

ABSTRACT
Herpes simplex virus (HSV) dramatically reorganizes the infected-cell nucleus, leading to the formation of prereplicative sites and replication compartments. This process is driven by the essential viral single-stranded DNA (ssDNA) binding protein ICP8, which can form double-helical filaments in the absence of DNA. In this paper, we show that two conserved motifs, FNF (F1142, N1143, and F1144) and FW (F843 and W844), are essential for ICP8 self-interactions, and we propose that the FNF motif docks into the FW region during filament formation. Mammalian expression plasmids bearing mutations in these motifs (FNF and FW) were unable to complement an ICP8-null mutant for growth and replication compartment formation. Furthermore, FNF and FW mutants were able to inhibit wild-type (WT) virus plaque formation and filament formation, whereas a double mutant (FNF-FW) was not. These results suggest that single mutant proteins are incorporated into nonproductive ICP8 filaments, while the double mutant is unable to interact with WT ICP8 and does not interfere with WT growth. Cells transfected with WT ICP8 and the helicase-primase (H/P) complex exhibited punctate nuclear structures that resemble prereplicative sites; however, the FNF and FW mutants failed to do so. Taken together, these results suggest that the FNF and FW motifs are required for ICP8 self-interactions and that these interactions may be important for the formation of prereplicative sites and replication compartments. We propose that filaments or other higher-order structures of ICP8 may provide a scaffold onto which other proteins can be recruited to form prereplicative sites and replication compartments.
IMPORTANCE For nuclear viruses such as HSV, efficient DNA replication requires the formation of discrete compartments within the infected-cell nucleus in which replication proteins are concentrated and assembled into the HSV replisome. In this paper, we characterize the role of filament formation by the single-stranded DNA binding protein ICP8 in the formation of prereplicative sites and replication compartments. We propose that ICP8 protein filaments generate a protein scaffold for other cellular and viral proteins, resulting in a structure that concentrates both viral DNA and replication proteins. Replication compartments may be similar to other types of cellular membraneless compartments thought to be formed by phase separations caused by low-affinity, multivalent interactions involving proteins and nucleic acids within cells. ICP8 scaffolds could facilitate the formation of replication compartments by mediating interactions with other components of the replication machinery.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a double-stranded DNA virus that replicates in the nucleus of an infected cell in large globular domains called replication compartments (1). These compartments are non-membrane-bound structures in the nucleus that serve to concentrate and compartmentalize viral and cellular molecules that are required for gene expression, DNA replication, and encapsidation (2–4). HSV-1 encodes seven essential replication proteins required for origin-dependent DNA replication: a single-stranded DNA (ssDNA) binding protein (ICP8), an origin binding protein (UL9), the helicase-primase complex (UL5-UL8-UL52 [UL5/8/52]), and the polymerase complex (UL30/42) (reviewed in reference 5). All seven replication proteins, as well as several cellular proteins, localize to replication compartments in HSV-infected cells (1, 6–12).
Replication compartments are formed in HSV-infected cells via an ordered sequence of events starting with the formation of ICP4/27 nucleoprotein complexes on viral genomes (13, 14). The first ICP8-containing structures are punctate foci called prereplicative sites that also contain the helicase-primase complex (UL5/8/52) and the origin binding protein UL9 (13, 15). UL30 and UL42 are recruited to prereplicative sites (16), and when replication is allowed to proceed, they coalesce with each other and with ICP4/27 foci to form mature replication compartments (17, 18). The absence of prereplicative sites in cells infected with an ICP8-null mutant virus has been taken as evidence that ICP8 is the nucleating factor required for their formation (2, 19, 20). During the formation of prereplicative sites and replication compartments, ICP8 is known to undergo conformational changes (21); however, it is not clear how these changes contribute to the formation of prereplicative sites and replication compartments.
ICP8 is a 128-kDa zinc metalloprotein that binds ssDNA cooperatively and forms thin nucleoprotein filaments on ssDNA (22, 23) and double-helical filaments in the absence of DNA (24, 25). The 60 C-terminal residues of ICP8 are required both for filament formation and for cooperative binding to ssDNA (26, 27). In 2005, the crystal structure of ICP8 lacking the C-terminal 60 amino acid residues was reported, revealing an N-terminal domain (residues 9 to 1038) that was described as containing head, neck, and shoulder regions (28). Based on the ICP8 structure, Mapelli et al. predicted putative ssDNA binding residues as well as residues that may be important for protein-protein interactions. For instance, they suggested that conserved residues F827, F843, W844, L857, and I865 within the head region form a hydrophobic pocket that could interact with another protein (28) (see Fig. 1A).
FIG 1.

