependent on SPA. Disulfide cross-linking based on ectopic Cys residues showed that the contacts between Gag proteins on the membrane are similar to the known contacts in virus-like particles. Taken together, we interpret these results to mean that Gag membrane interaction is cooperative in that it depends on the ability of Gag to multimerize.
IMPORTANCE The retroviral structural protein Gag has three major domains. The N-terminal MA domain interacts directly with the plasma membrane (PM) of cells. The central CA domain, together with immediately adjoining sequences, facilitates the assembly of thousands of Gag molecules into a lattice. The C-terminal NC domain interacts with the genome, resulting in packaging of viral RNA. For assembly in vitro with purified Gag, in the absence of membranes, binding of NC to nucleic acid somehow facilitates further Gag-Gag interactions that lead to formation of the Gag lattice. The contributions of MA-mediated membrane binding to virus particle assembly are not well understood. Here, we report that in the absence of nucleic acid, membranes provide a platform that facilitates Gag-Gag interactions. This study demonstrates that the binding of Gag, but not of MA, to membranes is cooperative and identifies SPA as a major factor that controls this cooperativity.
INTRODUCTION
Late in the retroviral life cycle, the structural protein Gag localizes to the inner leaflet of the plasma membrane (PM), where it assembles around the viral genomic RNA, leading to budding from the cell. Gag proteins include three major domains, the N-terminal matrix (MA) domain, which mediates membrane binding; the central capsid (CA) domain, which mediates Gag-Gag interactions during virion assembly; and the C-terminal nucleocapsid (NC) domain, which interacts specifically with the viral genomic RNA. Each of these domains is critical for the production of infectious virions.
Retroviral Gag proteins employ multiple signals to mediate membrane binding, including electrostatic interaction, fatty acid modification, recognition of specific lipid head groups, and protein multimerization (reviewed in reference 1). MA contains a basic patch of clustered lysine and arginine residues that mediate electrostatic interactions with the negatively charged inner leaflet of the plasma membrane (2). Electrostatic interaction is a major contributor to membrane association for Rous sarcoma virus (RSV) Gag, since mutations of basic amino acids in MA abrogate membrane association in vivo but are rescued by compensatory mutations (3) in nearby parts of the polypeptide. Also, RSV MA is stripped off liposome membranes in vitro by increasing NaCl concentrations (4). Both RSV Gag and HIV Gag are sensitive to the charged PM inner leaflet lipid l-α-phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] (5–13). HIV MA has a defined PI(4,5)P2 binding pocket (14), but RSV MA has no known pocket, consistent with the idea that sensitivity to PI(4,5)P2 is primarily due to electrostatics (4, 9). HIV Gag-membrane interaction is sensitive, not only to the lipid head groups, but also to the acyl chain composition (15), while RSV Gag-membrane interaction appears to be independent of the acyl chain type (16).
Multimerization is a strong contributor to membrane binding of Gag. Mutations that weaken HIV Gag assembly in cells also reduce Gag-membrane association (17). Artificial multimerization of MA, a model for the effect of Gag multimerization, greatly increases membrane binding (4, 18–20). In vitro, RSV MA dimerization increases membrane binding by as much as 10-fold (4), and HIV MA dimerization partially rescues myristoylation defects that block membrane association of monomeric MA (18). In cells, monomeric HIV MA is largely cytoplasmic, while dimerized MA localizes to the PM (18, 19). Monomeric and dimeric RSV MA are cytoplasmic, while MA that is artificially hexamerized by fusion to the Ccmk4 domain from the carboxysome shell protein (21, 22) localizes strongly to the PM (19).
Virion assembly is thought to begin with the dimerization of two Gag molecules, leading eventually to the formation of an incomplete lattice of Gag hexamers (reviewed in reference 23). The majority of contacts in the Gag hexameric lattice occur between CA domains, with some additional contacts immediately upstream and downstream of CA (23). Recently published 8-Å cryo-electron microscopy (EM) structures of immature virions clearly show many of the contacts in the immature Gag lattice of RSV (24) and HIV (25). In RSV, a stretch of polypeptide in the p10 domain immediately upstream of CA plays an essential role in the formation of the immature lattice. A helix near the C terminus of p10 loops into the N-terminal region of the CA hexamer, forming a critical contact with the neighboring CA N-terminal domain (NTD) (24, 26). Mutations in this sequence disrupt the formation of virus-like particles (VLPs) (26–29). Furthermore, a pair of ectopic cysteine residues positioned to capture, by disulfide bond formation, contacts between p10 and the N-terminal domain of CA in VLPs result in the formation of cross-linked CA-p10 hexamers in VLPs both in vitro and in vivo (26).
Between the CA and NC domains of some retroviral Gag proteins is a short cleavage product that is critical for virion assembly, named the spacer peptide (SP1 or p2) in HIV (30–32) or the spacer peptide assembly (SPA) domain in RSV (33), as reviewed in reference 23. The spacer peptide likely forms a six-helix bundle that extends below the CA lattice in the immature virion (34). In vitro and in vivo, mutations in SP1 (35) or SPA (33, 35) disrupt proper assembly. Alanine-scanning mutagenesis of RSV SPA revealed that the residues essential for in vitro assembly are positioned on the hydrophobic faces of the helices that are modeled to hold the six-helix bundle together (33).
Gag assembly occurs at the inner surface of the PM in most retrovirus genera, with the exception of beta-retroviruses, such as mouse mammary tumor virus (MMTV) and Mason-Pfizer monkey virus (M-PMV), where immature Gag particles are formed in the cytoplasm and transported to the PM. Assembly at the PM is dependent on efficient membrane association. For example, in HIV, a glycine-to-arginine mutation that prevents HIV Gag myristoylation blocks budding (17, 36–38). Mutations of basic amino acids, which disrupt membrane association, also result in a loss of VLP assembly (39, 40). Measured by Förster resonance energy transfer (FRET) and fluorescence correlation spectroscopy (FCS), RSV Gag is multimerized into low-molecular-weight complexes (41). By RNA-protein cross-linking in vivo, viral genomic RNA (vgRNA) was found to associate with HIV Gag molecules that were primarily monomeric or dimeric in the cytoplasm, although the PM was required for multimerization into higher-molecular-weight products (42).
Recent studies suggest that NC may contribute to the early steps of Gag membrane association. In solution, the HIV Gag protein was found to sample a wide range of conformations, but on average, it was compact (43). This finding contrasts with findings for RSV Gag and murine leukemia virus (MLV) Gag, which on average adopts an elongated conformation (16, 43, 44). Compared with HIV and MLV, RSV was highly flexible, a property that was inferred to be due at least in part to an unstructured region between the membrane binding domain (MBD)—the N-terminal end of MA—and p10 (16). Measurements of membrane-bound Gag conformations by neutron reflectometry (NR) showed that RSV and HIV Gag proteins do not extend far from the bilayer, consistent with a compact Gag structure (16, 45). The membrane-bound Gag conformation was interpreted to result from both the MA and NC domains simultaneously interacting with the bilayer (16, 45). In support of this model, removal of the NC domain significantly reduced the binding of RSV Gag to membranes (16).
The present study was undertaken to test two models of Gag membrane binding. Using purified RSV Gag and Gag C-terminal truncations, we showed that Gag membrane binding is not dependent on the C-terminal NC domain. In addition, we found that the SPA domain is critical in eliciting strong membrane binding. In cells, while the removal of NC and SPA resulted in a loss of PM localization, constructs containing an intact SPA partially rescued this defect. Based on surface plasmon resonance (SPR) of supported bilayers, as well as on disulfide bond formation between ectopic cysteine residues in Gag bound to liposomes in vitro, we infer that Gag membrane binding involves protein-protein interaction.
MATERIALS AND METHODS
DNA constructs, protein purification, and VLP assembly.
DNA constructs used for tissue culture were cloned into pEGFP using standard subcloning techniques, resulting in C-terminal truncations and cysteine mutants of Gag delta protease (referred to throughout as Gag) in the context of the Gag-green fluorescent protein (GFP) fusion protein (9). DNA constructs used for protein purification (diagrammed in Fig. 1; also see Fig. 4) were cloned into the pSUMO vector using standard subcloning techniques and as previously described (19). Protein purification was performed using bacterial expression, as previously described (19). Briefly, BL21 bacterial cells containing the DNA construct of interest were grown to an optical density at 600 nm (OD600) of ∼0.6, at which point protein expression was induced with the addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 0.5 mM. About 5 to 7 h postinduction, cells were harvested by centrifugation and frozen at −20°C. The frozen cell pellets were lysed by sonication in buffer [20 mM Tris-HCl (pH 8), 500 mM NaCl, 5% glycerol, 2 μM ZnCl2, 2 mM Tris-(2-carboxyethyl)phosphine (TCEP) reducing reagent, 2 mM PMSF (phenylmethylsulfonyl fluoride) protease inhibitor]. The lysed cells were cleared by centrifugation at 90,000 rpm in a Beckman TLA110 rotor for 45 min at 4°C. Nucleic acid was removed from the cleared lysates by adding polyethyleneimine (PEI) to a final concentration of 0.3%. Precipitated nucleic acid was removed by centrifugation at 10,000 rpm for 10 min in a Sorvall SA600 rotor. The protein of interest was precipitated from the lysate by addition of ammonium sulfate to ∼20% and 10,000-rpm centrifugation for 10 min in a Sorvall SA600 rotor. The ammonium sulfate-precipitated protein pellet was dissolved in buffer (20 mM Tris-HCl [pH 8], 5% glycerol, 50 mM NaCl, 2 μM ZnCl2, 1 mM TCEP). The protein was further purified by cation-exchange chromatography (HiTrap SP FF; GE Healthcare). The eluted protein was than purified using Ni2+ chromatography (HisTrap HP; GE Healthcare). The eluted SUMO-tagged protein was then dialyzed (20 mM Tris-HCl [pH 8], 5% glycerol, 500 mM NaCl, 0.5 mM TCEP, 2 μM ZnCl2) overnight in the presence of ULP1 protease (46) to cleave off the SUMO tag. The SUMO tag and ULP1 protease were removed from the dialyzed protein by Ni2+ chromatography. Purified protein at 3 to 5 mg/ml at a 260/280-nm absorption ratio of ∼0.58 in dialysis buffer was flash frozen and stored at −80°C.
