

ABSTRACT
African green monkeys (AGM) are natural hosts of simian immunodeficiency virus (SIV), and infection in these animals is generally nonpathogenic, whereas infection of nonnatural hosts, such as rhesus macaques (RM), is commonly pathogenic. CCR5 has been described as the primary entry coreceptor for SIV in vivo, while human-derived CXCR6 and GPR15 also appear to be used in vitro. However, sooty mangabeys that are genetically deficient in CCR5 due to an out-of-frame deletion are infectible with SIVsmm, indicating that SIVsmm can use alternative coreceptors in vivo. In this study, we examined the CCR5 dependence of SIV strains derived from vervet AGM (SIVagmVer) and the ability of AGM-derived GPR15 and CXCR6 to serve as potential entry coreceptors. We found that SIVagmVer replicated efficiently in AGM and RM peripheral blood mononuclear cells (PBMC) in the presence of the CCR5 antagonist maraviroc, despite the fact that maraviroc was capable of blocking the CCR5-tropic strains SIVmac239, SIVsmE543-3, and simian-human immunodeficiency virus SHIV-AD8 in RM PBMC. We also found that AGM CXCR6 and AGM GPR15, to a lesser extent, supported entry of pseudotype viruses bearing SIVagm envelopes, including SIVagm transmitted/founder envelopes. Lastly, we found that CCR5, GPR15, and CXCR6 mRNAs were detected in AGM and RM memory CD4+ T cells. These results suggest that GPR15 and CXCR6 are expressed on AGM CD4+ T cells and are potential alternative coreceptors for SIVagm use in vivo. These data suggest that the use of non-CCR5 entry pathways may be a common feature of SIV replication in natural host species, with the potential to contribute to nonpathogenicity in these animals.
IMPORTANCE African green monkeys (AGM) are natural hosts of SIV, and infection in these animals generally does not cause AIDS, whereas SIV-infected rhesus macaques (RM) typically develop AIDS. Although it has been reported that SIV generally uses CD4 and CCR5 to enter target cells in vivo, other molecules, such as GPR15 and CXCR6, also function as SIV coreceptors in vitro. In this study, we investigated whether SIV from vervet AGM can use non-CCR5 entry pathways, as has been observed in sooty mangabeys. We found that SIVagmVer efficiently replicated in AGM and RM peripheral blood mononuclear cells in the presence of the CCR5 antagonist maraviroc, suggesting that non-CCR5 entry pathways can support SIVagm entry. We found that AGM-derived GPR15 and CXCR6 support SIVagmVer entry in vitro and may serve as entry coreceptors for SIVagm in vivo, since their mRNAs were detected in AGM memory CD4+ T cells, the preferred target cells of SIV.
INTRODUCTION
Serological evidence of simian immunodeficiency virus (SIV) infection has been reported for approximately 40 different species of nonhuman primates (NHP). African NHP, such as African green monkeys (AGM) and sooty mangabeys (SM), are natural hosts of SIV, and infection in these animals is generally nonpathogenic despite high viral loads. Cross-species transmission of these viruses into nonnatural hosts, such as humans and rhesus macaques (RM), gives rise to pathogenic infections that generally result in peripheral CD4+ T cell loss and progression to AIDS (1, 2). Numerous studies have analyzed similarities and differences between SIV-infected natural and nonnatural hosts in an attempt to delineate the mechanisms underlying differences in pathogenicity. Similarities that have been identified include high plasma viral loads (3–6), a short in vivo life span of productively infected cells (7, 8), acute immune activation (9–12), and depletion of mucosal CD4+ T cells during acute infection (13, 14). Factors in nonnatural hosts that differ from natural host species and thus may contribute to disease progression include a loss of peripheral CD4+ T cells (15, 16), preferential depletion of Th17 cells (17–19), mucosal damage to the epithelial barrier of the gastrointestinal tract (20–22), translocation of microbial products from the intestinal lumen (23), and chronic generalized immune activation (24–28).
Another factor that may differentiate between SIV-infected natural and nonnatural hosts is target cell tropism, which is dependent on coreceptor use and coreceptor expression profiles. Various studies indicate that many natural host species have developed mechanisms to modulate SIV receptor (CD4) and/or coreceptor (i.e., CCR5) expression levels on CD4+ T cells. For example, AGM downregulate the CD4 molecule on their CD4+ T cells as these cells enter the memory pool, thus creating a subset of CD4-negative cells that are resistant to SIV infection but are able to carry out CD4+ T cell helper-like functions (29–31). Additionally, Pandrea et al. revealed that natural host species have substantially lower levels of CCR5 on CD4+ T cells from blood, lymph nodes, and mucosal tissues than those in nonnatural hosts, whereas CCR5 expression levels on CD8+ T cells are comparable between natural and nonnatural hosts (32). Lastly, a recent study reported that SIVsmm preferentially infects SM effector memory T cells compared to central memory T cells. Preferential targeting of effector memory T cells is likely a result of high levels of CCR5 expression on SM effector memory T cells relative to CCR5 expression levels on SM central memory T cells (33). Thus, it appears that natural hosts may have evolved multiple mechanisms to avoid disease progression, such as shifting the target cell tropism to more dispensable cell subsets while sparing critical cell subsets, such as central memory T cells, that play an important role in maintaining immune cell homeostasis (33, 34). However, the underlying mechanisms for how natural hosts exhibit high viral loads in the context of low CCR5 expression on CD4+ T cells remain unclear.
Cellular targeting by human immunodeficiency virus (HIV) or SIV replication is primarily dependent on receptor/coreceptor expression levels. HIV-1 gains entry into cells by binding to CD4 and a coreceptor, typically CCR5 or CXCR4. Unlike HIV-1, many SIV strains use CCR5 as a major entry coreceptor and rarely utilize CXCR4 for entry but are capable in vitro of utilizing a number of human alternative coreceptors, including GPR1, GPR15 (BOB), and CXCR6 (Bonzo/STRL33/CD186) (35–38). Although various strains of SIV are thought to use CCR5 as the major entry coreceptor in vivo, several exceptions have been reported. The first reported example is SIVrcm, isolated from red-capped mangabeys, which have a high allelic frequency (86.6%) of a CCR5-inactivating deletion (RCM-CCR5Δ24). SIVrcm utilizes CCR2b rather than CCR5 for entry (39). In addition, SIVsmm can utilize non-CCR5 entry pathways in sooty mangabeys in vivo. Thus, genetically CCR5-deficient SM carrying mutant SM CCR5 alleles (SM-CCR5Δ2 and SM-CCR5Δ24) that abrogate CCR5 surface expression are infectible with SIVsmm (39–41). Surprisingly, half of the animals carrying homozygous CCR5 mutant alleles were SIVsmm infected and exhibited robust viral replication (40) that was only slightly lower than viral loads from animals with wild-type CCR5 alleles. This finding revealed that SIVsmm can utilize non-CCR5 entry pathways in vivo and that these CCR5-independent pathways can sustain robust virus replication.
In the current study, we sought to determine if the use of non-CCR5 entry pathways can be observed in other natural host species, such as AGM. Studies indicate that there are four subspecies of AGM that harbor their own species-specific strains of SIVagm: SIVagmVer in vervet AGM (Chlorocebus pygerythrus), SIVagmGri in grivet AGM (Chlorocebus aethiops), SIVagmTan in tantalus monkeys (Chlorocebus tantalus), and SIVagmSab in sabaeus monkeys (Chlorocebus sabaeus) (42–46). Historically, SIVagm has been described as a CCR5-dependent virus in vivo, although in vitro studies reveal that some subspecies-specific strains of SIVagm can utilize non-CCR5 entry pathways, such as CXCR4, CXCR6 (Bonzo/STRL33/CD186), and GPR15 (BOB), when the human homologues are overexpressed in human cell lines (36, 37, 47). However, since these human coreceptors differ genetically from the AGM homologues, it has been difficult to determine the significance of these findings for ex vivo or in vivo SIVagm infection.
In the current study, we sought to determine if SIVagmVer is exclusively CCR5 dependent or is capable of using non-CCR5 entry pathways in AGM peripheral blood mononuclear cells (PBMC) in the presence of the CCR5 antagonist maraviroc. Additionally, we sought to determine if the AGM homologues of GPR15 and CXCR6 could support SIVagmVer entry in vitro, and if so, whether these alternative coreceptors were expressed on CD4+ T cells, thus potentially serving as entry coreceptors. To address this, we examined mRNA expression levels of CCR5, GPR15, and CXCR6 in CD4+ T cell subsets from uninfected and SIV-infected AGM and RM by using a quantitative reverse transcriptase PCR (qRT-PCR) assay.
MATERIALS AND METHODS
Ethics.
This study was carried out in strict accordance with the recommendations described in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, the Office of Animal Welfare, and the U.S. Department of Agriculture (48). All animals were housed and cared for in accordance with the American Association for Accreditation of Laboratory Animal Care standards in American Association for Accreditation of Laboratory Animal Care-accredited facilities. All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committees of the National Institute of Allergy and Infectious Diseases, under protocol LMM6. All procedures were carried out under ketamine anesthesia by trained personnel under the supervision of veterinary staff, and all efforts were made to ameliorate animal welfare and to minimize animal suffering in accordance with recommendations in the Weatherall report on the use of nonhuman primates (49). Animals were housed in adjoining individual primate cages allowing social interactions under controlled conditions of humidity, temperature, and light (12-h light–12-h dark cycles). Food and water were available ad libitum. Animals were monitored daily and fed commercial monkey chow, treats, and fruit twice daily by trained personnel.
Animals.
This study involved two SIV-naive vervet African green monkeys (AG5387 and AG5431), which were housed at the NIH Animal Center (Dickerson, MD). AG5387 and AG5431 were inoculated intrarectally with SIVagmVer90 weekly at escalating infectious doses until they became infected. SIVagm transmitted/founder (T/F) envelopes were generated from plasma collected at 1 week postinfection as described below. Blood and plasma were collected periodically, and plasma viral RNA levels were determined by qRT-PCR. This study involved 15 additional vervet AGM (5 uninfected animals and 10 SIVagmVer-infected animals) and 14 RM (5 uninfected animals and 9 SIVsmE660-infected animals). Descriptions of these animals along with their viral strains and viral loads (RNA copies per milliliter) are listed in Tables 1 and 2.
TABLE 1.
Plasma viral loads for vervet African green monkeys used in sorting experiments for CCR5, GPR15, and CXCR6 relative mRNA analysisa
| Monkey | Virus strain | Infectious dose (TCID50) | Plasma viral load (RNA copies/ml) |
|---|---|---|---|
| AG2 | SIVagmVer1 | 50 | 8.8 × 104 |
| AG8 | SIVagmVer1 | 50 | 1.4 × 103 |
| AG14 | SIVagmVer1 | 50 | 1.4 × 105 |
| AG18 | SIVagmVer1 | 50 | 8.1 × 104 |
| AG22 | SIVagmVer90 | 1,000 | 1.7 × 106 |
| AG28 | SIVagmVer9063 | 1,000 | 6.0 × 104 |
| AG37 | SIVagmVer90 | 1,000 | 1.3 × 105 |
| AG38 | SIVagmVer90 | 1,000 | 1.2 × 105 |
| AG5387 | SIVagmVer90 | 100 | 6.34 × 104 |
| AG5431 | SIVagmVer90 | 50,000 | 4.4 × 103 |
| AG33 | Uninfected | NA | NA |
| AG36 | Uninfected | NA | NA |
| AG39 | Uninfected | NA | NA |
| AG40 | Uninfected | NA | NA |
| AG5339 | Uninfected | NA | NA |
NA, not applicable.
