

ABSTRACT
The 1918-1919 influenza pandemic remains the single greatest infectious disease outbreak in the past century. Mouse and nonhuman primate infection models have shown that the 1918 virus induces overly aggressive innate and proinflammatory responses. To understand the response to viral infection and the role of individual 1918 genes on the host response to the 1918 virus, we examined reassortant avian viruses nearly identical to the pandemic 1918 virus (1918-like avian virus) carrying either the 1918 hemagglutinin (HA) or PB2 gene. In mice, both genes enhanced 1918-like avian virus replication, but only the mammalian host adaptation of the 1918-like avian virus through reassortment of the 1918 PB2 led to increased lethality. Through the combination of viral genetics and host transcriptional profiling, we provide a multidimensional view of the molecular mechanisms by which the 1918 PB2 gene drives viral pathogenicity. We demonstrate that 1918 PB2 enhances immune and inflammatory responses concomitant with increased cellular infiltration in the lung. We also show for the first time, that 1918 PB2 expression results in the repression of both canonical and noncanonical Wnt signaling pathways, which are crucial for inflammation-mediated lung regeneration and repair. Finally, we utilize regulatory enrichment and network analysis to define the molecular regulators of inflammation, epithelial regeneration, and lung immunopathology that are dysregulated during influenza virus infection. Taken together, our data suggest that while both HA and PB2 are important for viral replication, only 1918 PB2 exacerbates lung damage in mice infected with a reassortant 1918-like avian virus.
IMPORTANCE As viral pathogenesis is determined in part by the host response, understanding the key host molecular driver(s) of virus-mediated disease, in relation to individual viral genes, is a promising approach to host-oriented drug efforts in preventing disease. Previous studies have demonstrated the importance of host adaptive genes, HA and PB2, in mediating disease although the mechanisms by which they do so are still poorly understood. Here, we combine viral genetics and host transcriptional profiling to show that although both 1918 HA and 1918 PB2 are important mediators of efficient viral replication, only 1918 PB2 impacts the pathogenicity of an avian influenza virus sharing high homology to the 1918 pandemic influenza virus. We demonstrate that 1918 PB2 enhances deleterious inflammatory responses and the inhibition of regeneration and repair functions coordinated by Wnt signaling in the lungs of infected mice, thereby promoting virus-associated disease.
INTRODUCTION
Annual influenza epidemics cause significant morbidity and mortality. On occasion, novel influenza A strains emerge that are capable of producing severe respiratory disease on a global scale (1). The 1918-1919 pandemic was an unprecedented event, with an estimated 40 to 50 million deaths worldwide (2, 3). Sequence analysis indicates that the 1918 pandemic influenza virus is avian in origin (4–8) and 1918 viral genes are highly homologous to those of circulating avian influenza H1N1 viruses (9). In a recent study, a reconstructed virus comprised of avian influenza viral segments with high homology to the 1918 virus was shown to be pathogenic in ferrets and mice. Specific amino acid changes in PB2 and PA viral polymerase proteins, in combination with amino acid changes in the hemagglutinin (HA) viral glycoprotein, resulted in the “1918-like avian virus” efficiently transmitting between ferrets via the respiratory droplet route (9). Host-adaptive changes in polymerase and NP genes enhancing avian influenza virus replicative fitness are well documented (10, 11). In particular, PB2 627K has become a genomic signature of host adaptation, which was present in the 1918 pandemic influenza virus PB2 gene, though novel host adaptive mutations continue to be characterized in the ribonucleoprotein complex (12–14). These human-adaptive changes may confer efficient replication of avian influenza viruses in humans raising concerns about the pandemic potential of circulating avian influenza viruses. Influenza virus genes involved in host adaptation are key virulence determinants, although the importance of these genes in the context of a virus sharing close homology to 1918 is still unknown.
Two of the most well-characterized viral determinants of 1918 pathogenesis are HA and neuraminidase (NA) (15–17). In the genetic background of a human H1N1 influenza virus, 1918 HA and NA induced macrophages and neutrophils into the lung that play an important role in the control of virus replication (18, 19). The HA glycoprotein is required for influenza virus binding to sialic acid receptors on the host cell surface, whereas NA catalyzes these linkages to promote egress of progeny virions. The HA receptor-binding site is invariant in all avian HAs [with a preferential binding for α(2-3) sialic acids]; however, it varies in mammalian-adapted HA that exhibit preferential binding to α(2-6)-linked sialic acids (20–22). Genomic-based approaches have expanded on our understanding of HA-mediated virulence by demonstrating that, aside from the role HA plays in dictating viral tropism, HA promotes protracted proinflammatory responses involving acute phase signaling and protein ubiquitination pathways (17, 18).
In contrast to 1918, exchange of H5N1 HA and NA genes in the genetic background of a nonlethal avian virus did not increase pathogenicity but rather the substitution of the polymerase genes contribute to H5N1 pathogenesis (23–25). The PB2 gene, which encodes one of the three viral RNA polymerase subunits required for viral replication (26), is also a determinant of host range and virulence of avian influenza viruses (12, 27, 28). Characterization of the adaptation markers of PB2 has demonstrated crucial amino acid substitutions in the PB2, including E627K, protein of avian influenza viruses (29, 30), which play an important role in enhancing replication of avian viruses in mammalian cells and mouse lungs. On the other hand, insertion of PB2 from a low-pathogenicity avian influenza H1N1 virus into the genetic background of 1918 diminishes virulence in mice (31). While adaptive changes in HA have been described as a major virulence determinant and contributes to unabated proinflammatory responses elicited by the 1918 virus (18, 32), how PB2 contributes to changes in the host response remains to be addressed.
Here, we evaluate the global transcriptional responses to influenza viruses sharing close homology to the 1918 virus to evaluate the biological processes that drive disease severity. We use single-reassortant viruses in the backbone of the “1918-like avian virus” and apply genomic-based approaches to understand the impact of 1918 HA and 1918 PB2 on host transcriptional responses to influenza A virus (IAV) infection. We demonstrate that the 1918 PB2 gene enhances both the kinetics and the magnitude of the host response to viral infection, leading to the induction of strong inflammatory responses with increased cellular infiltration in the infected lung. Finally, we show that expression of 1918 PB2 results in the repression of Wnt signaling exacerbating inflammatory responses and impairing lung regenerative and repair functions.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic kidney (HEK293T) cells and Madin-Darby canine kidney (MDCK) cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum and minimal essential medium supplemented with 5% newborn calf serum, respectively. A549 cells and derived cell lines were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. All cells were maintained at 37°C in 5% CO2. With the exception of A/duck/Alberta/35/76 (DK/ALB), all viruses used in this study were generated by plasmid-driven reverse genetics and propagated as previously described (9, 33).
Mouse experiments.
Five- to six-week-old female BALB/c mice (Jackson Laboratory) were used for the experiments. Isoflurane-anesthetized mice were intranasally inoculated with 10-fold serial dilutions of 1918, 1918-like avian, 1918 PB2/avian, and 1918 HA/avian viruses. The dose required to kill 50% of mice (MLD50) was calculated using the Reed-Muench method (34). For analysis of virus growth and microarray profiling, mice were intranasally inoculated with 106 PFU of virus (n = 4/virus/time point) or PBS (n = 3/time point). At days 1, 2, and 4 after infection, lungs were harvested from the infected mice. Lungs were processed for RNA extraction for microarray studies and virus titer determination. Statistical analyses for virus lung titers were performed by using analysis of variance (ANOVA) in GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA); P values of <0.05 were considered significant.
All experiments with the 1918-like viruses were performed in a biosafety level 3 containment laboratory approved for such use by the Centers for Disease Control and Prevention (CDC) and the U.S. Department of Agriculture. The research program, procedures, occupational health plan, documentation, security, and facilities are reviewed annually by the University of Wisconsin—Madison Responsible Official and at regular intervals by the CDC and the Animal and Plant Health Inspection Service as part of the University of Wisconsin—Madison Select Agent Program. All animal experiments were performed in accordance with the University of Wisconsin—Madison's Regulations for Animal Care and Use and approved by the Animal Experiment Committee of the University of Wisconsin—Madison.
Mouse microarray experiments and data processing.
