

ABSTRACT
High-risk human papillomavirus 31 (HPV31)-positive cells exhibit constitutive activation of the ATM-dependent DNA damage response (DDR), which is necessary for productive viral replication. In response to DNA double-strand breaks (DSBs), ATM activation leads to DNA repair through homologous recombination (HR), which requires the principal recombinase protein Rad51, as well as BRCA1. Previous studies from our lab demonstrated that Rad51 and BRCA1 are expressed at high levels in HPV31-positive cells and localize to sites of viral replication. These results suggest that HPV may utilize ATM activity to increase HR activity as a means to facilitate viral replication. In this study, we demonstrate that high-risk HPV E7 expression alone is sufficient for the increase in Rad51 and BRCA1 protein levels. We have found that this increase occurs, at least in part, at the level of transcription. Studies analyzing protein stability indicate that HPV may also protect Rad51 and BRCA1 from turnover, contributing to the overall increase in cellular levels. We also demonstrate that Rad51 is bound to HPV31 genomes, with binding increasing per viral genome upon productive replication. We have found that depletion of Rad51 and BRCA1, as well as inhibition of Rad51's recombinase activity, abrogates productive viral replication upon differentiation. Overall, these results indicate that Rad51 and BRCA1 are required for the process of HPV31 genome amplification and suggest that productive replication occurs in a manner dependent upon recombination.
IMPORTANCE Productive replication of HPV31 requires activation of an ATM-dependent DNA damage response, though how ATM activity contributes to replication is unclear. Rad51 and BRCA1 play essential roles in repair of double-strand breaks, as well as the restart of stalled replication forks through homologous recombination (HR). Given that ATM activity is required to initiate HR repair, coupled with the requirement of Rad51 and BRCA1 for productive viral replication, our findings suggest that HPV may utilize ATM activity to ensure localization of recombination factors to productively replicating viral genomes. The finding that E7 increases the levels of Rad51 and BRCA1 suggests that E7 contributes to productive replication by providing DNA repair factors required for viral DNA synthesis. Our studies not only imply a role for recombination in the regulation of productive HPV replication but provide further insight into how HPV manipulates the DDR to facilitate the productive phase of the viral life cycle.
INTRODUCTION
Human papillomaviruses (HPVs) are small double-stranded DNA viruses approximately 8 kb in size that exhibit a preferential tropism for epithelial cells. High-risk mucosal HPV subtypes are the causative agents of cervical cancer and have been increasingly associated with anogenital, oropharyngeal, and head and neck cancers (1). The life cycle of HPV is intimately linked to the differentiation of its host cell, the keratinocyte (2). After exposure through a microwound in the stratified epithelium, HPV infects the actively dividing basal cells. Upon infection, viral genomes are amplified transiently to 50 to 100 copies per cell, which are subsequently maintained by replicating once per cell cycle, along with cellular DNA. As infected daughter cells migrate out of the basal stratum into the suprabasal cell layers to undergo differentiation, expression of viral E7 and E6 proteins prevents the normal exit from the cell cycle and promotes reentry of infected cells into S phase, providing a cellular environment conducive for viral DNA synthesis. Upon differentiation, the productive phase of the viral life cycle is induced, resulting in amplification of viral genomes to thousands of copies per cell, late gene expression, and virion assembly and release from the outermost surface of the epithelium (3).
Previous studies demonstrated that high-risk HPV31 promotes the constitutive activation of an ATM (ataxia telangiectasia-mutated kinase)-dependent DNA damage response and that ATM activity is necessary for productive viral replication (4). Activation of ATM is instrumental in the cellular response to certain types of genomic damage, particularly DNA double-strand breaks (DSBs), one of the most harmful types of DNA lesions if left unrepaired (5, 6). Phosphorylation of ATM sets in motion signaling events that temporarily stop progression of the cell cycle, activate downstream repair factors, and, if necessary, initiate apoptosis (7). Earlier studies demonstrated that although ATM kinase activity is critical for productive amplification of HPV31 genomes, episomal maintenance is not affected with inhibition of ATM in undifferentiated cells (4). These studies suggest that HPV induces ATM activation specifically for productive replication, although how HPV utilizes this activity for viral replication is unclear.
Previous studies by our lab and others demonstrated the recruitment of ATM-dependent DNA damage response factors (γH2AX, Chk2, 53BP1, MRN complex [Mre11, Rad50, Nbs1]) to sites of HPV DNA synthesis (8, 9). Furthermore, increased levels of the homologous recombination (HR) proteins Rad51 and BRCA1, as well as their colocalization to sites of viral DNA replication, were observed (8). Coupled with the detection of the DNA damage marker γH2AX and phosphorylated RPA32, a marker of DNA damage and/or perturbed replication (10–12), at HPV replication foci (8), these results suggest that HPV could utilize the ATM DNA damage response pathway to upregulate, activate, and recruit DNA repair factors to viral genomes to promote amplification through DNA repair mechanisms.
Restoration of proper cellular genomic structure from DNA double-strand breaks is facilitated through two principal pathways of repair that function at different stages of the cell cycle. Nonhomologous end joining (NHEJ) can occur throughout the cell cycle, while HR repair is found predominantly in the late S/G2 phase due to the requirement of a sister chromatid as a template for repair (13). Unlike NHEJ, which is capable of inadvertently removing large regions of DNA during the repair process, HR is generally viewed as an error-free system of DNA damage repair. Central to the HR repair process is the recombinase Rad51. Initial recognition and resection of DSBs by the MRN complex and the nuclease CtIP result in coating of single-stranded DNA (ssDNA) by the tripartite RPA complex (7, 14). RPA is then replaced by Rad51, resulting in the formation of a Rad51-ssDNA nucleofilament that facilitates strand annealing and invasion of the neighboring homologous template, leading to the displacement of the identical DNA strand (D-loop) (15).
The tumor suppressor BRCA1 (breast cancer 1, early onset) functions to maintain genomic stability through the assembly of multiple protein complexes involved in DNA repair, cell cycle arrest, and transcriptional regulation (16). BRCA1 is significantly involved at multiple steps along the HR repair pathway (17). Interactions of BRCA1 with the CtIP endonuclease and the MRN complex are important for the initial processing of DSB ends (18, 19), which stimulates HR repair while repressing NHEJ (20). In addition, the association of BRCA1 with PALB2 and BRCA2 aids in the recruitment of Rad51 to sites of DNA damage (21).
Mounting evidence indicates that viruses exploit the cellular DNA damage response in order to facilitate the replication of their own genetic material (22, 23), and recombination-dependent replication has been previously described as a viable and efficient method for production for some virus types (24). In fact, DNA viruses such as simian virus 40 (SV40) (25), herpes simplex virus 1 (HSV-1) (26–28), Epstein-Barr virus (EBV) (29, 30), and Kaposi's sarcoma-associated herpesvirus (KSHV) (31) have exhibited the use of replication-associated recombination in the synthesis of their genomes during infection. We recently demonstrated the importance of members of the DNA double-strand break recognition complex, MRN, in the HPV productive life cycle (32). MRN facilitates the activation of ATM in response to DSBs and also functions downstream of ATM to facilitate HR repair, as mentioned above. We found that disruption of the MRN complex through depletion of Nbs1 abrogates productive replication of HPV31 and blocks the recruitment of Mre11 and Rad50, as well as Rad51, to viral genomes (32). Given the importance of ATM, the MRN complex, Rad51, and BRCA1 to DSB repair through HR, these results suggest that recombination may play a role in the amplification of HPV genomes.
In this work, we investigate the mechanisms by which Rad51 and BRCA1 are regulated in HPV-infected cells, as well as whether they contribute to viral replication throughout the life cycle. We have found that E7 expression alone is sufficient to increase the protein levels of Rad51 and BRCA1. Importantly, we have demonstrated that Rad51 binds to HPV31 genomes, with binding per genome increasing during productive replication. Knockdown of Rad51 and BRCA1, as well as inhibition of Rad51 activity using a small-molecule inhibitor, is sufficient to block productive replication but not viral genome maintenance, similarly to previously published studies after loss of ATM and MRN activity (4, 32). Overall, these studies indicate that HPV may utilize ATM activity to facilitate recombination at viral genomes through Rad51 and BRCA1.
MATERIALS AND METHODS
Cell culture.
Primary human foreskin keratinocytes (HFKs) were isolated from neonatal foreskins as previously described and cultured in DermaLife keratinocyte growth medium (KGM; Lifeline Cell Technology) (33). CIN612 9E, HFK-31, HFK31-E6, HFK31-E7, HFK31-E6E7, and HFK31/DR-GFP cells were cultured in E medium supplemented with 5 ng/ml mouse epidermal growth factor (EGF; BD Biosciences) in the presence of mitomycin C-treated J2 3T3 fibroblast feeder cells, as described previously (33). 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% bovine growth serum (BGS; ThermoFisher Scientific). When necessary, J2 feeders were removed from HPV-positive cells by incubation with 1 mM EDTA in phosphate-buffered saline (PBS).