TABLE 2.
Plasma viral loads for rhesus macaques used in sorting experiments for CCR5, GPR15, and CXCR6 relative mRNA analysisa
| Monkey | Virus strain | Infectious dose (TCID50) | Plasma viral load (RNA copies/ml) |
|---|---|---|---|
| H846 | SIVsmE660 | 1,000 | 9.8 × 106 |
| H851 | SIVsmE660 | 1,000 | 7.8 × 105 |
| H852 | SIVsmE660 | 1,000 | 8.6 × 102 |
| H853 | SIVsmE660 | 1,000 | 2.6 × 105 |
| H862 | SIVsmE660 | 500 | 2.9 × 106 |
| H864 | SIVsmE660 | 500 | 1.1 × 104 |
| H870 | SIVsmE660 | 500 | 3.9 × 104 |
| H875 | SIVsmE660 | 500 | 1.3 × 105 |
| H876 | SIVsmE660 | 500 | 2.8 × 105 |
| H705 | Uninfected | NA | NA |
| H734 | Uninfected | NA | NA |
| H788 | Uninfected | NA | NA |
| DCRG | Uninfected | NA | NA |
| DCCW | Uninfected | NA | NA |
NA, not applicable.
Primary cells and cell lines. (i) Primary cells.
Peripheral blood mononuclear cells (PBMC) were isolated from EDTA-treated whole blood from vervet AGM, RM, and human samples by density centrifugation using lymphocyte separation medium (MP Biomedicals, LLC). PBMC suspensions were maintained in RPMI 1640 containing 10% fetal bovine serum (FBS) supplemented with 1% penicillin-streptomycin (Invitrogen) and 1% l-glutamine (Invitrogen) and activated with 5 μg/ml phytohemagglutinin (PHA; Roche) for 3 days at 37°C. The medium was supplemented with 10% interleukin-2 (IL-2; Advanced Biotechnologies) at 1 day poststimulation in preparation for SIV/HIV/simian-human immunodeficiency virus (SHIV) infections. Frozen PBMC samples from vervet AGM (n = 33) were used for genomic DNA isolation for the CCR5 sequencing analysis described below.
(ii) Cell lines.
TZM-bl indicator cells were obtained from the NIH AIDS reagent program and grown in Dulbecco's modified Eagle medium (DMEM) containing 10% FBS and supplemented with 0.5 mg/ml Geneticin sulfate (Corning) and 0.25 M HEPES (Life Technologies). 293T cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and grown in DMEM containing 10% FBS and supplemented with 1% penicillin-streptomycin (Invitrogen) and 1% l-glutamine (Invitrogen). Cells were incubated at 37°C in the presence of 5% CO2.
Viruses. (i) Replication-competent viruses.
SIVagmVer90 is an uncloned virus isolated from the mesenteric lymph node of a naturally infected vervet AGM, AGM90, through the coculturing of infected mononuclear cells with pigtail macaque (PTM) PBMC. Virus titers were determined by limiting dilution infection of CEMss cells. SIVagmVer9063 is an infectious molecular clone derived from the SIVagmVer90 primary isolate following passage in vivo through a PTM. In vivo experimental infections conducted by our lab indicated that SIVagmVer90 and SIVagmVer9063 result in nonpathogenic infections in AGM natural hosts and pathogenic infections in PTM (50). Additional strains used as controls in this study were infectious molecular clones SIVmac239 and SIVsmE543-3 and SHIV-AD8, an SIV/HIV chimeric virus primarily composed of the SIVmac239 genome containing the HIV-1 AD8 (R5 tropic) envelope gene (51), as well as the replication-competent HIV-1 strains BaL (R5 tropic) and NL4-3 (X4 tropic).
(ii) SIVagm envelope clones and generation/standardization of pseudotype viruses.
The SIVagmVer9063 rev and env genes were cloned from a plasmid containing the full-length viral genome by using the following primers: SIVagm90 RevEnv for (5′-CAC CAT GCC CCT AGG ACC AGA A-3′) and SIVagm90 Env rev (5′-CTA ATT AAG GAT TTC CTC AAG CCC-3′). SIVagm T/F envelopes were isolated from plasmas collected at 1 week postinfection from two AGM, AG5387 and AG5431, that had been inoculated intrarectally with uncloned SIVagmVer90. Briefly, viral RNA was extracted from plasma by use of a QIAamp viral RNA purification kit (Qiagen), and cDNA was synthesized from viral RNA by using the SuperScript III first-strand synthesis system (Life Technologies) and the primer SIVagm90 Env rev (5′-CTA ATT AAG GAT TTC CTC AAG CCC-3′) according to the manufacturer's instructions. PCR amplification was performed using 2 μl cDNA as a template and the SIVagm90 Env primer pair listed above under the following cycling conditions: 98°C for 30 s followed by 35 cycles of 98°C for 10 s, 60°C for 30 s, and 72°C for 90 s, with a final extension of 72°C for 10 min. PCR products were cloned into the pcDNA3.1 Directional TOPO expression vector (Invitrogen). Transformed colonies containing correct inserts were identified by sequence analysis. Expression plasmids containing the SIVagm rev and env genes were used to generate pseudotype viruses.
Pseudotype viruses were generated by cotransfecting 293T cells with a plasmid encoding the NL4-3-based env-deleted luciferase-expressing virus backbone (pNL4-3-luc-E−R+) (52) along with expression plasmids encoding the envelopes of SIVagmVer9063, SIVagm5387 T/F, SIVagm5431 T/F, and vesicular stomatitis virus glycoprotein (VSV-g), which served as a positive control. Cells were transfected using Fugene6 (Promega), and supernatants were collected at 2 days posttransfection, clarified by filtration, and stored at −80°C until use. Infectivity of pseudotype virions was determined using TZM-bl cells that stably express human CD4 and human CCR5. Briefly, TZM-bl cells seeded in a 96-well plate were infected with serially diluted pseudotype virus in the presence of 12.5 μg/ml DEAE-dextran (Sigma). Cells were lysed at 2 days postinfection with 1× cell culture lysis buffer (Promega). Cell lysate and luciferase substrate (Promega) were mixed, and luciferase activity was measured using a Mithras LB 940 luminometer (Berthold Technologies). Virus inocula were standardized on the basis of infectivity for subsequent use in SIVagm envelope-mediated entry assays.
SGA of SIVagm envelope genes.
Viral RNAs were extracted from the SIVagmVer90 stock inoculum and from plasmas collected at 1 week postinfection from AG5387 and AG5431 by using a QIAamp viral RNA minikit (Qiagen). cDNAs were synthesized using the SuperScript III first-strand synthesis system (Life Technologies) and the primer SIVagm Env rev2 (5′-AAG AGA CTG AGT TAC AAG CCA GCG-3′). Single-genome amplification (SGA) was performed as previously described (53). Briefly, cDNA was endpoint diluted in a 96-well plate such that approximately 30 PCR cycles yielded an amplification product. At these dilutions, most wells contained amplicons from a single cDNA molecule. PCR mixtures were composed of 1× Platinum Taq High Fidelity PCR buffer, 2 mM MgSO4, a 0.2 mM concentration of each deoxynucleoside triphosphate, 0.2 μM (each) primers, and 0.025 U/μl of Platinum Taq High Fidelity DNA polymerase in a 20-μl reaction volume for the first- and second-round reactions. First-round PCR mixtures contained 1 μl diluted cDNA (1:700 dilution for the SIVagmVer90 stock, 1:30 dilution for AG5387 plasma, and 1:60 dilution for AG5431 plasma) and the following primers: SIVagm Env for2 (5′-CAA CAG CAG ATG GAA TGA TAC ACC-3′) and SIVagm Env rev2 (5′-AAG AGA CTG AGT TAC AAG CCA GCG-3′). Second-round PCR mixtures contained 1 μl of first-round product and the following primers: SIVagm Env for1 (5′-CAT TTC CGT TGT GGT TGT CGT AG-3′) and SIVagm Env rev1 (5′-CCA TTC ATC CCA TTC GTC TCC-3′). The PCR conditions for the first-round reaction were as follows: 94°C for 2 min and then 35 cycles of 94°C for 15 s, 60°C for 30 s, and 68°C for 4 min, followed by a final extension of 68°C for 15 min. The PCR conditions for the second-round reaction followed the conditions above, with the exception that 45 cycles were performed. Amplicons were gel purified and sequenced by members of the Laboratory of Molecular Microbiology (LMM) Core, NIAID, NIH.
Cloning of vervet AGM and RM entry receptor/coreceptors.
Full-length receptors were cloned from AGM and RM genomic DNAs (CCR5, GPR15, and CXCR6) or AGM cDNAs (CD4 and CXCR4) extracted from AGM and RM PBMC. Genomic DNAs were extracted from PBMC by use of a QIAamp DNA Blood minikit (Qiagen) following the manufacturer's instructions. For cDNA synthesis, mRNAs were extracted from AGM PBMC by use of an AllPrep DNA/RNA minikit (Qiagen) and then reverse transcribed using a SuperScript III first-strand synthesis kit (Life Technologies) and an oligo(dT) primer. For CCR5 amplification, the following nested PCR was performed. First-round PCR was performed using 100 ng genomic DNA and primers RM-CCR5 for1 (5′-ATT CCC CCA ACA GAG CCA AG-3′) and RM-CCR5 rev1 (5′-TTC TCC CCA CAG CAA GAC AAA G-3′) under the following cycling conditions: 98°C for 30 s and then 10 cycles of 98°C for 10 s, 60°C for 30 s, and 72°C for 60 s, followed by 72°C for 10 min. Second-round PCR was performed using 5 μl of first-round PCR product with primers AGM-CCR5-D for (5′-CAC CAT GGA TTA TCA AGT GTC AAG TC-3′) and AGM-CCR5 rev (5′-TCA CAA GCC CAC AGA TGT TTC CTG-3′) under the cycling conditions described above, with the following modifications: 20 cycles and a 62°C annealing temperature were used. PCR products were gel extracted using a QIAquick gel extraction kit (Qiagen) and TA cloned using a pcDNA3.1 Directional TOPO expression kit (Invitrogen). Clones were confirmed through nucleotide sequence analysis (Eurofins).
For cloning of AGM and RM GPR15 and CXCR6, the following primers were used: AGM GPR15 for (5′-CAC CAT GGA CCC AGA AGA AAC TTC-3′), AGM GPR15 rev (5′-TTA GAG TGA CAC AGA CCT CTT CCT CC-3′), AGM CXCR6 for (5′-CAC CAT GGC AGA GTA TGA TCA CTA-3′), and AGM CXCR6 rev (5′-CTA TAA CTG GAA CAT GCT GGT G-3′). Cycling conditions were similar to those described above, with the following annealing temperatures: 64°C for GPR15 and 60°C for CXCR6. PCR products were gel purified and cloned by use of a pcDNA3.1 Directional TOPO expression kit (Invitrogen) as described above.