RNA was isolated from total lung homogenates, and fluorescently labeled probes were generated from each sample using Agilent One-Color LowInput Quick Amp labeling kit (Agilent Technologies). Individual cRNA samples were hybridized to oligonucleotide microarrays for gene expression profiling using a whole-mouse genome microarray kit (G4122A; Agilent Technologies), as previously described (17). RNA isolated from control animal lungs served as an uninfected reference. Quantitative reverse transcription-PCR was performed on mouse lung samples using a primer-probe set targeting the viral matrix sequence and confirmed all infected samples expressed matrix mRNA.
The primary transcriptomic data were extracted and quantile normalized using the “normalizeBetweenArrays” method available in the “limma” package of the R suite as previously described (35) and adjusted for batch effects using the ComBat software (36). Expressions across each sample were then normalized to the average expression of study and time-matched mocks. Differential expression of 1918 wild-type (WT) and 1918-like avian reassortant viruses was determined by comparing the average gene expression of influenza virus-infected replicates to the average of time-matched mock-infected samples applying a linear model fit using the “limma” package. Criteria for differential expression were an absolute log2-fold change of 3 and an adjusted P value of <0.05, calculated by Benjamini-Hochberg correction (1).
Functional enrichment analysis.
Functional analysis of differentially expressed (DE) genes was done using Ingenuity Pathway Analysis (IPA; Ingenuity Systems). IPA function enrichment was calculated using a right-tailed Fisher exact test with a threshold of significance set at P value of 0.05. Enrichment of diseases and biological functions and upstream regulators were based on activation |z-scores| >2. Analysis of upstream regulators was performed using the web-based software tool, Enrichr (37), which utilizes the ChIP-x Enrichment Analysis (ChEA) database (38) of experimentally validated putative targets for transcription factors in mammalian cells. Regulatory transcription factors were ranked based on the combined score Combined scores are derived by multiplying the P value determined by the Fisher exact test, and the computed z-score to assess the deviation from the expected rank. The Wikipathways and KEGG tools in Enrichr were also used for functional analysis.
Computational measurement of immune cell subsets.
To predict immune cell composition, we utilized microarray data available from Mouse GeneAtlas v3 (GEO accession number GSE10246) as previously described (35). A Fisher exact test was used to determine enrichment scores for each module relative to the expression of DE genes in our data set enrichment scores (ES) were reported as −log10 (P value), and the cutoff for significant enrichment was set at ES = 1.3. Specific immune cell dynamics where probed using the digital cell quantifier (DCQ) algorithm. Briefly, genome-wide gene expression for each infectious condition was normalized relative to the average of the time-matched mock and log2 transformation was applied. The data were transformed by dividing each gene expression entry by the standard deviation across test samples. Analysis of relative cell quantity was run with three repeats and a lambda minimum of 0.2 as previously described (39).
Luciferase reporter assay.
An A549-derived β-catenin reporter cell line was generated (A549-BAR), as previously described (40, 41). A549-BAR cells were transfected with 4 μg of plasmids expressing YFP, 1918 PB2, or avian PB2 using X-tremegene HP (Roche), followed by stimulation with 50 ng of recombinant Wnt3A (Peprotech)/ml for the indicated time points. Firefly and Renilla luciferase activity was assessed using the Dual-Glo luciferase assay system (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity and expressed as fold changes relative to mock-treated cells.
Quantitative PCR of gene expression.
Total RNA from A549 BAR cells was extracted using Qiazol reagent (Qiagen). The RNA was DNase treated and reverse transcribed using a QuantiTect reverse transcription kit (Qiagen). Real-time PCR was performed on the synthesized DNA using the SYBR Select master mix (Invitrogen) and a 7900HT real-time system (Applied Biosciences) according to the manufacturer's instructions. Primers sequences used for target gene amplification are available upon request. Samples were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and expressed as the fold change with respect to mock-treated vector control cells. Statistical analyses for gene expression were performed by using ANOVA with a Bonferroni correction in GraphPad Prism version 5.0; P values of <0.05 were considered significant.
Data dissemination.
The data used in this study are available via the following accession identifiers on the NCBI-GEO database: A/duck/Alberta/35/76 (DUCK), A/Brevig Mission/1/18 (1918 WT), 1918-like avian virus, 1918 HA/avian, and 1918 PB2/avian viruses with the associated mock-infected conditions (GSE70502); 1918 WT, 1918HA/K173, 1918PB1/K173, and 1918(3P+NP)/K173 viruses (GSE44595); and A/California/04/2009, mouse-adapted A/California/04/2009, A/New Jersey/8/76, A/Brisbane/59/2007, A/Mexico/4482/2009, and 1918 WT viruses (GSE36328).
RESULTS
The 1918 PB2 and HA genes enhance the replicative potential of a 1918-like avian virus.
In previous studies, a reconstructed 1918-like avian virus containing viral segments that share close homology to the 1918 virus was moderately pathogenic (i.e., a mouse 50% lethal dose [MLD50] of 5.5 log10 PFU) in mice compared to an authentic, low-pathogenicity avian influenza virus (A/duck/ALB/35/76; DK/ALB) (MLD50 of 6.8 log10 PFU) and the highly pathogenic 1918 pandemic virus (MLD50 of 2.7 log10 PFU) (9). To understand the impact of the HA and PB2 genes, which contribute to the host range, pathogenicity, and replication of the 1918 and H5N1 viruses (12, 23, 27, 42) on the virulence of the 1918-like avian virus, we constructed single-gene reassortant viruses composed of either the 1918 HA or PB2 gene in the genetic background of the 1918-like avian virus (Fig. 1A). Introduction of the 1918 PB2 gene (1918 PB2/avian) into the 1918-like avian virus backbone decreased the MLD50 (MLD50 of 1.3 log10 PFU). On the other hand, introduction of the 1918 HA gene (1918 HA/avian) into the avian backbone had no impact on pathogenicity (MLD50 of 5.5 log10 PFU) (Fig. 1B). These results suggest that in the mouse, mammalian adaptation conferred by 1918 PB2, but not HA, promotes pathogenesis of a virus closely resembling the 1918 pandemic strain.
FIG 1.
Pathogenicity of the 1918 and 1918-like reassortant viruses. The biological properties of the 1918 WT, 1918-like avian, 1918 PB2/avian, and 1918 HA/avian viruses were determined. (A) Schematic of 1918 viral gene reassortants. (B) Calculation of median lethal dose (MLD50) of 1918 PB2 and 1918 HA reassortant viruses. MLD50 values for 1918 WT and the 1918-like avian viruses were previously calculated and reported by Watanabe et al. (9). (C) Lung titers of C57/Bl6 mice intranasally inoculated with 104 PFU of virus. Viral titers were determined by plaque assay in MDCK cells. Represented are the mean lung titers (n = 4/virus) and their standard deviations. Asterisks indicate statistical significance: ***, P < 0.001; **, P < 0.01; and *, P < 0.05.
We then evaluated the viral growth kinetics in the lung after intranasal inoculation of mice with 1918 WT, 1918-like avian, 1918 PB2/avian, 1918 HA/avian, or DK/ALB (n = 4/virus; 106 PFU). Lung titers of 1918 WT, 1918-like avian, 1918 PB2/avian, and 1918 HA/avian viruses were significantly greater than those of the nonpathogenic avian strain, DK/ALB, at all time points. The growth of the 1918-like avian virus was three orders of magnitude lower than that of the 1918 WT virus (Fig. 1C). We observed a 10-fold increase in the lung viral titers after insertion of the 1918 HA into the avian backbone. Similarly, 1918 PB2 had a robust impact on viral replication as we observed viral growth comparable to that of the 1918 WT virus. Moreover, 3 of 4 mice infected with 1918 PB2/avian succumbed to viral infection by day 4 postinfection (p.i.), while all other mice survived viral challenge. Furthermore, mammalian adaptation of the 1918-like avian PB2 gene through substitution of 627 glutamic acid residue to lysine was sufficient to enhance the polymerase activity of the 1918-like avian virus polymerase complex in human epithelial cell lines (9) and increased viral virulence in infected mice (MLD50 of 1.4 log10 PFU). These results suggest that both the 1918 PB2 and the HA genes enhance the replicative potential of the 1918-like avian virus, but only the 1918 PB2 promotes viral pathogenicity in the backbone of an avian virus sharing high homology to 1918. Given the similarity in the viral replication of 1918 PB2/avian and 1918 WT, the enhanced lethality observed in 1918 PB2/avian-infected mice is likely independent of viral replication and suggests that the virus-host interactions that shape the host response are a major factor of viral pathogenicity.