ICP8 protein structure. (A) Ribbon diagram of ICP8 (PDB ID 1URJ) colored according to subdomain. The head, neck, and shoulder subdomains are colored red, yellow, and blue, respectively; the C-terminal domain is colored purple. The green spheres show the alpha-carbon of the FW residues within the head subdomain. The 60 C-terminal residues of ICP8 are not represented in the structure. (B) Schematic of the ICP8 protein showing the locations of the FW motif and the FNF motif. A double alanine substitution in F843 and W844 (FW) and a triple alanine substitution in F1142, N1143, and F1144 (FNF) were generated as described in Materials and Methods. (C) WT and mutant versions (FNF, FW, FW-FNF, and Δ60) of ICP8 were expressed and purified from Sf9 insect cells infected with recombinant baculoviruses as described in Materials and Methods. Purified proteins were resolved by 8% SDS-PAGE and were stained with Coomassie blue. Molecular mass markers (in kilodaltons) are shown on the left.
In this study, we identified two conserved motifs involved in filament formation, one within the C-terminal 60 amino acids (amino acids 1142 to 1144 [FNF]) and the other within the hydrophobic head region (F843 and W844), that had been predicted to participate in protein interactions (28). Mutant proteins containing alanine substitutions within these motifs failed to form filaments in solution and were unable to complement an ICP8-null mutant for viral growth and replication compartment formation. Furthermore, these mutants were unable to form prereplicative site-like structures in transfection-based assays. We propose that filaments of ICP8 may form a scaffold onto which other viral and cellular proteins can be recruited to assemble prereplicative sites and replication compartments.
MATERIALS AND METHODS
Cell culture and viruses.
African green monkey kidney (Vero) cells were purchased from the American Type Culture Collection and were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum (FBS) and 0.1% penicillin-streptomycin. The ICP8-complementing cell line S2 was generously provided by David Knipe (Harvard University, Boston, MA) (29). S2 cells were maintained in DMEM with 5% FBS and 0.1% penicillin-streptomycin and were kept under G418 selection (400 μg/ml). The HSV-1 strain KOS was used as the wild-type (WT) strain in all experiments. The ICP8-null virus HD-2, which contains an in-frame lacZ insertion and a deletion in the UL29 gene, was generously provided by David Knipe (Harvard Medical School, Cambridge, MA) (29).
DNA constructs and mutagenesis.
The ICP8 expression vector pSAK-ICP8 contains the full-length ICP8 gene under the control of the cytomegalovirus (CMV) promoter (30). ICP8 mutants were constructed in pSAK-ICP8 for mammalian expression and in pFastBac1 for the construction of recombinant baculoviruses. The ICP8Δ60NLS mutant was constructed by PCR amplification of the ICP8 gene in pSAK-ICP8 with primers that resulted in the deletion of the C-terminal 60 amino acids of ICP8 and the addition of the simian virus 40 (SV40) nuclear localization signal (NLS). The resulting PCR product was cloned into pSAK, which had been digested with HindIII and EcoRI. ICP8Δ60 was introduced into pFastBac1 using a similar strategy, except that no NLS was added. Alanine substitutions in the conserved motifs FNF (amino acids 1142 to 1144) and FW (amino acids 843 and 844) were made in pSAK-ICP8 and pFastBAC1-ICP8 by using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's suggested protocol. The FNF mutant (pSAK-ICP8 FNF) contains three alanine residues in place of FNF, and the FW mutant (pSAK-ICP8 FW) contains two alanine residues in place of FW. A double mutant containing both the FNF and FW mutations (pSAK-ICP8 FW-FNF) was constructed by adding the FW mutation to pSAK-ICP8 FNF and pFastBac1-ICP8 FNF using the QuikChange II XL site-directed mutagenesis kit.
Protein purification.
Recombinant baculoviruses bearing WT and mutant versions of ICP8 were generated in the Bac-to-Bac system (Invitrogen) as described previously (31). Sf9 insect cells were infected with recombinant baculoviruses bearing WT or mutant versions of ICP8 and were harvested at ∼48 to 50 h postinfection (hpi). Purification was carried out as described previously (32). Briefly, infected insect cell pellets were resuspended in lysis buffer, homogenized, and cleared by centrifugation at 30,000 rpm in a 70 Ti rotor for 40 min. The supernatant was applied to a HiLoad 16/10 SP Sepharose anion-exchange column and was eluted with a salt gradient. An additional purification step was performed for the FW mutant due to the presence of a contaminating nuclease. After SP-Sepharose chromatography, ICP8 protein-containing fractions were pooled and were further purified on a Superose 6 size exclusion column.
Purified proteins were resolved by denaturing 8% SDS-PAGE and were visualized by Coomassie blue staining. Fractions containing the highest purity were dialyzed against storage buffer (20 mM HEPES [pH 7.6], 1 mM dithiothreitol [DTT], 0.1 mM EDTA, 100 mM NaCl, and 5% glycerol) and were stored at −80°C. The UL5-UL8-UL52 complex was expressed in insect cells coinfected with recombinant baculoviruses bearing UL5, His-tagged UL8, and UL52. The heterotrimeric complex was purified on a HIS-Select nickel affinity column (Sigma) as described previously (33).
Electron microscopy.