Plasmids and chemicals.
The HPV31 E6-, HPV31 E7-, and HPV31 E6E7-pLSXN vectors have been previously described (34). The pBR322-HPV31 plasmid containing the wild-type HPV31 genome has been previously described (34, 35). The p1203 PML2d HPV16 plasmid containing the wild-type HPV16 genome was obtained from Addgene (plasmid no. 10869). The homologous recombination reporter vector pHPRT-DRGFP was previously described (36) and obtained from Addgene (plasmid no. 26476). The pHFUW-ISceI lentiviral plasmid was a kind gift from David M. Weinstock and has been described previously (37). Luciferase reporter constructs containing the wild-type proximal promoter of Rad51 or BRCA1 directly upstream of the firefly luciferase gene have been previously described and were a kind gift from Peter M. Glazer (38, 39). The promoterless pGL3-basic and pRL-SV40 (Renilla luciferase) reporter constructs were obtained from Promega. B02 was obtained from Calbiochem.
Generation of HPV31- and HPV16-positive HFKs.
HFKs stably maintaining HPV31 and HPV16 genomes were created as described previously (33). Briefly, the HPV31 and HPV16 genomes were excised from plasmid backbones using HindIII and BamHI, respectively. Excised genomes were religated using T4 DNA ligase (Life Technologies). Primary HFKs were transfected with 1 μg of ligated HPV genomes along with 1 μg of pSV2-Neo using FuGene 6 per the manufacturer's instructions (Promega). Stable transfectants were generated through 8 days of G418 selection (Sigma), and surviving populations were pooled and expanded for analysis.
Differentiation of keratinocytes.
For differentiation, 1.5% methylcellulose was used as described previously (33). Cells were harvested at 0 h, during log-phase growth (undifferentiated), as well as 24 and 48 h after suspension in methylcellulose. High-calcium medium was also used to induce differentiation, as previously described (4). Subconfluent cells were harvested as at T0, and the remaining plates of cells were serum starved overnight in basal keratinocyte growth medium (KGM; Lonza) with supplements for 16 h. Cells were then incubated in keratinocyte basal medium (KBM; Lonza) without supplements but with 1.8 mM CaCl2 (Sigma). Cells were allowed to differentiate for 48, 72, or 96 h after addition of high-calcium medium. DNA and protein were harvested at each time point, and viral genome amplification was measured by Southern blotting for each experiment to ensure activation of the productive phase of the viral life cycle.
Luciferase assays.
HFK or CIN612 9E cells (1 × 105) were seeded in 6-well plates. The next day, cells were transfected in triplicate with 500 ng of either pGL3-basic, Rad51WT-luc, or BRCA1WT-luc reporter plasmids along with 100 ng of the Renilla-luc plasmid (Promega) using polyethyleneimine (PEI) in Opti-MEM (Gibco). Sixteen hours later, transfection medium was replaced with fresh KGM for HFKs and E medium for CIN612 cells. Seventy-two hours later, cells were harvested using 1× passive lysis buffer per the manufacturer's instructions (Promega). Luciferase activity was determined using the Dual-Glo substrate kit per the manufacturer's instructions (Promega), and samples were read on the LMax luminometer (Molecular Devices) with parameters of a 2-s delay followed by a 10-s read. Promoter activity was determined by normalizing firefly luciferase activity to Renilla luciferase activity and calculating the fold change over pGL3-basic control values.
Lentivirus production.
Lentivirus was produced as previously described (40). Plasmids carrying the short hairpin RNA (shRNA) sequences for Rad51 (TRCN0000018876 and TRCN0000018879) or BRCA1 (TRCN0000009824 and TRCN0000039833) or a scramble nontarget control shRNA cloned into the pLKO.1-puro background were obtained from the UNC Lentiviral Core Facility (Chapel Hill, NC). Each of these plasmids (5 μg) was transiently transfected into 293T cells, along with 3.37 μg Gag-Pol-Tet-Rev plasmid DNA and 1.66 μg vesicular stomatitis virus G (VSV-G) plasmid DNA using polyethyleneimine (PEI) (VWR). Supernatants containing lentivirus were harvested 72 h posttransfection, sterile filtered, and stored at −80°C until used. I-SceI lentivirus was packaged according to the same procedure. CIN612 9E and HFK-31 cells were transduced with 5 ml viral supernatant in the presence of 4.8 μg/ml hexadimethrine bromide (Polybrene) (Sigma-Aldrich).
Southern blot analysis.
DNA was isolated and Southern blot analysis was performed as described previously (41). Briefly, cells were harvested in buffer containing 400 mM NaCl, 10 mM Tris (pH 7.5), and 10 mM EDTA. Cells were lysed by the addition of 30 μl of 20% SDS and subsequently treated with 15 μl of 10-mg/ml proteinase K overnight at 37°C. DNA was then extracted with phenol chloroform and precipitated using sodium acetate and ethanol. Five micrograms of resultant DNA was digested with BamHI (which does not cut the HPV31 genome) or HindIII (which linearizes the HPV31 genome) (New England BioLabs), separated on an 0.8% agarose gel for 15 h at 40 V, and subsequently transferred to a positively charged nylon membrane (Immobilon-Ny+; Millipore). 32P-labeled linearized HPV31 genome was used as a probe.
Western blot analysis.
Whole-cell lysates were prepared by lysing cell pellets in radioimmunoprecipitation assay (RIPA) buffer supplemented with Complete Mini protease inhibitor and PhosSTOP phosphatase inhibitor tablets (Roche). Total cellular protein concentrations were determined by the Bio-Rad protein assay (Bio-Rad). Equal amounts of total cellular protein were separated via SDS-PAGE and subsequently transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore). Proteins were visualized using primary antibodies as follows: Rad51, involucrin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz); BRCA1 (Calbiochem); cleaved caspase-7 D198, caspase-7 (Cell Signaling Technology); phospho-ATM S1981 (Abcam); ATM (Bethyl Laboratories). Secondary antibodies were as follows: horseradish peroxidase (HRP)-conjugated anti-rabbit (Cell Signaling Technology) and HRP-conjugated anti-mouse (GE Life Sciences). Clarity Western enhanced chemiluminescence (ECL) blotting substrate (Bio-Rad) was used to detect antibody binding.
Cell cycle analysis.
CIN612 9E cells transduced with shRNAs for Scramble, Rad51, or BRCA1 were harvested as at T0 or suspended in methylcellulose to induce differentiation. At the indicated time points, cells were fixed in 70% ice-cold ethanol and stored at 4°C overnight. The next day, cells were washed once with 1× PBS (Gibco), suspended in 1× PBS containing 10 μg/ml RNase A and 20 μg/ml propidium iodide, and incubated for 20 min at 37°C. Stained cells were analyzed for total DNA content using the CyAn (Beckman Coulter Inc.) flow cytometer. Cell cycle profiles were visualized, and percent values were calculated using FlowJo software v10.0.7 (FlowJo).
qPCR analysis.
Total cellular RNA was isolated from HFKs and HFK-31 and CIN612 9E cells, as well as HFK-E6, HFK-E7, and HFK-E6E7 cells using RNA Stat 60 (Tel-Test) followed by DNase digestion according to the manufacturer's instructions (Promega). cDNA was generated by reverse transcription using the iScript reverse transcription kit per the manufacturer's instructions (Bio-Rad). Fifty nanograms of cDNA was analyzed in triplicate reactions using quantitative PCR (qPCR) with 375 nM primers and iTaq Universal SYBR green Supermix (Bio-Rad) in a total reaction volume of 20 μl. Reactions were carried out in an Applied Biosystems QuantStudio 6 Flex real-time PCR thermal cycler (Life Technologies). Reaction profiles were set up as follows: initial denaturation at 95°C for 10 min followed by 40 cycles of 95°C for 15 s, 63°C for 1 min, and 72°C for 30 s. Melt curves were subsequently performed to ensure proper primer annealing. Relative transcript amounts were calculated using the threshold cycle method (ΔΔCT) with GAPDH as the reference gene and normalized to uninfected HFK samples. Gene-specific primers were as follows: Rad51 forward, 5′-TCTCTGGCAGTGATGTCCTGGA-3′; Rad51 reverse, 5′-TAAAGGGCGGTGGCACTGTCTA-3′; BRCA1 forward, 5′-CTGAAGACTGCTCAGGGCTATC-3′; BRCA1 reverse, 5′-AGGGTAGCTGTTAGAAGGCTGG-3′; GAPDH forward, 5′-CTGTTGCTGTAGCCAAATTCGT-3′; GAPDH reverse, 5′-ACCCACTCCTCCACCTTTGAC-3′.