For AGM CD4, cDNA was synthesized using the SuperScript III first-strand synthesis system (Life Technologies) and the primer AGM CD4 rev (5′-TGC CTC AAA TGG GGC TAC-3′) and then amplified using a nested PCR. First-round PCR was performed using 1 μl cDNA and primers AGM CD4 for1 (5′-CAG CAA GGC CAC AAT GAA C-3′) and AGM CD4 rev (5′-TGC CTC AAA TGG GGC TAC-3′) under the first-round cycle conditions described above, with an annealing temperature of 58°C. Second-round PCR was performed using 5 μl of first-round PCR product with primers AGM CD4 for2 (5′-CAC CCA GCA AGG CCA CAA TGA AC-3′) and AGM CD4 rev (5′-TGC CTC AAA TGG GGC TAC-3′) under the cycling conditions described above, for 35 cycles. PCR products were gel purified and cloned by use of a pcDNA3.1 Directional TOPO expression kit (Invitrogen) as described above.
Analysis of coreceptor use by SIVagm Env-pseudotyped reporter virus.
AGM- and RM-specific coreceptor functions were evaluated by infection of 293T target cells transiently expressing AGM CD4 and species-specific CCR5, GPR15, or CXCR6. Target cells were cotransfected with plasmids encoding AGM CD4 and a coreceptor of choice by using the Fugene6 (Promega) transfection reagent. At 1 day posttransfection, cells were washed and seeded into a 96-well plate (2 × 104 cells/well). The following day, target cells were infected with 100 50% tissue culture infective doses (TCID50) of SIVagm Env-pseudotyped reporter virus in the presence of DEAE-dextran (Sigma) and spin inoculated at 2,000 rpm for 20 min. At 2 days postinfection, cells were lysed using 1× cell culture lysis buffer, and luciferase was quantified using a Mithras LB 940 luminometer (Berthold Technologies) as described above.
Ex vivo SIV infections of AGM and RM PBMC.
PBMC were isolated from whole blood by density centrifugation as described above. PBMC were stimulated with 5 μg/ml PHA and incubated at 37°C for 3 days. IL-2 (final concentration, 10%) was added at 1 day post-PHA stimulation. PHA-stimulated cells were seeded in a 96-well plate at 1.5 × 105 cells/well. Maraviroc-treated cells were preincubated with the inhibitor 1 h prior to infection (final concentration, 15 μM); untreated cells were incubated at 37°C for 1 h prior to infection. Cells were then infected with 500 50% infective doses (ID50) of SIVagmVer90, spin inoculated at 2,000 rpm for 30 min, and incubated overnight at 37°C. The following day, cells were washed 3 times with complete medium to remove excess virus. After the final wash, cells were resuspended in medium containing 10% IL-2 and incubated at 37°C. Culture supernatant samples were collected every 3 days for up to 3 weeks and stored at −20°C. At the time of culture supernatant collection, fresh medium (with or without maraviroc) was added to cells. An RT assay was performed to measure viral activity levels in culture supernatants.
Cell sorting using a FACSAria flow cytometer.
PBMC were isolated from EDTA-treated AGM and RM whole-blood samples as described above. Cells were washed twice with phosphate-buffered saline (PBS) and incubated with LIVE/DEAD fixable aqua dead cell stain (Invitrogen) for 10 min at room temperature. Cells were then stained with the following fluorescently conjugated monoclonal antibodies: anti-CD3 (clone SP34-2 conjugated to Alexa 700; BD Biosciences), anti-CD4 (clone L200 conjugated to allophycocyanin [APC]; BD Bioscience), anti-CD8α (clone RPA-T8 conjugated to Pacific Blue; BD Biosciences), anti-CD8β (clone 2ST8.5H7 conjugated to phycoerythrin [PE]; BD Biosciences), anti-CD28 (clone 28.2 conjugated to ECD; Beckman Coulter), and anti-CD95 (clone DX2 conjugated to PE-Cy5; BD Bioscience). Cells were incubated at 4°C for 30 min, washed with PBS, and resuspended in medium containing 10% FBS. Cells were sorted using a FACSAria II flow cytometer (BD Biosciences) into the following subsets: CD4 naive (live, singlet, CD3+ CD4+ CD28+ CD95−), CD4 memory (live, singlet, CD3+ CD4+ CD28+ CD95+), CD8αα (live, singlet, CD3+ CD4− CD8αdim), and CD8αβ (live, singlet, CD3+ CD4− CD8α+ CD8β+).
mRNA extraction and cDNA synthesis.
Sorted cells were washed twice in PBS, and RNAs and genomic DNAs were extracted using an AllPrep DNA/RNA minikit (Qiagen) according to the manufacturer's instructions. RNAs were treated with recombinant DNase I (Roche) at 37°C for 20 min. The DNase I enzyme was inactivated by the addition of 8 mM EDTA and incubated at 75°C for 10 min. RNA cleanup was performed using an RNeasy minikit (Qiagen). RNAs were stored at −80°C until use. cDNAs were synthesized using the SuperScript III first-strand synthesis system (Life Technologies) with an oligo(dT) primer as described above.
qRT-PCR.
The following primer pairs were used to detect coreceptor and actin mRNA levels in sorted cell subsets: AGM CCR5 qPCR for1 (5′-GGT TTT GTG GGC AAC ATA CTG GTC-3′) and AGM CCR5 qPCR rev1 (5′-CGA TTG TCA GGA GGA TGA TGA AG-3′), which yield a 244-bp product; AGM GPR15 qPCR for1 (5′-CTG CCT CTG ACT TCA TTT TCC TTG-3′) and AGM GPR15 qPCR rev1 (5′-AGT GGA GTT GCC TTT TTC TCT GC-3′), which yield a 346-bp product; AGM X6 qPCR for2 (5′-GTT CTT GCC ACC CAG ATG ACA C-3′) and AGM X6 qPCR rev2 (5′-TGA CAA ACG CAT AGA GCA CAG G-3′), which yield a 304-bp product; and QuantumRNA β-actin internal standards (Life Technologies). PCR products from AGM PBMC were cloned and sequenced for validation prior to mRNA detection in sorted cell subsets. The PCR mixture contained forward and reverse primers (0.2 μM [each]) specific for CCR5, GPR15, CXCR6, or actin and iTaq Sybr green master mix (Bio-Rad). Coreceptor-specific master mix was added to a 96-well PCR plate along with 2 μl cDNA template. The following thermocycling conditions were used: 95°C for 30 s followed by 45 cycles of 95°C for 10 s and 60°C for 30 s (sample reads were taken at this step), followed by a melt cycle (65°C to 95°C, increasing by 0.5°C increments). qRT-PCR was performed using a CFX96 Touch real-time PCR detection system (Bio-Rad). qRT-PCR results were analyzed using CFX Manager software (Bio-Rad). Relative coreceptor mRNA levels were calculated using the ΔCT method, using a reference gene and the following equation: ratio (reference/target) = 2CT (reference) − CT (target). The actin gene was used as a reference gene in qRT-PCRs. In rare instances where sample duplicates yielded incongruent results, samples were excluded from analysis.
Statistical analyses.
All statistical analyses and graphic analyses were performed using GraphPad Prism6 (GraphPad Prism Software, La Jolla, CA). Coreceptor mRNA levels in T cell subsets were compared using the Wilcoxon test. Coreceptor mRNA levels in AGM and RM and in uninfected and SIV-infected animals were compared using the Mann-Whitney test.
Nucleotide sequence accession numbers.
All nucleotide sequences determined in this study were deposited in GenBank under the following accession numbers: KU382464 to KU382472.
RESULTS
Examination of CCR5 alleles in a vervet AGM colony.
Based upon the previous finding that SIVsmm can use CCR5-independent pathways in sooty mangabeys, we hypothesized that the use of non-CCR5 entry coreceptors might be a feature of SIV infection in natural host species. Therefore, we sought to examine if inactivating mutations in CCR5 may exist in another natural host species, the vervet AGM. We amplified and sequenced CCR5 genes from 33 vervet AGM originating from Kenya (via Germany) and Tanzania. We identified several synonymous point mutations as well as three nonsynonymous mutations, which resulted in an asparagine-to-aspartic-acid substitution at position 13 and two glutamine-to-lysine substitutions, at positions 93 and 188, consistent with the previously described vervet AGM CCR5 alleles (54). In our analysis, we did not identify any significant mutations or deletions that may have led to a dysfunctional protein. Our consensus vervet AGM CCR5 sequence differed from a published grivet AGM CCR5 sequence by three amino acids (98.2% homologous), and it shared 97.2% homology with human CCR5 and 98.9% homology with RM CCR5. Although it appeared that all the vervet AGM in our colony possessed wild-type CCR5 genes, CCR5 is known to be expressed at very low levels on CD4+ T cells of AGM. Therefore, we continued to evaluate the coreceptor usage of SIVagmVer in AGM PBMC.
SIVagmVer90 can utilize CCR5-independent pathways to enter AGM PBMC.
In this study, we sought to determine if SIVagm is capable of utilizing non-CCR5 entry pathways in AGM PBMC. We therefore evaluated whether PHA-stimulated AGM PBMC were infectible with uncloned SIVagmVer90 in the absence or presence of the CCR5 antagonist maraviroc. RT activity was measured in culture supernatants that were collected periodically for 3 weeks postinfection. For control purposes, we evaluated the inhibitory effect of maraviroc treatment of HIV-infected human PBMC by using the CCR5-tropic HIV-1 clone BaL. Untreated human PBMC infected with HIV-1 BaL exhibited a standard viral replication curve, whereas treatment of human PBMC with maraviroc completely inhibited HIV-1 BaL entry, consistent with a major role of CCR5 in entry of this virus (Fig. 1A, top panel). Conversely, the presence of maraviroc did not inhibit HIV-1 NL4-3 (X4-tropic) infection of human PBMC, since this virus uses the CXCR4 coreceptor, not CCR5, for entry (Fig. 1A, bottom panel).
FIG 1.

SIVagmVer90 uses non-CCR5 entry pathways in AGM PBMC. (A) Viral replication kinetics of HIV-1-infected human PBMC in the absence (black) or presence (red) of maraviroc (15 μM). (Top) R5-tropic HIV-1 BaL infection; (bottom) X4-tropic HIV-1 NL4-3 infection. (B) Viral replication kinetics of SIVagmVer90-infected AGM PBMC (n = 6) in the absence (black) or presence (red) of maraviroc. Data are mean RT activities ± standard errors of the means (SEM). (C) 293T target cells expressing constant levels of AGM CD4 and variable levels of AGM CCR5 were infected with an HIV (pNL4-3) reporter virus pseudotyped with the SIVagm9063 envelope in the absence (black) or presence (red) of maraviroc. Infection was measured by the number of relative light units (RLU) in cell lysates at 3 days postinfection. Data depict means and standard deviations (SD) for a representative experiment (n = 3).
Next, we infected PBMC from six juvenile vervet AGM (AG26, AG28, AG32, AG33, AG36, and AG39) with uncloned SIVagmVer90 in the absence or presence of maraviroc. One set of cells were pretreated with maraviroc prior to infection, and the remaining cells were left untreated. Cells were then infected with 500 TCID50 of SIVagmVer90, and culture supernatants were periodically collected and tested for RT activity as described above. As shown in Fig. 1B, the mean viral replication curves for both untreated and maraviroc-treated AGM PBMC from six animals were similar. Thus, maraviroc treatment did not significantly block entry or replication of SIVagm infection in AGM PBMC, indicating that this virus appears to enter AGM PBMC in a CCR5-independent manner. In addition to SIVagmVer90, we also tested the CCR5 dependence of an infectious molecular clone, SIVagmVer9063, derived from an SIVagmVer90-infected PTM at the terminal stages of AIDS (50). However, we found that SIVagmVer9063, like the parental SIVagmVer90 strain, was also capable of entering and efficiently replicating in AGM PBMC in the presence of maraviroc (data not shown). Thus, CCR5 independence was not apparently affected by macaque passage and adaptation.