Purified protein samples at a concentration of 0.3 mg/ml were incubated in a buffer containing 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 5 mM MgCl2 overnight at 4°C. Samples were diluted 1:10 in water, adsorbed on Formvar-carbon-coated 300-mesh copper grids, negatively stained with 2% uranyl acetate, and examined using a Hitachi H-7650 transmission electron microscope (TEM) at an accelerating voltage of 80 kV. Images were taken at magnifications of ×12,000 and ×60,000. For analysis of filament length, 0.15 mg/ml of WT ICP8 was incubated with an equal concentration of mutant protein or bovine serum albumin (BSA) and was incubated overnight under filament-forming conditions as described above. The lengths of all filaments detected in a randomly selected area of 100 μm2 were measured using ImageJ analysis.
IF analysis.
Immunofluorescence (IF) analysis was performed as described previously (34). Briefly, cells were grown on glass coverslips, washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde, and permeabilized using 1% Triton X-100. Cells were blocked in 3% normal goat serum and were incubated with the following antibodies: monoclonal mouse anti-ICP4 (dilution, 1:200; US Biological) and polyclonal rabbit anti-ICP8 clone 367 (dilution, 1:400; a gift from William Ruyechan [35]). Alexa Fluor-labeled secondary antibodies (dilution, 1:200; Molecular Probes) were used with fluorophores excitable at a wavelength of 488 or 594 nm. All images were taken using a Zeiss LSM 510 Meta confocal microscope and a Zeiss 63× objective lens (numerical aperture, 1.4). Images were processed and arranged using Adobe Photoshop CS3 and Illustrator CS3.
Transient complementation assays.
Vero cells were grown in a 35-mm dish to 70% confluence and were transfected with 1 μg of plasmid by using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's suggested protocol. At 24 h posttransfection, cells were infected with HD-2 at a multiplicity of infection (MOI) of 3 PFU/cell for 1 h at 37°C. After an adsorption period of 1 h, cells were washed twice with PBS, and the medium was replaced. At 24 hpi, cells and growth media were collected and were subjected to three cycles of freezing and thawing. Viral yields were determined by titration on S2 complementing cells. To test for replication compartment formation, cells were grown on coverslips to 70% confluence and were transfected with 250 ng of pSAK-ICP8, pSAK-ICP8 FNF, pSAK-ICP8 FW, pSAK-ICP8 FW-FNF, or pSAK-ICP8 Δ60 and 750 ng of empty vector (as carrier DNA). At 16 h posttransfection, cells were infected with HD-2 (MOI, 20), and at 6 hpi, cells were fixed and stained for IF analysis.
Plaque reduction assay.
Infectious DNA was prepared as described previously (36). Vero cells were grown in a 60-mm dish to 70% confluence and were transfected with 25 ng of infectious WT KOS DNA, along with 100 ng of a plasmid expressing a WT or mutant version of ICP8 (FNF, FW, or FW-FNF); the ICP8 plasmid was in an 80-fold molar excess of infectious DNA. In addition, 1.4 μg of carrier DNA (the pUC19 empty vector) was included in the transfection mixture. At 24 h posttransfection, cells were washed with PBS and were incubated for 4 days. Plaques were counted, and data are presented as the percentage of reduction in the number of plaques from that in the WT.
ssDNA binding assay.
The fluorescently labeled 50-nucleotide (nt) ssDNA substrate (F50nt5′cy3, TGCGGATGGCTTAGAGCTTAATTGCTGAATCTGGTGCTGTAGCTCAACAT) was purchased from Integrated DNA Technologies. Reaction mixtures (10 μl) containing 20 mM Tris-HCl (pH 7.5), 4% glycerol, 0.1 mg/ml BSA, 0.5 mM DTT, 5 mM MgCl2, and 100 nM fluorescently labeled substrate were incubated with 100 or 200 nM of WT or mutated ICP8 protein for 30 min at room temperature. Reactions were stopped by the addition of 2 μl of 6× loading buffer (40% sucrose), and mixtures were loaded onto 5% nondenaturing PAGE gels. Fluorescent signals were visualized using a Bio-Rad ChemiDoc MP imaging system.
Transfection assay.
Vero cells were grown on a glass coverslip in a 35-mm dish to 70% confluence and were transfected with 250 ng of WT or mutant ICP8 plasmid and 250 ng of each of the following plasmids: pCM-UL8, pCM-UL5b, and pCMV-UL52 (provided as a gift by Regina Heilbronn [37]). Cells were transfected with Lipofectamine Plus (Invitrogen) according to the manufacturer's suggested protocol. Twenty hours after transfection, cells were fixed and stained for IF analysis.
Surface plasmon resonance (SPR).
Protein-protein interactions were studied in real time using the Biacore T200 optical biosensor (GE Healthcare). Purified WT or mutant ICP8 proteins (FNF, FW, FW-FNF, and Δ60) were covalently immobilized by amine coupling through 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)–N-hydroxysuccinimide (NHS) on a CM5 sensor chip. The UL5/8/52 analyte in HSB buffer (10 mM HEPES [pH 7.4], 3 mM EDTA, 150 mM NaCl, and 0.05% surfactant P20) was injected over the sensor surface at a flow rate of 30 μl/min for 180 s (on rate), followed by HSB buffer at the same flow rate for another 180 s (off rate). The sensor chip was regenerated between injections by passing 10 mM NaOH at a flow rate of 50 μl/min for 120 s. Purified BSA (Sigma) was used as a negative control. The data were analyzed using Biacore T200 evaluation software and were fitted onto the 1:1 Langmuir binding model.