Measurement of protein half-life.
HFK or CIN612 9E cells (2 × 105) (plus J2 feeders) were seeded in 60-mm dishes. Forty-eight hours later, one culture dish was harvested for the 0-h point and the rest of the culture dishes were treated with 50 μg/ml cycloheximide (Sigma-Aldrich). Cells were harvested for total cellular protein at 2, 4, 6, 8, 12, and 24 h after cycloheximide addition. At harvest, 3T3-J2 feeder cells were removed with Versene prior to harvesting of CIN612 cells. Western blot analysis was performed as described above using 100 μg of lysate from HFKs and 50 μg of lysates from CIN612 cells.
ChIP.
At the indicated time points, CIN612 cells were cross-linked with 1% formaldehyde at 37°C for 10 min, subsequently quenched with 0.125 M glycine, and centrifuged at 240 × g for 4 min at room temperature. Cells were washed with 1× PBS and permeabilized in cell lysis buffer (10 mM EDTA, pH 8.0, 0.5 mM EGTA, 10 mM HEPES, pH 6.5, 1% Triton X-100, 1% NP-40) on ice for 10 min. Cells were washed in wash buffer (pH 8.0; 0.5 mM EGTA, 10 mM HEPES, pH 6.5), resuspended in 500 μl of sonication buffer (10 mM Tris, pH 8, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine) supplemented with 1× protease inhibitor cocktail (PIC) (Roche) and 1 mM phenylmethylsulfonyl fluoride (PMSF), and incubated for 20 min on ice. Sonication was performed with a Bioruptor-300 at 22 cycles of 30 s on and 30 s off. Four hundred micrograms of cleared chromatin was subjected to immunoprecipitation overnight with 10 μg of Rad51 (Santa Cruz) or IgG antibodies. One percent of the sample used for immunoprecipitation was saved as the input. The next day, samples were incubated with 25 μl of Protein AG UltraLink resin beads (Thermo Scientific). The beads were washed for 2 min with the following buffers: low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris [pH 8.1]) with 1× PIC freshly added, high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, 20 mM Tris [pH 8.1]), LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris [pH 8.1]), and two TE buffer (10 mM Tris [pH 8.1] and 1 mM EDTA) washes. DNA was eluted from the beads with 200 μl of elution buffer (1% SDS, 0.1 M NaHCO3) followed by reverse cross-linking with 4 μl of 5 M NaCl and 1 μl of RNase A (Sigma) incubated overnight at 65°C. Input chromatin was also subjected to similar treatment to reverse cross-link. DNA was harvested after proteinase K digestion using spin columns (Qiagen) and eluted with water. Input and immunoprecipitated DNAs were assayed in triplicate reactions by quantitative PCR (qPCR) in an Applied Biosystems QuantStudio 6 Flex real-time PCR thermal cycler (Life Technologies). Reaction profiles were set up as follows: initial denaturation at 95°C for 10 min followed by 40 cycles of 95°C for 15 s, 63°C for 1 min, and 72°C for 30 s. Melt curves were subsequently performed to ensure proper primer annealing. The following primer pairs specific to the HPV31 upstream regulatory region (URR) (bp 7708 to 7784) were used for qPCR: forward primer 5′ TCCTACACACCTTAAACTGCTTTTAGG 3′ and reverse primer 5′ GCAAAAGCCAGCACTGCAATC 3′. Chromatin immunoprecipitation (ChIP) signals were represented as a percentage of input chromatin calculated by normalization against signals of nonspecific IgG.
Homologous recombination reporter assay.
To generate the HFK31/DR-GFP cell line, the DR-GFP fragment was isolated from the pHPRT-DRGFP plasmid by digestion with SacI and KpnI (New England BioLabs) and subsequent gel purification. One microgram of isolated DR-GFP fragment was transfected into HFK-31 cells using FuGene 6 per the manufacturer's instructions (Promega), followed by stable selection using puromycin (Sigma) and 6-thioguanine (Sigma) as previously described (36). Surviving populations were pooled and expanded for analysis. For analysis of homologous recombination activity, 2 × 105 HFK31/DR-GFP cells were plated onto 100-mm tissue culture dishes seeded with mitomycin C-treated 3T3-J2 feeder cells in E medium supplemented with EGF. The following day, cells were transduced with I-SceI viral supernatant for 16 h. After transduction, viral supernatant was replaced with fresh E medium and mitomycin C-treated 3T3-J2 feeder cells on each culture plate. B02 was added to respective plates with the medium change, and plates were incubated at 37°C for 72 h. At harvest, feeder cells were removed using EDTA-PBS (Versene) and HFK31/DR-GFP cells were removed using trypsin and pelleted. Cells were then fixed in 3% paraformaldehyde (Sigma), analyzed on a CyAn (Beckman Coulter Inc.) flow cytometer, and visualized using Flowing software v2.5.1 (University of Turku, Finland) for percent green fluorescent protein (GFP) expression. Homologous repair frequencies were calculated by subtracting the percentage of cells expressing GFP from nonspecific homologous recombination events (i.e., without I-SceI virus treatment) and then determining percent GFP expression relative to I-SceI virus-treated cells without B02.
RESULTS
Homologous recombination factors Rad51 and BRCA1 are increased in HPV-positive cells.
Previous studies by our laboratory demonstrated that HPV31-positive CIN612 9E (referred to as CIN612) keratinocytes, which are derived from a cervical intraepithelial neoplasia grade 1 (CIN1) lesion, exhibit increased protein levels of the homologous recombination factors Rad51 and BRCA1 compared to uninfected keratinocytes derived from neonatal human foreskins (HFKs) (8). We also found that Rad51 and BRCA1 colocalize to sites of viral replication in undifferentiated cells, as well as to sites of productive viral replication upon differentiation, indicating that HPV may increase levels of these factors to drive viral replication. To determine if HPV31 lines generated in the laboratory exhibit a similar increase in Rad51 and BRCA1 protein levels, HFKs were transfected with either recircularized HPV31 (HFK-31) or HPV16 (HFK-16) genomes, along with a neomycin resistance cassette. Stable lines were generated through selection in neomycin. Similar to CIN612 cells, HFK-31 as well as HFK-16 cells exhibited increased Rad51 and BRCA1 protein levels compared to uninfected HFKs, across multiple HFK backgrounds (Fig. 1A). As shown in Fig. 1B, BRCA1 levels were similar between CIN612 and HFK-31 cells and were significantly increased over those in HFKs. Although HFK-31 and CIN612 cells also exhibited a significant increase in Rad51 protein levels (Fig. 1B), CIN612 cells consistently displayed higher levels of Rad51 than did HFK-31 cells. While we did observe some variation in Rad51 and BRCA1 protein levels between different HFK backgrounds, HPV31-positive cells (CIN612 and HFK-31) consistently exhibited significantly higher levels of Rad51 and BRCA1 protein, regardless of the background utilized (Fig. 1B).
FIG 1.