Since maraviroc was developed specifically to block human CCR5 (55, 56), the lack of effect in AGM PBMC could be due to inefficient activity against the AGM homologue. Therefore, we cloned CD4 and CCR5 from a vervet AGM and used these AGM-derived receptors to evaluate the effect of maraviroc in a transient expression system in 293T cells. Target cells were cotransfected with AGM CD4 and various amounts of a plasmid encoding AGM CCR5, ranging from 50 ng to 1 μg, and infected with pseudotype viruses carrying the SIVagmVer9063 envelope in the absence or presence of maraviroc. In target cells expressing low levels of AGM CCR5 (transfected with 50 ng plasmid), comparable to the physiological expression levels of CCR5 on primary AGM CD4+ T cells, 94% of virus entry was blocked in the presence of maraviroc compared to the level in infected untreated target cells (Fig. 1C). Taken together, these results suggest that maraviroc effectively blocks AGM CCR5 and that uncloned SIVagmVer90 and the infectious molecular clone SIVagmVer9063 can use non-CCR5 entry pathways to enter AGM target cells ex vivo.
Examining the CCR5 dependence of various SIVs in RM PBMC.
We next asked if non-CCR5 entry pathways could support SIV entry in PBMC from rhesus macaques, a nonnatural host of SIV, or whether non-CCR5 entry was specific to AGM coreceptors. We therefore evaluated whether PHA-stimulated RM PBMC were infectible with replication-competent SIVagmVer90 in the absence or presence of maraviroc, using SIVmac239, SIVsmE543-3, and SHIV-AD8 as controls. Similar to the results with AGM PBMC, we found that SIVagmVer90 effectively entered and replicated in RM PBMC in the presence of maraviroc. However, the viral replication curve generated for maraviroc-treated cells was slightly lower than the viral replication curve observed for untreated RM PBMC (Fig. 2A). Similar results were observed with SIVagmVer9063 and RM PBMC (data not shown). Notably, the entry and replication of SIVagmVer90 and SIVagmVer9063 in maraviroc-treated RM PBMC suggest that functional non-CCR5 entry pathways are present in RM PBMC. Conversely, infection of RM PBMC with SIVmac239 and SIVsmE543-3 resulted in substantial virus inhibition in the presence of maraviroc, although some breakthrough replication was observed in these cells at 15 days postinfection (Fig. 2B and C). This finding suggests that SIVmac239 and SIVsmE543-3 primarily depend on CCR5 for entry into RM PBMC. Lastly, SHIV-AD8 entry and replication were completely blocked in the presence of maraviroc, suggesting that this virus is strictly CCR5 dependent in RM PBMC, which is consistent with its HIV-1 R5-tropic envelope (Fig. 2D). Additionally, the complete inhibition of SHIV-AD8 in the presence of maraviroc indicates that maraviroc effectively blocks RM-derived CCR5. Collectively, these findings suggest that SIVagm can use CCR5-independent pathways to enter RM PBMC.
FIG 2.

SIVmac239, SIVsmE543-3, and SHIV-AD8 are primarily CCR5 dependent in RM PBMC. PHA-stimulated RM PBMC were infected with SIVagmVer90 (A), SIVmac239 (B), SIVsmE543-3 (C), or SHIV-AD8 (D) in the absence (black) or presence (red) of maraviroc (15 μM). Viral replication kinetics were determined by measuring the RT activity in culture supernatants over a 3-week period. Graphs depict mean RT activities ± SD for representative experiments (n = 3).
AGM- and RM-derived alternative coreceptors, CXCR6 and GPR15, can support SIVagm entry in vitro.
Our PBMC infection experiments revealed that SIVagmVer90 was capable of using CCR5-independent pathways to enter AGM and RM PBMC. We next sought to determine which non-CCR5 coreceptors could mediate entry of SIVagm in these cells. Several published studies revealed that in addition to using CCR5, SIVagm envelopes are capable of entering cells expressing human alternative coreceptors, such as the chemokine receptor CXCR6 and the orphan receptor GPR15, in vitro (36, 37). In order to determine if CXCR6 and GPR15 can support SIVagm entry, we cloned these receptors from a vervet AGM and an RM. Sequence analysis revealed that AGM and RM coreceptors are highly homologous, with four amino acid differences between AGM CCR5 and RM CCR5, at positions 13, 163, 198, and 348 (98.9% homologous); two amino acid differences between AGM GPR15 and RM GPR15, at positions 178 and 279 (99.4% homologous); and seven amino acid differences between AGM CXCR6 and RM CXCR6, at positions 10, 13, 31, 109, 123, 258, and 263 (97.9% homologous).
Additionally, we asked if the CCR5 independence exhibited by SIVagmVer90 was an inherent feature of the virus or an evolved feature, as seen by some HIV-1 strains that develop the use of the entry coreceptor CXCR4 (57, 58). Using SGA, we generated SIVagm T/F envelopes from plasmas collected at 1 week postinfection from two vervet AGM, AG5387 and AG5341, infected with SIVagmVer90 (via the intrarectal route). We isolated and sequenced approximately 20 rev/env amplicons from the virus stock and from the plasma of each animal. HIV Highlighter plots revealed that envelopes isolated from the uncloned SIVagmVer90 inoculum exhibited high sequence diversity. In contrast, SIVagm T/F envelopes from AG5387 and AG5431 had extremely low sequence diversity, which is consistent with an infection established by a single transmitted/founder variant (data not shown). Due to sequence diversity in the SIVagm T/F envelopes from each animal, we cloned the consensus envelopes from AG5387 and AG5431 into expression plasmids, denoted SIVagm5387 T/F and SIVagm5431 T/F. Next, we made pseudotype viruses carrying these envelopes and examined their coreceptor tropism in vitro. Briefly, HIV-1 (pNL4-3) reporter pseudotype viruses were generated in 293T cells containing one of the following envelope genes: that for SIVagmVer9063, SIVagm5387 T/F, SIVagm5431 T/F, or vesicular stomatitis virus glycoprotein (VSV-g). Pseudotype viruses were then used to infect 293T target cells expressing AGM-derived CD4 and the AGM- or RM-derived coreceptor CCR5, GPR15, or CXCR6.
As expected, control pseudotype viruses carrying VSV-g envelopes equally entered the various target cells examined. Our negative control consisted of 293T cells transfected with AGM CD4 alone, which did not mediate entry of viruses pseudotyped with SIVagm envelopes. AGM CCR5 efficiently mediated entry of viruses pseudotyped with the SIVagmVer9063 and SIVagm5387 T/F envelopes, whereas virus pseudotyped with the SIVagm5431 T/F envelope showed moderate use of this coreceptor. All SIVagm envelopes tested efficiently utilized AGM CXCR6 for entry into target cells; moreover, these SIVagm envelopes utilized AGM CXCR6 more efficiently than AGM CCR5 in this overexpression system. Additionally, all SIVagm envelopes examined modestly utilized AGM GPR15 for entry into target cells (Fig. 3A). Conversely, the SIVagmVer9063 and SIVagm5387 T/F envelopes used RM CCR5 more efficiently than RM CXCR6, while the SIVagm5431 T/F envelope utilized these coreceptors at relatively similar levels. Entry through RM GPR15 was considerably lower than entry through RM CCR5 and RM CXCR6 (Fig. 3B). Overall, these results indicate that various SIVagm envelopes can utilize the AGM- and RM-derived alternative coreceptors CXCR6 and GPR15 to various degrees, in addition to CCR5, in vitro.
FIG 3.
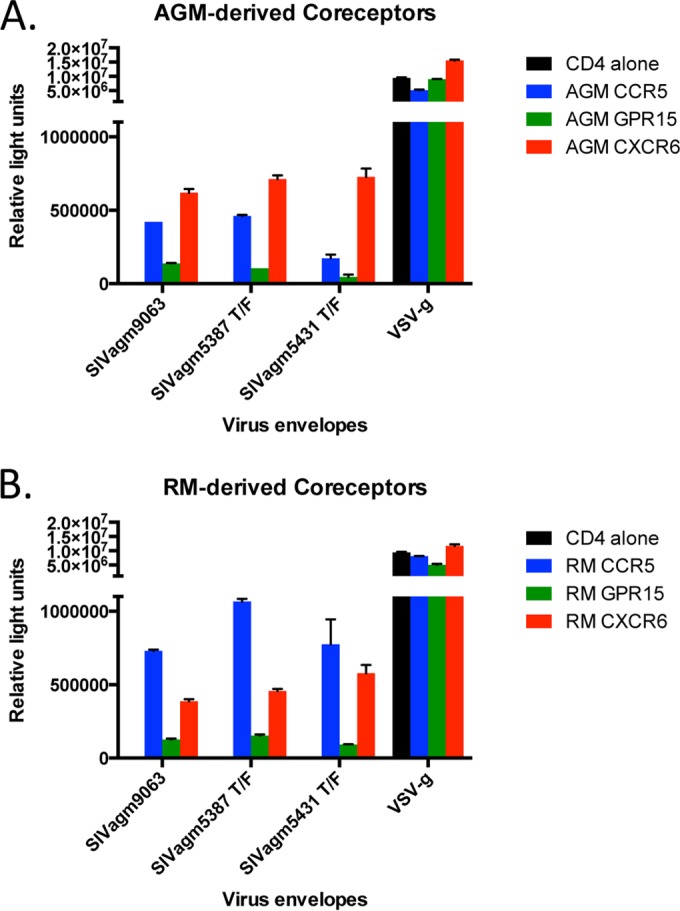
AGM and RM alternative coreceptors, GPR15 and CXCR6, can support entry by pseudotype viruses bearing SIVagm envelopes. Pseudotype reporter viruses bearing various SIVagm envelopes, including transmitted/founder (T/F) envelopes, were used to infect 293T target cells expressing AGM-derived (A) and RM-derived (B) coreceptors, i.e., CCR5, GPR15, and CXCR6. Pseudotype reporter viruses bearing vesicular stomatitis virus glycoprotein (VSV-g) served as a positive control. Infection was measured by the number of relative light units in cell lysates at 3 days postinfection. Graphs depict means and SD for representative experiments (n = 3).
Examination of relative mRNA levels for CCR5, GPR15, and CXCR6 in AGM CD4+ and CD8+ lymphocyte subsets.
CD4+ T cells are the primary target cells of HIV/SIV. In order to determine if GPR15 and/or CXCR6 could serve as a potential entry coreceptor for SIVagm in AGM PBMC, we next sought to determine whether these potential alternative coreceptors are expressed on AGM CD4+ T cells. Unfortunately, cross-reactive antibodies against AGM GPR15 and AGM CXCR6 are not commercially available, which prevents the examination of their protein expression levels on CD4+ T cells. Due to reagent limitations, we sought to characterize GPR15, CXCR6, and CCR5 mRNA profiles in AGM lymphocyte subsets by using qRT-PCR.