FIG 1.

Liposome binding analysis of purified Gag proteins. LUVs were prepared with the following compositions: POPC/POPS/Chol (34/30/36) and POPC/POPS/Chol/PI(4,5)P2 (32/30/36/2). (A) Schematic of all purified proteins used (left) and representative Coomassie-stained polyacrylamide gel of flotation results (right). The asterisks indicate the relative locations of the I475A mutations. MB, membrane-bound fractions; NMB, non-membrane-bound fractions. (B) The bars represent the average percentages of total protein associated with liposomes for no less than four reactions; the error bars represent standard deviations.
FIG 4.
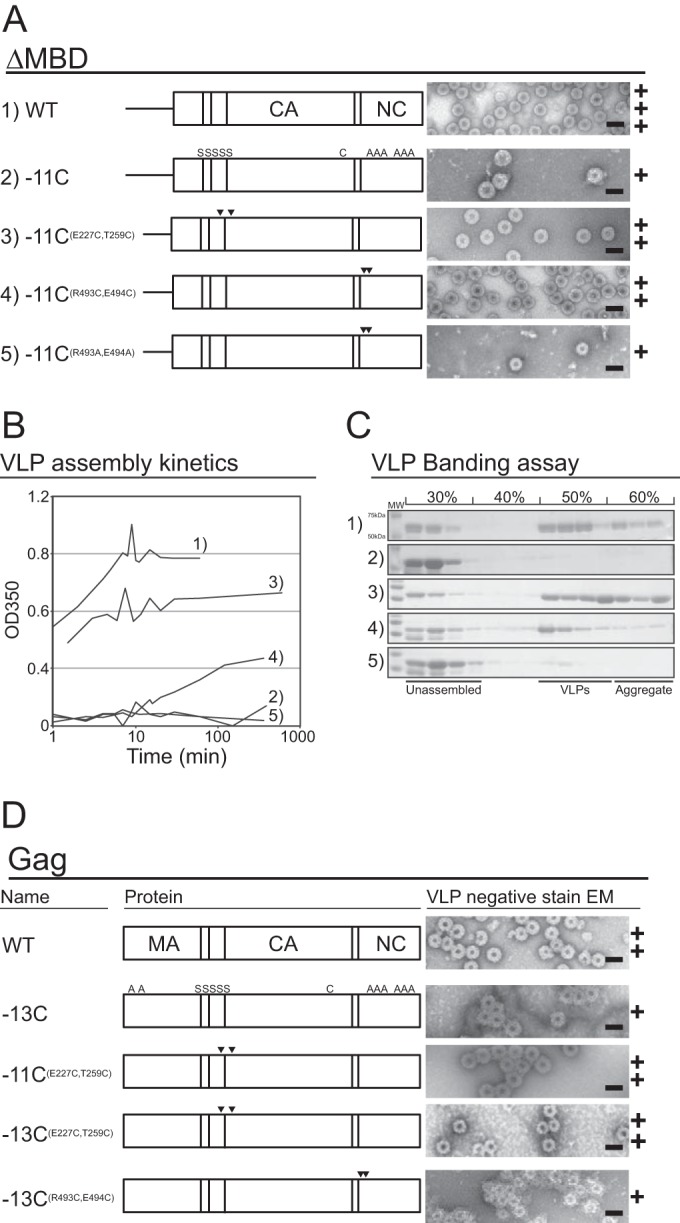
In vitro assembly of ΔMBD and Gag cysteine mutants. The numbers 1 to 5 in panels A to C correspond. In panels A and D, the letters S and A are minus cysteine, arrowheads are plus cysteine, and C corresponds to the endogenous cysteine in CA (25). (A) Schematic of purified ΔMBD proteins (left) and representative negative-stain EM images (right). (B) Assembly kinetics of ΔMBD constructs measured by light scattering. (C) VLP banding of ΔMBD constructs on a 30 to 60% (wt/wt) sucrose step gradient. Unassembled and aggregated protein fractions and VLPs are identified, and the molecular weight bands identified for sample 1 are the same for all samples. (D) Schematic of purified Gag proteins (left) and representative EM images (right). Scale bars, 100 nm. The plus symbols represent qualitative estimates of particle numbers per EM grid (+, tens; ++, hundreds; +++, thousands).
Liposome flotation and pelleting.
All the lipids used (Table 1) were assayed for phosphate to determine stock concentrations to within 1% (47) and assayed by thin-layer chromatography (TLC) to verify lipid quality. Lipids dissolved in chloroform were mixed at the ratios reported (in mol%). Lipid mixtures were exchanged from organic solvent into aqueous buffer (20 mM Tris-HCl [pH 8]) by rapid solvent exchange (RSE) (48) followed by extrusion (9), resulting in large unilamellar vesicles (LUVs) (∼100 nm). Flotation and pelleting were performed as previously described (15, 16, 19). Briefly, binding reaction mixtures contained 15 μg protein (precleared by high-speed centrifugation to remove any aggregates) and 50 μg liposomes in buffer with 150 mM NaCl in a final volume of 25 μl. The reaction mixtures were incubated at room temperature for 10 min. For flotations, the 25 μl binding reaction mixture was mixed with 75 μl of 67% sucrose to a final sucrose concentration of 50%, and 80 μl of the resulting mixture was transferred to a Beckman TLA100 ultracentrifuge tube. The 50% sucrose binding reaction mixture was overlaid with 120 μl 40% sucrose and 40 μl 4% sucrose. All sucrose solutions were prepared as weight/weight in 20 mM Tris-HCl (pH 8) and 150 mM NaCl. Sucrose gradients were ultracentrifuged in a Beckman TLA100 rotor at 90,000 rpm for 1 h at 4°C. Four 60-μl fractions were taken for each gradient and subjected to PAGE, Coomassie blue staining, and densitometry analysis. For pelleting, the 25-μl binding reaction mixture was mixed with 215 μl buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl), followed by ultracentrifugation as described for flotation. Following centrifugation, the supernatant was discarded, and the pellet was dissolved in 1× SDS-PAGE buffer and analyzed as described for flotation.
TABLE 1.
Lipids used in the study
| Name | Abbreviation | Chain |
|---|---|---|
| 1-Palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine | POPC | 16:0-18:1 |
| 1-Palmitoyl-2-oleoyl-sn-glycerol-3-phospho-l-serine | POPS | 16:0-18:1 |
| 1,2-Dioleoyl-sn-glycerol-3-phosphocholine | DOPC | 18:1-18:1 |
| 1,2-Dioleoyl-sn-glycerol-3-phospho-l-serine | DOPS | 18:1-18:1 |
| l-α-Phosphatidylinositol-4,5-bisphosphate | PI(4,5)P2 | 18:0-20:4a |
Most abundant acyl chain composition.
In vitro assembly.
In vitro assembly of purified Gag and GagΔMBD proteins (see Fig. 4) was performed as previously described (23, 49). Briefly, purified protein at 3 to 5 mg/ml was mixed with 1:10 mass of the 50-mer oligonucleotide (GT25), followed by a 1:5 dilution with assembly buffer (20 mM MES [morpholineethanesulfonic acid] [pH 6.5], 2 μM ZnCl2) to a final NaCl concentration of 100 mM. Assembly reaction mixtures were screened for VLPs by negative-stain (2% uranyl acetate) EM on a Morgagni 268 transmission electron microscope. Semiquantitative measurements of VLP assembly rates and efficiency were performed as previously described (50).
Assembly of full-length Gag frequently resulted in VLPs that appeared stuck together in large aggregates, which we assumed to be due to misassembled protein. This phenomenon prevented quantification of assembly. We hypothesize that at least some of the aggregation was a consequence of the interaction of the basic MA domain with nucleic acid, effectively bridging between VLPs, since no aggregation was observed for ΔMBD assembly. Increasing the NaCl from the standard 100 mM to 200 mM did not prevent aggregation, and VLP assembly was not detected at NaCl concentrations above 200 mM.
Oxidation.
Disulfide bond formation in VLPs and in membrane-bound Gag was achieved by oxidation of cysteines, as previously described (26). TCEP was removed from protein stocks by serial dilution and spin concentration with buffer (20 mM Tris-HCl [pH 8], 500 mM NaCl, 2 μM ZnCl2). Assembly and membrane binding reaction mixtures each contained 15 μg of protein and a final buffer of 20 mM MES, pH 6.5, 100 mM NaCl, and 2 μM ZnCl2 at a final volume of 25 μl. Membrane binding reaction mixtures also contained 50 μg lipid. All reaction mixtures were incubated for 1 h prior to oxidation. Chemically induced oxidation was performed by adding copper phenanthroline (Cu-Phen) (60 μM CuSO4, 267 μM O-phenanthroline), mixing by vortexing, and immediately quenching with 20 mM iodoacetamide and 3.7 mM neocuproine. All samples were analyzed by PAGE in nonreducing 4 to 20% TGX gradient gels (Bio-Rad) and by Coomassie staining.
Cell imaging and VLP release.
