

ABSTRACT
We have shown previously that herpes simplex virus 1 (HSV-1) lacking expression of the entire glycoprotein K (gK) or expressing gK with a 38-amino-acid deletion (gKΔ31–68 mutation) failed to infect ganglionic neurons after ocular infection of mice. We constructed a new model for the predicted three-dimensional structure of gK, revealing that the gKΔ31–68 mutation spans a well-defined β-sheet structure within the amino terminus of gK, which is conserved among alphaherpesviruses. The HSV-1(McKrae) gKΔ31–68 virus was tested for the ability to enter into ganglionic neuronal axons in cell culture of explanted rat ganglia using a novel virus entry proximity ligation assay (VEPLA). In this assay, cell surface-bound virions were detected by the colocalization of gD and its cognate receptor nectin-1 on infected neuronal surfaces. Capsids that have entered into the cytoplasm were detected by the colocalization of the virion tegument protein UL37, with dynein required for loading of virion capsids onto microtubules for retrograde transport to the nucleus. HSV-1(McKrae) gKΔ31–68 attached to cell surfaces of Vero cells and ganglionic axons in cell culture as efficiently as wild-type HSV-1(McKrae). However, unlike the wild-type virus, the mutant virus failed to enter into the axoplasm of ganglionic neurons. This work suggests that the amino terminus of gK is a critical determinant for entry into neuronal axons and may serve similar conserved functions for other alphaherpesviruses.
IMPORTANCE Alphaherpesviruses, unlike beta- and gammaherpesviruses, have the unique ability to infect and establish latency in neurons. Glycoprotein K (gK) and the membrane protein UL20 are conserved among all alphaherpesviruses. We show here that a predicted β-sheet domain, which is conserved among alphaherpesviruses, functions in HSV-1 entry into neuronal axons, suggesting that it may serve similar functions for other herpesviruses. These results are in agreement with our previous observations that deletion of this gK domain prevents the virus from successfully infecting ganglionic neurons after ocular infection of mice.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) encodes at least 26 tegument proteins and 11 virally encoded glycoproteins, as well as several nonglycosylated membrane-associated proteins. Viral glycoproteins gD, gB, gH, and gL serve critical roles in virion entry (1–5). Virion entry is initiated by the binding of glycoproteins gB and gC to glycosaminoglycan (GAG) moieties of cell surface proteoglycans (6). This initial attachment causes the interaction of gD with one or more of its specific receptors, including the herpesvirus entry mediator (HVEM) (HveA), nectin-1 (HVEC), and 3-O-sulfated HS. In addition, gB binds to PILR-α, NMHC-IIA, and myelin-associated glycoprotein (MAG) receptors (7). HSV-1 enters into neurons strictly via a pH-independent fusion of the viral envelope with neuronal plasma membranes (8–10), while it can enter a wide range of nonneuronal cells via either pH-independent or pH-dependent endocytosis (11). Fusion of the viral envelope with cellular, including neuronal, membranes causes deposition of the viral capsid into the cytoplasm, which is subsequently transported to the cell nucleus. Virus entry into all cells involves the coordinated functions of the glycoproteins gD, gB, gH, gL, and gC. Initial binding of gD to the nectin-1 receptor is thought to alter interactions of the gH/gL complex with gB, triggering gB-mediated fusion of the viral envelope with plasma membranes (reviewed in reference 12).
The UL20 and UL53 (gK) genes are highly conserved in all alphaherpesviruses and encode proteins of 222 and 338 amino acids, respectively, each with four membrane-spanning domains (13–17). HSV-1 gK is posttranslationally modified by N-linked carbohydrate addition at the amino terminus of gK, while the UL20 protein (UL20p) is not glycosylated (13, 15, 18). HSV-1 gK and UL20 functionally and physically interact, and these interactions are necessary for their coordinate intracellular transport, cell surface expression, and functions in virus-induced cell fusion, virus entry, virion envelopment, and egress from infected cells (16, 19–29). The gK/UL20 protein complex interacts with gB and gH and is required for gB-mediated cell fusion (30, 31). HSV-1 gK is a structural component of virions and functions in virion entry (26, 32). Deletion of amino acids 31 to 68 within the amino terminus of gK inhibits virus-induced cell-to-cell fusion and virus entry without drastically inhibiting virion envelopment and egress. Moreover, deletion of gK amino acids 31 to 68 inhibited virus-induced cell fusion caused by syncytial mutations in gK and entry into PILR-α-expressing Chinese hamster ovary cells (30, 33). We have shown that gK is essential for neuronal infection and virulence (34). Specifically, we have reported that gK-null virus was unable to infect axonal termini and egress from neuronal cell bodies (8). In addition, we have recently shown that the HSV-1(McKrae) gKΔ31–68 virus, specifying gK with a deletion of amino acids 31 to 68, was unable to efficiently infect mouse trigeminal ganglia after ocular infection of scarified mouse eyes (35). These results indicate that the amino terminus of gK plays a pivotal role in corneal infection and neuroinvasiveness.
HSV-1 as well as other viruses utilizes the intracellular microtubular network, which is utilized to move intracellular cargo in a retrograde manner toward the microtubule-organizing center (MTOC) and nucleus, as well as in an anterograde manner toward the cell periphery during maturation and cellular egress (36–40) (Fig. 1). Cellular cargo is transported in a retrograde manner toward the MTOC and the cell periphery in conjunction with the dynein and kinesin motors, respectively. Intracellular cargo can simultaneously bind to both dynein and kinesin and move bidirectionally along microtubules. This dynein/kinesin competition for intracellular cargo transport is highly coordinated within cells to maintain subcellular organization (39, 41). In a similar fashion, HSV-1, vaccinia virus, and adenovirus utilize the dynein-dynactin motor complex for intracellular transport (37, 42–48). Also, substantial evidence suggests that a number of other viruses, including African swine fever virus (ASFV) (49), canine parvovirus (CPV) (50), influenza virus X-31 (51), human foamy virus (HFV) (52), Mason-Pfizer monkey virus (M-PMV) (53), and rabies virus (RV) (54), utilize the microtubular network for their intracellular transport.
FIG 1.

Schematic representation of intracellular transport of virion capsids via the cellular microtubular network. The virus enters the cell via fusion or endocytosis and is transported by dynein motors toward the nucleus (retrograde transport), presumably by the interaction of dynein with one or more inner tegument proteins. Kinesin transports the cargo toward the cell membrane (anterograde transport).
After fusion of the HSV-1 envelope with the host plasma membrane, the tegumented capsids containing the viral genome are released into the cytosol. The majority of the outer tegument proteins remain at the plasma membrane along with viral glycoproteins, while inner tegument proteins, such as the UL36, UL37, and US3 proteins remain attached to virion capsids (55, 56). HSV-1 tegument proteins UL36 and UL37 are strong candidates for binding to the dynein motor (57, 58). The HSV-1 UL37 is a 120-kDa phosphorylated tegument protein expressed in both mature virions and light particles (59–62). UL36 (also called VP1/2) is the largest capsid-bound tegument protein encoded by the Herpesviridae, containing 3,164 amino acids (63). The UL36 and UL37 proteins remain with capsids undergoing retrograde transport in neuronal and epithelial cells, while the majority of the other tegument proteins are absent from capsids during transport (36, 64, 65). Also, a lack of either UL36 or UL37 causes failure of retrograde transport of capsids to the nucleus (66). Recent evidence indicates that pseudorabies virus (PRV) UL36 binds to dynein/dynactin, facilitating microtubule transport, neuroinvasion, and pathogenesis (67).