Coreceptor mRNA levels were examined in CD4+ and CD8+ T cell subsets from the 15 vervet AGM (5 uninfected animals and 10 SIVagm-infected animals) listed in Table 1. Notably, vervet AGM downregulate the CD4 molecule upon activation and thus exhibit a unique population of CD8αα cells, which appear to serve as helper T cells (29). Therefore, our analysis of AGM lymphocyte subsets included CD4 (naive and central memory), CD8αα, and CD8αβ T cells. Upon staining and examining sorted AGM PBMC, we found that the majority of AGM memory CD4+ T cells (CD95+) were comprised of central memory T cells (CD28+ CD95+). Although RM PBMC contained both CD4+ central memory (CD28+ CD95+) and effector memory (CD28− CD95+) T cell subsets, for the purpose of comparison, we analyzed only the CD4+ central memory T cells so the coreceptor mRNA levels could be compared directly. Throughout this study, central memory CD4+ T cells are referred to as memory CD4+ T cells. Among the AGM lymphocyte subsets examined, CCR5 mRNA levels were lowest in the naive CD4+ T cell subset and highest in the CD8αα and CD8αβ T cell subsets. In SIV-infected AGM, CCR5 mRNA levels were significantly higher in memory CD4+ T cells than in naive CD4+ T cells (P = 0.0078) (Fig. 4B, middle panel). A similar trend was also seen for uninfected AGM, although this difference did not reach statistical significance (Fig. 4B, left panel).
FIG 4.

Relative CCR5, GPR15, and CXCR6 mRNA levels in sorted AGM CD4+ and CD8+ T cell subsets from uninfected and SIV-infected vervet AGM. (A) Representative FACS plots for two uninfected vervet AGM, showing CCR5 protein expression levels on CD4+ (naive and memory) and CD8+ (CD8αα and CD8αβ) T cell subsets. SSC, side scatter. Quantitative RT-PCR was used to determine relative CCR5 (B), GPR15 (C), and CXCR6 (D) mRNA levels in sorted AGM CD4+ and CD8+ T cell subsets from uninfected (left panels; open symbols) and SIV-infected (middle panels; filled symbols) animals. The median coreceptor mRNA level is shown for each subset examined. For panels B to D, left and middle panels, P values were calculated using the Wilcoxon test to determine the level of significance of differences in coreceptor mRNA levels between various cell subsets. For panels B to D, right panels, P values were calculated using the Mann-Whitney test to determine the level of significance of differences in coreceptor mRNA levels between uninfected and SIV-infected AGM. P values that reached statistical significance (P ≤ 0.05) are displayed on graphs.
In order to verify that AGM CCR5 mRNA levels are a surrogate for AGM CCR5 protein expression, we utilized a commercially available human anti-CCR5 antibody that cross-reacts with AGM CCR5. Using flow cytometry, we analyzed AGM CCR5 expression levels on AGM CD4+ and CD8+ T cell subsets. Representative fluorescence-activated cell sorting (FACS) plots for two uninfected AGM revealed that CCR5 expression was extremely low on AGM CD4+ T cells (∼0.12% of naive CD4+ T cells expressed CCR5, and ∼3% of memory CD4+ T cells expressed CCR5) relative to CCR5 expression on AGM CD8+ T cell subsets (approximately 50% of CD8αα and CD8αβ cells expressed CCR5) (Fig. 4A). Importantly, our CCR5 protein expression results were consistent with the expression patterns of CCR5 mRNA data for CD4+ and CD8+ T cell subsets, suggesting that coreceptor mRNA levels detected using qRT-PCR likely reflect protein expression levels.
Next, we examined the GPR15 and CXCR6 mRNA levels in AGM CD4+ and CD8+ T cell subsets. GPR15 mRNA levels were extremely low in AGM naive CD4+ T cells, with undetectable levels in more than 50% of the AGM examined. GPR15 mRNA detection was significantly higher in memory CD4+ T cells than in naive CD4+ T cells (P = 0.031 for SIV-infected AGM) (Fig. 4C, middle panel); this trend was also seen in uninfected AGM, although it did not reach statistical significance (Fig. 4C, left panel). In contrast to the low CCR5 and GPR15 mRNA levels detected in AGM naive CD4+ T cells, CXCR6 mRNA was detected at relatively high levels in AGM naive CD4+ T cells, which were comparable to CXCR6 mRNA levels in AGM memory CD4+ T cells (Fig. 4D, left and middle panels). Interestingly, we found that CCR5, GPR15, and CXCR6 mRNA levels were substantially lower in memory CD4+ T cells from SIV-infected AGM than in those from uninfected animals (P = 0.008 for CCR5 [Fig. 4B, right panel]; P = 0.001 for GPR15 [Fig. 4C, right panel]; and P = 0.0753 for CXCR6 [Fig. 4D, right panel]). Taken together, these data indicate that in addition to CCR5 mRNA, GPR15 and CXCR6 mRNAs were also detected at relatively high levels in the memory CD4+ T cell subsets, suggesting that these receptors may be expressed in AGM CD4+ T cells and could potentially serve as entry coreceptors for SIVagm in vivo.
Examination of CCR5, GPR15, and CXCR6 mRNA levels in RM CD4+ and CD8+ lymphocyte subsets.
CCR5, GPR15, and CXCR6 mRNA levels were determined for RM CD4+ and CD8+ T cell subsets from 14 RM (5 uninfected animals and 9 SIVsmE660-infected animals) (Table 2). CCR5 mRNA levels were significantly higher in memory CD4+ T cells than in naive CD4+ T cells from uninfected RM (P = 0.0625) (Fig. 5B, left panel) and SIV-infected RM (P = 0.0039) (Fig. 5B, middle panel). Notably, our RM CCR5 mRNA data are consistent with the CCR5 expression data for RM CD4+ and CD8+ T cell subsets generated by flow cytometry. As shown in Fig. 5A, CCR5 expression was extremely low on naive CD4+ T cells (∼0.1% CCR5+ cells), moderate on memory CD4+ T cells (∼13% CCR5+ cells), and relatively high on CD8+ T cells (∼25% CCR5+ cells).
FIG 5.

Relative CCR5, GPR15, and CXCR6 mRNA levels in sorted RM CD4+ and CD8+ T cell subsets from uninfected and SIV-infected RM. (A) Representative FACS plots for two uninfected RM, showing CCR5 protein expression levels on CD4+ (naive and memory) and CD8+ T cell subsets. SSC, side scatter. Quantitative RT-PCR was used to determine relative CCR5 (B), GPR15 (C), and CXCR6 (D) mRNA levels in sorted RM CD4+ and CD8+ T cell subsets from uninfected (left panels; open symbols) and SIV-infected (middle panels; filled symbols) animals. The median coreceptor mRNA level is shown for each subset examined. For panels B to D, left and middle panels, P values were calculated using the Wilcoxon test to determine the level of significance of differences in coreceptor mRNA levels between various cell subsets. For panels B to D, right panels, P values were calculated using the Mann-Whitney test to determine the level of significance of differences in coreceptor mRNA levels between uninfected and SIV-infected RM. P values that reached statistical significance (P ≤ 0.05) are displayed on graphs.
Next, we examined GPR15 and CXCR6 mRNA levels in RM lymphocyte subsets. We found that GPR15 mRNA levels were substantially higher in memory CD4+ T cells than in naive CD4+ T cells for both uninfected and SIV-infected animals (P = 0.0625) (Fig. 5C, left and middle panels). On examining CXCR6 mRNA levels, we found that the CXCR6 mRNA level in RM naive CD4+ T cells was substantially higher than the CCR5 and GPR15 mRNA levels in this subset. However, CXCR6 mRNA levels in memory CD4+ T cells were considerably higher than those in naive CD4+ T cells for both uninfected and SIV-infected animals (P = 0.0625 and P = 0.020, respectively) (Fig. 5D).
Similar to our results for AGM, we found that CCR5, GPR15, and CXCR6 mRNA levels in memory CD4+ T cells from SIV-infected RM were significantly lower than the coreceptor mRNA levels in uninfected animals (P = 0.029 for CCR5 [Fig. 5B, right panel]; P = 0.004 for GPR15 [Fig. 5C, right panel]; and P = 0.019 for CXCR6 [Fig. 5D, right panel]). In summary, these data indicate that GPR15 and CXCR6 mRNAs, in addition to CCR5 mRNA, were detected at relatively high levels in the memory CD4+ T cell subset, suggesting that these receptors may be expressed in RM CD4+ T cells and could potentially serve as entry coreceptors.
CCR5, GPR15, and CXCR6 mRNA levels differ between AGM and RM memory CD4+ T cell subsets.
Next, we compared CCR5, GPR15, and CXCR6 mRNA levels between AGM and RM lymphocyte cell subsets. CCR5 mRNA levels were comparable in AGM and RM naive CD4+ T cells for both uninfected and SIV-infected animals (Fig. 6A). As expected, CCR5 mRNA levels were significantly higher in RM than in AGM memory CD4+ T cells for both uninfected and SIV-infected animals (P = 0.0079 and P = 0.0004, respectively). Conversely, CCR5 mRNA levels were significantly higher in AGM CD8+ T cells than in RM CD8+ T cells for SIV-infected animals (P = 0.0101) (Fig. 6A, right panel). Our CCR5 mRNA data generated by qRT-PCR are consistent with our CCR5 expression data generated by flow cytometry, which revealed that RM exhibit a higher percentage of CCR5-expressing memory CD4+ T cells than AGM do, whereas CCR5 expression on CD8+ T cells is relatively higher in AGM than in RM CD8+ T cells (Fig. 4A and 5A).
FIG 6.

Examination of relative coreceptor mRNA levels between AGM and RM T cell subsets from uninfected and SIV-infected animals. Quantitative RT-PCR was used to determine relative CCR5 (A), GPR15 (B), and CXCR6 (C) mRNA levels in AGM and RM CD4+ and CD8+ T cell subsets from uninfected (left panels; open symbols) and SIV-infected (right panels; filled symbols) animals. The median coreceptor mRNA level is shown for each subset examined. P values were calculated using the Mann-Whitney test to determine the level of significance of differences in coreceptor mRNA levels between AGM and RM. P values that reached statistical significance (P ≤ 0.05) are displayed on graphs.
Similar to our CCR5 mRNA findings, GPR15 mRNA levels were significantly higher in RM than in AGM memory CD4+ T cells for both uninfected and SIV-infected populations (P = 0.0317 and P < 0.0001, respectively). We also found a significantly higher level of GPR15 mRNA in RM naive CD4+ T cells than in AGM naive CD4+ T cells from SIV-infected animals (P = 0.0016), and a similar trend was seen for uninfected animals. Interestingly, we found comparable levels of AGM and RM CXCR6 mRNAs in each of the lymphocyte subsets examined, with the exception of memory CD4+ T cells. Significantly higher levels of CXCR6 mRNA were observed in SIV-infected RM memory CD4+ T cells than in AGM memory CD4+ T cells (P = 0.0101) (Fig. 6C, right panel). Overall, we found that CCR5, GPR15, and CXCR6 mRNA levels were substantially higher in RM memory CD4+ T cells than in AGM memory CD4+ T cells.
CCR5, GPR15, and CXCR6 mRNA levels correlate in RM memory CD4+ T cells but not in AGM memory CD4+ T cells.
Lastly, we examined if there were any associations between CCR5, GPR15, and CXCR6 mRNA levels detected in AGM and RM lymphocyte subsets. Interestingly, for SIV-infected RM memory CD4+ T cells, we found significant positive correlations in mRNA levels between CXCR6 and CCR5 (P = 0.004; r = 0.9500) and between CXCR6 and GPR15 (P = 0.0369; r = 0.7167) (Fig. 7B). We also found a positive trend between GPR15 and CCR5 mRNA levels (P = 0.0760; r = 0.6333) in this subset (Fig. 7B, bottom panel). These findings suggest that RM with low levels of CXCR6 mRNA in their memory CD4+ T cells also exhibit low levels of CCR5 and GPR15 mRNAs. Conversely, animals with high levels of CXCR6 mRNA in their memory CD4+ T cells also exhibit high levels of CCR5 and GPR15 mRNAs. Associations between coreceptor mRNA levels were not identified in RM naive CD4+ T cells or CD8+ T cells from SIV-infected animals (data not shown). Interestingly, we did not find any significant associations between CCR5, GPR15, and CXCR6 mRNA levels in AGM naive and memory CD4+ T cells (Fig. 7A) or CD8+ cell subsets (data not shown) from infected animals. Additionally, correlations were not observed between coreceptor mRNA levels in uninfected AGM and RM. Taken together, these findings suggest that CCR5, GPR15, and CXCR6 expression may be coordinately regulated in RM memory CD4+ T cells but not in AGM memory CD4+ T cells.