1918 PB2 enhances the kinetics and magnitude of the early host response to the 1918-like avian virus.
To evaluate distinctions in the host responses elicited by the 1918 WT and the 1918-like avian viruses, we profiled the global lung transcriptional changes in mice infected with viruses of high (1918 WT and 1918 PB2/avian), intermediate (1918-like avian and 1918HA/avian), and low (DK/ALB) pathogenicity. These profiles were visualized by multidimensional scaling (MDS) and a high degree of similarity between the 1918 WT, 1918-like avian, and 1918 HA/avian viruses at day 1 p.i. was revealed. On the other hand, the transcriptional signature of 1918 PB2/avian diverged from all other infection conditions beginning at day 1. However, at days 2 and 4 p.i., gene expression profiles induced by the highly pathogenic viruses were closely related and diverged from the profiles induced by viruses with intermediate pathogenicity (Fig. 2A).
FIG 2.

Global analysis of the transcriptional profile of 1918 and 1918-like avian virus infected lungs. (A) Multidimensional scaling (MDS) representation of the similarities in the lung transcriptional profiles elicited by viral infection over time. Each biological replicate is represented as a single point, where color denotes virus infection and shape denotes day postinfection. The quality of the representation is provided by the Kruskal stress value, with a low percentage of Kruskal stress (15.02%) suggesting a faithful two-dimensional representation of global transcriptional differences between viral strains. (B) Number of highly differentially expressed (DE) genes following virus infection relative to time-matched mocks. Differential gene expression cutoff was set to |log2 FC| >3 and a q-value < 0.05 calculated using a moderated t test with subsequent Benjamini-Hochberg correction. (C) Hierarchical clustering of the union (4,013 genes) of DE genes based on Euclidean distance reveals distinct modules of gene expression in influenza-infected lungs. A heat map represents the average expression intensities for each infectious condition and time point relative to the average of time-matched mock-infected samples. (D to F) Time- and strain-dependent distribution of DE genes following infections with influenza viruses. Venn diagrams show the overlap of DE genes at day 1 (D), day 2 (E), and day 4 (F) postinfection.
We detected robust changes in transcriptional responses early after infection with highly or intermediately pathogenic viruses (log fold change >|3|). The modest detectable changes in differentially expressed (DE) genes, and their decrease to baseline in DK/ALB-infected mice, were suggestive of the control and resolution of viral infection. Expression of 1918 PB2 had a stark impact on host gene expression during later time points given that we detected a >3-fold increase in the number of DE genes at the later time points of infection (Fig. 2B). By determining the intersection among DE genes for each condition at each time point (Fig. 2D to F), we noted a high degree of overlap in the gene expression promoted by 1918 PB2 expression at days 2 and 4 p.i. (Fig. 2E and F), supporting the observations made through the MDS analysis.
To further probe the consequences of 1918 PB2 on lung gene expression and the molecular pathways associated with enhanced viral pathogenesis, we clustered the 4,013 DE genes according to their expression pattern (Fig. 2C) and performed canonical pathway enrichment analysis on gene sets corresponding to each cluster (Table 1). Overall, the magnitude of gene expression was greater in response to viruses expressing the 1918 PB2 genes (1918 WT and 1918 PB2/avian) during the course of infection. In all infection conditions, we observed the upregulation of pattern recognition receptor-mediated antiviral responses and inflammatory responses (cluster 5). There was sustained expression of genes involved in antiviral and inflammatory responses (clusters 1, 4, and 5), and the late induction of DNA damage and cell cycle regulatory genes (cluster 3), as well as genes involved in wound healing (cluster 2), that was concomitant with increased virulence of the 1918 and the 1918-like viruses. Taken together, these data demonstrate that mammalian adaptation of PB2, rather than HA, affects both the kinetics and the magnitude of host gene expression culminating in the enhancement of the pathogenesis of a 1918-like avian virus.
TABLE 1.
Canonical pathways affected by 1918, 1918-like avian, and reassortant viruses
| Canonical pathway | P | Ratio |
|---|---|---|
| Cluster 1 “orange” (905 transcripts) | ||
| Communication between innate and adaptive immune cells | 1.60E–12 | 18/64 |
| Allograft rejection signaling | 3.78E–11 | 14/42 |
| Cross talk between dendritic cells and natural killer cells | 6.61E–10 | 16/69 |
| Autoimmune thyroid disease signaling | 2.27E–09 | 11/31 |
| GVHD signaling | 1.30E–07 | 10/35 |
| Cluster 2 “sky blue” (675 transcripts) | ||
| Glycolysis I | 1.82E–04 | 5/23 |
| Hepatic fibrosis/hepatic stellate cell activation | 1.91E–04 | 14/189 |
| Intrinsic prothrombin activation pathway | 4.03E–04 | 5/27 |
| TR/RXR activation | 1.31E–02 | 6/81 |
| Sucrose degradation V (mammalian) | 1.46E–02 | 2/8 |
| Cluster 3 “darkblue” (167 transcripts) | ||
| Cell cycle: G2/M DNA damage checkpoint regulation | 2.47E–12 | 10/49 |
| Mitotic roles of Polo-like kinase | 6.90E–10 | 9/61 |
| ATM signaling | 2.75E–07 | 7/59 |
| DNA damage-induced 14-3-3σ signaling | 2.32E–07 | 5/19 |
| GADD45 signaling | 1.06E–05 | 4/19 |
| Cluster 4 “black” (353 transcripts) | ||
| Granulocyte adhesion and diapedesis | 9.21E–13 | 18/151 |
| Interferon signaling | 6.53E–11 | 9/29 |
| Role of PRRs in recognition of bacteria and viruses | 2.84E–10 | 14/116 |
| Agranulocyte adhesion and diapedesis | 2.54E–09 | 15/161 |
| Dendritic cell maturation | 6.58E–09 | 14/147 |
| Cluster 5 “dark red” (103 transcripts) | ||
| LXR/RXR activation | 6.94E–05 | 5/108 |
| Role of PRRs in recognition of bacteria and viruses | 1.17E–03 | 4/116 |
| IL-10 signaling | 2.20E–03 | 3/65 |
| Role of hypercytokinemia/chemokinemia in the pathogenesis of influenza | 5.12E–03 | 2/27 |
| Role of RIG-I-like receptors in antiviral innate immunity | 6.30E–03 | 2/30 |
| Cluster 6 “yellow” (583 transcripts) | ||
| Calcium-induced T lymphocyte apoptosis | 1.64E–03 | 6/54 |
| B cell development | 1.88E–03 | 4/23 |
| Nur77 signaling in T lymphocytes | 4.81E–03 | 5/47 |
| Tight-junction signaling | 4.89E–03 | 10/161 |
| Autoimmune thyroid disease signaling | 5.77E–03 | 4/31 |
| Cluster 7 “red” (1,230 transcripts) | ||
| Retinol biosynthesis | 2.87E–04 | 7/29 |
| Tryptophan degradation X (mammalian, via tryptamine) | 6.05E–04 | 5/16 |
| Axonal guidance signaling | 6.39E–04 | 35/421 |
| Glutathione-mediated detoxification | 8.13E–04 | 6/25 |
| Ethanol degradation IV | 1.10E–03 | 5/18 |
1918 PB2 is a key driver of inflammatory responses enhancing immune cell infiltration and activation in the infected lung.
Previously, we reported that the early inflammatory response to IAV infection can severely impact the disease outcome (35). To better understand whether 1918 PB2 contributes to these detrimental responses, we focused on genes upregulated immediately following IAV infection at day 1 p.i. and whose expression was sustained throughout infection only in 1918 WT and 1918 PB2/avian infected mice (Fig. 2C; cluster 4, “black”). These genes were specifically involved in innate responses, the communication between innate and adaptive immune cells (P = 1.57E–6), and granulocyte/agranulocyte adhesion and diapedesis (Table 1). We integrated our early response profiles (day 1 p.i.) with those from previous studies of H1N1 infection to determine whether 1918 PB2 expression led to increased gene expression (17, 35). Approximately 20% of cluster 4 genes were significantly altered by 1918 PB2, including cytokines and chemokine genes and their receptors, such as Csf3, Il1b, Cxcl2, and Cxcr2, as well as the cytokine regulatory gene, Socs3 (Fig. 3A). We next evaluated potential regulators mediating the cluster 4 gene expression by utilizing ChIP enrichment analysis (ChEA). We identified Stat3, a crucial regulator of interleukin-6 (IL-6)-mediated inflammation and lung repair (43), as an upstream regulator of the response to 1918 WT and 1918-like avian viruses (Fig. 3B). Additionally, regulators of lymphocyte development (Lmo2, Runx1, and Gata2), differentiation (Stat4 and Stat5), or immune cell-specific activation (Irf8, Spi1, and Myb) were found to be critical regulators of the sustained inflammatory responses by 1918 PB2. Thus, we hypothesized that expression of 1918 PB2 could promote increased infiltration and activation of immune cells in the lung. Indeed, 1918 PB2/avian-infected lungs had a greater representation of genes involved in the recruitment, infiltration, and quantity of immune cells than that observed in 1918-like avian and 1918 HA/avian infected lungs (activation z-score > 2) on day 1 p.i. (Fig. 3C).