QT6 (quail) fibroblasts were maintained as previously described (9). Briefly, the cells were grown in Dulbecco modified Eagle medium supplemented with 5% fetal bovine serum, 5% NuSerum (BD Biosciences), 1% heat-inactivated chick serum, standard vitamins, l-glutamine, penicillin, and streptomycin. At 24 h postseeding, the cells were transfected with Lipofectamine 2000 (Invitrogen) and 2 μg of DNA per well in a six-well dish, according to the manufacturer's protocol.
For VLP release, ∼30 h posttransfection, extracellular and cellular fractions were harvested as previously described (9). Cellular and VLP-associated Gag proteins were assayed for with anti-CA antibody (1:1,000), followed by anti-rabbit antibody (1:20,000) (DyLight 800; Rockland). Membranes were imaged on a Licor Odyssey scanner and quantified using the densitometry function in ImageQuant 5.2. At least three transfections were performed for each construct. P values were calculated using Student's t test. For imaging of GFP fusion proteins, slides were fixed and mounted as previously described (19) and imaged on a spinning-disk confocal microscope (Andor Revolution). At least three transfections and 30 images were collected for each construct. The plasma membrane-to-cytoplasmic signal ratio was calculated by measuring the signal intensity using the ImageJ plot profile function (19). Two or three measurements were taken for each cell. The box plots represent at least 65 measurements per construct, where the horizontal line is the median, the box is the 25th to 75th percentile, and whiskers represent the largest and smallest values that are not outliers.
SPR measurements of protein binding to stBLMs.
Both the SPR method for quantifying intermolecular interactions (51) and the sparsely tethered bilayer lipid membrane (stBLM) model membrane system (52) are well established. stBLMs incorporate a single lipidic bilayer membrane of controlled composition that is tethered to a planar electrode—a 0.05-μm-thick Au film on a glass or Si substrate—via short ethylene oxide anchors (53), thereby providing a hydrated environment on both sides of the membrane. Thus, the bilayer retains its intrinsic fluidity (54), is virtually defect free (52), and is resilient to external manipulations, such as buffer exchanges, as monitored by electrochemical impedance spectroscopy (EIS) (55). In combination, SPR performed on stBLMs offers unique advantages for the study of viral assembly processes on membranes.
stBLMs were prepared as follows. The tether compound HC18 [Z20-(Z-octadec-9-enyloxy)-3,6,9,12,15,18,22-heptaoxatetracont-31-ene-1-thiol)] was synthesized and characterized as described previously (53). Lipid mixtures used to form stBLMs were mixed in chloroform and placed under a high vacuum overnight to remove the solvent. The resulting dry lipid films were hydrated in a high-salt aqueous buffer (1 M NaCl, 10 mM NaPO4 [pH 7.4]) at lipid concentrations of ∼5 mg/ml and subsequently sonicated until clear vesicle solutions were obtained. The vesicle solutions were extruded through polycarbonate membranes with a pore size of 100 nm to obtain uniform distribution of unilamellar liposomes.
Glass microscope slides were cleaned with sulfuric acid plus Nochromix (Godax Laboratories, Cabin John, MD), followed by ultrapure water (EMD Millipore) and pure ethanol (Pharmco-Aaper) rinses, and dried under nitrogen gas. The substrates were loaded into a magnetron sputtering instrument (ATC Orion; AJA International) and coated with an ∼2-nm Cr adhesion layer followed by an ∼45-nm Au layer. Self-assembled monolayers (SAMs) were prepared by overnight incubation of the Au-coated substrate in 0.2 mM ethanolic solution (final concentration) of the lipid tether compound HC18 and β-mercaptoethanol (β-ME) in molar ratios of 1:1. The vesicles were immediately incubated on the SAM-covered slides for at least 2 h, followed by a low-ionic-strength buffer (50 mM NaCl, 20 mM Tris, 2 mM dithiothreitol [DTT] [pH 8]) that completed stBLM formation. For SPR measurements, stBLMs were assembled on a custom-built SPR instrument (SPR Biosystems, Germantown, MD) in the Kretschmann configuration (56). To confirm bilayer quality, EIS characterization was performed before each SPR experiment (55), and the resonance angle at the neat bilayer was measured for reference. Thereafter, increasing concentrations of protein were added. Protein adsorption induces changes in the refractive index near the bilayer-buffer interface, resulting in a shift of the resonance angle. The software package SPR Aria (SPR Biosystems) was used for real-time data recording at 25°C. Time courses of the SPR response, R(t), at each protein concentration were monitored until equilibrated to determine the equilibrium value, Req. The Hill equation, , with the coefficient N (with N = 1, corresponding to the Langmuir adsorption isotherm), was used for modeling the concentration-dependent protein load on the membrane, thereby quantifying the protein affinity in terms of the dissociation constant, Kd, and the equilibrium surface density of bound protein, R∞ (51).
RESULTS
Multimerization of a membrane binding protein such as Gag should increase its membrane affinity if the binding sites on the monomers face in the same direction toward the bilayer. Experimentally, using artificially dimerized or hexamerized MA fusion proteins, we showed previously that increasing the multimerization state of MA dramatically increased its binding to liposomes (4, 18, 19). More recently, we reported that purified RSV Gag in solution was monomeric and flexible and that at 150 mM NaCl it bound to membranes significantly better than did the isolated MA domain or a truncated Gag missing NC (16). The membrane associations of hexamerized MA and of Gag were quantitatively similar (19). These results suggested a model in which the strong interaction of Gag with membranes is due to protein multimerization. On the other hand, neutron reflectometry measurements of both RSV (16) and HIV (45) Gag membrane binding showed that both the MA and NC domains interact simultaneously with the membrane at 50 mM NaCl. This result suggests a model in which simultaneous membrane interaction of the two Gag domains potentiates a tighter interaction, as might be expected.
Liposome binding of purified RSV Gag and Gag truncations.
To determine if NC plays a role in RSV Gag membrane association at physiological ionic strengths, we purified a series of Gag C-terminal-truncation proteins (Fig. 1A) and tested their membrane binding by liposome flotation at 150 mM NaCl to liposomes with the compositions POPC/POPS/Chol (34/30/36) and POPC/POPS/Chol/PI(4,5)P2 (32/30/36/2) (lipid names are shown in Table 1). The truncation sites for this first group of proteins are delineated by the natural Gag-processing sites. In a second group of proteins, we extended the C terminus to include several residues of the NC domain (MASP+n), resulting in proteins that include the entire SPA domain, which extends several residues into NC (33, 57). In the context of this series of proteins, we also tested the membrane binding of a protein with an SPA point mutation known to disrupt in vitro Gag assembly (33).
C-terminal truncation of RSV Gag resulted in a dramatic decrease in membrane association, with GagΔNC (ΔNC), GagΔNCΔSP (MACA), and MA itself binding to POPC/POPS/Chol liposomes and POPC/POPS/Chol plus 2% PIP2 liposomes to approximately the same extent (Fig. 1A). That is, liposomes with PIP2 supported about 30% binding and those without PIP2 supported less than 10% binding. However, compared with ΔNC, proteins with an extension of four (MASP+4), six (MASP+6), or eight (MASP+8) amino acids into NC increased binding to PIP2-containing liposomes significantly (to 67%, 62%, and 80%, respectively). MASP+8 bound nearly as well to PIP2-containing membranes as Gag. On the other hand, binding to liposomes without PIP2 was not increased. Taken together, these data suggest that the major contribution of the RSV NC domain to membrane interaction is not due to the basic parts of NC, but instead, at least under these conditions, stems from an intact SPA domain, which extends into the first few residues of the N-terminal end of NC. This function of SPA is apparent only in the presence of PIP2 and presumably is to transiently stabilize Gag oligomerization.
The SPA domain is predicted to form an α-helix that, in the context of immature virus assembly, forms a six-helix bundle below the C-terminal domain of CA (CACTD) lattice (33, 34). A single isoleucine-to-alanine mutation (I475A) in SPA abrogates both six-helix-bundle formation (33) and in vitro assembly of VLPs (33). To test if the robust membrane association observed for the MASP+n constructs is dependent on the ability of SPA to undergo homotypic interactions, we tested the effect of the I475A mutation in the context of MASP+6 and MASP+8 (Fig. 1B). The mutation reduced membrane association of MASP+6 and MASP+8 by roughly one-half, measured with membranes containing PI(4,5)P2. We interpret this result to mean that the effect of SPA on membrane association is based on protein-protein interactions that increase membrane binding.
Previously, we reported that hexameric MA (MA-Ccmk4), when generated by in vitro translation in a reticulocyte extract, associated with membranes at levels similar to those of RSV Gag (19). Similarly, when fused to GFP, the same hexameric protein localized strongly to the PM in cells, quantitatively similar to the PM localization of Gag-GFP. We have now compared the membrane association of purified MA-Ccmk4, known to be hexameric in solution (19), with the membrane binding of purified RSV Gag. As observed for protein synthesized by in vitro translation, Gag and MA-Ccmk4 were found to associate with liposomes at similar levels, independently of PIP2 (Fig. 1B). Therefore, while Gag and MA-Ccmk4 do not require PIP2 for robust membrane association, MASP+n proteins do. We speculate that the multivalent PIP2 can stabilize transient MASP+n oligomers that bind to the membrane. On the other hand, monovalent PS in the absence of PIP2 cannot offer such stabilization, so that membrane-bound MASP+n oligomers that dissociate into monomers show an increased off rate from the membrane, leading to the observed reduction in affinity. In contrast, protein-protein interactions between full-length Gag or MA-Ccmk4, which is already oligomeric in solution, are more resilient and do not require such stabilization when bound to the membrane. This model would predict that Gag association with the membrane is cooperative in nature, while the membrane association of shorter constructs, such as the MA domain, is not.