RESULTS
The FNF and FW motifs of ICP8 are required for filament formation.
The HSV ssDNA binding protein ICP8 has been reported to form double-helical protein filaments in vitro in the absence of DNA (24, 25, 38). ICP8 homologs BALF2 from Epstein-Barr virus (EBV) and ORF6 from Kaposi's sarcoma-associated herpesvirus (KSHV) also form double-helical protein filaments (26, 39), suggesting that filament formation is a common property of herpesvirus ssDNA binding proteins. Removal of the 60 C-terminal amino acids from ICP8, BALF, or ORF6 has been reported to eliminate filament formation and to reduce cooperative binding to ssDNA (26, 27, 40). These observations suggest that the 60 C-terminal residues of ICP8 and its homologs include residues important for protein-protein interactions that mediate filament formation and cooperative binding (26–28, 40). ICP8 has emerged as the nucleating factor for replication compartment formation (2, 19, 20), and we are interested in the relationship between filament formation in vitro and replication compartment formation in HSV-infected cells.
Sequence alignments of ICP8 homologs from the Alphaherpesvirinae subfamily of Herpesviridae revealed a conserved region within the C-terminal 60 amino acids that was predicted to be involved in filament formation (28). Within this region, the FNF motif (F1142, N1443, and F1144) was predicted to play a role in ICP8-ICP8 interactions (28). Furthermore, ICP8 with a mutation in the FNF region (F1144C) was previously reported to be unable to complement the growth of an ICP8-null virus or to localize to replication compartments (41). The FNF motif was predicted to dock into a conserved hydrophobic region (F827, F843, W844, L857, and I865) in the head region of a neighboring ICP8 molecule (28) (Fig. 1A and B); however, these predictions have never been directly tested, nor has the F1144C mutation been studied for its ability to form filaments in vitro. In order to test these predictions, we introduced a triple alanine mutation into the FNF motif and a double alanine mutation into the FW motif (residues F843 and W844) (Fig. 1B). We also reconstructed the previously engineered 60-amino-acid C-terminal deletion for comparison with the alanine substitution mutations.
WT and mutant proteins were purified from insect cells infected with recombinant baculoviruses (Fig. 1C) and were tested for their abilities to form filaments in vitro. ICP8 proteins were incubated under conditions previously shown to promote filament formation and were subjected to analysis by electron microscopy as described in Materials and Methods. As anticipated, WT ICP8 was able to form a double-helical filament (Fig. 2A and B), while ICP8Δ60 was unable to do so (Fig. 2F). Neither the FNF nor the FW mutant protein was able to form filaments under these conditions; instead, these proteins formed small clusters that could represent nonspecific aggregates, possibly monomers or dimers (Fig. 2C to E). These observations suggest that intact FNF and FW motifs in ICP8 may be required for interactions between ICP8 molecules that lead to filament formation.
FIG 2.

Filament formation for WT and mutant versions of ICP8 as visualized by electron microscopy. (A and B) WT ICP8 formed double-helical filaments after overnight incubation at a concentration of 0.15 mg/ml at 4°C in a buffer containing 5 mM MgCl2. (C to F) The FNF (C), FW (D), FW-FNF (E), and Δ60 (F) ICP8 mutants were unable to form filaments. All samples were negatively stained with 2% uranyl acetate and were visualized by electron microscopy at an accelerating voltage of 80 kV. Bars, 100 nm (magnification, ×60,000) and 500 nm (×12,000).
ICP8 mutants that fail to form filaments are unable to complement the growth of an ICP8-null virus.
In order to test whether mutants that fail to form filaments retain the ability to complement the growth of an ICP8-null mutant virus, we introduced each of the mutant constructs into a mammalian expression vector under the control of the human CMV (HCMV) promoter. For ICP8Δ60, which lacks an NLS, an SV40 NLS was added to ensure that ICP8Δ60 would localize to the nucleus (42, 43). The abilities of WT and mutant versions of ICP8 to complement the growth of an ICP8-null virus (HD-2) (29) were measured by transfecting Vero cells with WT or mutant versions of the ICP8 expression plasmids, followed by infection with HD-2. Progeny virus was harvested 24 h postinfection, and viral yields were determined by titration on an ICP8-complementing cell line (S2) (29). None of the mutants were able to complement HD-2, and viral yields were comparable to those in cells transfected with an empty vector (Table 1). We conclude that the ICP8 mutants that fail to form filaments in vitro are also defective in viral growth.
TABLE 1.
Mutants that disrupt ICP8 filament formation fail to complement the growth of the HD-2 virus
| Expression plasmid transfected | % Complementation (avg ± SEMa) |
|---|---|
| ICP8 | 100 ± 0 |
| FW | −0.1 ± 0.13 |
| FNF | 0.6 ± 0.34 |
| FW-FNF | 0.02 ± 0.20 |
| Δ60 | −0.2 ± 0.11 |
| Empty vector | 0 ± 0 |
From 4 independent experiments.