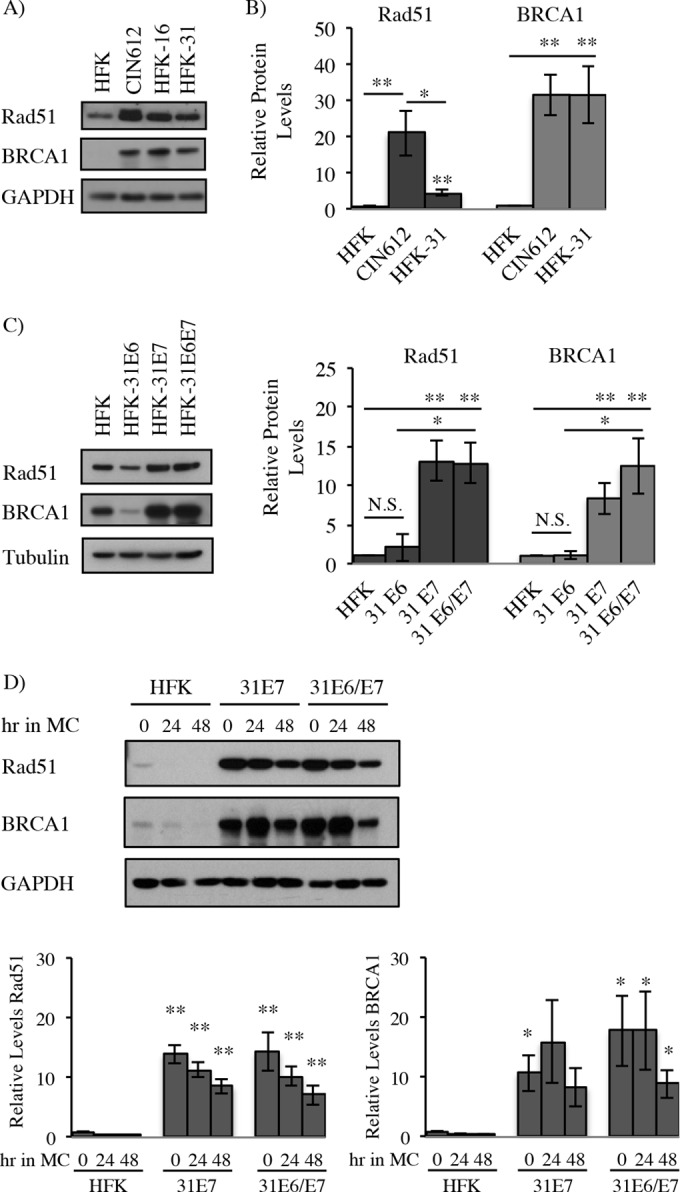
High-risk HPV-positive keratinocytes exhibit higher levels of Rad51 and BRCA1 proteins in an E7-dependent manner. (A) Whole-cell lysates were isolated from uninfected primary human foreskin keratinocytes (HFKs) and CIN612 9E cells, as well as HFKs stably maintaining HPV31 (HFK-31) or HPV16 (HFK-16) genomes. Immunoblotting was performed using antibodies specific to Rad51 and BRCA1, with GAPDH serving as a loading control. (B) Densitometry was performed across at least three independent experiments examining Rad51 and BRCA1 protein levels by immunoblotting in HFKs and CIN612 and HFK-31 cells using ImageJ software. Protein levels were normalized to the GAPDH loading control. Error bars represent means ± standard errors. *, P < 0.05; **, P < 0.01. (C) Western blot analysis of Rad51 and BRCA1 protein levels in whole-cell lysates from HFKs stably transduced with HPV31 E6-, E7-, or E6/E7-expressing retroviral vectors. Tubulin was used as a loading control. Densitometry was performed across at least four independent experiments using ImageJ software. Protein levels were normalized to the respective loading control. Error bars represent means ± standard errors. *, P < 0.05; **, P < 0.01; N.S., not significant. (D) Whole-cell lysates were harvested from control HFKs as well as HFKs stably expressing HPV31 E7 or E6/E7 at T0 (undifferentiated) or after differentiation in methylcellulose (24 and 48 h). Western blot analysis was performed using antibodies to Rad51 and BRCA1 with GAPDH as a loading control. Densitometry was performed using ImageJ software. Protein levels were normalized to the GAPDH loading control. Densitometry was performed across at least four independent experiments. Error bars represent means ± standard errors. *, P < 0.05; **, P < 0.01.
A recent study by Park et al. demonstrated that expression of HPV16 E7 in oral keratinocytes is sufficient to increase protein levels of Rad51 (42). To determine if HPV31 E7 is also capable of increasing the levels of Rad51, as well as BRCA1, we examined HFKs retrovirally transduced and stably expressing HPV31 E6, E7, or E6/E7 in combination. As shown in Fig. 1C, expression of E7 alone was sufficient to significantly increase protein levels of Rad51 and BRCA1 compared to HFKs, as well as E6-expressing cells, and this effect was maintained in E6/E7-expressing cells. In contrast, Rad51 and BRCA1 levels in E6-expressing cells were on average more similar to those found in HFKs (Fig. 1C). As shown in Fig. 1D, levels of Rad51 and BRCA1 were maintained in E7- and E6/E7-expressing cells upon differentiation induced by suspension in methylcellulose, which is commonly used to study the productive phase of the viral life cycle. Similar results were observed with HFKs expressing HPV16 E7 (data not shown). These results suggest that E7 is a key factor in maintaining the increased Rad51 and BRCA1 levels observed in HPV-positive cells.
HPV31-positive cells exhibit increased transcript levels of Rad51 and BRCA1.
Our studies demonstrate that HPV31-positive cells maintain elevated levels of Rad51 and BRCA1 protein. To determine if HPV regulates Rad51 and BRCA1 at the level of transcription, we first examined the promoter activity of Rad51 and BRCA1 using a luciferase reporter system. Expression vectors containing the proximal promoter sequence of either Rad51 or BRCA1 directly upstream of the firefly luciferase gene were transiently transfected into undifferentiated HFKs or HPV31-positive CIN612 cells. Forty-eight hours posttransfection, cells were harvested and luciferase activity was measured. As shown in Fig. 2A, the promoter activity of Rad51 and BRCA1 was significantly higher in CIN612 cells than in uninfected cells, exhibiting an approximately 7.5-fold increase in Rad51 promoter activity and an approximately 9-fold increase in BRCA1 promoter activity. These results suggest that HPV31 may increase Rad51 and BRCA1 protein levels through increased transcription.
FIG 2.

Rad51 and BRCA1 promoter activity and transcript levels are increased in HPV31-positive cells. (A) HFKs and HPV31-positive CIN612 cells were transiently transfected with luciferase reporter constructs containing either Rad51 or BRCA1 proximal promoter sequences, along with a Renilla luciferase-expressing control vector. Luciferase assays were performed 72 h posttransfection. Luciferase activity was normalized to Renilla luciferase activity, and values were calculated as fold change over luciferase activity in uninfected HFKs. Values are averages from five independent experiments ± standard errors of the means. *, P ≤ 0.03. (B and C) RNA was extracted from uninfected HFKs (B and C), HPV31-positive CIN612 cells (B), and HFK-31 cells (C). Quantitative reverse transcription-PCR analysis was performed using primers specific to Rad51 and BRCA1. Fold change was calculated using the 2−ΔΔCT method. Shown is the fold change in message levels relative to that in uninfected HFKs, which is set to 1. Values are averages from five independent experiments ± standard errors of the means. *, P = 0.003; **, P = 0.0003. (D) Quantitative reverse transcription-PCR was used to examine transcript levels of Rad51 and BRCA1 in primary HFKs and HFKs that stably express HPV31 E6, E7, or E6/E7. Expression levels are shown relative to uninfected HFKs and were calculated using GAPDH as the reference gene. Shown is the relative fold change in gene expression over seven independent experiments, across three HFK backgrounds. Data are means ± standard errors. *, P ≤ 0.02; **, P ≤ 0.01.
To examine whether increased promoter activity correlated with increased transcription, we examined Rad51 and BRCA1 mRNA levels in CIN612 cells compared to HFKs. As shown in Fig. 2B, CIN612 cells exhibited a significant increase in transcript levels of Rad51 and BRCA1 compared to uninfected HFKs, across multiple HFK backgrounds. Rad51 and BRCA1 transcript levels were also significantly increased in HFK-31 cells compared to the matched HFKs, across several HFK backgrounds (Fig. 2C). To determine if these results could be attributed to the expression of E7, we examined Rad51 and BRCA1 message levels in the HPV31 E6, E7, and E6/E7 HFK lines. As shown in Fig. 2D, transcript levels of Rad51 and BRCA1 significantly increased upon expression of E7, as well as E6/E7 in combination, compared to HFKs, as well as E6-expressing cells. Interestingly, despite similar levels of Rad51 and BRCA1 protein being observed in E6-expressing cells compared to HFKs (Fig. 1B), the mRNA levels of Rad51 and BRCA1 were significantly increased compared to the HFK control (Fig. 2D). This observation suggests the possibility that the regulation of Rad51 and BRCA1 levels in HPV31-positive cells is not due simply to increased transcript levels.
HPV31 increases the half-life of Rad51 and BRCA1 proteins.
The expression of E7 is well known to lead to the degradation of key cellular proteins necessary for progression of the viral life cycle (i.e., Rb, p107, and p130); however, other proteins have been reported to have increased half-lives due to the presence of E7 (i.e., p53) (43). E6 is best known for targeting proteins for degradation (i.e., p53, BAX, and PDZ domain-containing proteins) through interaction with the E6AP ubiquitin ligase (44). To determine if HPV31 has an effect on the half-life of Rad51 and BRCA1, we inhibited de novo protein synthesis by treating HFKs and CIN612 cells with cycloheximide for the indicated times (Fig. 3). As shown in Fig. 3, the half-lives of both Rad51 and BRCA1 proteins were significantly extended in CIN612 cells compared to HFKs, across multiple HFK backgrounds. These data support the possibility that Rad51 and BRCA1 are regulated in a transcriptional as well as posttranscriptional manner. The finding that E7 expression significantly increases protein levels of Rad51 and BRCA1 while E6 does not suggests that E7 may impart an additional level of regulation through enhancing protein stability.