To assess if the MASP+n proteins behave in vivo as they do in vitro, we created C-terminally fused GFP versions of a subset of the constructs shown in Fig. 1A. These DNAs were transfected into quail QT6 cells, and then the cells were examined by fluorescence microscopy. MACA-GFP and MASP+8(I475A)-GFP were both cytoplasmic, while MASP+8-GFP (and MASP+6-GFP [data not shown]) were partially localized to the PM (Fig. 2). Compared with Gag, which exhibited punctate fluorescence at the PM (9), MASP+8-GFP exhibited diffuse fluorescence similar to that of RSV MA-Ccmk4 (19). Quantification of the PM and cytoplasmic signal intensities of each construct confirmed the visual impression that MASP+8-GFP is enriched at the PM compared with MACA-GFP and MASP+8(I475A)-GFP (Fig. 2B). However, the ratio between signals observed at the PM and in the cytoplasm for MASP+8-GFP was significantly less than that for Gag-GFP (with medians of 1.8 and 3.5, respectively).
FIG 2.

Cellular localization and VLP release of Gag C-terminal-truncation proteins. All proteins were C-terminally GFP fused. (A) (Top) Representative confocal images of cellular Gag-GFP fluorescence. (Bottom) The orange lines correspond to the signal intensity plot used to quantify the ratio of plasma membrane to cytoplasmic localized protein in panel B. (B) Box plots of no less than 65 plot profile scans for each construct analyzed, as described in Materials and Methods. (C) Percent VLP release compared to Gag (normalized to 100%) as determined by Western blot quantification of supernatant and cytoplasmic Gag. The error bars represent the standard deviations of no less than three replicates (t test; *, P < 0.01).
We also measured the release of VLPs from transfected cells. MASP+8-GFP VLPs were not found in significant numbers in the medium (Fig. 2C). VLPs of MASP+6, i.e., protein lacking the GFP domain, also were not found in the medium, ruling out the possibility that the GFP domain prevents VLP formation (data not shown). We interpret these data to mean that, while the NC domain is partially dispensable for PM localization in vivo, it is required for the release of VLPs. In summary, these results show that efficient RSV Gag membrane association in vitro does not require the NC domain when PI(4,5)P2 is present and suggest that membrane association is enhanced by protein-protein interactions contributed by the SPA domain.
Surface plasmon resonance measurements of membrane binding.
We quantified the affinities of RSV MA and Gag binding to membrane bilayers in stBLMs at an NaCl concentration of 50 mM using SPR. The binding of MA and full-length Gag to DOPC/DOPS (70/30) membranes was different in several respects. First, while MA was removed completely by subsequent rinses with protein-free buffer (Fig. 3A), full-length Gag remained partially bound to the membrane surface (Fig. 3B). Second, unlike that for MA, the SPR response for Gag did not follow a Langmuir model, suggesting collective adsorption that is more appropriately described by the Hill equation (see above) with N greater than 1. Such behavior is characteristic of cooperativity and suggests that Gag oligomerization stabilizes the interaction of the protein with the membrane in a way that is absent for MA. Third, the measured membrane affinity of Gag was accordingly much greater than the affinity of MA (the Kd was approximately nanomolar versus micromolar) (Fig. 3C, black curves), confirming and quantifying the observations in liposome binding experiments.
FIG 3.
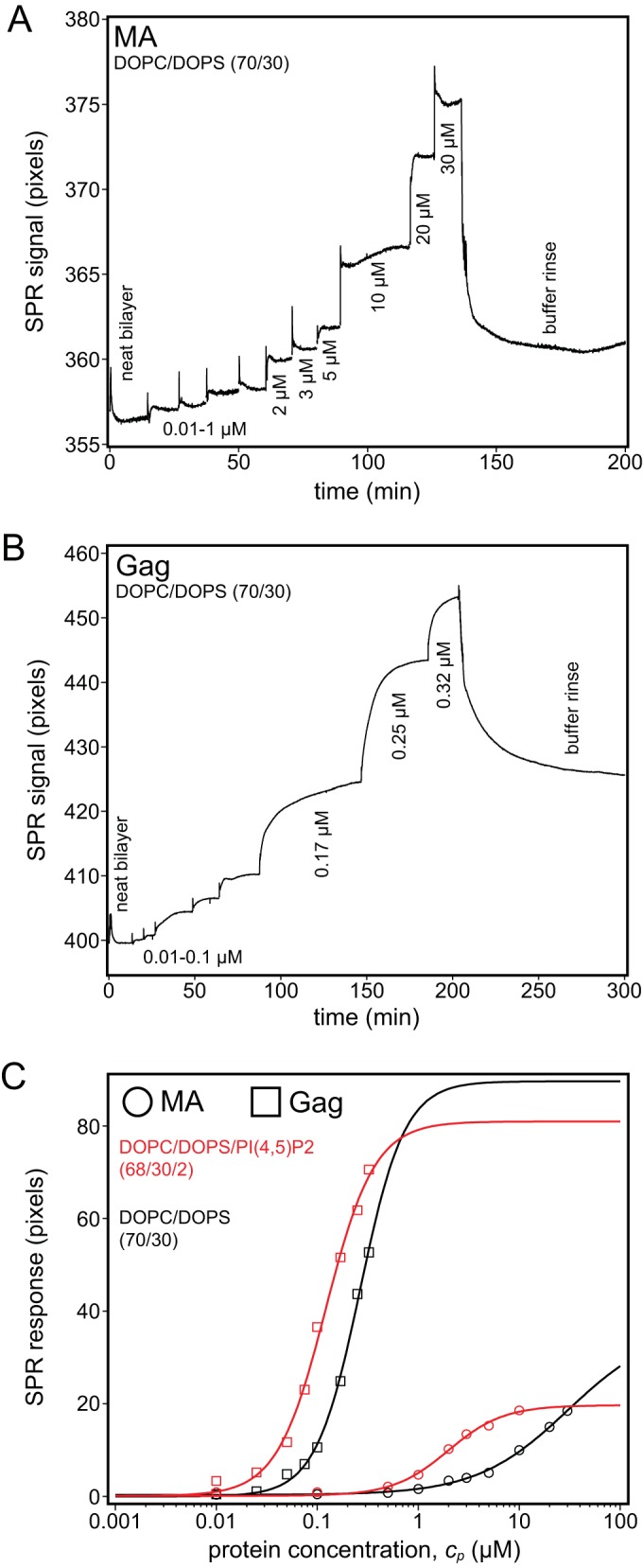
Surface plasmon resonance of Gag and the Gag MA domain. Protein binding affinities for stBLMs were quantified in 50 mM NaCl (pH 8) at a fixed PS concentration of 30%. (A and B) Exemplary SPR binding curves (raw data) of MA (A) and Gag (B) to DOPC/DOPS (70/30) stBLMs in which neat bilayers were exposed to increasing protein concentrations, followed by a final rinse. The plateaus correspond to equilibrium responses at given protein concentrations. Gag remained bound to the membrane after the buffer rinse, while MA was almost completely removed. (C) Gag and MA binding to DOPC/DOPS stBLMS either with (red) (2 mol%) or without (black) PI(4,5)P2. The MA data in panel A are well described by a Langmuir isotherm (Hill equation with N equal to 1). In contrast, the Gag data in panel B could be fitted only with Hill exponents where N was greater than 1 (provided in Table 2), which indicates cooperative membrane binding. Measurements were repeated no less than two times.
We also quantified Gag and MA binding to stBLMs containing 2% PI(4,5)P2, in the background of the same PS composition, DOPC/DOPS/PI(4,5)P2 (68/30/2). PI(4,5)P2 increased the affinity of MA almost by an order of magnitude but had a more modest effect on Gag affinity (Fig. 3C and Table 2). Significantly, PI(4,5)P2 did not increase the cooperativity of the binding event, at least at the ionic strength used in these measurements. We interpret these results to mean that Gag membrane binding is dominated by lateral protein-protein interactions rather than by lipid-protein interactions, as in the case of MA. Finally, incorporation of 30% cholesterol did not enhance the affinity of either Gag or MA (Table 2). We note that RSV MA association with model membranes shows trends for lipid composition similar to those for unmyristoylated HIV-1 MA but with higher affinity and protein load on the membrane surface. Conversely, myristoylated HIV-1 MA shows higher affinity to stBLMs than RSV MA and is more sensitive to the cholesterol content of the membrane (Marilia Barros, Frank Heinrich, Siddhartha A. K. Datta, Alan Rein, Ioannis Karageorgos, Hirsh Nanda, and Mathias Lösche, submitted for publication).
TABLE 2.
SPR results
| Protein | Lipid | NaCl (mM) | Kd (μM) | Hill coefficient (N) | R∞d |
|---|---|---|---|---|---|
| Gag | PC/PS (70/30) | 50 | 0.24 ± 0.03a | 2 | 75 |
| PC/PS/Chol (50/30/20) | 50 | 0.2 ± 0.01b | 3 | 90 | |
| PC/PS/PIP2 (68/30/2) | 50 | 0.13 ± 0.01b | 2 | 91 | |
| MA | PC/PS (70/30) | 50 | 24 ± 4b | 1 | 40 |
| PC/PS/Chol (50/30/20) | 50 | 22c | 1 | 49 | |
| PC/PS/PIP2 (68/30/2) | 50 | 3.9 ± 0.5b | 1 | 31 |
Average and standard deviation of three measurements.
Average and range of two measurements.
One measurement.
R∞, the equilibrium surface density of protein, is shown as the average if applicable.
Assembly of Gag cysteine mutants.