Resolving the structures of membrane proteins is challenging mainly due to difficulties in overexpression, purification, and reassembly of membrane proteins into membrane-mimetic systems for structural analysis. Consequently, <2% of structures available in the Protein Data Bank (PDB) are membrane proteins. Computational methods, such as homology-based modeling, are instrumental in shedding light on the secondary and tertiary structures of membrane proteins (68). Homology modeling is a well-established technique to study protein functions and mechanisms (69, 70). For example, it can be used to investigate the pandemic potential of mutant influenza viruses and rational antiviral drug and vaccine design (71), find important amino acid residues through computer-guided mutations (72), and elucidate protein-protein interactions that are important for host-pathogen interactions (73).
Here, we generated a new model for the predicted three-dimensional (3D) structure of gK by assembling individual domains modeled separately into a full-chain model. This gK model reveals highly conserved domains among alphaherpesviruses, including a β-sheet structure that spans the gKΔ31–68 deletion. We show that the HSV-1(McKrae) gKΔ31–68 virus is unable to enter into neuronal axons in cell culture, in agreement with in vivo ocular infections of mice, which indicated that the virus was not transported into trigeminal ganglionic neurons (35). Based on the conservation of gK predicted domains among alphaherpesviruses, we suggest that gK may function to facilitate entry of other alphaherpesviruses into neurons.
MATERIALS AND METHODS
Cell lines and viruses.
African green monkey kidney (Vero) cells were obtained from the American Type Culture Collection (Manassas, VA) and were maintained in Dulbecco's modified Eagle's medium (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal calf serum and antibiotics.
HSV-1(F), VC1, wild-type (WT) McKrae, McKrae gKΔ31–68, and McKrae D4V5 viruses were used in this study. VC1 was engineered to express gK with the V5 epitope inserted in frame immediately after amino acid 68 of gK and a 3×FLAG epitope inserted in frame at the amino terminus of UL20, as described earlier (26). HSV-1(McKrae ΔgK31–68) was engineered to express gK lacking 38 amino acids immediately after the gK signal sequence (35), and gKΔ31–68-D4V5 (D4V5) was constructed by insertion of the V5 epitope tag at the carboxyl terminus of gK of HSV-1(McKrae gKΔ31–68) using double-red mutagenesis implemented on the viral genome cloned as an bacterial artificial chromosome, as described previously (74).
Virus entry proximity ligation assay (VEPLA).
Proximity ligation assay (PLA) was performed as we have described earlier (75). Briefly, Vero cells were grown on 8-well chamber slides (Nunc Lab-Tek II chamber slide system) and infected with strain F virus at a multiplicity of infection (MOI) of 10. The virus was allowed to attach to confluent monolayer of Vero cells at 4°C for 1 h and shifted to 37°C to allow virus entry, and the chamber slides were removed at time zero, 30 min, 2 h, 3 h, 6 h, 9 h, 12 h, and 16 h and fixed with ice-cold methanol for 10 min at −20°C. Mouse antidynein antibody against intermediate-chain I (Abcam) and rabbit anti-UL37 antibody (a gift from Frank J. Jenkins, University of Pittsburgh Cancer Institute) were used for dynein/UL37 detection. Mouse anti-gD antibody (Virusys, Inc.) and rabbit anti-nectin-1 antibody (Santa Cruz) were used as positive control for gD/nectin-1 interaction in Vero cells. Mouse antidynein antibody and rabbit anti-gM antibody (a gift from Joel Baines, Louisiana State University) were used as a negative control for detecting dynein/gM interaction. Mouse monoclonal antibody against α-tubulin (fluorescein isothiocyanate [FITC]) (Abcam), was used for microtubule detection. Duolink in situ PLA probes, (anti-rabbit plus and anti-mouse minus) were added to the samples (1:5 dilution) and incubated at 37°C for 1 h, followed by ligation at 37°C for 30 min after washing with Duolink in situ wash buffer A. Amplification solution (40 to 50 μl) was added, and slides were incubated for 1.5 h at 37°C. Texas Red-labeled oligonucleotide detection probes (Olink Bioscience) were used. The slides were subsequently washed with Duolink in situ wash buffer B twice for 10 min each and once with 0.1× Duolink buffer B, followed by mounting with mounting medium containing DAPI (4′,6′-diamidino-2-phenylindole) (Duolink II), and stored at −20°C with protection from light until confocal images were taken. The images were taken using a 60× objective on an Olympus Fluoview FV10i confocal laser scanning microscope.
Quantification of PLA signal.
The quantification of the fluorescent signals was done using the SlideBook5 digital microscopy imaging software (Intelligent Imaging Innovation, Denver, CO) (kind help was provided by Masami Yoshimura, Department of Comparative Biomedical Sciences, Louisiana State University). The fluorescent images were imported as TIF files into the SlideBook software. In order to quantify the PLA signals, segment mask was done for both the PLA signals and DAPI-stained nucleus. Both the size (number of pixels) and average intensity of PLA signals were measured. The sum intensity (average intensity × number of pixels) of the PLA signals was divided by the area (in pixels) of the nucleus. This value was calculated for WT McKrae gD/nectin, McKrae gKΔ31–68 gD/nectin-1, WT McKrae UL37/dynein, and gKΔ31–68 UL37/dynein. Unpaired Student t test results showed that the two-tailed P value for WT McKrae and McKrae gKΔ31–68 gD/nectin-1 is 0.0738 (P > 0.05), which is not statistically significant, and the two-tailed P value for WT McKrae UL37/dynein and gKΔ31–68 UL37/dynein is 0.0372 (P < 0.05), which is considered to be statistically significant. The efficiency of virus entry for the data shown in Fig. 7 was calculated as E = number fluorescent spots × intensity (UL37/dynein PLA)/number of spots × intensity (gD/nectin-1 PLA); E (gKΔ31–68)/E (McKrae) = 0.58.
FIG 7.
McKrae gKΔ31–68 virions enter more slowly than parental McKrae virions in Vero cells. VEPLA was utilized to detect wild-type McKrae and gKΔ31–68 entry into Vero cells. Confluent Vero cell monolayers were infected with either McKrae or gKΔ31–68 viruses at an MOI of 10, and capsids that entered the cytoplasm and virions bound to cell surfaces were detected at 1 h postinfection with anti-UL37/antidynein and anti-gD/anti-nectin-1 antibodies, respectively. The PLA signals were quantified using SlideBook 5 digital imaging software. The efficiency of virus entry was calculated as E = number of fluorescent spots × intensity (UL37/dynein PLA)/number of spots × intensity (gD/nectin-1 PLA); E (gKΔ31–68)/E (McKrae) = 0.58.
Infection of DRG neurons in cell culture.