FIG 7.

CCR5, GPR15, and CXCR6 mRNA levels may be coordinately regulated in RM memory CD4+ T cells. Comparison of CCR5 and CXCR6 mRNA levels (top panels), CXCR6 and GPR15 mRNA levels (middle panels), and GPR15 and CCR5 mRNA levels (bottom panels) in SIV-infected AGM (A) and RM (B) memory CD4+ T cells as determined by qRT-PCR. The P value and Spearman's rank correlation coefficient (r) were calculated for each coreceptor pair and are displayed in each graph.
DISCUSSION
African NHP are natural hosts of SIV, and despite high viral loads, infection in these animals is generally nonpathogenic. Investigation of the numerous ways in which natural hosts remain asymptomatic in the face of SIV infection may shed light on novel approaches to treating HIV-infected humans. Previous studies suggested that a variety of factors may contribute to the nonpathogenicity seen in SIV-infected natural hosts, including (i) attenuation of chronic immune activation (9, 12, 59), (ii) maintenance of the mucosal gut epithelial barrier (14, 60), (iii) a lack of microbial translocation (23), (iv) downregulation of the CD4 receptor on potential target cells, thus creating a subset of cells that are SIV resistant (29–31), and (v) low CCR5 expression on CD4+ T cells from natural hosts compared to CCR5 expression on CD4+ T cells from nonnatural hosts (32). In this study, we propose that in addition to the aforementioned factors, SIV isolated from natural host species may be able to utilize CCR5-independent entry pathways. Expanded coreceptor utilization would likely affect target cell tropism and potentially affect infection outcomes. We hypothesize that broad coreceptor usage by SIV may contribute to reduced pathogenicity if potential alternative coreceptors are highly expressed on expendable cell subsets (i.e., effector and/or effector memory T cells) and are expressed at low levels on critical cell subsets, such as central memory T cells. Since our results suggest that CCR5 is not coordinately regulated with GPR15 or CXCR6 levels in AGM CD4+ T cells, the use of alternative coreceptors could potentially spare critical CCR5-expressing subsets in this species. This differs from the coordinate regulation of these coreceptors in rhesus macaque CD4+ T cells, and this difference between species could play a role in pathogenicity.
It has largely been accepted that CCR5 is the primary entry coreceptor used by SIV in vivo, although several exceptions have been reported (39–41, 61). Interestingly, a number of studies revealed that various strains of SIV were able to infect and replicate in CCR5-null human PBMC as well as in CEMx174 cells, which lack CCR5 expression (62–64). These findings indicate that coreceptors other than CCR5 can support SIV entry and replication ex vivo. Additionally, the notion that CCR5-independent pathways can support and sustain SIV infection in vivo was demonstrated in a study where genetically CCR5-deficient SM were identified that were SIVsmm positive and exhibited robust plasma viral loads (40). This finding raised many questions regarding SIVsmm coreceptor usage and target cell tropism in SM natural hosts and prompted us to investigate whether the use of CCR5-independent pathways is unique to SM or is a common feature attributed to other natural host species.
In the current study, we examined the CCR5 dependence and alternative coreceptor use of SIV isolated from vervet AGM. Interestingly, we found that SIVagmVer90 was able to efficiently replicate in AGM PBMC in a CCR5-independent manner, which is consistent with a study reporting that SIVsmm primary isolates from SM natural hosts were able to enter and replicate in maraviroc-treated SM PBMC (40, 65). Our finding supports the notion that SIVagmVer, like SIVsmm, may utilize non-CCR5 entry coreceptors in vivo, and it supports our hypothesis that the use of CCR5-independent pathways may be a common feature attributed to SIV strains in natural host species. Conversely, we found that entry and replication of SIVmac239, SIVsmE543-3, and SHIV-AD8 were significantly inhibited in maraviroc-treated RM PBMC. This finding suggests that either (i) the latter viruses are primarily CCR5 dependent in RM PBMC or (ii) non-CCR5 entry coreceptors are not expressed or functional for use by SIV in RM PBMC. To address the latter point, we investigated whether SIVagm could utilize CCR5-independent entry pathways in RM PBMC, and we found that SIVagmVer90 replicated efficiently in maraviroc-treated RM PBMC, confirming that non-CCR5 entry pathways are available and functional for SIV use in these cells. Therefore, the inability of SIVmac239, SIVsmE543-3, and SHIV-AD8 to efficiently enter and replicate in maraviroc-treated RM PBMC may be due to virus-dependent factors that prevent the use of RM-specific non-CCR5 coreceptors. Based on these findings, we speculate that SIV coreceptor use, which likely affects target cell tropism, may differ between SIV-infected natural (AGM) and nonnatural (RM) hosts.
Next, we investigated which alternative coreceptors could support SIV entry in AGM PBMC. It has been reported for many years that SIV envelope proteins isolated from various strains of SIV (i.e., SIVsmm and SIVmac) have promiscuous coreceptor tropism in vitro and are capable of utilizing various chemokine and orphan receptors for entry, such as APJ, GPR1, GPR15 (BOB), and CXCR6 (Bonzo/STRL33/CD186) (35–38, 66). However, the in vivo significance of alternative coreceptor use remains unclear. Although in vitro alternative coreceptor use has been studied extensively, a caveat to many of these studies was the use of human-derived alternative coreceptors, thus making it unclear if species-matched alternative coreceptors could also support SIV entry. Recently, a number of studies began to address this issue and have shown, particularly in the case of SM, that SM-derived GPR15 and CXCR6 mediate entry of pseudotype viruses bearing SIVsmm envelopes (65, 67). Likewise, we found that AGM-derived GPR15 and CXCR6 supported entry of SIVagm-pseudotyped viruses to various degrees. Interestingly, SIVagmVer transmitted/founder envelopes were able to utilize AGM GPR15 and AGM CXCR6 for entry into target cells, suggesting that these alternative coreceptors not only support replication of SIVagm but may mediate transmission of the virus in vervet AGM. The use of AGM-derived GPR15 and CXCR6 combined with the relatively high levels of GPR15 and CXCR6 mRNAs in memory CD4+ T cells strongly suggests that these alternative coreceptors may serve as potential alternative coreceptors for SIVagm entry in vivo.
In contrast to our finding that SIVagm could utilize CCR5-independent pathways in AGM PBMC, we found that SIVmac239, SIVsmE543-3, and SHIV-AD8 were primarily CCR5 dependent in RM PBMC. Interestingly, SIVmac239 has been shown to efficiently utilize human-derived alternative coreceptors, such as GPR1, GPR15, and CXCR6, for entry in vitro (37). However, a recent study examined the role of GPR15 use by SIVmac239 in CCR5-null human PBMC and found that a large percentage of SIVmac239-infected cells did not express GPR15. Based on this finding, it was concluded that GPR15 does not play a significant role as an entry coreceptor for SIVmac239 (64). Additionally, studies have reported that RM CXCR6 does not mediate efficient entry of multiple SIVmac derivatives, while human CXCR6 and SM CXCR6 mediate efficient entry of these viruses (65, 68, 69). The inability of RM CXCR6 to mediate efficient entry of SIVmac239 was mapped to a single amino acid substitution, S30R. Pohlmann et al. found that a serine at position 30 is critical for CXCR6 coreceptor function (68). Interestingly, human CXCR6, SM CXCR6, and AGM CXCR6 (vervet, grivet, and sabaeus subspecies) contain a serine at position 30, whereas RM and PTM CXCR6 sequences have an arginine at this position. We observed that RM CXCR6 supported entry of pseudotype viruses bearing SIVagm envelopes, albeit less efficiently than entry though RM CCR5 and AGM CXCR6. Our findings suggest that although GPR15 and CXCR6 mRNAs were detected in RM CD4+ T cell subsets, a combination of virus-dependent factors and host factors may prevent these alternative coreceptors from supporting SIVmac infection in vivo. Overall, these findings highlight the importance of studying species-matched alternative coreceptors and their corresponding viruses.
Lastly, we also characterized the mRNA expression profiles of CCR5, GPR15, and CXCR6 in AGM and RM CD4+ and CD8+ T cell subsets. To our knowledge, this study describes for the first time the CCR5, GPR15, and CXCR6 mRNA levels in primary vervet AGM CD4+ and CD8+ T cell subsets. Previous reports indicate that CCR5 expression levels are substantially lower on CD4+ T cells from natural hosts than on those from nonnatural hosts (32). Our CCR5 mRNA data corroborate these studies, as CCR5 mRNA levels were significantly lower in AGM memory CD4+ T cells than in RM memory CD4+ T cells. GPR15 and CXCR6 expression levels have been defined for primary human cells and, to a lesser extent, for primary cells from nonhuman primates. Farzan et al. detected GPR15 mRNA in RM PBMC, and a number of studies have reported that GPR15 mRNA and/or protein was detected in human colon and lymphoid tissues, particularly in human CD4+ and CD8+ T cell subsets, with naive T cells being largely GPR15 negative (35, 64, 70). Our analysis of GPR15 mRNA in primary AGM and RM T cell subsets is consistent with published reports. Among human cells, CXCR6 expression has been detected in human PBMC, T cells, and NK cells, and at low levels in monocytes (36, 71). However, there are conflicting reports regarding CXCR6 expression on naive and memory T cell subsets. Littman et al. detected CXCR6 expression exclusively on human memory (CD45RO+) T cells, while Lee et al. found CXCR6 expression exclusively on human naive CD4+ T cells (CD45RA+ CD62L+) (69, 71). Interestingly, in AGM and RM primary cells, we detected relatively high levels of CXCR6 mRNA in both naive and memory CD4+ T cell subsets as well as in AGM and RM CD8+ T cell subsets. Lastly, previous studies have reported that CCR5 and CXCR6 are coordinately regulated on the memory subset of human T cells, where most CCR5+ cells also express CXCR6 (71). Similarly, we found strong positive correlations between CCR5, CXCR6, and GPR15 mRNA levels in RM memory CD4+ T cells, particularly between CCR5 and CXCR6, suggesting that these coreceptors may be coexpressed on RM CD4+ T cells. Interestingly, we did not observe any correlations between CCR5, GPR15, and CXCR6 mRNA levels in any of the AGM CD4+ and CD8+ T cell subsets examined, suggesting that these coreceptors are not coordinately regulated in AGM CD4+ T cells.