FIG 3.

1918 PB2 promotes proinflammatory responses associated with increased lymphocyte infiltration into the lung. (A) Meta-analysis of early lung transcriptional responses following H1N1 virus infection. A heat map represents the relative expression of 57 genes within the “black” gene cluster (cluster 4) (derived from Fig. 2C) that display increased expression when 1918 PB2 is expressed in infected lungs at day 1 p.i. Viruses expressing the 1918 PB2 gene are located on the left side of the heat map. Viruses which contain PB2 segments that differ from 1918 PB2 are clustered in the right side of the heat map. Statistical significance cutoff was set to a P value of <0.05 as was determined by one-tailed t test. (B) Transcription factors associated with the regulation of gene expression of transcripts associated with the “black” gene cluster (cluster 4). Regulators were identified using ChIP-X enrichment analysis (ChEA). Bar graphs represent the combined scores for each regulator (bar length) and the number of target genes associated with each regulator. The PubMed Identifier (PMID) for the specific gene sets associated with each transcriptional regulator is shown. (C) Gene set enrichment of biological functions pertaining to immune cell regulation derived from IPA. A bubble plot representation of significant enrichment scores (activation z-score > 2) in at least one infectious condition at day 1 p.i. is shown. Crosses signify a lack of significant enrichment. Bubble diameter represents the −log10 P value as determined by Fisher exact test.
To dissect the dynamics of the immune cell infiltration associated with 1918 PB2 expression, we used an integrative statistical approach that identifies genes overexpressed in distinct immune cells allowing us to infer specific cellular infiltration into the lung (35, 44, 45). Using this approach, we captured an enrichment of macrophage (Fig. 4A, left) and granulocyte-specific (Fig. 4A, right) genes in all infection conditions throughout the course of infection. There was unique enrichment of NK and T cells in the lung of mice infected with 1918 PB2 expressing viruses (Fig. 4A, right). To better understand the contribution of these predicted cell types toward 1918 pathogenesis, we used a second algorithm for immune cell deconvolution, a digital cell quantifier (DCQ). The DCQ is a previously validated algorithm that leverages cell type-specific whole-genome transcriptional expression profiles derived from 207 immune cells (Immgen; reviewed in reference 46) to infer changes in immune cell quantities based on cell-specific surface markers (39). We used the DCQ to derive the relative quantity of predicted cells within the infected lung and infer their activation states. Specifically, we focused on the immune subsets inferred through both of our enrichment approaches. Like our previous analysis, the DCQ revealed a general increase in granulocytes, dendritic cells, and monocytes/macrophages throughout the course of all infections (data not shown). However, we were able to capture a distinct recruitment of classical Ly6C+ inflammatory monocytes (MO.6C+II- and MO.6C+II-.BL) (Fig. 4B), as well as enhancement of different macrophage subsets infiltrating the lung (Fig. 4C) by 1918 PB2 expressing viruses. In particular, we observed a specific increase in activated macrophages (MF.11.cloSER.Salm3.SI and MF.103-11+.Salm3.SI) that corresponded with the enhanced morbidity of the 1918 PB2/avian virus.
FIG 4.
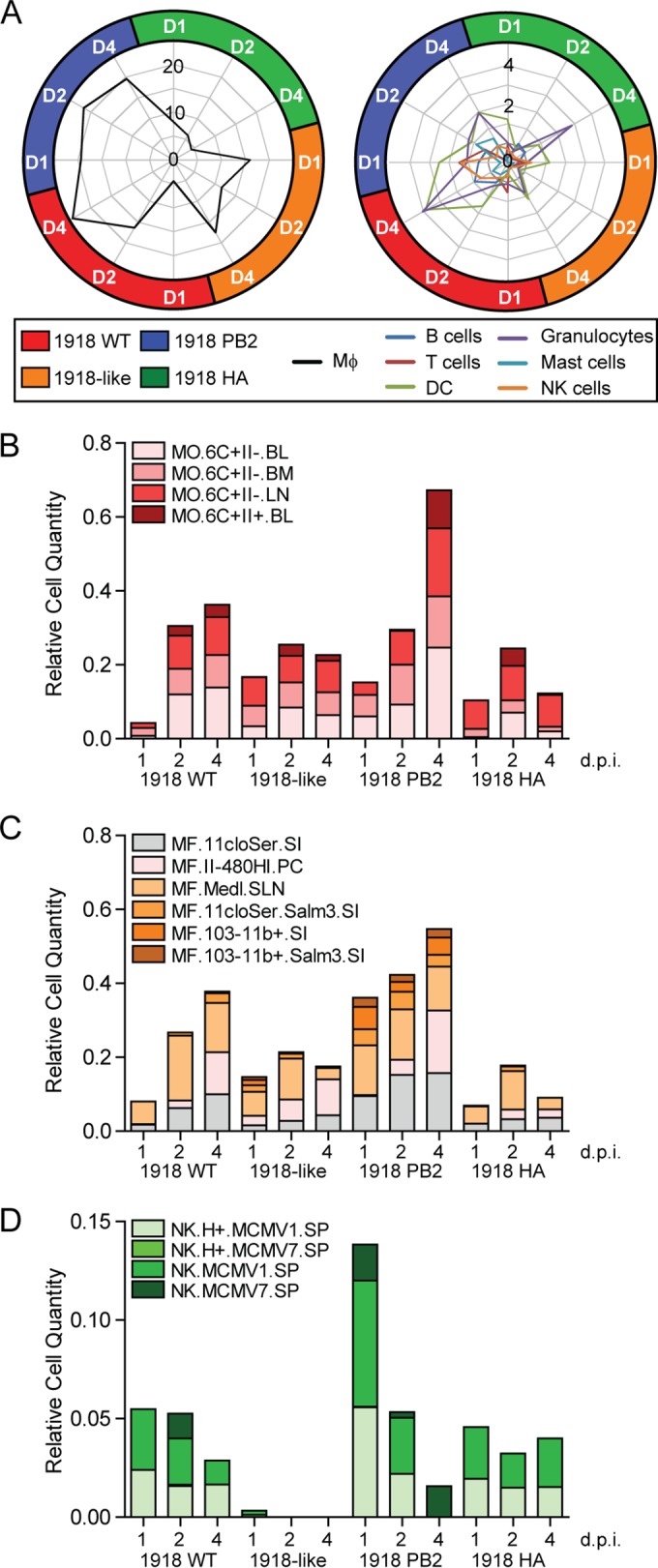
1918 PB2 promotes the recruitment of distinct monocytes, macrophages, and activated NK cells, contributing to pulmonary inflammation. (A) Computational deconvolution of immune cell subsets using GeneAtlas v3. Represented in the radar plot are the −log10 (P value) enrichment scores for DE genes corresponding to distinct cell subsets as determined by Fisher exact test. The threshold of significant enrichment was set at 1.3 −log10 P value. (B to D) Immune cell dynamics in infected lungs as predicted by digital cell quantifier (DCQ). Bar graphs show the relative cell quantities for monocytes (B), macrophages (C), and NK cells (D) after influenza virus infection at the indicated time points. Distinct colors have been assigned to each immune cell type from the Immgen compendium. Each cell type name is followed by the tissue from which it was previously isolated, abbreviated as follows: BL, blood; BM, bone marrow; LN, lymph nodes; SI, small intestine; PC, peritoneal cavity; SLN, subcutaneous LN; and SP, spleen.