Embryonic day 18 (E18) Sprague-Dawley rat dorsal root ganglia (DRGs) in specialized medium including nerve growth factor (NGF) were obtained from BrainBits Inc. and seeded on poly-d-lysine-coated 8-well culture slides (catalog number 354632; Becton Dickinson, Inc.) per the manufacturer's instructions. Neuronal cell cultures obtained from DRGs were highly enriched in neurons. The DRG is surrounded by a connective tissue capsule and is histologically composed of neuronal cell bodies, which are surrounded by supportive cells (satellite cells). Satellite cells provide electrical insulation for the pseudounipolar neurons in the ganglia. Neuronal extensions of these DRG cultures are practically devoid of DRG-associated fibroblast or glial cells. Consequently, the presence of fibroblast and epithelial cells surrounding neuronal extensions was very sparse, constituting less than one percent of the neuronal axonal projects in any microscope field examined. Cultures contained glial cells, while they were largely devoid of fibroblast and epithelial cells (not shown). Maintenance tissue culture medium consisted of neural basal medium with B-27 supplement at the manufacturer's recommended concentration (Invitrogen, Grand Island, NY). Medium was supplemented with 50 ng/ml neural growth factor 2.5s (Invitrogen), 2% normal rat serum (Invitrogen), 1% GlutaMAX (Invitrogen), and 0.2% Primocin (InvivoGen, San Diego, CA). The ganglia were monitored regularly for axonal growth and cultured with neural basal medium supplemented with neuronal growth factors. A healthy extension of axons was observed at 7 days postseeding, and the ganglia were ready to be infected. The medium was then removed, and 200,000 PFU of either HSV-1(McKrae) or gKΔ31–68 mutant virus was added to the ganglia. The virus was removed after 1 h, and the slides were fixed with ice-cold methanol for 10 min at −20°C. PLA assay was performed on these slides as described above.
Immunogold labeling for TEM.
Purified virions immobilized on 400-mesh Butvar/carbon-coated nickel grids (Electron Microscopy Sciences, Inc., Hatfield, PA) were used to detect the presence of gK and gD on the virions using immunogold labeling, as we have described previously (26). Briefly, the grids were incubated with 5 μl of mouse anti-V5 antibody (Invitrogen) and anti-gD antibody (Virusys) at a dilution of 1:10,000 in 1% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 30 min to detect gK and gD, respectively, followed by 30 min of incubation with 5 μl of goat anti-mouse IgG (whole molecule)–10-nm colloidal gold (Sigma-Aldrich) at a 1:80 dilution in 1% BSA in TBS. A 2% solution of sodium phosphotungstate (pH 6.8) was added as a final step for contrast purposes. Grids were desiccated and visualized by transmission electron microscopy (TEM). The number of virions scanned for each panel ranged from 40 to 65. Not all virions were positive for immune gold particles. On average, 10% of virions did not show the presence of any gold particles.
Virus purification.
The virus was purified as we have described earlier (26). Briefly, supernatants and cells from 10 T-150 flasks of Vero cells infected with YE102-VC1(gK-EK-V5;UL20-3×FLAG), gKΔ31–68, and gKΔ31–68-D4V5 viruses were collected at 36 h postinfection (hpi) and purified by 50 to 20% discontinuous iodixanol gradients twice, followed by a 20% iodixanol cushion. The resulting pellet was resuspended in 250 μl of NP-40 lysis buffer (Invitrogen) and used for immunoblot assay.
Immunoprecipitation and immunoblot assays.
The HSV-1(F) virus-infected lysate was immunoprecipitated with protein G magnetic Dynabeads bound to dynein antibody according to the manufacturer's instructions (Invitrogen). The protein was eluted from the magnetic beads in 40 μl of elution buffer and used for immunoblot assays. Immunoblot assays were carried out using anti-gB (Virusys), anti-UL37, goat anti-mouse–horseradish peroxidase (HRP) (Abcam), and goat anti-rabbit–HRP (Abcam).
Protein modeling of gK.
The gK sequence was divided into domains, and each part was modeled separately. Atomic structures were built using Modeler (76) from template-target alignments calculated by HHpred for transmembrane domains and by eThread (77) for the N terminus and domain II. Subsequently, the individual components were assembled into a full-chain model using Chimera (78) according to the current understanding of the orientation of the domains with respect to each other and the lipid bilayer. Loops and gaps in the alignments were constructed by Modeler. Finally, the model of gK was embedded in a POPC membrane (140 Å by 50 Å) composed of 170 lipid molecules using VMD (79) and PyMOL (PyMOL Molecular Graphics System, version 1.2r3pre; Schrödinger, LLC); the entire system comprises 18,887 nonhydrogen atoms.
RESULTS
Molecular modeling of the effect of the gKΔ31–68 mutation on the predicted structure of gK.
We have reported previously that the gKΔ31–68 domain deletion did not adversely affect infectious virus production, although it inhibited the ability of syncytial mutations in gB to cause extensive virus-induced cell fusion. In contrast, the smaller amino acid deletion gKΔ31–48 and the larger gKΔ31–117 mutation inhibited infectious virus production drastically (30). To better understand the structural aspects of gK, we constructed the 3D structure of gK by modeling individual domains separately, followed by assembly of a full-chain model according to the experimentally derived topography of gK in membranes (16), as described in Materials and Methods. The model revealed that the gKΔ31–68 deletion spanned a well-defined predicted β-sheet structure within the amino terminus of gK that when deleted did not appreciably change the overall structure of the remaining amino terminus of gK. Also, the model predicted a prominent 41-amino-acid alpha-helical domain spanning gK domain II located intracellularly (Fig. 2).
FIG 2.

(A) Predicted structures of gK and gKΔ31–68. Transmembrane and intracellular domains are shown in cyan and green, respectively. (B) The 3D structure predicts a prominent β-sheet structure after the signal sequence. (C) Structure of the amino terminus of gKΔ31–68 (gK without 38 amino acids [aa]).
Glycoprotein gKΔ31–68 is incorporated into virions.
We have shown that gK is expressed on virion particles (32) and that insertion of the V5 epitope tag at the carboxyl terminus of gK, as well as within selected internal sites of gK, did not adversely affect overall viral replication and infectious virus production (16). To detect the presence of gKΔ31–68 within virions, we generated the HSV-1(McKrae) gKΔ31–68-D4V5 (D4V5) mutant virus expressing a V5 epitope tag inserted at the carboxyl terminus of gK using double-red mutagenesis, as we have described previously (35) (see Materials and Methods). The presence of gK on virion particles was initially assessed using colloidal gold immunoelectron microscopy (Fig. 3A). An average of 50 virion particles for each virus were inspected for the presence of gold particles. V5-tagged gK was readily detected on most virion envelopes (>45 out of 50 visualized virions contained gold particles) for recombinant viruses VC1 (containing a V5 epitope tag within the amino terminus of gK) and D4V5, while none of the gKΔ31–68 virions contained gold particles. Similarly, the anti-gD monoclonal antibody readily detected gD on all virions (Fig. 3A). The presence of gK on virions was confirmed in immunoblots of extracts from purified VC1 and D4V5 virion extracts probed with anti-V5 antibody. VC1 gK protein species migrated with apparent molecular masses ranging from 38 to 55 kDa, while D4V5 gK protein species migrated with apparent molecular masses ranging from 45 to 55 kDa. There were no gK protein species detected in HSV-1(McKrae) gKΔ31–68 virion extracts (Fig. 3B). The V5-tagged gK was also detected on the surface of infected cells via fluorescence-activated cell sorting (FACS) (not shown).