If membrane binding of Gag is a cooperative process involving protein-protein interactions, we hypothesized that these interactions would be the same as in the Gag lattice of authentic immature virus particles and Gag VLPs assembled in vitro. To test this hypothesis, we employed two Gag protein constructs with a pair of ectopic Cys residues positioned to form disulfide-cross-linked Gag hexamers. In order to lower nonspecific disulfide cross-linking in these experiments, versions of Gag were used in which most of the endogenous Cys residues had been replaced with Ala or Ser (26).
Before testing the effect of membrane binding on Gag multimerization, we characterized the effect on in vitro assembly of removal of endogenous, and addition of ectopic, Cys residues. A Gag protein, ΔMBD-11CE227C T259C, lacking the membrane binding domain and all but one natural Cys residue but containing the two ectopic Cys residues E227C and T259C (formerly called E51C and T20C, respectively [26]), was shown previously to form regular spherical VLPs in vitro (26). When these VLPs were oxidized, the p10 and CA domains of neighboring proteins became cross-linked, resulting in covalently linked dimeric to hexameric protein complexes easily visible on nonreducing SDS-PAGE (26). Similarly, based on a model for the SPA putative six-helix bundle (34), we predicted that VLPs assembled from the Gag-11C protein carrying two Cys residues in SP (R493C and E494C) should also lead to multimeric complexes upon oxidation.
Negative-stain electron microscopy confirmed that all the Gag mutants were able to assemble into particles, but at very different efficiencies (Fig. 4A to C). Removal of 11 cysteines from ΔMBD (ΔMBD-11C) resulted in severe impairment of VLP assembly. From kinetic analysis based on light scattering (50), ΔMBD assembly reached completion in ∼10 min, while ΔMBD-11C assembly was hardly evident (Fig. 4B) and led to no significant accumulation of VLPs in isopycnic sucrose gradient sedimentation (Fig. 4C). Addition of the two Cys residues E227C and T259C largely rescued this defect. Thus, ΔMBD-11CE227C T259C showed assembly kinetics similar to those of ΔMBD and produced approximately equal numbers of VLPs in the sucrose gradients. For the Gag construct carrying the pair of ectopic Cys residues in SP (ΔMBD-11CR493C E494C), partial rescue of assembly occurred, although the rate of assembly was significantly lower than for ΔMBD or ΔMBD-11CE227C T259C. We conclude that the impaired assembly of ΔMBD-11C is at least partially rescued by the addition of either of two pairs of cysteines that are positioned to enhance Gag lattice formation.
We next sought to determine if full-length Gag containing the cysteine mutations behaves the same as the ΔMBD protein with the same mutations. As previously reported (16), in vitro assembly of VLPs by RSV Gag is less efficient than assembly of ΔMBD. In addition to the presence of large amounts of aggregated protein in the assembly reaction mixture (see Materials and Methods), the Gag VLPs tended to aggregate into clumps. Therefore, we were unable to carry out either kinetic measurements or sedimentation assays. However, we did observe regular VLPs for all Gag constructs screened by negative-stain EM. The MBD of Gag contains two natural cysteine residues (C12 and C16), which were mutated to alanine in the context of the Gag-11C construct. This protein, called Gag-13C, formed few VLPs, similar to ΔMBD-11C (Fig. 4D). Compared with Gag-13C, significantly more VLPs were observed for Gag-11CE227C T259C, Gag-13CE227C T259C, and Gag-13CR493C E494C. Thus, ectopic Cys residues promote in vitro assembly in both the context of ΔMBD and the context of full-length Gag.
Membrane localization and budding of Gag cysteine mutants in vivo.
In cells, RSV Gag-GFP is strongly localized to the PM, with a pattern typified by many fluorescent puncta when observed by confocal microscopy (9). For all retroviruses, these puncta are generally considered to be assembly or budding sites. Gag multimerization is a critical component of the ability of Gag to bind stably to the PM. RSV MA and HIV MA with and without GFP are not localized to the PM (18, 19, 58–62), unless these proteins are artificially dimerized (for HIV MA) or hexamerized (for RSV MA) (19). Cytoplasmic HIV Gag is largely in a monomer-dimer equilibrium, reaching higher multimeric states only at the PM (42). Recently, though, a study by Hendrix et al. identified concentration-dependent slowly diffusing cytoplasmic HIV Gag complexes (63). RSV Gag has been reported to be in multimeric states greater than dimeric before reaching the PM (41). These observations suggest that Gag multimerization is critical for PM association and that, at least in part, higher-order Gag multimerization occurs at the PM.
To further test the hypothesis that Gag membrane binding is cooperative, we expressed C-terminally GFP-tagged Gag cysteine mutants in cells (Fig. 5). Replacing the two natural Cys residues in the MBD with Ala (GagC12A C16A) did not disrupt PM localization (Fig. 5A) and only slightly reduced virion release (Fig. 5C). Both Gag-11C and Gag-13C appeared to be concentrated in the nucleus (Fig. 5A). RSV Gag is known to traffic through the nucleus before reaching the PM, due to its two nuclear localization sequences (NLS) in MA and NC and its nuclear export signal (NES) in p10 (58, 64–66). Finding Gag-11C and Gag-13C in the nucleus was unexpected, since neither of these constructs has mutations that disrupt the NES. Both PM localization and virion release were partially rescued by the ectopic Cys residues for Gag-13CE227C T259C and for Gag-13CR493C E494C compared with Gag-13C (Fig. 5A to C). When considered with the VLP assembly data (Fig. 4), overall, these results suggest that removal of cysteines greatly reduces Gag's ability to assemble in vivo and that this reduction is partially counteracted by the addition of cysteines positioned to enhance Gag-Gag interactions.
FIG 5.
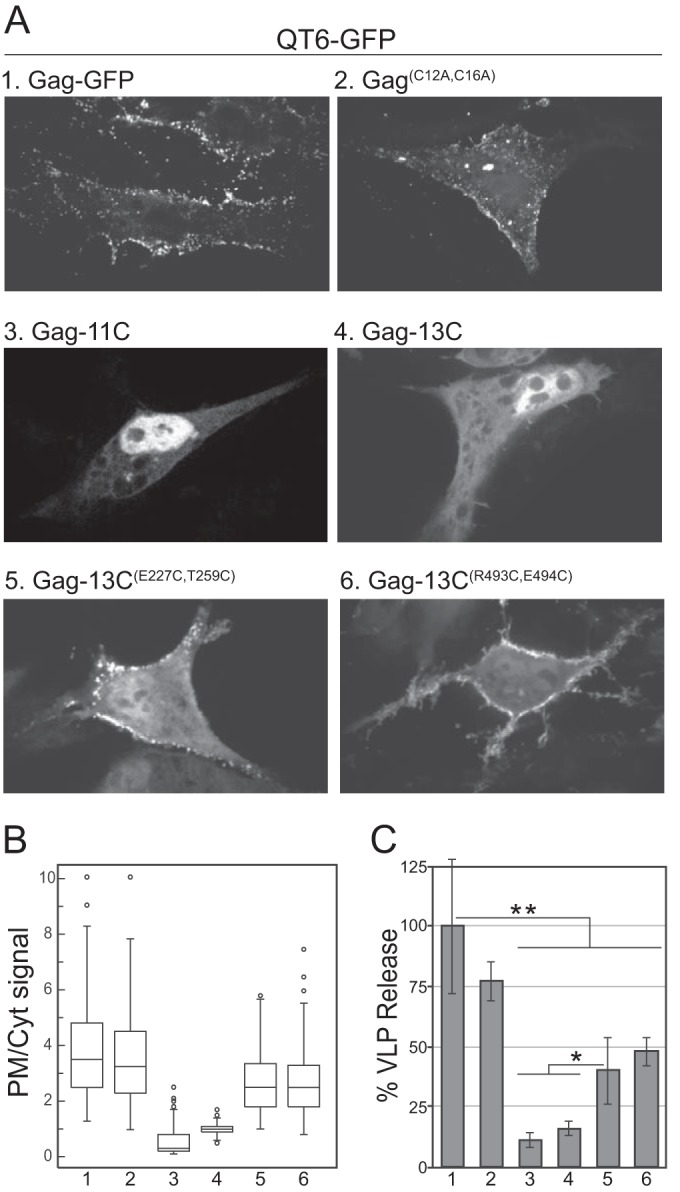
Cellular localization and VLP release of Gag cysteine mutants. All constructs were C-terminally fused with GFP and expressed in QT6 cells. (A) Representative confocal images. (B) Box plots of no less than 65 plot profile scans for each construct analyzed. The numbers at the bottom correspond to the construct numbers in panel A. For samples 3 and 4, the plot profile scans include the nucleus, where Gag was significantly enriched. (C) Percent VLP release compared to Gag as determined by Western blot analysis. The bars represent the averages of no less than three replicates (t test; **, P < 0.01; *, P < 0.05).
Cross-linking of Gag bound to membranes in vitro.