Next, we examined the effect of 1918 PB2 expression in T cell and NK cell dynamics. Although no notable changes in T helper cells were captured throughout the course of infection (data not shown), we were able to unveil a robust activation of CD8+ T cell responses, which had not been captured initially through our enrichment analysis of either 1918-like avian or 1918 HA/avian-infected cells (Fig. 4A, right). Examination of NK cell-specific dynamics demonstrated that 1918 PB2 expression impacts both cell quantity and the distinct activation state throughout the course of infection. Specifically, 1918 PB2 expression was associated with a greater representation of host genes which overlapped with those observed in virus-stimulated NK cells (NK.MCMV7.SP) (Fig. 4D). Severe lung pathology caused by the 1918 virus has been largely attributed to increased immune cell infiltrate in the lung (47), which accompanies protracted inflammatory responses. Our results suggest that 1918 PB2 exacerbates pulmonary inflammation and tissue damage by promoting both the recruitment and activation of inflammatory monocytes, macrophages, and activated NK cells (48, 49).
Loss of Wnt signaling in the lung results in increased viral pathogenesis.
Having characterized the inflammatory responses enhanced by 1918 PB2, we next explored specific biological processes inhibited by viral infection. Overall, we observed an increase in the repression of genes associated with adaptive immune responses (predominantly major histocompatibility complex class II genes), epithelial barrier integrity (cluster 6), and lung metabolic processes (cluster 7) in infected lungs (Fig. 2C and Table 1). To identify specific regulators inhibiting these cellular processes, we utilized ChEA (38) and ranked the predicted transcription factors (TFs) controlling the expression of the union of downregulated transcripts. Among the top-ranked TFs, we identified regulators that are crucial for the maintenance of lung homeostasis (Wt1) (50), inflammation-promoted epithelial regeneration (Yap1) (51), members of the polycomb repressive complex (PRC2) (Suz12 and Ezh2) (52), and PRC2 regulatory proteins (Jarid2 and Mtf2) (53, 54) (Fig. 5A). Additionally, we identified the LEF/TCF member, Tcf3, which in concert with β-catenin regulates the expression of the Wnt/β-catenin signaling pathway effector genes (Fig. 5A). Given that several of the enriched TFs regulate Wnt gene expression and downstream Wnt effector functions (55–57), we hypothesized that expression of 1918 PB2 leads to dysregulation of Wnt signaling pathways.
FIG 5.

Downregulation of Wnt signaling-associated genes contributes to viral pathogenesis. Functional analysis of downregulated genes following viral infection demonstrates the inhibition of Wnt signaling genes. (A) Ranking of the top transcriptional regulators of downregulated genes in the infected lung during the course of infection. Regulators were identified using ChEA. Bar graphs represent the combined scores for each regulator (bar length) and the number of target genes associated with each regulator. The PMID for the specific gene sets associated with each transcriptional regulator is shown. (B) IPA-generated regulatory network analysis of the top-ranked TFs. Each node represents a transcription factor or regulatory cytokine. Arrows indicate the relationship between TFs. Color indicates the predicted (blue) or observed (green) inhibition of molecule activity. (C) Canonical pathways associated with Wnt genes. Pathway enrichment was reported as (−log10 P value). Bubbles represent significant pathway enrichment, as determined by Fisher exact test. (D) Heat map of expression intensities of Wnt signaling-associated genes.
We first evaluated the relationship between Wnt ligands and the ChEA-identified transcriptional regulators by constructing a regulatory network using Ingenuity Pathway Analysis (IPA). Most of the top-ranking TFs were found to be directly linked to Wnt or indirectly through Wnt-associated TFs within the network (Fig. 5B). We then examined the impact of 1918 PB2 expression on the activation state of these transcriptional regulators, taking into account the global changes in gene expression and their directionality. Expression of 1918 PB2 led to loss of Tcf3 activity, as observed in 1918 PB2/avian day 1 p.i. profiles. Similar inferences were made based on day 4 p.i. 1918 WT-infected lung transcriptional profiles, but not from profiles expressing the avian PB2 homologue (data not shown).
Expression of 1918 PB2 resulted in the downregulation of transcripts associated with both canonical (Wnt/β-catenin) and noncanonical (Wnt/PCP) Wnt signaling (Fig. 5C). Significant Wnt-associated pathway enrichment was evident within the first 48 h after infection with viruses expressing the 1918 PB2 gene (1918 WT and 1918 PB2/avian) and to a lesser extent in the 1918-like avian virus-infected lungs during late acute infection. Not only did the expression of 1918 PB2 result in the differential expression of an increased number of genes encoding Wnt ligands (Wnt2, Wnt3A, and Wnt10), receptors (Lrp4/6 and Fzd2), inhibitors (Sox9), and adaptor molecules, but 1918 PB2 also affected the magnitude of gene expression during the course of infection compared to 1918-like avian and 1918 HA/avian infection (Fig. 5D).
To determine whether expression of PB2, in the absence of viral infection, is sufficient to perturb the Wnt/β-catenin signaling cascade, we examined the effect of PB2 expression on the transcriptional activation of a β-catenin responsive promoter (40). In human lung epithelial cells, transient expression of either the 1918 PB2 or the 1918-like avian PB2 gene resulted in a significant inhibition of luciferase activity relative to that observed upon expression of yellow fluorescent protein (YFP) in the presence of Wnt3A stimulation. The inhibition of β-catenin activity was comparable between both the 1918 and the 1918-like PB2 expressing cells at both 12 and 24 h poststimulation (Fig. 6A). We further evaluated the inhibitory effect of PB2 by examining the endogenous mRNA levels of known Wnt-targeted genes, TROY/TNFRSF19, Axin2, Nkd1, and Lef1 in Wnt3A-stimulated cells (Fig. 6B). Consistent with our previous observation, PB2 expression led to marked decreases in transcript levels within 12 h of cytokine stimulation (Fig. 6B, left), which were still noticeable at 24 h poststimulation (Fig. 6B, right). There were significant differences in the inhibition of TROY, Axin2, and NKD1 gene expression between the two PB2 proteins at 12 h poststimulation, with 1918 PB2 eliciting a more robust significant inhibition. The integrity of the lung epithelium is essential for normal lung function and Wnt/β-catenin signaling plays an important role in lung development and repair following injury (58). Thus, by directly repressing the canonical Wnt signaling cascade, 1918 PB2 abrogates the signaling pathways that coordinate lung repair.
FIG 6.

The PB2 gene inhibits Wnt-mediated signal transduction. Expression of 1918 PB2 or the1918-like avian PB2 gene results in the inhibition of Wnt signaling in vitro. (A) Inhibition of β-catenin promoter activation in A549 cells. A549-BAR reporter cells were transfected with 4 μg of 1918 PB2, avian PB2, or YFP expression constructs. Cells were stimulated with Wnt3A (50 ng/ml) or vehicle control 48 h posttransfection. Firefly luciferase and renilla luciferase activity was measured at the indicated time points. Firefly luciferase activity was normalized to the renilla luciferase activity. Bar graphs represent the change in firefly luciferase activity relative to time-matched, vehicle-treated cells. Asterisks indicate statistical significance: ***, P < 0.001; **, P < 0.01; and *, P < 0.05). (B) Regulation of endogenous Wnt-mediated gene expression by PB2 genes. A549-BAR cells were transiently transfected with a 1918 PB2, avian PB2, or YFP expression construct as previously indicated. At 24 h posttransfection, fresh culture medium was added to cells, which were incubated for an additional 24 h prior to stimulation with Wnt3A (50 ng/ml) or vehicle control. Total RNA was harvested at the indicated time points and the expression of target genes was quantified by quantitative PCR. Bar graphs represent the fold change in gene expression normalized to GAPDH and time-matched, vehicle-treated cells. Asterisks indicate statistical significance: ***, P < 0.001; **, P < 0.01; and *, P < 0.05.
Network analysis identifies the relationship between inflammatory and epithelial repair responses disrupted by 1918 PB2.
To put the relationships between the biological processes affected by 1918 PB2 into perspective, we used an integrative network approach that evaluated the connections between the coexpressed genes derived from day 1 p.i. 1918 PB2/avian-infected lung transcriptional profiles. The top-scored network generated from the molecular network analysis was enriched with genes involved primarily in cell-to-cell signaling interaction, inflammatory responses, and infectious disease as expected from our previous observations (Fig. 3). Within the manually curated subnetworks, we identified a network associated with cellular development, growth, and proliferation in which Wnt3A was among the highly connected nodes (hubs) (Fig. 7A). Hubs exert regulatory influence within the network by controlling information flow between different regions of the network. Interestingly, we also identified Jnk as a hub, associating the Wnt signaling with lung inflammatory responses and immune cell infiltration into the lung (Fig. 7A).