FIG 3.

Detection of gKΔ31–68 on virion particles. (A) VC1(F), McKrae gKΔ31–68, and McKrae D4V5 extracts from purified virions were reacted with anti-V5 antibody to detect V5-tagged gKs and visualized by immunogold transmission electron microscopy. Approximately 50 virion images were scanned for the presence of immunogold particles on virion envelopes. Detection of gD was used for positive-control purposes, while the parent gKΔ31–68 without a V5 tag on its gK was used as negative control. (B) Extracts from purified virions were electrophoretically separated and immunoblotted with anti-V5 antibody to detect V5-tagged gKs. The presence of gK species was detected in both VC1 (tagged with V5 within the amino terminus of gK) and D4V5 (tagged with V5 at its carboxyl terminus), while no gK was detected in the untagged parental virus gKΔ31–68.
The UL37 protein interacts with cytoplasmic dynein.
UL36 and UL37 are associated with capsids undergoing retrograde transport in neuronal and epithelial cells. To test whether the UL37 protein interacts with dynein, cell extracts from infected cells were immunoprecipitated with antidynein antibody, and the samples were immunoblotted and probed with both anti-UL37 and anti-gB antibodies. Dynein immunoprecipitates contained the UL37 protein, detected as a protein species migrating with molecular mass of 120 kDa (Fig. 4, band marked with an asterisk), while dynein did not coprecipitate gB, which was used as a negative control. Additional negative controls included lysates from uninfected Vero cells (Fig. 4).
FIG 4.
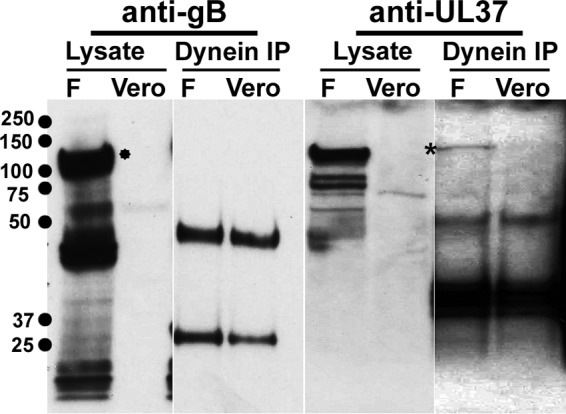
Dynein interacts with UL37. Lysates from cells infected with HSV-1(F) were subjected to immunoprecipitation as described in Materials and Methods. Negative controls included detection of gB (*) in dynein immunoprecipitates and uninfected cell extracts.
Detection of UL37 interaction with cytoplasmic dynein and development of the VEPLA.
Cell surface-bound virions were detected by monitoring interactions of virion gD with its cognate receptor nectin-1 using PLA. Similarly, PLA was utilized to detect UL37-dynein interactions to monitor viral capsids that have entered the cytoplasm and bound dynein as the prerequisite step for microtubular loading and retrograde transport (Fig. 5). Bright red fluorescent signals were obtained when PLA detected colocalization of gD with nectin-1 immediately after adsorption of the virus for 1 h at 4°C (zero time point) on Vero cells. In contrast, PLA with antibodies against dynein and UL37 did not detect colocalization of UL37 and dynein at the zero time point (Fig. 6). UL37-dynein colocalization was readily visible as early as 30 min after virus entry, visualized as numerous bright red fluorescent spots in the cytoplasm of infected cells. Glycoprotein M (gM) did not colocalize with dynein at any time point tested (negative control). UL37-dynein colocalization was detected at all times postinfection tested except the zero time point. The distribution and density of fluorescent spots revealing colocalization of UL37 with dynein changed at 6 hpi, assuming a perinuclear distribution at 9 hpi. The overall number and intensity of fluorescent spots dissipated after 12 hpi, and they were undetected at 16 hpi (Fig. 6).
FIG 5.

Schematic representation of the virus entry proximity ligation assay (VEPLA). Antibodies to UL37 and dynein are reacted with secondary antibodies linked to proximity probe primers, that after annealing to connector primers and amplification by a rolling-circle mechanism are detected by a DNA-intercalating fluorescent dye. A similar strategy was used to detect gD binding to nectin-1, representing virions attached to cell surfaces (not shown).
FIG 6.
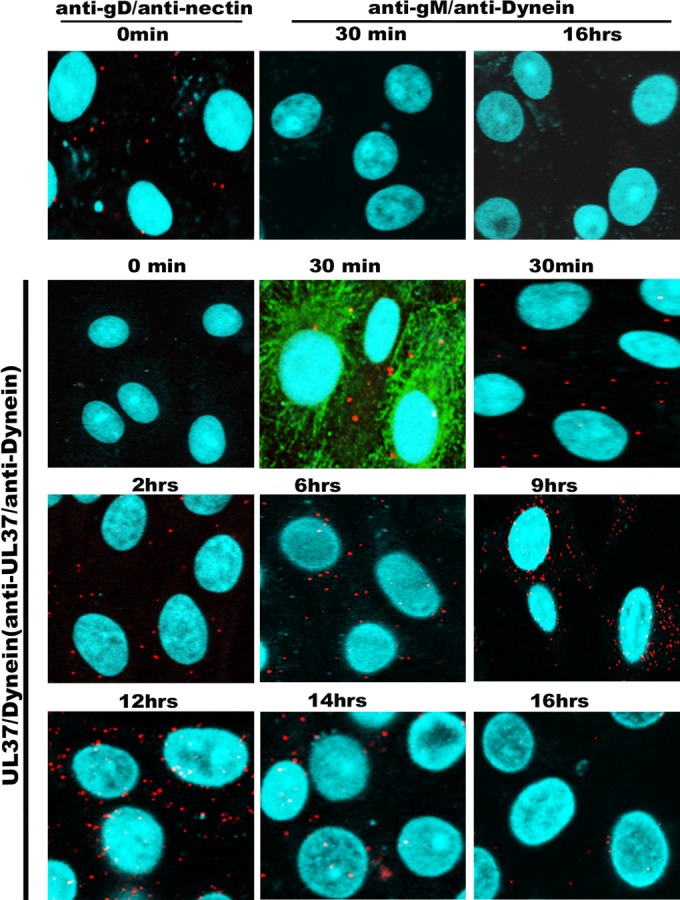
Kinetics of virus entry into Vero cells. Confluent monolayers of Vero cells seeded in microscopy chamber slides were infected at an MOI of 10 with wild-type virus HSV-1(F) and tested by VEPLA for cell surface-bound virions (anti-gD/anti-nectin-1 antibodies) and cytoplasmic capsids at different times postinfection at 37°C (anti-UL37/antidynein). Anti-gM and antidynein antibodies were utilized as negative controls. The microtubules were visualized using mouse monoclonal antibody against α-tubulin (FITC), shown in green. DAPI (blue) was used for visualization of the nucleus, and the PLA signals were seen as red spots.