FIG 3.

The half-life of Rad51 and BRCA1 protein is extended in HPV31-positive cells. (A) Uninfected HFKs and HPV31-positive CIN612 cells were treated with 50 μg/ml cycloheximide (CHX) over a period of 24 h. Whole-cell lysates were prepared at the indicated time points, and Western blot analysis was performed using antibodies to Rad51 and BRCA1. GAPDH served as a loading control. One hundred micrograms of protein was used for the HFK samples, while 50 μg was used for the CIN612 cells. (B) Graphed are the relative protein levels at each time point, with T0 values for HFK and CIN612 cells each set to 100. Densitometry was performed across nine independent experiments using ImageJ software. Protein levels were normalized to the respective GAPDH loading control. Error bars represent means ± standard errors. *, P ≤ 0.05; **, P ≤ 0.01. Results were confirmed through digital imaging of Western blots using the Bio-Rad ChemiDoc MP system. Densitometry was performed using Image Lab software (data not shown).
Rad51 and BRCA1 are required for productive viral replication.
The increase in Rad51 and BRCA1 in HPV31-positive cells and their localization to viral replication foci suggest that they may be important for viral replication. To test this, we utilized small hairpin RNAs (shRNAs) containing a control sequence (Scramble; shScram) or sequences that target Rad51 (shRad51) or BRCA1 (shBRCA1). The efficiency of Rad51 and BRCA1 knockdown is shown in Fig. 4A. We first examined the effect of transiently knocking down Rad51 and BRCA1 expression on the ability of viral episomes to be maintained in undifferentiated cells. CIN612 cells were left untreated or were transduced with lentivirus particles containing a scramble control shRNA (shScram), Rad51 shRNA #1, or BRCA1 shRNA #1 (Fig. 4B). Since stable knockdown of Rad51 and BRCA1 proved to be toxic to HPV31-positive cells, we analyzed episome copy number 4 days posttransduction. Despite localization to viral genomes, transient knockdown of Rad51 or BRCA1 had little to no effect on the ability of viral episomes to be maintained (Fig. 4B). To determine if Rad51 and BRCA1 were required for productive replication upon differentiation, CIN612 cells were left untreated or transduced with lentiviruses containing shScram, shRad51, or shBRCA1. Forty-eight hours posttransduction, undifferentiated cells were either harvested (T0) or differentiated in methylcellulose for 24 and 48 h to activate the productive phase of the viral life cycle (Fig. 4C and D). As shown in Fig. 4C, while knockdown of Rad51 had no detectable effect on episome maintenance in undifferentiated cells, productive replication was significantly decreased upon differentiation. Similarly, knockdown of BRCA1 did not affect episome copy number in undifferentiated cells, while productive replication was significantly decreased upon differentiation (Fig. 4D). These results were confirmed using a second shRNA to target Rad51 and BRCA1 (data not shown). Importantly, levels of the differentiation-specific marker involucrin were not affected by knockdown of Rad51 and BRCA1, indicating that the defect in viral genome amplification was not an indirect effect of blocking cellular differentiation (Fig. 4C and D). Overall, these results indicate that Rad51 and BRCA1 are required for productive replication, potentially through mediating HR at sites of viral replication.
FIG 4.

Knockdown of Rad51 and BRCA1 decreases HPV31 genome amplification upon differentiation. (A) HPV31-positive CIN612 cells were transduced with lentivirus containing control scramble shRNA sequences (scScram) or two shRNA sequences for Rad51 (shRad51) or BRCA1 (shBRCA1) to analyze efficiency of knockdown. Western blot analysis was performed using antibodies to Rad51 and BRCA1. GAPDH served as a loading control. (B) CIN612 cells were transduced with shScram, shRad51 #1, or shBRCA1 #1 and grown as a monolayer for 96 h. DNA was harvested at the indicated times, digested with BamHI (noncutter) or HindIII (which linearizes the viral genome), and examined by Southern blotting for changes in viral episome levels using the HPV31 genome as a probe. (C and D) CIN612 cells were left untreated (UT) or transiently transduced with lentivirus particles containing a control shRNA (shScram), Rad51 shRNA #1, or BRCA1 shRNA #1 for 48 h. At this time, either DNA and protein were harvested as a T0 (undifferentiated) sample or cells were suspended in methylcellulose to induce differentiation for 24 and 48 h. DNA harvested at each time point was analyzed by Southern blotting using the HPV31 genome as a probe. Whole-cell lysates harvested at the indicated time points were analyzed by immunoblotting to demonstrate cellular differentiation (involucrin) and reduced Rad51 or BRCA1 protein in shRNA-transduced cells. GAPDH was used as a loading control. Fold change in episome copy number was determined by performing densitometry of episomal bands from four independent experiments using ImageJ software. Shown is the fold change relative to T0 shScram, which is set to 1. Error bars represent means ± standard errors. *, P ≤ 0.05. WB, Western blot.
HPV requires infected cells to be maintained in a G2/M-like phase upon differentiation in order to properly amplify viral DNA and produce infectious virions (45). In addition to DNA repair through HR, BRCA1 is important in regulating cell cycle checkpoints in response to DNA damage (16). To ensure that the block in productive replication observed with BRCA1 as well as Rad51 knockdown was not due to an effect on the cell cycle, we analyzed DNA content by flow cytometry. As shown in Fig. 5, untreated CIN612 cells, as well as CIN612 cells expressing shScram, exhibited a shift in the cell cycle to the G2/M peak upon differentiation in methylcellulose. However, knockdown of Rad51 and BRCA1 had no significant effect on the G2/M shift compared to the Scramble control after 24 or 48 h of differentiation in methylcellulose (P > 0.05). These results indicate that the lack of viral genome amplification observed in CIN612-shRad51 and CIN612-shBRCA1 cells is not due to an indirect effect on the cell cycle and further suggest a direct role for these factors in facilitating viral replication.
FIG 5.

Cell cycle distribution is not affected by knockdown of Rad51 or BRCA1. CIN612 cells were transduced with control shRNA (shScram) or Rad51 (shRad51) or BRCA1 (shBRCA1) shRNA containing lentiviral particles and either harvested at T0 (undifferentiated) or differentiated in methylcellulose for 24 and 48 h. Cells from each time point were ethanol fixed, stained with propidium iodide (20 μg/ml), and analyzed for total cellular DNA content by flow cytometry. Graphed values are the averages of three independent experiments. Error bars represent means ± standard errors. Statistics were performed using a two-tailed t test comparing shScram to shRad51 and shBRCA1 for the G2/M phase at each time point, with a P value of >0.05. N.S., not significant; MC, methylcellulose.
Inhibition of Rad51 function prevents productive viral replication.
The ability of Rad51 to bind single-stranded DNA (ssDNA) is central to its role in HR (46). Single-stranded DNA is generated by resection of DSBs or by stalled replication forks, resulting in the binding of the tripartite RPA complex. RPA is ultimately replaced by Rad51, which forms nucleofilaments that mediate strand invasion into homologous sequences. We previously demonstrated that the RPA32 subunit localizes to HPV31 genomes (8), suggesting that resection occurs on viral DNA. In addition, RPA32 is phosphorylated on Ser33 at viral genomes upon differentiation, indicative of DNA damage and/or replication perturbation (10–12). To determine if Rad51 is bound to viral DNA, rather than just localized to sites of HPV replication, we performed chromatin immunoprecipitation (ChIP) analysis on CIN612 cells that were undifferentiated or differentiated in high-calcium medium for 72 h to induce the productive phase of the viral life cycle. As shown in Fig. 6A, we found that Rad51 is bound to the upstream regulatory region (URR) of HPV31 genomes in undifferentiated cells. Interestingly, upon differentiation, Rad51 binding to HPV DNA increased per viral genome, suggesting the formation of Rad51 nucleofilaments. To confirm the importance of Rad51 activity in productive viral replication, we utilized the Rad51-specific small-molecule inhibitor B02, which blocks Rad51 binding to DNA and inhibits DNA strand exchange into homologous sequences (47, 48). As demonstrated in Fig. 6B, CIN612 cells differentiated for 72 h in high-calcium medium with dimethyl sulfoxide (DMSO) resulted in amplification of viral genomes, consistent with activation of productive replication. However, treatment of CIN612 cells with increasing concentrations of B02 resulted in a significant dose-dependent decrease in viral genome amplification (Fig. 6B and C). B02 treatment did not affect the levels of Rad51 until the highest concentration (30 μM) (Fig. 6B), which corresponded with an increase in cellular toxicity (data not shown). Similarly to knockdown of Rad51 protein levels, there was no appreciable effect on the differentiation-specific marker involucrin upon treatment with B02 (Fig. 6B), indicating that differentiation was not affected. In addition, we also measured the effect of B02 treatment on the productive replication of HFKs stably maintaining HPV31 genomes (HFK-31) upon differentiation in high-calcium medium (Fig. 7). Similarly to treatment of CIN612 cells, a dose-dependent effect on productive replication was also observed in HFK-31 cells upon exposure to increasing concentrations of B02 (Fig. 7A and B). To confirm that HR activity is targeted by B02 treatment, we utilized HFK-31 cells containing a chromosomally integrated HR reporter (DR-GFP), which measures the efficiency of recombination of two GFP alleles after generation of a double-strand break by the I-SceI restriction enzyme (Fig. 7C). As shown in Fig. 7D, treatment of HFK-31 cells with increasing concentrations of B02 resulted in a dose-dependent decrease in HR activity. These results further support the idea that the ability of Rad51 to bind viral DNA and form nucleofilaments is important for efficient viral genome synthesis. Overall, our findings indicate that HPV increases levels of Rad51 and BRCA1 to drive HR at viral genomes for efficient DNA synthesis upon differentiation.