FIG 7.

Network analysis of the host responses affected by 1918 PB2 after IAV infection. Identification of disease and biological functions associated with Wnt signaling. (A) Molecular network analysis was performed using IPA. Networks were limited to 70 focus molecules. Within these networks, we identified a network containing 37 focus genes that is presented as a graph indicating the molecular relationships between inflammatory and Wnt-associated genes. Genes and gene products are represented as nodes (circles), and the biological relationship between two nodes is represented by edges (lines). Node color (pink or blue) indicates up- or downregulation, respectively. Uncolored nodes represent genes that are relevant to the network but were not identified as DE within the data set. Labels indicate gene set overlay and the most enriched biological functions associated with the identified network. (B) Heat map of gene product expression intensities of nodes contained within the network. The results represent the average log2-fold change in expression throughout the course of infection with 1918 WT, 1918-like/avian, 1918 PB2/avian, and 1918 HA/avian viruses. (C) Schematic of proposed model of 1918 PB2 dysregulation of inflammatory responses and Wnt-mediated signaling leading to viral pathogenesis.
1918 PB2 enhanced both the upregulation of genes highly connected to Jak2, an adaptor molecule of IL-6 family cytokine signaling, as well as the downregulation of genes downstream of Wnt3a (Fig. 7B). Given that in vitro studies have shown that canonical Wnt signaling inhibits IAV replication and controls IAV-induced inflammatory responses (59), our data suggest that in the context of an in vivo infection, the PB2 gene also inhibits the activation of the Wnt/β-catenin signaling pathway. The enhanced viral replication and detrimental inflammatory responses promoted by 1918 PB2, coupled with the loss of the regeneration and repair functions coordinated by Wnt, drive pathogenic IAV disease (Fig. 7C).
DISCUSSION
Viral virulence determinants, and their adaptive changes, promoting protracted inflammatory responses during 1918 influenza virus infection is an area of ongoing investigation. In this study, we examined host transcriptional profiles induced by a 1918-like avian virus sharing high homology to the pandemic 1918 virus and reassortant viruses carrying either the 1918 PB2 or HA genes to better understand the contribution of these host-adaptive genes to pathogenesis of the 1918 virus. Our results indicate that although the 1918 HA and PB2 genes both enhanced the replication of the 1918-like avian virus, only 1918 PB2 impacted viral virulence. Expression of 1918 HA had less impact on the morbidity caused by a 1918-like avian virus. This is in contrast to results obtained in reassortant studies that used a contemporary human H1N1 virus (A/Kawasaki/173/2001; K173) as a backbone. 1918 HA enhanced the replication of K173 in murine lungs and caused increased morbidity (MLD50 = 4.8 log10 PFU) compared to the parental virus (MLD50 = >6.5 log10 PFU) (17). One key difference is that the present study examines the contribution of HA and PB2 in the background of a virus with increased viral replication capacity and that is more pathogenic (1918-like avian; MLD50 = 5.5 log10 PFU) than either K173 or DK/ALB in mice. In the context of an intermediately pathogenic 1918-like avian virus, the effect of mammalian adaptation through lysine substitution of the 627 glutamic acid residue of PB2 is necessary and sufficient to promote virulence in mice. Although 1918 HA alone did not impact the 1918-like avian virus virulence in mice, it has been demonstrated that both the 1918-derived HA and PB2 act synergistically to regulate the transmissibility of the 1918 virus (9, 60). Despite having a replicative efficiency that was comparable to the 1918 virus, the 1918 PB2/avian virus displayed increased virulence in mice over the 1918 WT virus. It is likely that the enhanced lethality observed in 1918 PB2/avian-infected mice is attributed not only to increased viral replication (Fig. 1C) but also to the potential interactions between the 1918 PB2 polymerase subunit and the viral proteins encoded by the 1918-like avian virus (61), as well as specific viral and host protein interactions that are also necessary drivers of viral pathogenesis (62). Genome-wide transcriptional analyses provide an unbiased view of the regulatory processes that are perturbed during the course of viral infection. Global transcriptional profiling of infected lungs demonstrated that infection with a virus possessing 1918 PB2 led to increased dysregulation of host responses to viral infection. Concomitant with pathogenic 1918 PB2-expressing viruses was the elevated expression of genes involved in inflammatory responses that was consistent with the recruitment of leukocytes and lymphocytes into the infected lung (Fig. 3). Previously, we showed that unabated antiviral and inflammatory responses associated with the recruitment of proinflammatory monocytes and macrophages results in increased viral virulence (35). Here, we show that in addition to the recruitment of granulocytes and agranulocytes into the lung, 1918 PB2 promotes the infiltration and activation of NK cells following infection. Although NK cells are crucial mediators of the antiviral responses to influenza virus infection, their excessive activation contributes to lung inflammation and tissue damage (48).
Disruption of lung function, airway epithelial integrity, and airway remodeling has been associated with influenza virus disease severity. Our gene network analysis revealed that the function of the 1918 PB2 gene as a virulence factor is associated with inhibition of canonical and noncanonical Wnt signaling pathways. We observed a greater repression of Wnt genes involved in the maintenance of lung homeostasis and repair in lungs infected with the 1918 WT virus or the 1918 PB2 reassortant virus than in lungs infected with the 1918-like avian virus. In the presence of 1918 PB2, there was decreased expression of Wnt, Notch, and Shh genes encoding developmental regulators that have been implicated in lung disease. Decreased Wnt/β-catenin signaling and Notch signaling are hallmarks of chronic obstructive pulmonary disease (63). Furthermore, Wnt/β-catenin activation ameliorates emphysema in an experimental murine model (64). While the exact mechanism by which these developmental factors contribute to the regeneration of the adult lung are not clearly understood, it has been proposed that Wnt pathways are important for the regulation of lung epithelial progenitor cell maintenance. Additionally, active repression of canonical and noncanonical Wnt signaling in the infected lung results in the loss of negative regulation of IL-6-mediated regenerative inflammatory responses (65), thereby enhancing tissue damage (51). Furthermore, evidence supports an important role for Wnt in shaping noxious inflammatory responses (66). Thus, by both promoting increased cellular infiltration and downregulating Wnt inhibitory signals, 1918 PB2 contributes to the unabated inflammatory responses that are associated with severe IAV disease.
Previous studies using primary epithelial cells and cell lines have shown that Wnt receptor signaling modulates antiviral responses to influenza virus infection (59, 67). These studies demonstrated direct interactions between the IAV polymerase and matrix proteins and various proteins in the Wnt signaling network (59), potentially leading to the loss of downstream effector functions. Also, PB2 has been previously implicated in the negative regulation of MAVS-mediated antiviral responses (68–70). Thus, the direct repression of Wnt/β-catenin signaling by PB2 serves as an additional strategy for immune evasion. However, the relevance of these observations in the context of an in vivo infection had yet to be addressed. Through a combination of viral genetics and host transcriptional profiling, we provide a multidimensional view on the molecular mechanisms involved in the response to influenza virus infection and show the relationship between 1918 PB2 and the balanced inflammatory/Wnt signaling pathways in vivo that when perturbed during influenza virus infection lead to increased morbidity and immunopathogenesis.
ACKNOWLEDGMENTS
This project was funded by Public Health Service grants U19AI109761, R01AI080598, and P51OD010425 from the National Institutes of Health and by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health; by the Japan Initiative for Global Research Network on Infectious Diseases from the Ministry of Education, Culture, Sports, Science, and Technology; and by grants-in-aid from the Ministry of Health and by the ERATO Japan Science and Technology Agency.
The findings and conclusions in this report are those of the authors and do not necessarily reflect the views of the funding agencies.
We thank Thomas J. Hope at Northwestern University for kindly providing the YFP expression vector and Marcus J. Korth for a thoughtful review of the manuscript.