The FW and FNF mutants exhibit a transdominant phenotype.
If, as we predict, the FNF motif docks into the hydrophobic head region of a neighboring ICP8 molecule, the FNF and FW motif mutants would be expected to exert a transdominant inhibitory effect on ICP8 filament formation when coexpressed with WT ICP8. We predicted that a mixture of mutant and WT ICP8 proteins would generate a heterogeneous population of dimeric or higher-order structures that could prevent ICP8 filament extension and inhibit infection. In the experiment for which results are shown in Fig. 3A, Vero cells were transfected with mutant or WT versions of ICP8 along with infectious DNA isolated from cells infected with WT HSV-1 (KOS). The FNF and FW mutants displayed reductions in plaque formation of almost 60% and 50%, respectively. According to this scenario, we also predicted that a double mutant bearing mutations in both the FNF and FW motifs would be unable to form mixed oligomers with WT ICP8. To test this hypothesis, we constructed an ICP8 mutant containing both the FW and FNF mutations. The FW-FNF mutant was tested for transdominance as described above. In contrast to the single mutants, the FW-FNF double mutant had no effect on plaque formation (Fig. 3A). We conclude that the FNF and FW mutants are transdominant inhibitors of infection, possibly as a result of their nonproductive incorporation into ICP8 filaments. Presumably, the double mutant does not inhibit the growth of WT virus because it lacks the ability to form ICP8-ICP8 interactions and therefore is not able to be incorporated into a growing ICP8 filament.
FIG 3.

The FNF and FW mutants exhibit a transdominant phenotype. (A) A plaque reduction assay was conducted by transfecting Vero cells with 100 ng of a plasmid expressing a WT or mutant version of ICP8 (FNF, FW, or FW-FNF) and 25 ng of KOS infectious DNA. At 4 days posttransfection, plaques were counted. (B) Purified ICP8 protein was incubated with the FNF, FW, or FW-FNF mutant or with BSA at equal concentrations (0.15 mg/ml) and was incubated overnight at 4°C in a buffer containing 5 mM MgCl2. Electron microscopy was used to image the samples, and the filaments in a 100-μm2 area on the grid were measured and counted using ImageJ software analysis. Filament lengths were categorized as either <200 nm, 200 to 400 nm, or >400 nm.
To test whether the presence of mutant proteins could affect the length of ICP8 filaments formed in vitro, a mixture of WT and mutant proteins in a 1:1 ratio was examined for filament formation. In order to include the double mutant protein in this analysis, the FW-FNF mutant was also introduced into a recombinant baculovirus, and purified protein was obtained from infected insect cells (Fig. 1C). Like the single mutants, the double mutant is incapable of filament formation (Fig. 2). Samples were visualized by electron microscopy, and the lengths of filaments were measured using ImageJ software analysis (Fig. 3B). The limiting size for observing a filament is 50 nm; therefore, filaments over this size were counted and grouped into three categories: below 200 nm, between 200 and 400 nm, or above 400 nm in length. The mixture of WT and FW-FNF mutant proteins exhibited a distribution of filaments in the three categories similar to that in WT ICP8 alone; however, the mixture of WT and FNF mutant proteins exhibited few filaments with lengths above 400 nm and a higher number of smaller structures. In the mixture containing WT and FW proteins, very few structures were observed, indicating that the presence of this mutant protein was even more deleterious to WT filament formation than the presence of the FNF mutant. It will be of interest to determine why the FW mutant has an even more dramatic effect on mixed filament formation than the FNF mutant; however, at this point, we can conclude that both mutants interfere with the ability of WT ICP8 to form filaments in vitro and establish productive infection in Vero cells. The observation that the double mutant alleviated the transdominant phenotype of the single motif mutants is consistent with the notion that the FNF motif docks at the FW region.
Mutant proteins retain the ability to bind ssDNA, but with reduced cooperativity.
The loss of filament formation may represent disruption of interaction domains responsible for ICP8-ICP8 interactions or destabilization of mutant proteins. To rule out global misfolding, we used an electrophoretic mobility shift assay (EMSA) to test whether these mutants can still bind to ssDNA. In this assay, WT or mutant ICP8 proteins were incubated with a Cy3-labeled 50-nt oligomer, and the reaction products were resolved by 5% nondenaturing PAGE. Figure 4 shows that the WT and mutant proteins all bind to ssDNA in a concentration-dependent fashion; however, differences in the shift patterns were observed. WT ICP8 exhibited two predominant shifted bands, the slower of which may represent the ability of the WT protein to bind cooperatively to the 50-nt oligomer. All mutant proteins were able to shift ssDNA; however, none of the nucleoprotein complexes corresponded to the slowest species seen in the WT ICP8 samples. These results suggest that, as reported previously for ICP8Δ60 (27), the FNF, FW, and FW-FNF mutant proteins have lost their ability to bind cooperatively to ssDNA; however, the ability of all mutants to bind ssDNA indicates that the mutant proteins are not globally misfolded.
FIG 4.