This observation suggests that CCR5, GPR15, and CXCR6 may have differential expression patterns on AGM and RM CD4+ T cells. We propose that AGM natural hosts may benefit from the uncoordinated expression of CCR5, GPR15, and CXCR6 on CD4+ T cells, combined with the ability of SIV to use non-CCR5 entry pathways. As previously mentioned, natural hosts have a small proportion of CD4+ CCR5+ T cells. The notion that non-CCR5-expressing CD4+ T cells can support SIV replication could contribute to high viral loads in the context of low CCR5 expression on CD4+ T cells in natural hosts. Paiardini et al. reported that SM central memory CD4+ T cells are relatively spared from SIVsmm infection due to restricted CCR5 expression, which likely contributes to the preservation of immune cell homeostasis in these animals (33). It is plausible that AGM developed similar mechanisms to restrict CCR5 expression on critical cell subsets required for maintaining CD4+ T cell homeostasis. This theory is consistent with a model that uncouples high viral load production, which perhaps occurs in dispensable cell subsets that express CCR5, GPR15, CXCR6, or an unidentified alternative coreceptor, and the preservation of CD4+ T cell homeostasis in AGM natural hosts. However, we speculate that viral replication and CD4+ T cell homeostasis may be coupled in SIV-infected nonnatural hosts (i.e., RM), which may contribute to pathogenicity in these animals. Taken together, our findings suggest that the use of non-CCR5 entry coreceptors, such as GPR15 and CXCR6, may be a common feature attributed to SIV strains in natural host species, which may affect target cell tropism and contribute to nonpathogenicity in these animals.
ACKNOWLEDGMENTS
We thank Heather Cronise-Santis, Joanna Swerczek, and Richard Herbert of NIHAC for excellent care of the study animals. We also thank Yoshi Nishimura, Malcolm Martin, Eri Miyagi, Klaus Strebel, Katie Sheehan Wetzel, and Ron Collman for infectious viruses (SHIV-AD8 and HIV-1) and CD4 primers.
Funding for this study was provided in part by the Division of Intramural Research, NIAID, NIH.
Funding Statement
The content of this publication does not necessarily reflect the views or policies of DHHS, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
REFERENCES
- 1.Klatt NR, Silvestri G, Hirsch V. 2012. Nonpathogenic simian immunodeficiency virus infections. Cold Spring Harb Perspect Med 2:a007153. doi: 10.1101/cshperspect.a007153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.VandeWoude S, Apetrei C. 2006. Going wild: lessons from naturally occurring T-lymphotropic lentiviruses. Clin Microbiol Rev 19:728–762. doi: 10.1128/CMR.00009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lifson JD, Nowak MA, Goldstein S, Rossio JL, Kinter A, Vasquez G, Wiltrout TA, Brown C, Schneider D, Wahl L, Lloyd AL, Williams J, Elkins WR, Fauci AS, Hirsch VM. 1997. The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. J Virol 71:9508–9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staprans SI, Dailey PJ, Rosenthal A, Horton C, Grant RM, Lerche N, Feinberg MB. 1999. Simian immunodeficiency virus disease course is predicted by the extent of virus replication during primary infection. J Virol 73:4829–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rey-Cuillé MA, Berthier JL, Bomsel-Demontoy MC, Chaduc Y, Montagnier L, Hovanessian AG, Chakrabarti LA. 1998. Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease. J Virol 72:3872–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broussard SR, Staprans SI, White R, Whitehead EM, Feinberg MB, Allan JS. 2001. Simian immunodeficiency virus replicates to high levels in naturally infected African green monkeys without inducing immunologic or neurologic disease. J Virol 75:2262–2275. doi: 10.1128/JVI.75.5.2262-2275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon SN, Dunham RM, Engram JC, Estes J, Wang Z, Klatt NR, Paiardini M, Pandrea IV, Apetrei C, Sodora DL, Lee HY, Haase AT, Miller MD, Kaur A, Staprans SI, Perelson AS, Feinberg MB, Silvestri G. 2008. Short-lived infected cells support virus replication in sooty mangabeys naturally infected with simian immunodeficiency virus: implications for AIDS pathogenesis. J Virol 82:3725–3735. doi: 10.1128/JVI.02408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandrea I, Ribeiro RM, Gautam R, Gaufin T, Pattison M, Barnes M, Monjure C, Stoulig C, Dufour J, Cyprian W, Silvestri G, Miller MD, Perelson AS, Apetrei C. 2008. Simian immunodeficiency virus SIVagm dynamics in African green monkeys. J Virol 82:3713–3724. doi: 10.1128/JVI.02402-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, Francella N, Sidahmed A, Smith AJ, Cramer EM, Zeng M, Masopust D, Carlis JV, Ran L, Vanderford TH, Paiardini M, Isett RB, Baldwin DA, Else JG, Staprans SI, Silvestri G, Haase AT, Kelvin DJ. 2009. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest 119:3556–3572. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Estes JD, Gordon SN, Zeng M, Chahroudi AM, Dunham RM, Staprans SI, Reilly CS, Silvestri G, Haase AT. 2008. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J Immunol 180:6798–6807. doi: 10.4049/jimmunol.180.10.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacquelin B, Mayau V, Targat B, Liovat A-S, Kunkel D, Petitjean G, Dillies M-A, Roques P, Butor C, Silvestri G, Giavedoni LD, Lebon P, Barre-Sinoussi F, Benecke A, Müller-Trutwin MC. 2009. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest 119:3544–3555. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, Silvestri G, Muller-Trutwin M, Vasile-Pandrea I, Apetrei C, Hirsch V, Lifson J, Brenchley JM, Estes JD. 2010. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol 84:7886–7891. doi: 10.1128/JVI.02612-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon SN, Klatt NR, Bosinger SE, Brenchley JM, Milush JM, Engram JC, Dunham RM, Paiardini M, Klucking S, Danesh A, Strobert EA, Apetrei C, Pandrea IV, Kelvin D, Douek DC, Staprans SI, Sodora DL, Silvestri G. 2007. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J Immunol 179:3026–3034. doi: 10.4049/jimmunol.179.5.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandrea IV, Gautam R, Ribeiro RM, Brenchley JM, Butler IF, Pattison M, Rasmussen T, Marx PA, Silvestri G, Lackner AA, Perelson AS, Douek DC, Veazey RS, Apetrei C. 2007. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J Immunol 179:3035–3046. doi: 10.4049/jimmunol.179.5.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC. 2004. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med 200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picker LJ, Hagen SI, Lum R, Reed-Inderbitzin EF, Daly LM, Sylwester AW, Walker JM, Siess DC, Piatak M, Wang C, Allison DB, Maino VC, Lifson JD, Kodama T, Axthelm MK. 2004. Insufficient production and tissue delivery of CD4+ memory T cells in rapidly progressive simian immunodeficiency virus infection. J Exp Med 200:1299–1314. doi: 10.1084/jem.20041049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, Scheinberg P, Price DA, Hage CA, Kholi LM, Khoruts A, Frank I, Else J, Schacker T, Silvestri G, Douek DC. 2008. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 112:2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Favre D, Lederer S, Kanwar B, Ma Z-M, Proll S, Kasakow Z, Mold J, Swainson L, Barbour JD, Baskin CR, Palermo R, Pandrea I, Miller CJ, Katze MG, McCune JM. 2009. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog 5:e1000295. doi: 10.1371/journal.ppat.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cecchinato V, Trindade CJ, Laurence A, Heraud JM, Brenchley JM, Ferrari MG, Zaffiri L, Tryniszewska E, Tsai WP, Vaccari M, Parks RW, Venzon D, Douek DC, O'Shea JJ, Franchini G. 2008. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol 1:279–288. doi: 10.1038/mi.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bjarnason I, Sharpstone DR, Francis N, Marker A, Taylor C, Barrett M, Macpherson A, Baldwin C, Menzies IS, Crane RC, Smith T, Pozniak A, Gazzard BG. 1996. Intestinal inflammation, ileal structure and function in HIV. AIDS 10:1385–1391. doi: 10.1097/00002030-199610000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Kotler DP, Gaetz HP, Lange M, Klein EB, Holt PR. 1984. Enteropathy associated with the acquired immunodeficiency syndrome. Ann Intern Med 101:421–428. doi: 10.7326/0003-4819-101-4-421. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Estes JD, Duan L, Jessurun J, Pambuccian S, Forster C, Wietgrefe S, Zupancic M, Schacker T, Reilly C, Carlis JV, Haase AT. 2008. Simian immunodeficiency virus-induced intestinal cell apoptosis is the underlying mechanism of the regenerative enteropathy of early infection. J Infect Dis 197:420–429. doi: 10.1086/525046. [DOI] [PubMed] [Google Scholar]
- 23.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371. [DOI] [PubMed] [Google Scholar]
- 24.Meythaler M, Martinot A, Wang Z, Pryputniewicz S, Kasheta M, Ling B, Marx PA, O'Neil S, Kaur A. 2009. Differential CD4+ T-lymphocyte apoptosis and bystander T-cell activation in rhesus macaques and sooty mangabeys during acute simian immunodeficiency virus infection. J Virol 83:572–583. doi: 10.1128/JVI.01715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silvestri G, Sodora DL, Koup RA, Paiardini M, O'Neil SP, McClure HM, Staprans SI, Feinberg MB. 2003. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 18:441–452. doi: 10.1016/S1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 26.Chakrabarti LA, Lewin SR, Zhang L, Gettie A, Luckay A, Martin LN, Skulsky E, Ho DD, Cheng-Mayer C, Marx PA. 2000. Normal T-cell turnover in sooty mangabeys harboring active simian immunodeficiency virus infection. J Virol 74:1209–1223. doi: 10.1128/JVI.74.3.1209-1223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornfeld C, Ploquin MJY, Pandrea I, Faye A, Onanga R, Apetrei C, Poaty-Mavoungou V, Rouquet P, Estaquier J, Mortara L, Desoutter J-F, Butor C, Le Grand R, Roques P, Simon F, Barre-Sinoussi F, Diop OM, Müller-Trutwin MC. 2005. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J Clin Invest 115:1082–1091. doi: 10.1172/JCI23006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onanga R, Souquiere S, Makuwa M, Mouinga-Ondéme A, Simon F, Apetrei C, Roques P. 2006. Primary simian immunodeficiency virus SIVmnd-2 infection in mandrills (Mandrillus sphinx). J Virol 80:3301–3309. doi: 10.1128/JVI.80.7.3301-3309.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beaumier CM, Harris LD, Goldstein S, Klatt NR, Whitted S, McGinty J, Apetrei C, Pandrea I, Hirsch VM, Brenchley JM. 2009. CD4 downregulation by memory CD4+ T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nat Med 15:879–885. doi: 10.1038/nm.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murayama Y, Mukai R, Inoue-Murayama M, Yoshikawa Y. 1999. An African green monkey lacking peripheral CD4 lymphocytes that retains helper T cell activity and coexists with SIVagm. Clin Exp Immunol 117:504–512. doi: 10.1046/j.1365-2249.1999.00999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murayama Y, Amano A, Mukai R, Shibata H, Matsunaga S, Takahashi H, Yoshikawa Y, Hayami M, Noguchi A. 1997. CD4 and CD8 expressions in African green monkey helper T lymphocytes: implication for resistance to SIV infection. Int Immunol 9:843–851. doi: 10.1093/intimm/9.6.843. [DOI] [PubMed] [Google Scholar]