We have shown previously that the gKΔ31–68 virus enters Vero cells with slower entry kinetics than the wild-type virus (26, 32). Virus entry proximity ligation assay (VEPLA) performed at 1 hpi revealed that both the wild-type virus and the gKΔ31–68 virus attached equally well to Vero cell surfaces. In agreement with previous findings, VEPLA revealed a marked reduction in gKΔ31–68 virion entry in comparison to that of the wild-type virus, as evidenced by the significant reduction in the number and relative intensity of fluorescent spots produced by colocalization of UL37 with dynein. The efficiency of virus entry was calculated as E = number fluorescent spots × intensity (UL37/dynein PLA)/number of spots × intensity (gD/nectin-1 PLA); E (gKΔ31–68)/E (McKrae) = 0.58 (Fig. 7).
The amino terminus of HSV-1 gK is required for the entry of the virus into the DRG axons.
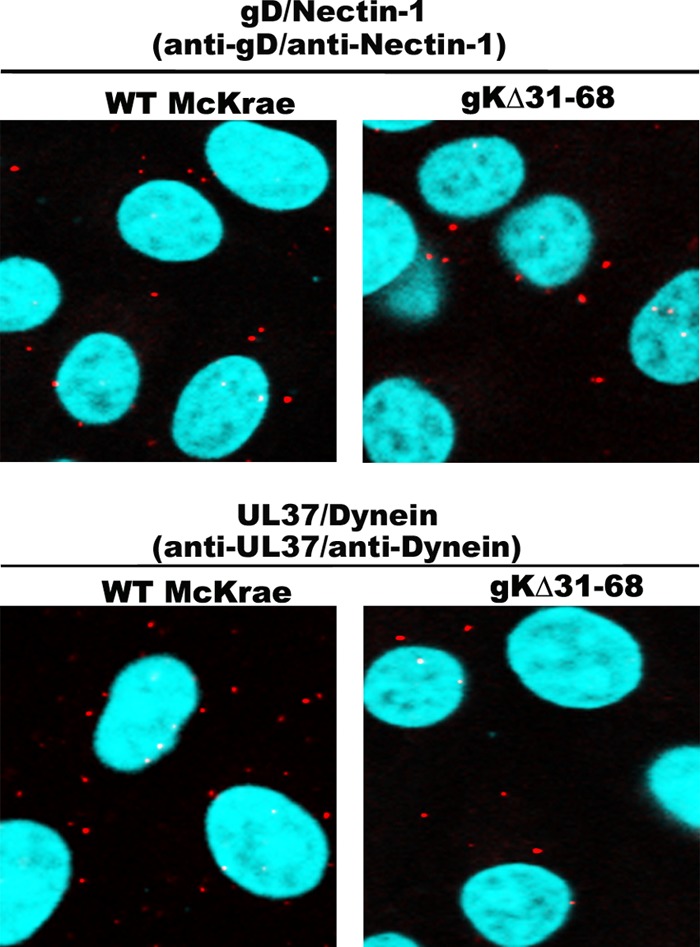
Recently, we showed that gK, specifically the amino-terminal 38 amino acids of gK, which are deleted in the gKΔ31–68 mutant virus, is required for the replication and efficient spread of the virus to the trigeminal ganglion after ocular infection of mice (8, 34). In addition, lack of gK caused an inability of the virus to infect axonal termini separated from neuronal somata in specialized microfluidic devices harboring purified ganglionic neurons (8). To directly test the ability of the gKΔ31–68 virus to enter axonal termini of ganglionic rat neurons in cell culture, purified rat dorsal root ganglia (DRGs) seeded on fluorescence microscopy slides were infected with either the HSV-1(McKrae) virus originally isolated from patients suffering from keratitis (80) or the gKΔ31–68 virus, and VEPLA was performed at 1 h postinfection (hpi). Both viruses attached to neuronal surfaces equally well, as evidenced by the number of fluorescent spots produced by colocalization of viral gD with the nectin-1 receptor. In contrast, there were no fluorescent spots detected on neuronal axons infected with the gKΔ31–68 virus, while numerous spots were detected on neuronal axons infected with the McKrae wild-type strain. Both viruses appeared to infect equally well glia cells surrounding neuronal axons, as well as neuronal somata (Fig. 8).
FIG 8.
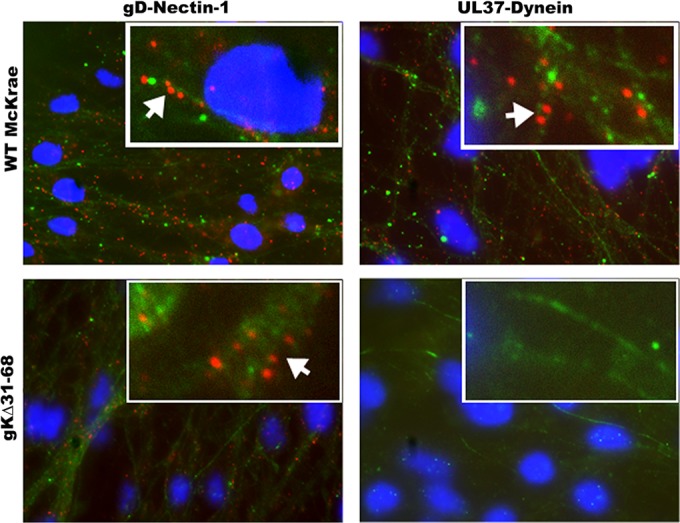
The McKrae gKΔ31–68 mutant virus is unable to enter DRG axons. VEPLA was utilized to asses McKrae and gKΔ31–68 virus entry into DRG neurons in cell culture. Embryonic day 18 (E8) rat DRGs were collected and seeded on polylysine-coated microscopy slides. DRGs were infected with WT HSV-1 strain McKrae or gKΔ31–68 at 6 days postseeding. The top and bottom left panels show interaction between gD and nectin-1 on DRG axons infected with WT McKrae and gKΔ31–68 mutant viruses, respectively, as seen by the presence of red spots along the axons (arrows). The top right panel shows the interaction between UL37 and dynein in DRG axons infected with WT McKrae virus. There was no interaction detected between UL37 and dynein in DRG axons infected with gKΔ31–68 (bottom right panel). Neurofilament marker (green) and DAPI (blue) were used to identify axons and the nuclei of glial cells, respectively.
DISCUSSION
Viral glycoprotein K (gK) and its interacting partner UL20 are highly conserved among all neurotropic alphaherpesviruses, while beta- and gammaherpesviruses do not specify gK or UL20 orthologues. Therefore, our working hypothesis is that gK and UL20 function to facilitate successful infection of neurons. Here, we show for the first time that a deletion of a predicted β-sheet structure within the amino terminus of HSV-1 gK, conserved among alphaherpesviruses, prevents the virus from entering into ganglionic axons, in agreement with previous findings that this gK deletion caused the inability of mutant viruses to infect ganglionic neurons and establish latency after ocular infection of mice (35).