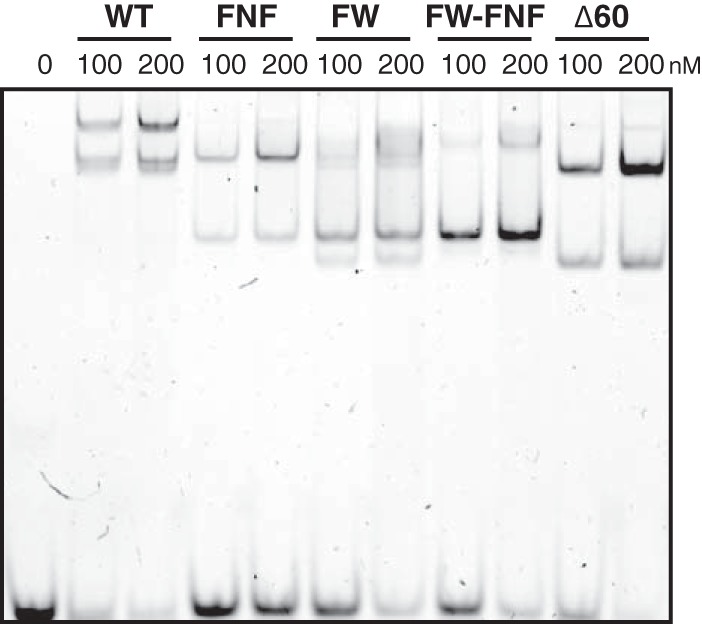
The ICP8 FNF, FW, FW-FNF, and Δ60 mutants retain the ability to interact with ssDNA, albeit with reduced cooperativity. WT ICP8 or the FNF, FW, FW-FNF, or Δ60 mutant was incubated at a concentration of either 100 nM or 200 nM with a 5′-fluorescein-labeled 50-nt ssDNA substrate at 100 nM for 30 min, and the complexes were separated on 5% nondenaturing polyacrylamide gels. Both WT ICP8 and the mutants were able to bind DNA, although there was a difference in the sizes of the nucleoprotein complexes formed. WT ICP8 forms a supershifted complex that is absent in the mutants, suggesting a lack of cooperative binding in the mutants. The signal was detected using the Bio-Rad ChemiDoc MP imaging system.
ICP8 mutants defective in filament formation are unable to generate prereplicative site-like structures and replication compartments.
As described in the introduction, ICP8 is required for the formation of replication compartments (2). The earliest detectable prereplicative sites contain ICP8, helicase-primase, and UL9 (2, 13, 15, 34). During the course of HSV infection, prereplicative site foci grow and coalesce into replication compartments. A transfection-based assay that mimics prereplicative site formation has been developed (13, 44). In cells transfected with ICP8 alone, a diffuse pattern is observed throughout the nucleus; however, in cells transfected with WT ICP8 plus the three subunits of the helicase-primase complex (UL5, UL8, and UL52), punctate foci are observed that resemble prereplicative sites and have been called prereplicative site-like structures (9, 13, 44). We asked whether ICP8 mutants that fail to form filaments in vitro are able to form prereplicative site-like structures in transfected cells. Vero cells transfected with WT or mutant versions of ICP8 displayed diffuse nuclear staining (Fig. 5A to E); however, in cells cotransfected with WT ICP8 and helicase-primase, prereplicative site-like structures were observed (Fig. 5F). In cells transfected with mutant ICP8 and the same cocktail of plasmids encoding the three helicase-primase subunits, ICP8 staining remained diffuse (Fig. 5G to J). Although we have not monitored the transfection efficiencies of the helicase-primase subunits in this experiment due to the poor quality of anti-helicase-primase antibodies, we observed that 48% of the WT ICP8-positive cells exhibited prereplicative site-like structures, suggesting that almost half of the cells in this experiment received all four plasmids. It is reasonable to conclude that transfection efficiencies with mutant ICP8 and helicase-primase plasmids are comparable. Thus, we conclude that ICP8 mutants that fail to form filaments are defective in the formation of prereplicative site-like foci.
FIG 5.

The ICP8 FNF, FW, FW-FNF, and Δ60NLS mutants do not form prereplicative site-like structures. Vero cells were transfected with plasmids expressing WT ICP8 (A and F), ICP8 FNF (B and G), ICP8 FW (C and H), ICP8 FW-FNF (D and I), or ICP8Δ60NLS (E and J). For panels F to J, plasmids expressing UL5, UL8, and UL52 (the helicase-primase complex [H/P]) were added. At 20 h posttransfection, cells were fixed and were stained with an anti-ICP8 primary antibody and an Alexa Fluor 488-labeled secondary antibody. Images were taken with a Zeiss LSM 510 confocal microscope.
In order to test the behavior of the ICP8 mutants in the context of viral infection, Vero cells were transfected with either WT or mutant versions of ICP8 followed by infection with HD-2 (MOI, 20 for 6 h). We reasoned that if mutant ICP8 was capable of assembling either prereplicative sites or replication compartments, these structures would be observed under these conditions. The immediate early protein ICP4 was used as a marker to identify infected cells, and transfected cells were identified on the basis of ICP8 staining. Cells that expressed both ICP8 and ICP4 were analyzed. WT ICP8 was able to complement HD-2 for replication compartment formation (Fig. 6A to C); however, the four ICP8 mutants did not support the formation of prereplicative sites or replication compartments in HD-2-infected cells (Fig. 6D to O). Thus, these results indicate that the ability to form filaments in vitro correlates with the formation of prereplicative sites and replication compartments.