REFERENCES
- 1.Neumann G, Kawaoka Y. 2011. The first influenza pandemic of the new millennium. Influenza Other Respir Viruses 5:157–166. doi: 10.1111/j.1750-2659.2011.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson NP, Mueller J. 2002. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull Hist Med 76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- 3.Taubenberger JK, Morens DM. 2006. 1918 Influenza: the mother of all pandemics. Emerg Infect Dis 12:15–22. doi: 10.3201/eid1209.05-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabadan R, Levine AJ, Robins H. 2006. Comparison of avian and human influenza A viruses reveals a mutational bias on the viral genomes. J Virol 80:11887–11891. doi: 10.1128/JVI.01414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reid AH, Fanning TG, Janczewski TA, Lourens RM, Taubenberger JK. 2004. Novel origin of the 1918 pandemic influenza virus nucleoprotein gene. J Virol 78:12462–12470. doi: 10.1128/JVI.78.22.12462-12470.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reid AH, Taubenberger JK, Fanning TG. 2004. Evidence of an absence: the genetic origins of the 1918 pandemic influenza virus. Nat Rev Microbiol 2:909–914. doi: 10.1038/nrmicro1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith GJ, Bahl J, Vijaykrishna D, Zhang J, Poon LL, Chen H, Webster RG, Peiris JS, Guan Y. 2009. Dating the emergence of pandemic influenza viruses. Proc Natl Acad Sci U S A 106:11709–11712. doi: 10.1073/pnas.0904991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Worobey M, Han GZ, Rambaut A. 2014. Genesis and pathogenesis of the 1918 pandemic H1N1 influenza A virus. Proc Natl Acad Sci U S A 111:8107–8112. doi: 10.1073/pnas.1324197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe T, Zhong G, Russell CA, Nakajima N, Hatta M, Hanson A, McBride R, Burke DF, Takahashi K, Fukuyama S, Tomita Y, Maher EA, Watanabe S, Imai M, Neumann G, Hasegawa H, Paulson JC, Smith DJ, Kawaoka Y. 2014. Circulating avian influenza viruses closely related to the 1918 virus have pandemic potential. Cell Host Microbe 15:692–705. doi: 10.1016/j.chom.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen GW, Chang SC, Mok CK, Lo YL, Kung YN, Huang JH, Shih YH, Wang JY, Chiang C, Chen CJ, Shih SR. 2006. Genomic signatures of human versus avian influenza A viruses. Emerg Infect Dis 12:1353–1360. doi: 10.3201/eid1209.060276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabriel G, Dauber B, Wolff T, Planz O, Klenk HD, Stech J. 2005. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc Natl Acad Sci U S A 102:18590–18595. doi: 10.1073/pnas.0507415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J Virol 67:1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG. 2005. Characterization of the 1918 influenza virus polymerase genes. Nature 437:889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- 14.Danzy S, Studdard LR, Manicassamy B, Solorzano A, Marshall N, Garcia-Sastre A, Steel J, Lowen AC. 2014. Mutations to PB2 and NP proteins of an avian influenza virus combine to confer efficient growth in primary human respiratory cells. J Virol 88:13436–13446. doi: 10.1128/JVI.01093-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pappas C, Aguilar PV, Basler CF, Solorzano A, Zeng H, Perrone LA, Palese P, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. Single gene reassortants identify a critical role for PB1, HA, and NA in the high virulence of the 1918 pandemic influenza virus. Proc Natl Acad Sci U S A 105:3064–3069. doi: 10.1073/pnas.0711815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, Garcia-Sastre A. 2005. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe T, Tisoncik-Go J, Tchitchek N, Watanabe S, Benecke AG, Katze MG, Kawaoka Y. 2013. 1918 Influenza virus hemagglutinin (HA) and the viral RNA polymerase complex enhance viral pathogenicity, but only HA induces aberrant host responses in mice. J Virol 87:5239–5254. doi: 10.1128/JVI.02753-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 19.Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, Van Rooijen N, Katz JM, Basler CF. 2005. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79:14933–14944. doi: 10.1128/JVI.79.23.14933-14944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matrosovich MN, Gambaryan AS, Teneberg S, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Karlsson KA. 1997. Avian influenza A viruses differ from human viruses by recognition of sialyloligosaccharides and gangliosides and by a higher conservation of the HA receptor-binding site. Virology 233:224–234. doi: 10.1006/viro.1997.8580. [DOI] [PubMed] [Google Scholar]
- 21.Gambaryan AS, Tuzikov AB, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Bovin NV, Matrosovich MN. 1997. Specification of receptor-binding phenotypes of influenza virus isolates from different hosts using synthetic sialylglycopolymers: non-egg-adapted human H1 and H3 influenza A and influenza B viruses share a common high binding affinity for 6′-sialyl(N-acetyllactosamine). Virology 232:345–350. doi: 10.1006/viro.1997.8572. [DOI] [PubMed] [Google Scholar]
- 22.Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. 1988. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333:426–431. doi: 10.1038/333426a0. [DOI] [PubMed] [Google Scholar]
- 23.Hatta M, Gao P, Halfmann P, Kawaoka Y. 2001. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293:1840–1842. doi: 10.1126/science.1062882. [DOI] [PubMed] [Google Scholar]
- 24.Hatta M, Hatta Y, Kim JH, Watanabe S, Shinya K, Nguyen T, Lien PS, Le QM, Kawaoka Y. 2007. Growth of H5N1 influenza A viruses in the upper respiratory tracts of mice. PLoS Pathog 3:1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fornek JL, Gillim-Ross L, Santos C, Carter V, Ward JM, Cheng LI, Proll S, Katze MG, Subbarao K. 2009. A single-amino-acid substitution in a polymerase protein of an H5N1 influenza virus is associated with systemic infection and impaired T-cell activation in mice. J Virol 83:11102–11115. doi: 10.1128/JVI.00994-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato A, Mizumoto K, Ishihama A. 1985. Purification and enzymatic properties of an RNA polymerase-RNA complex from influenza virus. Virus Res 3:115–127. doi: 10.1016/0168-1702(85)90002-4. [DOI] [PubMed] [Google Scholar]
- 27.Neumann G, Kawaoka Y. 2006. Host range restriction and pathogenicity in the context of influenza pandemic. Emerg Infect Dis 12:881–886. doi: 10.3201/eid1206.051336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q, Huang J, Chen Y, Chen H, Li Q, He L, Hao X, Liu J, Gu M, Hu J, Wang X, Hu S, Liu X. 2014. Virulence determinants in the PB2 gene of a mouse-adapted H9N2 virus. J Virol 89:877–882. doi: 10.1128/JVI.01775-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabriel G, Czudai-Matwich V, Klenk HD. 2013. Adaptive mutations in the H5N1 polymerase complex. Virus Res 178:53–62. doi: 10.1016/j.virusres.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 30.Manz B, Schwemmle M, Brunotte L. 2013. Adaptation of avian influenza A virus polymerase in mammals to overcome the host species barrier. J Virol 87:7200–7209. doi: 10.1128/JVI.00980-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi L, Davis AS, Jagger BW, Schwartzman LM, Dunham EJ, Kash JC, Taubenberger JK. 2012. Analysis by single-gene reassortment demonstrates that the 1918 influenza virus is functionally compatible with a low-pathogenicity avian influenza virus in mice. J Virol 86:9211–9220. doi: 10.1128/JVI.00887-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG. 2006. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443:578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reed LJ, M H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]
- 35.Josset L, Belser JA, Pantin-Jackwood MJ, Chang JH, Chang ST, Belisle SE, Tumpey TM, Katze MG. 2012. Implication of inflammatory macrophages, nuclear receptors, and interferon regulatory factors in increased virulence of pandemic 2009 H1N1 influenza A virus after host adaptation. J Virol 86:7192–7206. doi: 10.1128/JVI.00563-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson WE, Li C, Rabinovic A. 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 37.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A. 2010. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26:2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altboum Z, Steuerman Y, David E, Barnett-Itzhaki Z, Valadarsky L, Keren-Shaul H, Meningher T, Mendelson E, Mandelboim M, Gat-Viks I, Amit I. 2014. Digital cell quantification identifies global immune cell dynamics during influenza infection. Mol Syst Biol 10:720. doi: 10.1002/msb.134947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biechele TL, Moon RT. 2008. Assaying beta-catenin/TCF transcription with beta-catenin/TCF transcription-based reporter constructs. Methods Mol Biol 468:99–110. doi: 10.1007/978-1-59745-249-6_8. [DOI] [PubMed] [Google Scholar]