We generated a new model for the predicted three-dimensional structure of gK by assembling individual domains modeled separately into a full-chain model. This methodology has been successfully applied to derive protein models of other viral membrane proteins. For example, modeling of the structure of the H7N9 hemagglutinin (HA) helped predict the potential of the Chinese A/Hangzhou/1/2013 strain by predicting that the H7H9 strain could bind to human sialic acid receptors. The accuracy of the results of this study was confirmed after the crystal structure of the H7N9 HA protein was resolved later, leading to an almost identical structure with a root mean square deviation (RMSD) of only 0.721 Å over 305 atoms, while in vitro results showed binding capabilities similar to the predictions (71). Importantly, computer-generated protein models can be used not only to predict the overall function of a protein but also to elucidate the molecular mechanism of such function. For example, an X-ray structure of a four-helix bundle in the H protein in parainfluenza virus type 5 (PIV5) was used to model the H protein in the stalk region of the canine distemper virus to investigate the conformational changes in viral fusion (F) protein required for cell entry. Combining in vitro experiments and computational structure modeling ultimately led to the discovery of a new model for activation of the fusion machinery utilized by morbilliviruses (73).
To overcome the difficulty in predicting the three-dimensional structures of multiple membrane-spanning proteins such as gK and UL20, we predicted the structures of each gK domain (I to IV) separately and assembled them into a full-chain structure. This gK model produced a number of important structural features, including a prominent β-sheet structure spanning the gKΔ31–68 deletion in the amino terminus of gK and a 41-amino-acid α-helical structure spanning gK domain II located intracellularly (Fig. 2). These predicted domains are conserved among alphaherpesviruses such as HSV-2 and others (Fig. 9 and unpublished data). Syncytial mutations within the carboxyl terminus of gB do not cause fusion in the presence of the gKΔ31–68 deletion (30). In addition, we have shown that the amino-terminal 82-amino-acid domain of gK when expressed separately binds gB and complements the inability of the gKΔ31–68 mutation to support gB-mediated membrane fusion. These results suggest that gB-mediated membrane fusion is regulated by interactions between the amino termini of gB and gK (30, 31).
FIG 9.

Prediction of the three-dimensional structures of the amino termini of gK specified by HSV-1, HSV-2, VZV, and monkey B virus. The distal side of the amino terminus of gK is colored yellow and is retained for comparison purposes as background on all other frames. (A) HSV-1 gK. (B) HSV-2 gK. (C) VZV gK. (D) monkey B virus gK.
We have shown previously that gK is incorporated into virions and functions in virus entry (26, 32). Furthermore, the gKΔ31–68 deletion did not negatively affect infectious virus production (30). Similarly, the D4V5 virus replicated efficiently in Vero cells, achieving titers at 24 hpi that were similar to those of the parental gKΔ31–68 virus (not shown). The gKΔ31–68 deletion includes two N-glycosylation sites located within the amino terminus of gK. The D4V5 gKΔ31–68 gK migrated with a molecular mass of 45 to 55 kDa despite lacking both N-glycosylation sites. It has been shown that gK migrates anomalously in SDS-PAGE due to its high hydrophobicity and ability to multimerize (16, 18, 23). Characteristically, boiling of gK SDS-PAGE samples prevents gK from entering into gels. Heating up to 45°C for 15 min is typically utilized to allow gK to enter into separating gels. Therefore, it is likely that the observed gKΔ31–68 molecular mass range of 45 to 55 kDa is due to the overall gKΔ31–68 structure, which causes anomalous migration in SDS-PAGE.
Biochemical analysis and live imaging have strongly suggested that the viral VP1/2 (UL36) and UL37 proteins interact with dynein (66). In agreement with these findings, we detected interactions of UL37 with dynein via PLA. PLA is a relatively simple tool to detect potential protein-protein interactions. This technique has been utilized for detecting infectious agents and proteins with a sensitivity rated higher than even that of PCR (81). Interestingly, PLA detected UL37-dynein interactions throughout the course of infection of Vero cells. It is possible that UL37 may bind dynein to prevent dynein interference and ensure efficient utilization of kinesin-mediated anterograde transport during virion cytoplasmic envelopment and egress.
Most virus entry assays depend on the expression of viral proteins after deposition of viral DNA into the nuclei of infected cells through detection of either viral antigens or marker genes, such as green fluorescence protein expressed from the viral genome, or the induction of marker genes such as β-galactosidase under HSV-1 promoter control responding to expression of HSV-1 immediate early proteins (33, 82, 83). However, these assays measure the overall successful rate of infection and not necessarily the ability of virions to enter into the cytoplasm of infected cells, since entering capsids may not be efficiently transported to the nucleus. Detection of capsids in the cytoplasm of infected cells can be achieved by high-power confocal microscopy and transmission electron microscopy, with great difficulty in obtaining semiquantitative results. We describe here the development of the virus entry proximity ligation assay (VEPLA) as a general method to efficiently visualize and quantify the relative efficiency of virus entry into the cytoplasm of infected cells in a time-dependent manner by monitoring both enveloped virions attached to cell surfaces and capsids in the cytoplasm that interact with dynein prior to loading onto the microtubular network for retrograde transport.
VEPLA was utilized to show that the gKΔ31–68 mutation prevented virions from entering into neuronal axons, in agreement with our published results that this mutation caused the inability to infect ganglionic neurons after ocular infection of mice (35). It is likely that inhibition of virus entry reflects the inability of gB to cause fusion of viral envelopes with axonal membranes in the presence of the gKΔ31–68 mutation. This is supported by our previous results that the amino terminus of gK spanning the gKΔ31–68 mutation interacts with the amino terminus of gB (30). Alternatively, the amino terminus of gK may enable binding of virions to gK-specific receptors that are required for virus entry. HSV-1 and HSV-2, as well as all alphaherpesviruses, infect neuronal endings embedded into the epidermis and mucosal surfaces, including the highly innervated corneal epithelium. Prediction of the three-dimensional structures of gK specified by alphaherpesviruses shows remarkable primary and predicted tertiary structures (unpublished data). Of particular interest to this study is conservation of predicted structural domains within the amino termini of gK specified by HSV-1, HSV-2, varicella-zoster virus (VZV), and monkey B virus (Fig. 9). The β-sheet structure contained within the gKΔ31–68 deletion is conserved among these viruses as well as other alphaherpesviruses (not shown), suggesting that they may play a conserved role in infection of neuronal axons.
Understanding of the role of gK in neuronal entry will enable the production of new ways to ameliorate these infections, as well as assist in the development of safe live-attenuated vaccines and viral vectors to combat HSV and other infectious diseases. In support of this prediction, we have recently shown that the HSV-1 (VC2) vaccine strain containing the gKΔ31–68 mutation protected mice against lethal intravaginal challenge with either virulent HSV-1 or HSV-2 in mice (84).
ACKNOWLEDGMENTS
We acknowledge assistance provided by undergraduate students and administrative staff of the Division of Biotechnology and Molecular Medicine and the Department of Biological Sciences, College of Basic Sciences, and the assistance of Yeeun Kim from the Department of Biological Sciences with the schematic representations.
Funding Statement
The funders had no role in study design, data collection and interpretation, or decision to submit the work for publication.