FIG 6.

Inhibition of Rad51 DNA binding activity leads to a defect in productive replication. (A) Chromatin immunoprecipitation (ChIP) for Rad51 on the HPV31 upstream regulatory region (URR) was performed in CIN612 cells at time zero (undifferentiated) or after 72 h of differentiation in high-calcium medium (Ca). Data for ChIP signals (y axis) from three independent experiments were normalized to 1% of input used. Error bars represent means ± standard errors. *, P < 0.001. (B) DNA and protein were harvested from CIN612 cells at T0 (undifferentiated), as well as after differentiation in high-calcium medium for 72 h in the presence of DMSO (DM) or increasing concentrations of the Rad51 inhibitor B02. DMSO was added at a volume equal to that of the 30 μM concentration of B02. DNA was digested with BamHI (noncutter) or HindIII to linearize the viral genome and then analyzed by Southern blotting for effects on viral copy number using the HPV31 genome as a probe. Western blot analysis was performed to examine the effect of B02 treatment on Rad51 levels and differentiation (involucrin). (C) Effects on episomal copy number were determined by performing densitometry of episomal bands from three independent experiments using ImageJ software. Graphed is the average fold change relative to DMSO (DM)–72-h Ca, which is set to 1. Error bars represent means ± standard errors. *, P < 0.01. Statistics were assayed using a two-tailed t test. Ca, calcium; WB, Western blot.
FIG 7.

Productive replication is abrogated in HFK-31 cells treated with an inhibitor of Rad51 activity, which reduces HR activity. (A) DNA was harvested from HFK-31 cells at T0 as well as after differentiation in high-calcium medium for 72 h in the presence of DMSO (DM) or increasing concentrations of the Rad51 inhibitor B02. DMSO was added at a volume equal to that of the 30 μM concentration of B02. DNA was digested with BamHI (noncutter) or HindIII to linearize the viral genome and then analyzed by Southern blotting to examine episome copy number using the HPV31 genome as a probe. Western blot analysis was performed to examine the effect of B02 treatment on Rad51 levels and differentiation (involucrin). (B) Fold change in episome copy number in response to B02 treatment was determined by performing densitometry of episomal bands from three independent experiments using ImageJ software. Graphed is the average fold change relative to DMSO (DM)–72-h Ca, which is set to 1. Error bars represent means ± standard errors. Statistics were assayed using a two-tailed t test. *, P < 0.01. (C) Schematic of the DR-GFP reporter used to monitor HR activity. SceGFP is a modified GFP gene containing an I-SceI restriction site and in-frame termination codons. The iGFP fragment can be used to repair the I-SceI-induced DSB through HR, resulting in GFP expression. (D) HFK-31 cells containing a chromosomally integrated homologous recombination reporter cassette (DR-GFP) were transduced with I-SceI-containing lentivirus particles to induce a DSB within the DR-GFP reporter cassette. DMSO (DM) or increasing concentrations of B02 were added 24 h after transduction with I-SceI. Forty-eight hours after addition of B02, cells were fixed and analyzed for GFP expression by flow cytometry as a measurement of HR repair. Shown is the fold change in percent GFP relative to the untreated (UT) sample. Graphed values are averages of values from five independent experiments, with error bars representing means ± standard errors. Statistics were assayed using a two-tailed t test. *, P < 0.05. Ca, calcium; WB, Western blot.
DISCUSSION
The importance of Rad51 and BRCA1 in homologous recombination is well established (7, 17). Rad51 is central to recombination through its ability to coat single-stranded DNA (ssDNA) and catalyze homologous pairing and strand exchange (46). BRCA1 functions in two distinct steps in HR repair: (i) 5′-3′ resection of DSBs to generate 3′ overhangs and (ii) interaction with the PALB2-BRCA2 complex to load Rad51 onto resected ssDNA. In this study, we have found that Rad51 is bound to HPV genomes and that binding per viral genome increases upon differentiation. These results suggest that extensive resection occurs on viral DNA during productive replication, prompting Rad51 nucleofilament assembly, an early step in HR. In support of this, use of an inhibitor that blocks Rad51 binding to DNA abrogated productive replication. In addition to DNA repair, BRCA1 is involved in multiple cellular functions, including transcription and cell cycle checkpoint activation (16). However, no significant effects on the cell cycle were observed upon BRCA1 or Rad51 knockdown. The localization of BRCA1 to HPV replication foci, coupled with the binding of Rad51 to viral genomes and the requirement for BRCA1 and Rad51 in productive replication, supports a role for BRCA1 and Rad51 in facilitating recombination at HPV genomes during productive replication.
Recombination can be stimulated by resection of DSBs, as well as ssDNA generated by replication fork stalling, although how HPV utilizes recombination pathways for replication is unclear. Productive replication results in viral genome amplification to thousands of copies per cell, and DSBs could result if replication proceeds through a nick or gap in the viral DNA. Indeed, the localization of multiple effectors of the ATM-dependent DNA damage response to viral DNA foci suggests that DNA lesions form that may stimulate recombination for repair (8, 9). In support of this, multiple components of the ATM response, in addition to Rad51 and BRCA1, are required for productive replication, including the MRN complex (Mre11, Rad50, Nbs1) and Chk2 (4, 32). ATM can stimulate recombination through two homology-directed pathways: Rad51-dependent strand invasion (referred to as HR) and single-strand annealing (SSA), both of which rely on a resection intermediate (49). SSA, which is inhibited by Rad51 assembly on ssDNA, has recently been implicated in recombination-dependent replication of HSV-1 (26). The recruitment of Rad51 and BRCA1 to viral genomes suggests that HPV utilizes ATM activity to repair replication-associated DSBs through HR. As HR involves the use of the sister chromatid as a template for repair (50) and is generally considered an error-free process, HPV may preferentially use this method of repair for high-fidelity genome amplification upon differentiation. In addition, productive replication is thought to occur in a G2/M-like environment (45), at a time when HR is most active (45).
End resection is a key step in the choice of DSB repair pathway utilized, promoting pathways that use homology while suppressing error-prone NHEJ (20). Previous studies from our lab indicate that DNA-activated protein kinase (DNA-PK), a key factor in classic NHEJ, does not localize to viral genomes (8), suggesting that HR factors are specifically recruited. ATM and BRCA1 each play essential roles in directing repair through recombination by initiating resection of DSBs. ATM accomplishes this through phosphorylation of the endonuclease CtIP (18), while BRCA1 antagonizes the resection inhibitor 53BP1 (51–53) and recruits CtIP to the MRN resection complex (54–56). We previously demonstrated that Mre11's nuclease activity is required for productive replication and that the localization of Rad51 to HPV31 genomes in differentiating cells requires an intact MRN complex (32). The increased binding of Rad51 per viral genome upon differentiation suggests that MRN-dependent resection may be required for Rad51 loading on viral DNA. Recent studies by Langsfeld et al. indicate that Rad51 binding to viral genomes is dependent on the activity of the Sirtuin 1 deacetylase (57). Interestingly, in previous studies we found that 53BP1 localization to HPV31 genomes increases upon differentiation (8), which in theory should restrict HR at viral genomes. However, the requirement of Rad51 and BRCA1 for productive replication argues against this. Recent studies have shown that BRCA1 spatially excludes 53BP1 from the proximity of DSBs during S phase (53). BRCA1 may serve the same function on HPV genomes during productive replication. Future studies will focus on determining if and how HPV utilizes ATM activity and BRCA1 to influence the DSB repair pathway of choice, as has recently been shown for SV40 (58).