Using the Gag cysteine mutants described above, we tested the ability of membranes to facilitate Gag-Gag multimerization in vitro, as determined by SDS-PAGE in the absence of reducing agents. The predicted disulfide bonds between multimeric Gag proteins are shown in Fig. 6A. Prior to assessing disulfide cross-linking by nonreducing SDS-PAGE, we showed by liposome-pelleting assays, with the same lipid compositions shown in Fig. 1, that the three mutant proteins with ectopic Cys residues (Fig. 4D) bound liposomes similarly to Gag and Gag-13C and that PI(4,5)P2 modestly stimulated binding (data not shown). Oxidation of VLPs made either with the purified N-terminally truncated protein ΔMBD-11CE227C T259C or with ΔMBD-11CR493C E494C resulted in the same banding pattern previously reported for ΔMBD-11CE227C T259C (reference 26 and data not shown). For the full-length Gag proteins, we used Gag-13C, which contains only one endogenous cysteine located in the C-terminal domain of CA (C431), as a negative control. This protein showed only minor numbers of cross-linked bands after binding to liposomes and oxidation (Fig. 6B). Because Gag-13C disulfide cross-linking was observed only in the presence of membranes and because C431 is buried in the CA lattice (26), the minor cross-linked product is likely to represent a form of membrane-bound Gag not consistent with a regular Gag lattice. Compared with Gag-13C, Gag-13CR493C E494C showed significant disulfide cross-linking (Fig. 6B). In the absence of the oligonucleotide GT25 used to promote assembly (Fig. 6B, lane 4), some Gag multimers were observed, but significantly fewer than with the oligonucleotide. Six distinct bands were evident both for assembly reactions in the absence of liposomes (+GT25 [Fig. 6B, lane 5]) and in the presence of liposomes (+LUV [Fig. 6B, lane 6]). This banding pattern is consistent with that observed for purified ΔMBDE227C T259C hexamers (25). Thus, we interpret the result to mean that the Gag-Gag contacts occurring on the membrane are the same as those contacts that occur in the immature Gag lattice. In addition, these data further support the model in which the SPA domain forms a six-helix bundle (24, 33, 34).
FIG 6.
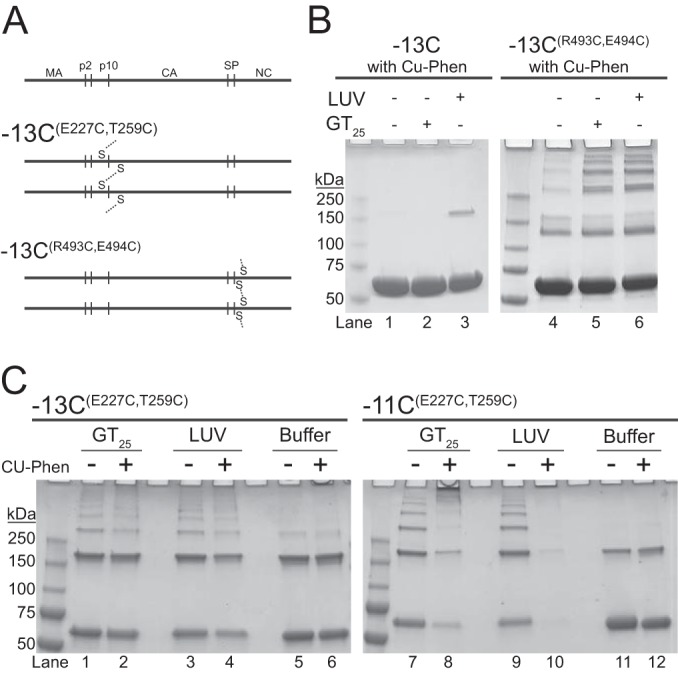
Disulfide cross-linking of membrane-bound Gag. (A) Schematic representation of disulfide bond formation by oxidation. For −13CE227 T259, cross-linking occurs between cysteine-227 and cysteine-259 of a neighboring Gag protein. For −13CR493C E494C, cross-linking occurs between cysteine-493 and cysteine-494 of a neighboring Gag. (B and C) Representative nonreducing Coomassie-stained gels of Gag cross-linking. Monomeric Gag is the first band, corresponding to the midpoint between the 50-kDa and 75-kDa mass marker bands. (B) Cu-Phen induced disulfide cross-linking of the control proteins, −13C (left) and −13CR493C E494C (right). Lanes 1 to 3 and 4 to 6 correspond to reactions with buffer only, plus GT25, and plus LUV, respectively. (C) Cross-linking of −13CE227 T259 (left) and −11CE227 T259 (right). Each condition was tested without (−) and with (+) Cu-Phen under the following conditions; plus GT25, plus LUV, and buffer only. LUVs were composed of POPC/POPS/Chol/PI(4,5)P2 (32/30/36/2).
For Gag-13CE227C T259C, disulfide cross-linking in the presence of GT25 but in the absence of liposomes was indistinguishable from the cross-linking observed in the presence of liposomes but in the absence of oligonucleotide (Fig. 6C, left). The addition of an oxidizing agent (Cu-Phen) did not significantly increase the formation of Gag multimers, suggesting that oxidation occurred efficiently in the presence of ambient oxygen. The residues C12 and C16 in the MBD of Gag-11CE227C T259C significantly increased the formation of multimers larger than hexamers in the presence of Cu-Phen (Fig. 6C, right). The low abundance of protein in lane 10 (Fig. 6C) is likely due to the formation of products too large to migrate into the gel. Because we did not observe these large products for Gag-13CE227C T259C or Gag-13CR493C E494C, we hypothesize that the cysteines in the MBD tie together disulfide-cross-linked Gag hexamers. Gag-13CE227C T259C and Gag-11CE227C T259C both exhibited Gag dimers, consistent with the previously described dimer composed of antiparallel Gag molecules (67), which are dead-end products that cannot form a Gag lattice. In the absence of GT25, the E227C T259C mutants showed less background (buffer only) than the R493C E494C mutants. This result may reflect the fact that the R493C and E494C residues are located in an unfolded region of Gag, unlike E227C and T259C. Taken together, and in summary, we interpret these results to mean that LUVs promote Gag multimerization as efficiently as oligonucleotides, and we hypothesize that the Gag-Gag contacts are the same, though this cannot be proven at this point.
DISCUSSION
We have shown that RSV Gag membrane binding involves protein-protein interactions. Both biochemically, by liposome binding in vitro, and by cellular localization in vivo, stable Gag-membrane interactions depended on a functional SPA domain, which was inferred previously to form a six-helix bundle in the Gag lattice (33, 34, 57). In SPR measurements with supported bilayers at 50 mM NaCl, Gag bound membranes 2 orders of magnitude more tightly than did MA, and unlike for MA, this binding showed the characteristics of cooperativity. In vitro assembly of a panel of Gag and GagΔMBD mutant proteins revealed that removal of cysteine residues nearly abrogated assembly, but this defect could be rescued by the addition of ectopic cysteine residues positioned near either the N- or C terminus of CA to promote Gag-Gag contacts in the Gag lattice. Oxidation experiments with these ectopic cysteine mutants implied that the interactions between Gag proteins bound to a membrane are the same as or similar to the contacts between Gag proteins in immature VLPs.
The discrepancy in RSV MA-membrane affinities determined by SPR here and by liposome flotation previously remains to be clarified. Dalton et al. (4) originally estimated a Kd of ∼1 mM for liposomes composed only of PC and PS, without cholesterol or PI(4,5)P2 and at 75 mM NaCl. This estimated affinity was also bolstered by electrostatic calculations. In contrast, our present SPR results show a Kd of ∼20 μM under similar conditions. Dalton et al. argued that the number of MA binding sites on liposomes was in considerable excess over protein, even at low concentrations of lipid, a condition that is required in order for the measurements to be interpretable. This critical assumption may have been overly optimistic. If the MA binding sites on liposomes were fewer than estimated by those authors, their flotation data would not lead to the affinity they estimated. In addition, flotation assays may underestimate protein binding due to dilution of lipids in the sucrose gradient (68).
The prevailing model for the SPA domain of RSV Gag (23, 33, 34, 57), and the similar SP1 domain of HIV Gag (34, 35), is that this segment coalesces to form a six-helix bundle tying together Gag molecules in the lattice (34). However, the functional boundaries of SPA are not accurately defined. Originally, Keller et al., using a baculovirus expression system and EM as the readout, assigned 24 residues to SPA—8 at the end of CA, 12 in SP, and 4 (AVVN) at the beginning of NC (57). A peptide comprising these residues plus the next four residues in NC (RERD) was shown to form a hexamer at high concentrations in solution (33), and modeling suggested that the RD pair could form a salt bridge that would stabilize the bundle. Molecular-dynamics simulations suggested that all 8 residues in NC are important for stability of the bundle (69). We have shown here that truncated Gag proteins ending with the 4 (AVVN), 6 (AVVNRE), or 8 (AVVNRERD) N-terminal residues of NC are similar in their membrane binding to a Gag construct that includes the entire NC domain. This suggests that the RERD sequence is dispensable for formation of the six-helix bundle and provides the first biochemical evidence, albeit indirect, that the six-helix bundle can form in a Gag molecule in the absence of the majority of NC.
The signaling lipid PI(4,5)P2, which is concentrated in the inner leaflet of the PM (70), enhances PM localization of RSV Gag in cells (11), although it is less effective in this enhancement than for HIV Gag (9, 13). PI(4,5)P2 also promotes liposome binding of RSV Gag in vitro (9, 13, 16, 19). The experiments presented here show that an RSV Gag mutant missing most of NC but including an intact SPA domain bound to liposomes at 150 mM NaCl almost as well as full-length Gag, but only in the presence of 2% PI(4,5)P2 in addition to 30% PS. By SPR, PI(4,5)P2 increased the affinity of Gag for membranes by a modest 2-fold at 50 mM NaCl. Gag proteins that do not include an intact SPA, i.e., MA, MA-CA, and ΔNC, did not show a strong PI(4,5)P2 effect in liposome binding, suggesting that PI(4,5)P2 in part acts by enhancing SPA-induced oligomerization of Gag. However, by SPR, the affinity of MA for membranes was increased 5-fold by PI(4,5)P2. We speculate that this discrepancy for MA, when SPR and liposome flotation are compared, results from the different ionic strengths used in the two assays. Due to protein aggregation, we were unable to measure Gag binding to supported bilayers at the more physiologically relevant ionic strength of 150 mM NaCl.