REFERENCES
- 1.Cai WH, Gu B, Person S. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol 62:2596–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desai PJ, Schaffer PA, Minson AC. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J Gen Virol 69:1147–1156. doi: 10.1099/0022-1317-69-6-1147. [DOI] [PubMed] [Google Scholar]
- 3.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol 65:1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 6.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. doi: 10.1016/S0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 7.Chowdhury S, Naderi M, Chouljenko VN, Walker JD, Kousoulas KG. 2012. Amino acid differences in glycoproteins B (gB), C (gC), H (gH) and L (gL) are associated with enhanced herpes simplex virus type-1 (McKrae) entry via the paired immunoglobulin-like type-2 receptor alpha. Virol J 9:112. doi: 10.1186/1743-422X-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.David AT, Saied A, Charles A, Subramanian R, Chouljenko VN, Kousoulas KG. 2012. A herpes simplex virus 1 (McKrae) mutant lacking the glycoprotein K gene is unable to infect via neuronal axons and egress from neuronal cell bodies. mBio 3:e00144–12. doi: 10.1128/mBio.00144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicola AV, Hou J, Major EO, Straus SE. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol 79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qie L, Marcellino D, Herold BC. 1999. Herpes simplex virus entry is associated with tyrosine phosphorylation of cellular proteins. Virology 256:220–227. doi: 10.1006/viro.1999.9673. [DOI] [PubMed] [Google Scholar]
- 11.Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol 79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debroy C, Pederson N, Person S. 1985. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 145:36–48. doi: 10.1016/0042-6822(85)90199-0. [DOI] [PubMed] [Google Scholar]
- 14.MacLean CA, Efstathiou S, Elliott ML, Jamieson FE, McGeoch DJ. 1991. Investigation of herpes simplex virus type 1 genes encoding multiply inserted membrane proteins. J Gen Virol 72:897–906. doi: 10.1099/0022-1317-72-4-897. [DOI] [PubMed] [Google Scholar]
- 15.Ramaswamy R, Holland TC. 1992. In vitro characterization of the HSV-1 UL53 gene product. Virology 186:579–587. doi: 10.1016/0042-6822(92)90024-J. [DOI] [PubMed] [Google Scholar]
- 16.Foster TP, Alvarez X, Kousoulas KG. 2003. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J Virol 77:499–510. doi: 10.1128/JVI.77.1.499-510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melancon JM, Foster TP, Kousoulas KG. 2004. Genetic analysis of the herpes simplex virus type 1 (HSV-1) UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J Virol 78:7329–7343. doi: 10.1128/JVI.78.14.7329-7343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutchinson L, Goldsmith K, Snoddy D, Ghosh H, Graham FL, Johnson DC. 1992. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J Virol 66:5603–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dietz P, Klupp BG, Fuchs W, Kollner B, Weiland E, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein K requires the UL20 gene product for processing. J Virol 74:5083–5090. doi: 10.1128/JVI.74.11.5083-5090.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster TP, Melancon JM, Baines JD, Kousoulas KG. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J Virol 78:5347–5357. doi: 10.1128/JVI.78.10.5347-5357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 1997. The UL20 gene product of pseudorabies virus functions in virus egress. J Virol 71:5639–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melancon JM, Fulmer PA, Kousoulas KG. 2007. The herpes simplex virus UL20 protein functions in glycoprotein K (gK) intracellular transport and virus-induced cell fusion are independent of UL20 functions in cytoplasmic virion envelopment. Virol J 4:120. doi: 10.1186/1743-422X-4-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster TP, Chouljenko VN, Kousoulas KG. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J Virol 82:6310–6323. doi: 10.1128/JVI.00147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster TP, Melancon JM, Kousoulas KG. 2001. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18–29. doi: 10.1006/viro.2001.1004. [DOI] [PubMed] [Google Scholar]
- 25.Foster TP, Melancon JM, Olivier TL, Kousoulas KG. 2004. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J Virol 78:13262–13277. doi: 10.1128/JVI.78.23.13262-13277.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jambunathan N, Chowdhury S, Subramanian R, Chouljenko VN, Walker JD, Kousoulas KG. 2011. Site-specific proteolytic cleavage of the amino terminus of herpes simplex virus glycoprotein K on virion particles inhibits virus entry. J Virol 85:12910–12918. doi: 10.1128/JVI.06268-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster TP, Kousoulas KG. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J Virol 73:8457–8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutchinson L, Roop-Beauchamp C, Johnson DC. 1995. Herpes simplex virus glycoprotein K is known to influence fusion of infected cells, yet is not on the cell surface. J Virol 69:4556–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jayachandra S, Baghian A, Kousoulas KG. 1997. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J Virol 71:5012–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chouljenko VN, Iyer AV, Chowdhury S, Chouljenko DV, Kousoulas KG. 2009. The amino terminus of herpes simplex virus type 1 glycoprotein K (gK) modulates gB-mediated virus-induced cell fusion and virion egress. J Virol 83:12301–12313. doi: 10.1128/JVI.01329-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J Virol 84:8596–8606. doi: 10.1128/JVI.00298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foster TP, Rybachuk GV, Kousoulas KG. 2001. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J Virol 75:12431–12438. doi: 10.1128/JVI.75.24.12431-12438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chowdhury S, Chouljenko VN, Naderi M, Kousoulas KG. 2013. The amino terminus of herpes simplex virus 1 glycoprotein K is required for virion entry via the paired immunoglobulin-like type-2 receptor alpha. J Virol 87:3305–3313. doi: 10.1128/JVI.02982-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.David AT, Baghian A, Foster TP, Chouljenko VN, Kousoulas KG. 2008. The herpes simplex virus type 1 (HSV-1) glycoprotein K (gK) is essential for viral corneal spread and neuroinvasiveness. Curr Eye Res 33:455–467. doi: 10.1080/02713680802130362. [DOI] [PubMed] [Google Scholar]
- 35.Saied AA, Chouljenko VN, Subramanian R, Kousoulas KG. 2014. A replication competent HSV-1(McKrae) with a mutation in the amino-terminus of glycoprotein K (gK) is unable to infect mouse trigeminal ganglia after cornea infection. Curr Eye Res 39:596–603. doi: 10.3109/02713683.2013.855238. [DOI] [PubMed] [Google Scholar]
- 36.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci U S A 102:5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greber UF, Way M. 2006. A superhighway to virus infection. Cell 124:741–754. doi: 10.1016/j.cell.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 38.Hammonds TR, Denyer SP, Jackson DE, Irving WL. 1996. Studies to show that with podophyllotoxin the early replicative stages of herpes simplex virus type 1 depend upon functional cytoplasmic microtubules. J Med Microbiol 45:167–172. doi: 10.1099/00222615-45-3-167. [DOI] [PubMed] [Google Scholar]
- 39.Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol 136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dohner K, Nagel CH, Sodeik B. 2005. Viral stop-and-go along microtubules: taking a ride with dynein and kinesins. Trends Microbiol 13:320–327. doi: 10.1016/j.tim.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Kramer T, Enquist LW. 2013. Directional spread of alphaherpesviruses in the nervous system. Viruses 5:678–707. doi: 10.3390/v5020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res 143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 43.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 44.Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. 2008. Transport and egress of herpes simplex virus in neurons. Rev Med Virol 18:35–51. doi: 10.1002/rmv.560. [DOI] [PubMed] [Google Scholar]