FIG 6.

ICP8 mutants are unable to complement HD-2 for replication compartment formation. Vero cells were transfected with either WT ICP8 (A to C), the FNF (D to F), FW (G to I), or FW-FNF (J to L) mutant, or ICP8Δ60NLS (M to O). At 16 h posttransfection, cells were infected with an ICP8-null virus (MOI, 20). At 6 h postinfection, cells were fixed and were stained for ICP8 and ICP4 with Alexa Fluor 488- and 594-labeled secondary antibodies, respectively. The immediate early protein ICP4 was used as a marker for infection. Only cells that contained both ICP4 and ICP8 were selected for analysis. Images were taken with a Zeiss LSM 510 confocal microscope.
The HSV-1 helicase-primase complex (UL5/8/52) interacts with ICP8 mutants.
The ability of prereplicative site-like structures to form in cells transfected with four viral proteins may reflect the fact that ICP8 can interact directly with the helicase-primase holoenzyme (45, 46). The inability of mutant ICP8 to form prereplicative site-like structures and replication compartments might suggest that mutant ICP8 proteins do not interact with the helicase-primase, To test this hypothesis, we used surface plasmon resonance to measure the binding of purified WT or mutant (FNF, FW, or FW-FNF) ICP8 protein to helicase-primase. WT and mutant ICP8 proteins were immobilized on a biosensor chip, and UL5/8/52 was injected over the chip at various concentrations (25, 50, 100, 200, and 400 nm). Figure 7 shows that both WT and mutant forms of ICP8 could interact with UL5/8/52. A negative control, BSA, exhibited no binding (data not shown). The binding data were analyzed at various concentrations of UL5/8/52 and were globally fitted using a 1:1 Langmuir binding model. A summary of the binding parameters is shown under each sensorgram (Fig. 7). WT ICP8 and the FW mutant interact with UL5/8/52 with comparable affinities, with dissociation constants (KD) of 7.6 × 10−8 M and 9.9 × 10−8 M, respectively. The FNF and FW-FNF mutants displayed slightly weaker binding affinities to helicase-primase (KD, 1.8 × 10−7 M and 1.2 × 10−7 M, respectively). The fact that all three mutants bind helicase-primase with comparable affinities indicates that the inability of these mutant proteins to form prereplicative sites and replication compartments is not due to loss of interaction with helicase-primase.
FIG 7.

The helicase-primase complex (UL5/8/52) interacts with WT ICP8 and the FNF, FW, and FW-FNF mutant proteins. SPR sensorgrams at increasing concentrations of UL5/8/52 (25, 50, 100, 200, and 400 nM) were generated at room temperature using a Biacore T200 system. The colored lines represent the binding of the analyte (UL5/8/52) at different concentrations to the specified ligand. The black lines represent the data fitted using a 1:1 Langmuir binding model, which was used to calculate the kinetic parameters (ka, association rate; kd, dissociation rate) of the reactions as summarized under each sensogram.
DISCUSSION
In this paper, we report that two motifs in the ICP8 protein, the FNF motif at residues 1142 to 1144 in the C terminus and the FW motif at residues 843 and 844 in the so-called hydrophobic head region, are important for filament formation and for the progression of HSV infection. Recombinant mutant proteins bearing alanine substitutions in the FNF and FW motifs are unable to assemble into filaments in vitro. In addition, the FNF and FW mutants exert a dominant negative inhibitory effect on viral growth and on the ability of WT ICP8 to form filaments in vitro. On the other hand, a double mutant bearing both the FNF and FW mutations had no effect on WT growth or filament formation. These results suggest that the single mutants are capable of creating nonproductive oligomers that interfere with the ability of WT ICP8 to form filaments, whereas the double mutant lacks the ability to form ICP8-ICP8 interactions and therefore could not be incorporated into a growing WT ICP8 filament. This result supports our hypothesis that the FNF motif is able to dock into the hydrophobic head region containing the FW motif. Mutants that fail to form filaments in vitro also fail to form prereplicative site-like structures in cells transfected with ICP8 and the three subunits of the helicase-primase. The ability of these ICP8 mutants to interact with the helicase-primase complex (Fig. 7) indicates that the failure to form prereplicative site-like structures is not due to loss of this interaction. These mutants are unable to complement the ICP8-null virus for growth or for the formation of prereplicative sites and replication compartments. Taken together, these results suggest that the FNF and FW motifs define ICP8 self-interaction sites required for filament formation and for the reorganization of the nucleus of the infected cell by ICP8 to form prereplicative sites and replication compartments.