- 41.Davidson KC, Adams AM, Goodson JM, McDonald CE, Potter JC, Berndt JD, Biechele TL, Taylor RJ, Moon RT. 2012. Wnt/β-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc Natl Acad Sci U S A 109:4485–4490. doi: 10.1073/pnas.1118777109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Chen H, Jiao P, Deng G, Tian G, Li Y, Hoffmann E, Webster RG, Matsuoka Y, Yu K. 2005. Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J Virol 79:12058–12064. doi: 10.1128/JVI.79.18.12058-12064.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kida H, Mucenski ML, Thitoff AR, Le Cras TD, Park KS, Ikegami M, Muller W, Whitsett JA. 2008. GP130-STAT3 regulates epithelial cell migration and is required for repair of the bronchiolar epithelium. Am J Pathol 172:1542–1554. doi: 10.2353/ajpath.2008.071052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, Saijo K, Glass CK, Hume DA, Kellie S, Sweet MJ. 2008. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res 4:5. doi: 10.1186/1745-7580-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrison J, Josset L, Tchitchek N, Chang J, Belser JA, Swayne DE, Pantin-Jackwood MJ, Tumpey TM, Katze MG. 2014. H7N9 and other pathogenic avian influenza viruses elicit a three-pronged transcriptomic signature that is reminiscent of 1918 influenza virus and is associated with lethal outcome in mice. J Virol 88:10556–10568. doi: 10.1128/JVI.00570-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shay T, Kang J. 2013. Immunological Genome Project and systems immunology. Trends Immunol 34:602–609. doi: 10.1016/j.it.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdul-Careem MF, Mian MF, Yue G, Gillgrass A, Chenoweth MJ, Barra NG, Chew MV, Chan T, Al-Garawi AA, Jordana M, Ashkar AA. 2012. Critical role of natural killer cells in lung immunopathology during influenza infection in mice. J Infect Dis 206:167–177. doi: 10.1093/infdis/jis340. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J, Matsuoka M, Cantor H, Homer R, Enelow RI. 2008. Cutting edge: engagement of NKG2A on CD8+ effector T cells limits immunopathology in influenza pneumonia. J Immunol 180:25–29. doi: 10.4049/jimmunol.180.1.25. [DOI] [PubMed] [Google Scholar]
- 50.Marcet-Palacios M, Ulanova M, Duta F, Puttagunta L, Munoz S, Gibbings D, Radomski M, Cameron L, Mayers I, Befus AD. 2007. The transcription factor Wilms tumor 1 regulates matrix metalloproteinase-9 through a nitric oxide-mediated pathway. J Immunol 179:256–265. doi: 10.4049/jimmunol.179.1.256. [DOI] [PubMed] [Google Scholar]
- 51.Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, Sandborn WJ, Hardiman G, Raz E, Maehara Y, Yoshimura A, Zucman-Rossi J, Guan KL, Karin M. 2015. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 519:57–62. doi: 10.1038/nature14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vizán P, Beringer M, Ballaré C, Di Croce L. 2015. Role of PRC2-associated factors in stem cells and disease. FEBS J 282:1723–1735. doi: 10.1111/febs.13083. [DOI] [PubMed] [Google Scholar]
- 53.Kinkel SA, Galeev R, Flensburg C, Keniry A, Breslin K, Gilan O, Lee S, Liu J, Chen K, Gearing LJ, Moore DL, Alexander WS, Dawson M, Majewski IJ, Oshlack A, Larsson J, Blewitt ME. 2015. Jarid2 regulates hematopoietic stem cell function by acting with polycomb repressive complex 2. Blood 125:1890–1900. doi: 10.1182/blood-2014-10-603969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, Krogan NJ, Reiter JF, Stanford WL. 2010. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 6:153–166. doi: 10.1016/j.stem.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim MS, Yoon SK, Bollig F, Kitagaki J, Hur W, Whye NJ, Wu YP, Rivera MN, Park JY, Kim HS, Malik K, Bell DW, Englert C, Perantoni AO, Lee SB. 2010. A novel Wilms tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J Biol Chem 285:14585–14593. doi: 10.1074/jbc.M109.094334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, Shao DD, Schumacher SE, Weir BA, Vazquez F, Cowley GS, Root DE, Mesirov JP, Beroukhim R, Kuo CJ, Goessling W, Hahn WC. 2012. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151:1457–1473. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng AS, Lau SS, Chen Y, Kondo Y, Li MS, Feng H, Ching AK, Cheung KF, Wong HK, Tong JH, Jin H, Choy KW, Yu J, To KF, Wong N, Huang TH, Sung JJ. 2011. EZH2-mediated concordant repression of Wnt antagonists promotes β-catenin-dependent hepatocarcinogenesis. Cancer Res 71:4028–4039. doi: 10.1158/0008-5472.CAN-10-3342. [DOI] [PubMed] [Google Scholar]
- 58.Rock J, Konigshoff M. 2012. Endogenous lung regeneration: potential and limitations. Am J Respir Crit Care Med 186:1213–1219. doi: 10.1164/rccm.201207-1151PP. [DOI] [PubMed] [Google Scholar]
- 59.Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Hoeven N, Pappas C, Belser JA, Maines TR, Zeng H, Garcia-Sastre A, Sasisekharan R, Katz JM, Tumpey TM. 2009. Human HA and polymerase subunit PB2 proteins confer transmission of an avian influenza virus through the air. Proc Natl Acad Sci U S A 106:3366–3371. doi: 10.1073/pnas.0813172106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Zhang Q, Gao Y, He X, Kong H, Jiang Y, Guan Y, Xia X, Shu Y, Kawaoka Y, Bu Z, Chen H. 2012. Key molecular factors in hemagglutinin and PB2 contribute to efficient transmission of the 2009 H1N1 pandemic influenza virus. J Virol 86:9666–9674. doi: 10.1128/JVI.00958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bortz E, Westera L, Maamary J, Steel J, Albrecht RA, Manicassamy B, Chase G, Martinez-Sobrido L, Schwemmle M, Garcia-Sastre A. 2011. Host- and strain-specific regulation of influenza virus polymerase activity by interacting cellular proteins. mBio 2:e00151-11. doi: 10.1128/mBio.00151-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uhl FE, Vierkotten S, Wagner DE, Burgstaller G, Costa R, Koch I, Lindner M, Meiners S, Eickelberg O, Konigshoff M. 2015. Preclinical validation and imaging of Wnt-induced repair in human 3D lung tissue cultures. Eur Respir J 46:1150–1166. doi: 10.1183/09031936.00183214. [DOI] [PubMed] [Google Scholar]
- 64.Kneidinger N, Yildirim AO, Callegari J, Takenaka S, Stein MM, Dumitrascu R, Bohla A, Bracke KR, Morty RE, Brusselle GG, Schermuly RT, Eickelberg O, Konigshoff M. 2011. Activation of the WNT/β-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med 183:723–733. doi: 10.1164/rccm.200910-1560OC. [DOI] [PubMed] [Google Scholar]
- 65.Kuncewitch M, Yang WL, Jacob A, Khader A, Giangola M, Nicastro J, Coppa GF, Wang P. 2015. Stimulation of Wnt/β-catenin signaling pathway with Wnt agonist reduces organ injury after hemorrhagic shock. J Trauma Acute Care Surg 78:793–800. doi: 10.1097/TA.0000000000000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suryawanshi A, Manoharan I, Hong Y, Swafford D, Majumdar T, Taketo MM, Manicassamy B, Koni PA, Thangaraju M, Sun Z, Mellor AL, Munn DH, Manicassamy S. 2015. Canonical Wnt signaling in dendritic cells regulates Th1/Th17 responses and suppresses autoimmune neuroinflammation. J Immunol 194:3295–3304. doi: 10.4049/jimmunol.1402691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hillesheim A, Nordhoff C, Boergeling Y, Ludwig S, Wixler V. 2014. β-Catenin promotes the type I IFN synthesis and the IFN-dependent signaling response but is suppressed by influenza A virus-induced RIG-I/NF-κB signaling. Cell Commun Signal 12:29. doi: 10.1186/1478-811X-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Graef KM, Vreede FT, Lau YF, McCall AW, Carr SM, Subbarao K, Fodor E. 2010. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol 84:8433–8445. doi: 10.1128/JVI.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patel D, Schultz LW, Umland TC. 2013. Influenza A polymerase subunit PB2 possesses overlapping binding sites for polymerase subunit PB1 and human MAVS proteins. Virus Res 172:75–80. doi: 10.1016/j.virusres.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 70.Varga ZT, Grant A, Manicassamy B, Palese P. 2012. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol 86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]