- 32.Pandrea I, Apetrei C, Gordon S, Barbercheck J, Dufour J, Bohm R, Sumpter B, Roques P, Marx PA, Hirsch VM, Kaur A, Lackner AA, Veazey RS, Silvestri G. 2007. Paucity of CD4+ CCR5+ T cells is a typical feature of natural SIV hosts. Blood 109:1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A, Vinton C, Gordon SN, Bosinger SE, Francella N, Hallberg PL, Cramer E, Schlub T, Chan ML, Riddick NE, Collman RG, Apetrei C, Pandrea I, Else J, Münch J, Kirchhoff F, Davenport MP, Brenchley JM, Silvestri G. 2011. Low levels of SIV infection in sooty mangabey central memory CD4+ T cells are associated with limited CCR5 expression. Nat Med 17:830–836. doi: 10.1038/nm.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brenchley JM, Silvestri G, Douek DC. 2010. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity 32:737–742. doi: 10.1016/j.immuni.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farzan M, Choe H, Martin K, Marcon L, Hofmann W, Karlsson G, Sun Y, Barrett P, Marchand N, Sullivan N, Gerard N, Gerard C, Sodroski J. 1997. Two orphan seven-transmembrane segment receptors which are expressed in CD4-positive cells support simian immunodeficiency virus infection. J Exp Med 186:405–411. doi: 10.1084/jem.186.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng HK, Unutmaz D, KewalRamani VN, Littman DR. 1997. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 388:296–300. doi: 10.1038/40894. [DOI] [PubMed] [Google Scholar]
- 37.Edinger AL, Hoffman TL, Sharron M, Lee B, O'Dowd B, Doms RW. 1998. Use of GPR1, GPR15, and STRL33 as coreceptors by diverse human immunodeficiency virus type 1 and simian immunodeficiency virus envelope proteins. Virology 249:367–378. doi: 10.1006/viro.1998.9306. [DOI] [PubMed] [Google Scholar]
- 38.Alkhatib G, Liao F, Berger EA, Farber JM, Peden KW. 1997. A new SIV co-receptor, STRL33. Nature 388:238. doi: 10.1038/40789. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z, Kwon D, Jin Z, Monard S, Telfer P, Jones MS, Lu CY, Aguilar RF, Ho DD, Marx PA. 1998. Natural infection of a homozygous delta24 CCR5 red-capped mangabey with an R2b-tropic simian immunodeficiency virus. J Exp Med 188:2057–2065. doi: 10.1084/jem.188.11.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riddick NE, Hermann EA, Loftin LM, Elliott ST, Wey WC, Cervasi B, Taaffe J, Engram JC, Li B, Else JG, Li Y, Hahn BH, Derdeyn CA, Sodora DL, Apetrei C, Paiardini M, Silvestri G, Collman RG. 2010. A novel CCR5 mutation common in sooty mangabeys reveals SIVsmm infection of CCR5-null natural hosts and efficient alternative coreceptor use in vivo. PLoS Pathog 6:e1001064. doi: 10.1371/journal.ppat.1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palacios E, Digilio L, McClure HM, Chen Z, Marx PA, Goldsmith MA, Grant RM. 1998. Parallel evolution of CCR5-null phenotypes in humans and in a natural host of simian immunodeficiency virus. Curr Biol 8:943–946. doi: 10.1016/S0960-9822(07)00378-8. [DOI] [PubMed] [Google Scholar]
- 42.Jin MJ, Hui H, Robertson DL, Müller MC, Barré-Sinoussi F, Hirsch VM, Allan JS, Shaw GM, Sharp PM, Hahn BH. 1994. Mosaic genome structure of simian immunodeficiency virus from West African green monkeys. EMBO J 13:2935–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukasawa M, Miura T, Hasegawa A, Morikawa S, Tsujimoto H, Miki K, Kitamura T, Hayami M. 1988. Sequence of simian immunodeficiency virus from African green monkey, a new member of the HIV/SIV group. Nature 333:457–461. doi: 10.1038/333457a0. [DOI] [PubMed] [Google Scholar]
- 44.Allan JS, Short M, Taylor ME, Su S, Hirsch VM, Johnson PR, Shaw GM, Hahn BH. 1991. Species-specific diversity among simian immunodeficiency viruses from African green monkeys. J Virol 65:2816–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirsch VM, McGann C, Dapolito G, Goldstein S, Ogen-Odoi A, Biryawaho B, Lakwo T, Johnson PR. 1993. Identification of a new subgroup of SIVagm in tantalus monkeys. Virology 197:426–430. doi: 10.1006/viro.1993.1606. [DOI] [PubMed] [Google Scholar]
- 46.Johnson PR, Fomsgaard A, Allan J, Gravell M, London WT, Olmsted RA, Hirsch VM. 1990. Simian immunodeficiency viruses from African green monkeys display unusual genetic diversity. J Virol 64:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandrea I, Kornfeld C, Ploquin MJY, Apetrei C, Faye A, Rouquet P, Roques P, Simon F, Barre-Sinoussi F, Müller-Trutwin MC, Diop OM. 2005. Impact of viral factors on very early in vivo replication profiles in simian immunodeficiency virus SIVagm-infected African green monkeys. J Virol 79:6249–6259. doi: 10.1128/JVI.79.10.6249-6259.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 49.The Royal Society. 12 December 2006. The use of non-human primates in research. https://royalsociety.org/topics-policy/publications/2006/weatherall-report/.
- 50.Hirsch VM, Dapolito G, Johnson PR, Elkins WR, London WT, Montali RJ, Goldstein S, Brown C. 1995. Induction of AIDS by simian immunodeficiency virus from an African green monkey: species-specific variation in pathogenicity correlates with the extent of in vivo replication. J Virol 69:955–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishimura Y, Shingai M, Willey R, Sadjadpour R, Lee WR, Brown CR, Brenchley JM, Buckler-White A, Petros R, Eckhaus M, Hoffman V, Igarashi T, Martin MA. 2010. Generation of the pathogenic R5-tropic simian/human immunodeficiency virus SHIVAD8 by serial passaging in rhesus macaques. J Virol 84:4769–4781. doi: 10.1128/JVI.02279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 53.Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BTM, Sharp PM, Shaw GM, Hahn BH. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuhmann SE, Madani N, Diop OM, Platt EJ, Morvan J, Müller-Trutwin MC, Barré-Sinoussi F, Kabat D. 2001. Frequent substitution polymorphisms in African green monkey CCR5 cluster at critical sites for infections by simian immunodeficiency virus SIVagm, implying ancient virus-host coevolution. J Virol 75:8449–8460. doi: 10.1128/JVI.75.18.8449-8460.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AIM, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, van der Ryst E. 2005. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med 11:1170–1172. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 56.Wood A, Armour D. 2005. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog Med Chem 43:239–271. doi: 10.1016/S0079-6468(05)43007-6. [DOI] [PubMed] [Google Scholar]
- 57.Blaak H, van't Wout AB, Brouwer M, Hooibrink B, Hovenkamp E, Schuitemaker H. 2000. In vivo HIV-1 infection of CD45RA(+)CD4(+) T cells is established primarily by syncytium-inducing variants and correlates with the rate of CD4(+) T cell decline. Proc Natl Acad Sci U S A 97:1269–1274. doi: 10.1073/pnas.97.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scarlatti G, Tresoldi E, Björndal A, Fredriksson R, Colognesi C, Deng HK, Malnati MS, Plebani A, Siccardi AG, Littman DR, Fenyö EM, Lusso P. 1997. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat Med 3:1259–1265. doi: 10.1038/nm1197-1259. [DOI] [PubMed] [Google Scholar]
- 59.Onanga R, Kornfeld C, Pandrea I, Estaquier J, Souquiere S, Rouquet P, Mavoungou VP, Bourry O, M'Boup S, Barre-Sinoussi F, Simon F, Apetrei C, Roques P, Müller-Trutwin MC. 2002. High levels of viral replication contrast with only transient changes in CD4(+) and CD8(+) cell numbers during the early phase of experimental infection with simian immunodeficiency virus SIVmnd-1 in Mandrillus sphinx. J Virol 76:10256–10263. doi: 10.1128/JVI.76.20.10256-10263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, Barclay GR, Smedley J, Pung R, Oliveira KM, Hirsch VM, Silvestri G, Douek DC, Miller CJ, Haase AT, Lifson J, Brenchley JM. 2010. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog 6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milush JM, Reeves JD, Gordon SN, Zhou D, Muthukumar A, Kosub DA, Chacko E, Giavedoni LD, Ibegbu CC, Cole KS, Miamidian JL, Paiardini M, Barry AP, Staprans SI, Silvestri G, Sodora DL. 2007. Virally induced CD4+ T cell depletion is not sufficient to induce AIDS in a natural host. J Immunol 179:3047–3056. doi: 10.4049/jimmunol.179.5.3047. [DOI] [PubMed] [Google Scholar]
- 62.Forte S, Harmon M-E, Pineda MJ, Overbaugh J. 2003. Early- and intermediate-stage variants of simian immunodeficiency virus replicate efficiently in cells lacking CCR5. J Virol 77:9723–9727. doi: 10.1128/JVI.77.17.9723-9727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Owen SM, Masciotra S, Novembre F, Yee J, Switzer WM, Ostyula M, Lal RB. 2000. Simian immunodeficiency viruses of diverse origin can use CXCR4 as a coreceptor for entry into human cells. J Virol 74:5702–5708. doi: 10.1128/JVI.74.12.5702-5708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kiene M, Marzi A, Urbanczyk A, Bertram S, Fisch T, Nehlmeier I, Gnirss K, Karsten CB, Palesch D, Münch J, Chiodi F, Pöhlmann S, Steffen I. 2012. The role of the alternative coreceptor GPR15 in SIV tropism for human cells. Virology 433:73–84. doi: 10.1016/j.virol.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 65.Elliott STC, Wetzel KS, Francella N, Bryan S, Romero DC, Riddick NE, Shaheen F, Vanderford T, Derdeyn CA, Silvestri G, Paiardini M, Collman RG. 2015. Dualtropic CXCR6/CCR5 simian immunodeficiency virus (SIV) infection of sooty mangabey primary lymphocytes: distinct coreceptor use in natural versus pathogenic hosts of SIV. J Virol 89:9252–9261. doi: 10.1128/JVI.01236-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edinger AL, Hoffman TL, Sharron M, Lee B, Yi Y, Choe W, Kolson DL, Mitrovic B, Zhou Y, Faulds D, Collman RG, Hesselgesser J, Horuk R, Doms RW. 1998. An orphan seven-transmembrane-domain receptor expressed widely in the brain functions as a coreceptor for human immunodeficiency virus type 1 and simian immunodeficiency virus. J Virol 72:7934–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elliott STC, Riddick NE, Francella N, Paiardini M, Vanderford TH, Li B, Apetrei C, Sodora DL, Derdeyn CA, Silvestri G, Collman RG. 2012. Cloning and analysis of sooty mangabey alternative coreceptors that support simian immunodeficiency virus SIVsmm entry independently of CCR5. J Virol 86:898–908. doi: 10.1128/JVI.06415-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pohlmann S, Lee B, Meister S, Krumbiegel M, Leslie G, Doms RW, Kirchhoff F. 2000. Simian immunodeficiency virus utilizes human and sooty mangabey but not rhesus macaque STRL33 for efficient entry. J Virol 74:5075–5082. doi: 10.1128/JVI.74.11.5075-5082.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharron M, Pohlmann S, Price K, Lolis E, Tsang M, Kirchhoff F, Doms RW, Lee B. 2000. Expression and coreceptor activity of STRL33/Bonzo on primary peripheral blood lymphocytes. Blood 96:41–49. [PubMed] [Google Scholar]
- 70.Kim SV, Xiang WV, Kwak C, Yang Y, Lin XW, Ota M, Sarpel U, Rifkin DB, Xu R, Littman DR. 2013. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 340:1456–1459. doi: 10.1126/science.1237013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR. 2000. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol 165:3284–3292. doi: 10.4049/jimmunol.165.6.3284. [DOI] [PubMed] [Google Scholar]