One possible mechanism that might explain how PI(4,5)P2 promotes multimerization is that the lipid increases the dwell time of individual proteins on the membrane, thus reducing the off rate, effectively increasing the probability that two membrane-bound Gag molecules associate with each other. A precedent for this idea is the report that the dwell time of the PI(3,4,5)P3-specific PH-PLHC domain, measured by total internal reflection fluorescence (TIRF) microscopy on supported bilayers, increases in the presence of PI(3,4,5)P3 and that the protein's surface diffusion rate is at the same order of magnitude as the diffusion rate of the lipid itself (71, 72). In other words, in that system, the protein-membrane interaction is so tight that the protein does not let go of the lipid on this time scale. We are in the process of testing this model for RSV Gag. We note that this mechanism does not necessarily rely on a PI(4,5)P2-specific binding pocket, as found for HIV MA (14). It could be that electrostatic interactions alone are sufficient for the implied tight binding of a protein to the lipid.
The role of the RSV NC domain in membrane binding of Gag is uncertain. First, while we have shown here that liposome binding of Gag is nearly as efficient in the absence as in the presence of NC, as long as the entire SPA domain is present, previously published data were interpreted to mean that NC is essential for PM binding in vivo (11). In regard to this point, we note that the SPA domain extends at least 4 residues into the N terminus of NC, and hence, complete deletion of NC also abrogates SPA function (33, 57). Nevertheless, from the results presented here, we cannot entirely rule out a role for the NC domain in mediating Gag-membrane association. SPR measurements of purified MASP+6 and MASP+8 showed slow binding kinetics that we were unable to fit into a model (data not shown). In addition, MASP+6 and MASP+8 did not form VLPs in vivo or in vitro under a range of assembly conditions (data not shown). These results are consistent with previous in vitro assembly experiments demonstrating that NC-mediated dimerization is critical for the initiation of virion formation (73). In one model of HIV Gag-membrane interaction, derived from NR measurements, if NC is not occupied by nucleic acid, it freely interacts with a negatively charged membrane. This interaction would result in a Gag configuration that is unfavorable for multimerization. Applied to cells, NC in this model would not only contribute to the recruitment of Gag to the PM, but also would play a role in regulating assembly. From all of our results taken together, we conclude that while an intact SPA domain is critical for membrane binding and apparently is required for cooperativity, the NC domain of RSV Gag is clearly indispensable for virion assembly.
Gag-GFP localization in cells was altered for mutants with either 11 or 13 of the endogenous cysteines knocked out. These mutant proteins accumulated in the nucleus, while wild-type Gag-GFP at steady state was primarily found in PM puncta, which presumably represent assembly and budding sites. This effect of Cys mutations is difficult to interpret. In principle, such a phenotype could result from misfolding of the Gag protein. However, the fact that either of two pairs of ectopic Cys residues partially restored normal localization argues against misfolding. RSV Gag contains two NLS and one NES (58, 64–66), and the latter overlaps a critical assembly determinant in p10 (26). It is well established that upon treatment with leptomycin B, an inhibitor of CRM 1-dependent nuclear export, RSV Gag becomes localized almost exclusively to the nucleus, implying that Gag normally is trafficked through the nucleus before returning through the cytoplasm to assemble at the PM (74). According to one model, RSV Gag enters the nucleus in order to pick up its vgRNA (74). The observations that removal of endogenous cysteines results in in vitro assembly defects and in retention of Gag in the nucleus and that ectopic Cys residues reverse this effect suggest that nuclear export of Gag depends on the ability of Gag to multimerize. However, it remains unclear if nuclear trafficking of RSV Gag is essential for the production of infectious virions. RSV Gag chimeras containing the HIV MA domain do not traffic through the nucleus, and yet their infectivity is not compromised (75).
From disulfide cross-linking, it appears that membrane binding of Gag in the absence of nucleic acid results in Gag-Gag contacts similar to those found in the immature virion. However, the detailed nature of the membrane-promoted Gag lattice is unknown. Presumably, it must differ somehow from the Gag lattice in immature virions, since Gag mutants with an intact SPA domain but without an NC domain neither form budding VLPs in cells nor assemble into VLPs in vitro. Preliminary experiments suggest that it may be possible to reconstitute a bilayer around Gag VLPs assembled in vitro, and thus, it may also be possible to examine by electron cryotomography an NC-truncated Gag protein bound to membranes.
ACKNOWLEDGMENTS
Helpful discussions with Frank Heinrich are gratefully acknowledged.
This work was supported by the NIH (grants R01GM107013 to Volker M. Vogt and R01GM101647 to Mathias Lösche). Imaging data were acquired through the Cornell University Biotechnology Resource Center, with NIH 1S10OD010605 funding for the shared Andor Revolution Spinning Disk Confocal Microscope.
REFERENCES
- 1.Dick RA, Vogt VM. 2014. Membrane interaction of retroviral Gag proteins. Front Microbiol 5:187. doi: 10.3389/fmicb.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray P, Li Z, Wang J, Tang C, Honig B, Murray D. 2005. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure 13:1521–1531. doi: 10.1016/j.str.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 3.Callahan E, Wills J. 2000. Repositioning basic residues in the M domain of the Rous sarcoma virus gag protein. J Virol 74:11222–11229. doi: 10.1128/JVI.74.23.11222-11229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalton A, Murray P, Murray D, Vogt V. 2005. Biochemical characterization of Rous sarcoma virus MA protein interaction with membranes. J Virol 79:6227–6238. doi: 10.1128/JVI.79.10.6227-6238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol(4,5)bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ono A, Waheed AA, Joshi A, Freed EO. 2005. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: use of a novel Gag multimerization assay. J Virol 79:14131–14140. doi: 10.1128/JVI.79.22.14131-14140.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J Virol 82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A 107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan J, Dick RA, Vogt VM. 2011. Rous sarcoma virus gag has no specific requirement for phosphatidylinositol-(4,5)-bisphosphate for plasma membrane association in vivo or for liposome interaction in vitro. J Virol 85:10851–10860. doi: 10.1128/JVI.00760-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alfadhli A, Still A, Barklis E. 2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol 83:12196–12203. doi: 10.1128/JVI.01197-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nadaraia-Hoke S, Bann DV, Lochmann TL, Gudleski-O'Regan N, Parent LJ. 2013. Alterations in the MA and NC domains modulate phosphoinositide-dependent plasma membrane localization of the Rous sarcoma virus Gag protein. J Virol 87:3609–3615. doi: 10.1128/JVI.03059-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inlora J, Chukkapalli V, Derse D, Ono A. 2011. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type 1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of HIV-1 Gag. J Virol 85:3802–3810. doi: 10.1128/JVI.02383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inlora J, Collins DR, Trubin ME, Chung JY, Ono A. 2014. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. mBio 5:e02202. doi: 10.1128/mBio.02202-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saad J, Miller J, Tai J, Kim A, Ghanam R, Summers M. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A 103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dick RA, Goh SL, Feigenson GW, Vogt VM. 2012. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc Natl Acad Sci U S A 109:18761–18766. doi: 10.1073/pnas.1209408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dick RA, Datta SA, Nanda H, Fang X, Wen Y, Barros M, Wang YX, Rein A, Vogt VM. 2015. Hydrodynamic and membrane binding properties of purified Rous sarcoma virus Gag protein. J Virol 89:10371–10382. doi: 10.1128/JVI.01628-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Carroll IP, Soheilian F, Kamata A, Nagashima K, Rein A. 2013. Elements in HIV-1 Gag contributing to virus particle assembly. Virus Res 171:341–345. doi: 10.1016/j.virusres.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalton AK, Ako-Adjei D, Murray PS, Murray D, Vogt VM. 2007. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J Virol 81:6434–6445. doi: 10.1128/JVI.02757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dick RA, Kamynina E, Vogt VM. 2013. Effect of multimerization on membrane association of Rous sarcoma virus and HIV-1 matrix domain proteins. J Virol 87:13598–13608. doi: 10.1128/JVI.01659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller H, Kräusslich HG, Schwille P. 2013. Multimerizable HIV Gag derivative binds to the liquid-disordered phase in model membranes. Cell Microbiol 15:237–247. doi: 10.1111/cmi.12064. [DOI] [PubMed] [Google Scholar]
- 21.Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, Yeates TO. 2005. Protein structures forming the shell of primitive bacterial organelles. Science 309:936–938. doi: 10.1126/science.1113397. [DOI] [PubMed] [Google Scholar]
- 22.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bush DL, Vogt VM. 2014. In vitro assembly of retroviruses. Annu Rev Virol 1:561–580. doi: 10.1146/annurev-virology-031413-085427. [DOI] [PubMed] [Google Scholar]