- 45.Dohner K, Wolfstein A, Prank U, Echeverri C, Dujardin D, Vallee R, Sodeik B. 2002. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol Biol Cell 13:2795–2809. doi: 10.1091/mbc.01-07-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leopold PL, Kreitzer G, Miyazawa N, Rempel S, Pfister KK, Rodriguez-Boulan E, Crystal RG. 2000. Dynein- and microtubule-mediated translocation of adenovirus serotype 5 occurs after endosomal lysis. Hum Gene Ther 11:151–165. doi: 10.1089/10430340050016238. [DOI] [PubMed] [Google Scholar]
- 47.Suomalainen M, Nakano MY, Keller S, Boucke K, Stidwill RP, Greber UF. 1999. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol 144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanderson CM, Hollinshead M, Smith GL. 2000. The vaccinia virus A27L protein is needed for the microtubule-dependent transport of intracellular mature virus particles. J Gen Virol 81:47–58. doi: 10.1099/0022-1317-81-1-47. [DOI] [PubMed] [Google Scholar]
- 49.Alonso C, Miskin J, Hernaez B, Fernandez-Zapatero P, Soto L, Canto C, Rodriguez-Crespo I, Dixon L, Escribano JM. 2001. African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light-chain dynein. J Virol 75:9819–9827. doi: 10.1128/JVI.75.20.9819-9827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suikkanen S, Aaltonen T, Nevalainen M, Valilehto O, Lindholm L, Vuento M, Vihinen-Ranta M. 2003. Exploitation of microtubule cytoskeleton and dynein during parvoviral traffic toward the nucleus. J Virol 77:10270–10279. doi: 10.1128/JVI.77.19.10270-10279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. 2003. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U S A 100:9280–9285. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petit C, Giron ML, Tobaly-Tapiero J, Bittoun P, Real E, Jacob Y, Tordo N, De The H, Saib A. 2003. Targeting of incoming retroviral Gag to the centrosome involves a direct interaction with the dynein light chain 8. J Cell Sci 116:3433–3442. doi: 10.1242/jcs.00613. [DOI] [PubMed] [Google Scholar]
- 53.Sfakianos JN, LaCasse RA, Hunter E. 2003. The M-PMV cytoplasmic targeting-retention signal directs nascent Gag polypeptides to a pericentriolar region of the cell. Traffic 4:660–670. doi: 10.1034/j.1600-0854.2003.00125.x. [DOI] [PubMed] [Google Scholar]
- 54.Raux H, Flamand A, Blondel D. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J Virol 74:10212–10216. doi: 10.1128/JVI.74.21.10212-10216.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maurer UE, Sodeik B, Grunewald K. 2008. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc Natl Acad Sci U S A 105:10559–10564. doi: 10.1073/pnas.0801674105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J Virol 79:3200–3205. doi: 10.1128/JVI.79.5.3200-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radtke K, Dohner K, Sodeik B. 2006. Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cell Microbiol 8:387–400. doi: 10.1111/j.1462-5822.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- 58.Antinone SE, Shubeita GT, Coller KE, Lee JI, Haverlock-Moyns S, Gross SP, Smith GA. 2006. The herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J Virol 80:5494–5498. doi: 10.1128/JVI.00026-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Albright AG, Jenkins FJ. 1993. The herpes simplex virus UL37 protein is phosphorylated in infected cells. J Virol 67:4842–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol 81:11790–11797. doi: 10.1128/JVI.01113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J Virol 83:6610–6623. doi: 10.1128/JVI.02655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J Virol 83:105–116. doi: 10.1128/JVI.01032-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibson W, Roizman B. 1972. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J Virol 10:1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Copeland AM, Newcomb WW, Brown JC. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J Virol 83:1660–1668. doi: 10.1128/JVI.01139-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Antinone SE, Smith GA. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J Virol 84:1504–1512. doi: 10.1128/JVI.02029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6:e1000991. doi: 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koehler Leman J, Ulmschneider MB, Gray JJ. 2015. Computational modeling of membrane proteins. Proteins 83:1–24. doi: 10.1002/prot.24703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramachandran S, Dokholyan N. 2012. Homology modeling: generating structural models to understand protein function and mechanism, p 97–116. In Dokholyan NV. (ed), Computational modeling of biological systems. Springer-Verlag, New York, NY. [Google Scholar]
- 70.Nayeem A, Sitkoff D, Krystek S. 2006. A comparative study of available software for high-accuracy homology modeling: from sequence alignments to structural models. Protein Sci 15:808–824. doi: 10.1110/ps.051892906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rajapaksha H, Petrovsky N. 2014. In silico structural homology modelling and docking for assessment of pandemic potential of a novel H7N9 influenza virus and its ability to be neutralized by existing anti-hemagglutinin antibodies. PLoS One 9:e102618. doi: 10.1371/journal.pone.0102618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morita E, Sandrin V, McCullough J, Katsuyama A, Baci Hamilton I, Sundquist Wesley I. 2011. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 9:235–242. doi: 10.1016/j.chom.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ader N, Brindley MA, Avila M, Origgi FC, Langedijk JPM, Örvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J Biol Chem 287:16324–16334. doi: 10.1074/jbc.M112.342493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 75.Jambunathan N, Chouljenko D, Desai P, Charles AS, Subramanian R, Chouljenko VN, Kousoulas KG. 2014. Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J Virol 88:5927–5935. doi: 10.1128/JVI.00278-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fiser A, Sali A. 2003. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 77.Brylinski M, Lingam D. 2012. eThread: a highly optimized machine learning-based approach to meta-threading and the modeling of protein tertiary structures. PLoS One 7:e50200. doi: 10.1371/journal.pone.0050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 79.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 80.Wang H, Davido DJ, Morrison LA. 2013. HSV-1 strain McKrae is more neuroinvasive than HSV-1 KOS after corneal or vaginal inoculation in mice. Virus Res 173:436–440. doi: 10.1016/j.virusres.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gustafsdottir SM, Nordengrahn A, Fredriksson S, Wallgren P, Rivera E, Schallmeiner E, Merza M, Landegren U. 2006. Detection of individual microbial pathogens by proximity ligation. Clin Chem 52:1152–1160. doi: 10.1373/clinchem.2005.065847. [DOI] [PubMed] [Google Scholar]
- 82.Simpson SA, Manchak MD, Hager EJ, Krummenacher C, Whitbeck JC, Levin MJ, Freed CR, Wilcox CL, Cohen GH, Eisenberg RJ, Pizer LI. 2005. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J Neurovirol 11:208–218. doi: 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- 83.Reinhard H, Le VT, Ohlin M, Hengel H, Trilling M. 2011. Exploitation of herpesviral transactivation allows quantitative reporter gene-based assessment of virus entry and neutralization. PLoS One 6:e14532. doi: 10.1371/journal.pone.0014532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stanfield BA, Stahl J, Chouljenko VN, Subramanian R, Charles AS, Saied AA, Walker JD, Kousoulas KG. 2014. A single intramuscular vaccination of mice with the HSV-1 VC2 virus with mutations in the glycoprotein K and the membrane protein UL20 confers full protection against lethal intravaginal challenge with virulent HSV-1 and HSV-2 strains. PLoS One 9:e109890. doi: 10.1371/journal.pone.0109890. [DOI] [PMC free article] [PubMed] [Google Scholar]