Our studies support a role for HR during productive replication, potentially for repair of replication-associated DNA breaks. However, increasing evidence supports a role for HR proteins, including Rad51, BRCA1, and BRCA2, as well as resection enzymes (CtIP and MRN), in replication fork stabilization independently of DSB repair (59–64). In response to replication stress, BRCA1 and BRCA2 stabilize Rad51 at stalled forks, protecting nascent strands against nucleolytic degradation (59–61). Fork restart requires Rad51, which catalyzes Holliday junction formation by invading a homologous molecule (65). HPV16 E7 induces replication stress due to unscheduled entry into the cell cycle, resulting in a paucity of nucleotides to support DNA synthesis (66). Upon differentiation, E7 pushes postmitotic cells back into the cell cycle to provide cellular factors required for productive replication (67). Whether cell cycle reentry upon differentiation also induces replication stress is unclear but could potentially impact viral genome amplification if cellular substrates are not immediately available. Alternatively, viral genome amplification could in itself lead to replication stress due to the high levels of cellular substrates required to meet the demand for increased viral DNA synthesis. HPV may utilize ATM activity and HR factors to preserve replication fork integrity, preventing the collapse of replication forks into DSBs. Understanding how HR factors are utilized during viral replication will be an important area of future study.
Our studies indicate that the transient loss of Rad51 and BRCA1 expression does not impact episomal copy number in undifferentiated cells. Although we cannot rule out a role for these HR factors in long-term episomal maintenance, these results mirror our previous studies in which inhibition of ATM activity (4), as well as disruption of the MRN complex (32), had minimal effect on maintenance replication but was specifically required for productive viral replication. Furthermore, our previous studies demonstrated that knockdown of Nbs1 affected only the localization of Rad51 to viral genomes upon differentiation (32). This may suggest that in undifferentiated cells, Rad51 is not localized to viral genomes for repair but may be present at single-stranded DNA formed through transient uncoupling of the replication fork from the viral E1 helicase (59, 68). These results also argue that perhaps there are inherent differences in the way HPV replicates and utilizes DNA repair pathways during different phases of the viral life cycle. In support of this, Flores and Lambert provided evidence that HPV16 and HPV31 undergo a shift in the mode of replication upon differentiation (69). In undifferentiated cells, HPV replicates bidirectionally; however, two-dimensional (2D) gel analysis revealed that productively replicating genomes exhibit branched structures, including X structures and simple Y arcs, consistent with strand invasion (Holliday junction formation) and unidirectional (rolling-circle) replication, respectively (70, 71). These studies offer support for our findings that recombination is important for successful viral genome amplification and suggest that productive replication may occur through rolling-circle replication and concatemer formation. If viral concatemers are indeed formed during productive replication, recombination may additionally be required to resolve concatemers into individual genomes.
We have found that Rad51 and BRCA1 are increased in HPV31-positive cells at the level of transcription and protein stability. Increased levels of Rad51 have been documented in a wide range of human tumors, including HPV-positive head and neck cancer cell lines, as well as HPV-positive cervical cancer lines (42, 72). Moreover, increased expression of Rad51 is correlated with increased genomic instability through an elevation in spontaneous and aberrant HR (73, 74). Although we can detect HR activity in HPV31-positive cell lines using an HR-specific reporter assay, whether this activity is increased relative to uninfected cells is currently unclear. How HPV infection affects DSB repair at cellular sites of damage is also unknown. HPV may increase expression of Rad51 and BRCA1 to drive productive replication, but in turn, this increase could be detrimental to the genomic stability of HPV-infected cells and contribute to cancer progression. Alternatively, HPV may sequester these factors for viral replication, preventing HR repair of cellular DSBs, resulting in increased genomic instability.
Our studies indicate that both HPV31 E6 and E7 are able to facilitate an increase in Rad51 and BRCA1 transcript levels, though the mechanism remains unclear. Both Rad51 and BRCA1 are regulated in a manner dependent on E2F transcription factors (38, 39, 75–77). The ability of E7 to target Rb and the related pocket proteins p130 and p107 for degradation, in turn resulting in constitutive activation of E2F transcription factors, may facilitate increased expression of Rad51 and BRCA1. E6 has also been shown to induce expression of E2F-responsive genes (78). In addition, p53 can repress Rad51 expression (79), and E6's ability to promote p53 degradation may account for the increase in Rad51 and BRCA1 transcripts that we observed. While E7 was also able to significantly increase Rad51 and BRCA1 protein levels compared to uninfected HFKs, this was not consistently observed for E6, despite the increase in Rad51 and BRCA1 transcript levels. These data suggest an additional level of regulation for the increased levels of Rad51 and BRCA1 protein observed in HPV-positive cells. In support of this, we have found that the protein half-life of Rad51 and BRCA1 is increased in HPV31-positive cells, although the mechanism is unclear. HPV31 induces an ATM- as well as ATR-dependent DNA damage response (4), which may in turn stabilize Rad51 and BRCA1 through phosphorylation (7, 16). In addition, HPV16 E7 has been shown to bind BRCA1 (80), which may be sufficient to protect BRCA1 from degradation. Whether E7 also binds Rad51 is unknown. While HPV16 E6 also interacts with BRCA1 (80), E6 has been shown to bind to the ubiquitin ligase HERC2 (81), which can target BRCA1 for degradation (82). Understanding the regulation of Rad51 and BRCA1 gene expression and protein stability in HPV-positive cells is an important area for future study.
In summary, we have demonstrated that Rad51 and BRCA1 are required for productive replication. We have found that Rad51 is bound to viral genomes and that Rad51 activity is required for successful amplification of viral genomes upon differentiation, suggesting that HPV replication is driven through recombination. In addition, we have shown that E7 increases Rad51 and BRCA1 levels in a transcriptional and potentially posttranscriptional manner. In addition to ensuring cell cycle reentry upon differentiation, E7 may contribute to productive replication by maintaining the expression of repair factors that are required for viral DNA synthesis. Overall, these studies provide a deeper understanding of how HPV utilizes the DNA damage response to regulate productive viral replication.
ACKNOWLEDGMENTS
We thank David Weinstock, Peter Glazer, and Jeremy Stark for their generous gifts of reagents.
This project was supported by NIH grant 1R01CA181581 and American Cancer Society grant A14-0113 (to C.A.M.) and NIH grant 5T32 AI007419 (to W.H.C.).