- 24.Schur FK, Dick RA, Hagen WJ, Vogt VM, Briggs JA. 2015. The structure of immature virus-like Rous sarcoma virus Gag particles reveals a structural role for the p10 domain in assembly. J Virol 89:10294–10302. doi: 10.1128/JVI.01502-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schur FK, Hagen WJ, Rumlova M, Ruml T, Müller B, Kräusslich HG, Briggs JA. 2015. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 A resolution. Nature 517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 26.Phillips JM, Murray PS, Murray D, Vogt VM. 2008. A molecular switch required for retrovirus assembly participates in the hexagonal immature lattice. EMBO J 27:1411–1420. doi: 10.1038/emboj.2008.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell S, Vogt VM. 1997. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: identification of the p10 domain as a morphological determinant in the formation of spherical particles. J Virol 71:4425–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson MC, Scobie HM, Ma YM, Vogt VM. 2002. Nucleic acid-independent retrovirus assembly can be driven by dimerization. J Virol 76:11177–11185. doi: 10.1128/JVI.76.22.11177-11185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joshi SM, Vogt VM. 2000. Role of the Rous sarcoma virus p10 domain in shape determination of gag virus-like particles assembled in vitro and within Escherichia coli. J Virol 74:10260–10268. doi: 10.1128/JVI.74.21.10260-10268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiegers K, Rutter G, Kottler H, Tessmer U, Hohenberg H, Kräusslich HG. 1998. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J Virol 72:2846–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gross I, Hohenberg H, Wilk T, Wiegers K, Grattinger M, Müller B, Fuller S, Kräusslich HG. 2000. A conformational switch controlling HIV-1 morphogenesis. EMBO J 19:103–113. doi: 10.1093/emboj/19.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Accola MA, Hoglund S, Göttlinger HG. 1998. A putative alpha-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 Gag precursor is crucial for viral particle assembly. J Virol 72:2072–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bush DL, Monroe EB, Bedwell GJ, Prevelige PE, Phillips JM, Vogt VM. 2014. Higher-order structure of the Rous sarcoma virus SP assembly domain. J Virol 88:5617–5629. doi: 10.1128/JVI.02659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J 26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Datta SA, Temeselew LG, Crist RM, Soheilian F, Kamata A, Mirro J, Harvin D, Nagashima K, Cachau RE, Rein A. 2011. On the role of the SP1 domain in HIV-1 particle assembly: a molecular switch? J Virol 85:4111–4121. doi: 10.1128/JVI.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bryant M, Ratner L. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A 87:523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Göttlinger HG, Sodroski JG, Haseltine WA. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A 86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Dou J, Ding L, Spearman P. 2007. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J Virol 81:12899–12910. doi: 10.1128/JVI.01280-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ono A, Orenstein JM, Freed EO. 2000. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol 74:2855–2866. doi: 10.1128/JVI.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freed EO, Englund G, Martin MA. 1995. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol 69:3949–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larson DR, Ma YM, Vogt VM, Webb WW. 2003. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J Cell Biol 162:1233–1244. doi: 10.1083/jcb.200303200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kutluay SB, Bieniasz PD. 2010. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog 6:e1001200. doi: 10.1371/journal.ppat.1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Datta SA, Curtis JE, Ratcliff W, Clark PK, Crist RM, Lebowitz J, Krueger S, Rein A. 2007. Conformation of the HIV-1 Gag protein in solution. J Mol Biol 365:812–824. doi: 10.1016/j.jmb.2006.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Datta SA, Zuo X, Clark PK, Campbell SJ, Wang YX, Rein A. 2011. Solution properties of murine leukemia virus gag protein: differences from HIV-1 gag. J Virol 85:12733–12741. doi: 10.1128/JVI.05889-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Datta SA, Heinrich F, Raghunandan S, Krueger S, Curtis JE, Rein A, Nanda H. 2011. HIV-1 Gag extension: conformational changes require simultaneous interaction with membrane and nucleic acid. J Mol Biol 406:205–214. doi: 10.1016/j.jmb.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. 2004. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics 5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 47.Kingsley PB, Feigenson GW. 1978. The synthesis of a predeuterated phospholipid: 1,2-dimyristoyl-sn-glycero-3-phosphocholine-d72. Chem Phys Lipids 24:135–147. [Google Scholar]
- 48.Buboltz JT, Feigenson GW. 1999. A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim Biophys Acta 1417:232–245. doi: 10.1016/S0005-2736(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 49.Campbell S, Vogt VM. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J Virol 69:6487–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu F, Joshi SM, Ma YM, Kingston RL, Simon MN, Vogt VM. 2001. Characterization of Rous sarcoma virus Gag particles assembled in vitro. J Virol 75:2753–2764. doi: 10.1128/JVI.75.6.2753-2764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schasfoort RBM, Tudos AJ. 2008. Handbook of surface plasmon resonance. Royal Society of Chemistry, Cambridge, United Kingdom. [Google Scholar]
- 52.McGillivray DJ, Valincius G, Vanderah DJ, Febo-Ayala W, Woodward JT, Heinrich F, Kasianowicz JJ, Lösche M. 2007. Molecular-scale structural and functional characterization of sparsely tethered bilayer lipid membranes. Biointerphases 2:21–33. doi: 10.1116/1.2709308. [DOI] [PubMed] [Google Scholar]
- 53.Budvytyte R, Valincius G, Niaura G, Voiciuk V, Mickevicius M, Chapman H, Goh HZ, Shekhar P, Heinrich F, Shenoy S, Lösche M, Vanderah DJ. 2013. Structure and properties of tethered bilayer lipid membranes with unsaturated anchor molecules. Langmuir 29:8645–8656. doi: 10.1021/la401132c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shenoy S, Moldovan R, Fitzpatrick J, Vanderah DJ, Deserno M, Lösche M. 2010. In-plane homogeneity and lipid dynamics in tethered bilayer lipid membranes (tBLMs). Soft Matter 2010:1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valincius G, McGillivray DJ, Febo-Ayala W, Vanderah DJ, Kasianowicz JJ, Lösche M. 2006. Enzyme activity to augment the characterization of tethered bilayer membranes. J Phys Chem B 110:10213–10216. doi: 10.1021/jp0616516. [DOI] [PubMed] [Google Scholar]
- 56.Shenoy S, Shekhar P, Heinrich F, Daou MC, Gericke A, Ross AH, Lösche M. 2012. Membrane association of the PTEN tumor suppressor: molecular details of the protein-membrane complex from SPR binding studies and neutron reflection. PLoS One 7:e32591. doi: 10.1371/journal.pone.0032591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keller PW, Johnson MC, Vogt VM. 2008. Mutations in the spacer peptide and adjoining sequences in Rous sarcoma virus Gag lead to tubular budding. J Virol 82:6788–6797. doi: 10.1128/JVI.00213-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheifele LZ, Garbitt RA, Rhoads JD, Parent LJ. 2002. Nuclear entry and CRM1-dependent nuclear export of the Rous sarcoma virus Gag polyprotein. Proc Natl Acad Sci U S A 99:3944–3949. doi: 10.1073/pnas.062652199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ding L, Derdowski A, Wang JJ, Spearman P. 2003. Independent segregation of human immunodeficiency virus type 1 Gag protein complexes and lipid rafts. J Virol 77:1916–1926. doi: 10.1128/JVI.77.3.1916-1926.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ono A, Demirov D, Freed EO. 2000. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J Virol 74:5142–5150. doi: 10.1128/JVI.74.11.5142-5150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol 73:4136–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fogarty KH, Berk S, Grigsby IF, Chen Y, Mansky LM, Mueller JD. 2014. Interrelationship between cytoplasmic retroviral Gag concentration and Gag-membrane association. J Mol Biol 426:1611–1624. doi: 10.1016/j.jmb.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hendrix J, Baumgartel V, Schrimpf W, Ivanchenko S, Digman MA, Gratton E, Kräusslich HG, Müller B, Lamb DC. 2015. Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J Cell Biol 210:629–646. doi: 10.1083/jcb.201504006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butterfield-Gerson KL, Scheifele LZ, Ryan EP, Hopper AK, Parent LJ. 2006. Importin-beta family members mediate alpharetrovirus gag nuclear entry via interactions with matrix and nucleocapsid. J Virol 80:1798–1806. doi: 10.1128/JVI.80.4.1798-1806.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gudleski N, Flanagan JM, Ryan EP, Bewley MC, Parent LJ. 2010. Directionality of nucleocytoplasmic transport of the retroviral gag protein depends on sequential binding of karyopherins and viral RNA. Proc Natl Acad Sci U S A 107:9358–9363. doi: 10.1073/pnas.1000304107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scheifele LZ, Ryan EP, Parent LJ. 2005. Detailed mapping of the nuclear export signal in the Rous sarcoma virus Gag protein. J Virol 79:8732–8741. doi: 10.1128/JVI.79.14.8732-8741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nandhagopal N, Simpson AA, Johnson MC, Francisco AB, Schatz GW, Rossmann MG, Vogt VM. 2004. Dimeric Rous sarcoma virus capsid protein structure relevant to immature Gag assembly. J Mol Biol 335:275–282. doi: 10.1016/j.jmb.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 68.Hamard-Peron E, Muriaux D. 2011. Retroviral matrix and lipids, the intimate interaction. Retrovirology 8:15. doi: 10.1186/1742-4690-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goh BC, Perilla JR, England MR, Heyrana KJ, Craven RC, Schulten K. 2015. Atomic modeling of an immature retroviral lattice using molecular dynamics and mutagenesis. Structure 23:1414–1425. doi: 10.1016/j.str.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ziemba BP, Falke JJ. 2013. Lateral diffusion of peripheral membrane proteins on supported lipid bilayers is controlled by the additive frictional drags of (1) bound lipids and (2) protein domains penetrating into the bilayer hydrocarbon core. Chem Phys Lipids 172-173:67–77. doi: 10.1016/j.chemphyslip.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knight JD, Lerner MG, Marcano-Velazquez JG, Pastor RW, Falke JJ. 2010. Single molecule diffusion of membrane-bound proteins: window into lipid contacts and bilayer dynamics. Biophys J 99:2879–2887. doi: 10.1016/j.bpj.2010.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma YM, Vogt VM. 2002. Rous sarcoma virus Gag protein-oligonucleotide interaction suggests a critical role for protein dimer formation in assembly. J Virol 76:5452–5462. doi: 10.1128/JVI.76.11.5452-5462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parent LJ. 2011. New insights into the nuclear localization of retroviral Gag proteins. Nucleus 2:92–97. doi: 10.4161/nucl.2.2.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baluyot MF, Grosse SA, Lyddon TD, Janaka SK, Johnson MC. 2012. CRM1-dependent trafficking of retroviral Gag proteins revisited. J Virol 86:4696–4700. doi: 10.1128/JVI.07199-11. [DOI] [PMC free article] [PubMed] [Google Scholar]