REFERENCES
- 1.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Longworth MS, Laimins LA. 2004. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev 68:362–372. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moody CA, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 4.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agarwal S, Tafel AA, Kanaar R. 2006. DNA double-strand break repair and chromosome translocations. DNA Repair (Amst) 5:1075–1081. doi: 10.1016/j.dnarep.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 6.Helleday T, Lo J, van Gent DC, Engelward BP. 2007. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillespie KA, Mehta KP, Laimins LA, Moody CA. 2012. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol 86:9520–9526. doi: 10.1128/JVI.00247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakakibara N, Chen D, Jang MK, Kang DW, Luecke HF, Wu SY, Chiang CM, McBride AA. 2013. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog 9:e1003777. doi: 10.1371/journal.ppat.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, Borowiec JA. 2014. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol 206:493–507. doi: 10.1083/jcb.201404111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, Zou L. 2013. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep 3:1651–1662. doi: 10.1016/j.celrep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vassin VM, Wold MS, Borowiec JA. 2004. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol Cell Biol 24:1930–1943. doi: 10.1128/MCB.24.5.1930-1943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothkamm K, Kruger I, Thompson LH, Lobrich M. 2003. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moynahan ME, Jasin M. 2010. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumann P, Benson FE, West SC. 1996. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87:757–766. doi: 10.1016/S0092-8674(00)81394-X. [DOI] [PubMed] [Google Scholar]
- 16.Savage KI, Harkin DP. 2015. BRCA1, a ‘complex' protein involved in the maintenance of genomic stability. FEBS J 282:630–646. doi: 10.1111/febs.13150. [DOI] [PubMed] [Google Scholar]
- 17.Prakash R, Zhang Y, Feng W, Jasin M. 2015. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7:a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Shi LZ, Wong CC, Han X, Hwang PY, Truong LN, Zhu Q, Shao Z, Chen DJ, Berns MW, Yates JR III, Chen L, Wu X. 2013. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet 9:e1003277. doi: 10.1371/journal.pgen.1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L, Nievera CJ, Lee AY, Wu X. 2008. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem 283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 20.Chapman JR, Taylor MR, Boulton SJ. 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 21.Sy SM, Huen MS, Chen J. 2009. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A 106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weitzman MD, Lilley CE, Chaurushiya MS. 2010. Genomes in conflict: maintaining genome integrity during virus infection. Annu Rev Microbiol 64:61–81. doi: 10.1146/annurev.micro.112408.134016. [DOI] [PubMed] [Google Scholar]
- 23.Turnell AS, Grand RJ. 2012. DNA viruses and the cellular DNA-damage response. J Gen Virol 93:2076–2097. doi: 10.1099/vir.0.044412-0. [DOI] [PubMed] [Google Scholar]
- 24.Lo Piano A, Martinez-Jimenez MI, Zecchi L, Ayora S. 2011. Recombination-dependent concatemeric viral DNA replication. Virus Res 160:1–14. doi: 10.1016/j.virusres.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Boichuk S, Hu L, Hein J, Gjoerup OV. 2010. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol 84:8007–8020. doi: 10.1128/JVI.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK. 2012. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog 8:e1002862. doi: 10.1371/journal.ppat.1002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkinson DE, Weller SK. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol 78:4783–4796. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilkinson DE, Weller SK. 2003. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life 55:451–458. doi: 10.1080/15216540310001612237. [DOI] [PubMed] [Google Scholar]
- 29.Kudoh A, Iwahori S, Sato Y, Nakayama S, Isomura H, Murata T, Tsurumi T. 2009. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in Epstein-Barr virus replication compartments. J Virol 83:6641–6651. doi: 10.1128/JVI.00049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dheekollu J, Deng Z, Wiedmer A, Weitzman MD, Lieberman PM. 2007. A role for MRE11, NBS1, and recombination junctions in replication and stable maintenance of EBV episomes. PLoS One 2:e1257. doi: 10.1371/journal.pone.0001257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dheekollu J, Chen HS, Kaye KM, Lieberman PM. 2013. Timeless-dependent DNA replication-coupled recombination promotes Kaposi's sarcoma-associated herpesvirus episome maintenance and terminal repeat stability. J Virol 87:3699–3709. doi: 10.1128/JVI.02211-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anacker DC, Gautam D, Gillespie KA, Chappell WH, Moody CA. 2014. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J Virol 88:8528–8544. doi: 10.1128/JVI.00517-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson R, Laimins LA. 2005. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol Med 119:157–169. [DOI] [PubMed] [Google Scholar]
- 34.Longworth MS, Laimins LA. 2004. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol 78:3533–3541. doi: 10.1128/JVI.78.7.3533-3541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hubert WG, Laimins LA. 2002. Human papillomavirus type 31 replication modes during the early phases of the viral life cycle depend on transcriptional and posttranscriptional regulation of E1 and E2 expression. J Virol 76:2263–2273. doi: 10.1128/jvi.76.5.2263-2273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. 2001. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fung H, Weinstock DM. 2011. Repair at single targeted DNA double-strand breaks in pluripotent and differentiated human cells. PLoS One 6:e20514. doi: 10.1371/journal.pone.0020514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bindra RS, Glazer PM. 2007. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene 26:2048–2057. doi: 10.1038/sj.onc.1210001. [DOI] [PubMed] [Google Scholar]
- 39.Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, Bristow RG, Classon MK, Glazer PM. 2005. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res 65:11597–11604. doi: 10.1158/0008-5472.CAN-05-2119. [DOI] [PubMed] [Google Scholar]
- 40.Mighty KK, Laimins LA. 2011. p63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J Virol 85:8863–8869. doi: 10.1128/JVI.00750-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fehrmann F, Klumpp DJ, Laimins LA. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol 77:2819–2831. doi: 10.1128/JVI.77.5.2819-2831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park JW, Nickel KP, Torres AD, Lee D, Lambert PF, Kimple RJ. 2014. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother Oncol 113:337–344. doi: 10.1016/j.radonc.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roman A, Munger K. 2013. The papillomavirus E7 proteins. Virology 445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howie HL, Katzenellenbogen RA, Galloway DA. 2009. Papillomavirus E6 proteins. Virology 384:324–334. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banerjee NS, Wang HK, Broker TR, Chow LT. 2011. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J Biol Chem 286:15473–15482. doi: 10.1074/jbc.M110.197574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morrical SW. 2015. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb Perspect Biol 7:a016444. doi: 10.1101/cshperspect.a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang F, Mazina OM, Zentner IJ, Cocklin S, Mazin AV. 2012. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J Med Chem 55:3011–3020. doi: 10.1021/jm201173g. [DOI] [PubMed] [Google Scholar]
- 48.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. 2011. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 6:628–635. doi: 10.1021/cb100428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gunn A, Bennardo N, Cheng A, Stark JM. 2011. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. J Biol Chem 286:42470–42482. doi: 10.1074/jbc.M111.309252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson RD, Jasin M. 2001. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans 29:196–201. doi: 10.1042/bst0290196. [DOI] [PubMed] [Google Scholar]
- 51.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. 2012. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 125:3529–3534. doi: 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. 1998. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem 273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 55.Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, Chen PL, Sharp ZD, Lee WH. 1999. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 285:747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 56.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. 2007. Human CtIP promotes DNA end resection. Nature 450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langsfeld ES, Bodily JM, Laimins LA. 2015. The deacetylase sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and Rad51 to viral genomes. PLoS Pathog 11:e1005181. doi: 10.1371/journal.ppat.1005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sowd GA, Mody D, Eggold J, Cortez D, Friedman KL, Fanning E. 2014. SV40 utilizes ATM kinase activity to prevent non-homologous end joining of broken viral DNA replication products. PLoS Pathog 10:e1004536. doi: 10.1371/journal.ppat.1004536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. 2010. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yeo JE, Lee EH, Hendrickson EA, Sobeck A. 2014. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum Mol Genet 23:3695–3705. doi: 10.1093/hmg/ddu078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. 2003. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. 2009. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 28:2601–2615. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. 2011. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng S, Schmidt-Grimminger DC, Murant T, Broker TR, Chow LT. 1995. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev 9:2335–2349. doi: 10.1101/gad.9.19.2335. [DOI] [PubMed] [Google Scholar]
- 68.Hashimoto Y, Puddu F, Costanzo V. 2012. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol 19:17–24. doi: 10.1038/nsmb.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flores ER, Lambert PF. 1997. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J Virol 71:7167–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Preiser PR, Wilson RJ, Moore PW, McCready S, Hajibagheri MA, Blight KJ, Strath M, Williamson DH. 1996. Recombination associated with replication of malarial mitochondrial DNA. EMBO J 15:684–693. [PMC free article] [PubMed] [Google Scholar]
- 71.Backert S. 2002. R-loop-dependent rolling-circle replication and a new model for DNA concatemer resolution by mitochondrial plasmid mp1. EMBO J 21:3128–3136. doi: 10.1093/emboj/cdf311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. 2002. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res 62:219–225. [PubMed] [Google Scholar]
- 73.Richardson C, Stark JM, Ommundsen M, Jasin M. 2004. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 23:546–553. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- 74.Klein HL. 2008. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bindra RS, Glazer PM. 2006. Basal repression of BRCA1 by multiple E2Fs and pocket proteins at adjacent E2F sites. Cancer Biol Ther 5:1400–1407. doi: 10.4161/cbt.5.10.3454. [DOI] [PubMed] [Google Scholar]
- 76.Iwanaga R, Komori H, Ohtani K. 2004. Differential regulation of expression of the mammalian DNA repair genes by growth stimulation. Oncogene 23:8581–8590. doi: 10.1038/sj.onc.1207976. [DOI] [PubMed] [Google Scholar]
- 77.Wang A, Schneider-Broussard R, Kumar AP, MacLeod MC, Johnson DG. 2000. Regulation of BRCA1 expression by the Rb-E2F pathway. J Biol Chem 275:4532–4536. doi: 10.1074/jbc.275.6.4532. [DOI] [PubMed] [Google Scholar]
- 78.Shai A, Brake T, Somoza C, Lambert PF. 2007. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res 67:1626–1635. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, Ashworth A, Silva A. 2006. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep 7:219–224. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y, Fan S, Meng Q, Ma Y, Katiyar P, Schlegel R, Rosen EM. 2005. BRCA1 interaction with human papillomavirus oncoproteins. J Biol Chem 280:33165–33177. doi: 10.1074/jbc.M505124200. [DOI] [PubMed] [Google Scholar]
- 81.Vos RM, Altreuter J, White EA, Howley PM. 2009. The ubiquitin-specific peptidase USP15 regulates human papillomavirus type 16 E6 protein stability. J Virol 83:8885–8892. doi: 10.1128/JVI.00605-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu W, Sato K, Koike A, Nishikawa H, Koizumi H, Venkitaraman AR, Ohta T. 2010. HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res 70:6384–6392. doi: 10.1158/0008-5472.CAN-10-1304. [DOI] [PubMed] [Google Scholar]