Previously, several functions have been assigned to the C terminus of ICP8: an NLS was mapped to the C-terminal 28 residues (43), and the region between amino acid residues 1080 and 1135 was implicated in interactions with viral or cellular factors required for replication compartment formation (41). However, no detailed biochemical analysis of mutations in this region has ever been performed. In this study, we extend the characterization of this region and show that in addition to affecting replication compartment formation, the FNF motif is involved in ICP8 self-interactions that are required for filament formation.
ICP8 is known to form two types of filaments in vitro: double-helical filaments that form in the absence of DNA (25) and a single-helical filament on ssDNA (22). A double-helical filament might be expected to be stabilized by two types of interactions: longitudinal interactions between adjacent subunits in each filament and lateral interactions between adjacent filaments. The inability of the FNF and FW mutants to form filaments and the transdominant inhibition of filament formation when these mutants are mixed with WT ICP8 are consistent with a role for the FNF and FW motifs in longitudinal interactions in filament formation. The simplest explanation for these results is that the FNF motif docks at the FW motif in an adjacent ICP8 molecule to form a linear filament.
Spatial organization within cells is critical for the control of the diverse metabolic activities necessary for cell survival, and subcellular compartmentalization provides boundaries that facilitate the regulation of biological processes. Some processes are regulated by the sequestration of components away from one another, while in other cases, it is beneficial to concentrate components that catalyze essential reactions. Although some cellular compartments are defined by membrane enclosure, it is becoming increasingly clear that many important subcellular compartments are not membrane limited. For instance, in the nucleus, many varieties of membraneless suborganelles are known to play important roles in the spatial and temporal organization of macromolecules, including promyelocytic leukemia (PML) nuclear bodies, speckles, and nucleoli (47). The mechanisms by which proteins and nucleic acids are recruited to form these phase-separated states are poorly understood; however, it appears that their formation may be caused by low-affinity, multivalent interactions between components (48–51). Proteins and nucleic acids in these suborganelles remain dynamic and mobile, facilitating rapid responses to various types of stimuli.
Viruses that replicate in the nucleus have evolved surprisingly efficient mechanisms to form compartments within the densely crowded nuclear environment, a function critical for the establishment of productive infection. The formation of viral subassemblies, such as prereplicative sites and replication compartments, may be driven by biophysical principles similar to those that influence the assembly of phase-separated membraneless organelles. In this paper, we have established that the ability of ICP8 protein to form filaments in solution is correlated with the formation of prereplicative site-like structures and replication compartments. Interestingly, mutant ICP8 proteins that fail to form filaments but can still interact with helicase-primase are unable to form prereplicative sites or replication compartments. These results suggest that the interaction between ICP8 and helicase-primase is not sufficient for the formation of prereplicative sites or replication compartments. We propose that the first step in prereplicative site formation requires dynamic interactions between ICP8 monomers that facilitate the formation of a scaffold on which prereplicative sites and replication compartments can form. Subsequent steps in the formation of prereplicative sites may involve interactions between ICP8 and helicase-primase, while the formation of large globular replication compartments appears to require active polymerase and UL42 (2, 7, 13, 16, 44). Interactions among the HSV replication proteins may resemble the low-affinity, multivalent interactions that drive phase separations and the assembly of membraneless globular domains. It will be important to determine whether the dynamic nature of ICP8 interactions with itself and with other viral and cellular proteins contributes to the formation of phase-separated states within cells.
ACKNOWLEDGMENTS
We thank the members of our laboratory, as well as Ann Cowen and John Carson, for helpful discussions on experimental design and manuscript preparation. We thank Jack Griffith and Oya Bermek for helpful suggestions. We thank Maya Yankova and Steve King for assistance with electron microscopy and George Korza for assistance with SPR experiments. We also thank Na Zhang for assistance in cloning pFastBac1-ICP8 FNF and pFastBac1-ICP8Δ60.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Quinlan MP, Chen LB, Knipe DM. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857–868. doi: 10.1016/0092-8674(84)90035-7. [DOI] [PubMed] [Google Scholar]
- 2.de Bruyn Kops A, Knipe DM. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857–868. doi: 10.1016/0092-8674(88)90141-9. [DOI] [PubMed] [Google Scholar]
- 3.Lamberti C, Weller SK. 1996. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology 226:403–407. doi: 10.1006/viro.1996.0668. [DOI] [PubMed] [Google Scholar]
- 4.Phelan A, Dunlop J, Patel AH, Stow ND, Clements JB. 1997. Nuclear sites of herpes simplex virus type 1 DNA replication and transcription colocalize at early times postinfection and are largely distinct from RNA processing factors. J Virol 71:1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weller SK, Coen DM. 2012. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol 4:a013011. doi: 10.1101/cshperspect.a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olivo PD, Nelson NJ, Challberg MD. 1989. Herpes simplex virus type 1 gene products required for DNA replication: identification and overexpression. J Virol 63:196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lukonis CJ, Weller SK. 1996. Characterization of nuclear structures in cells infected with herpes simplex virus type 1 in the absence of viral DNA replication. J Virol 70:1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilcock D, Lane DP. 1991. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 349:429–431. doi: 10.1038/349429a0. [DOI] [PubMed] [Google Scholar]
- 9.Wilkinson DE, Weller SK. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol 78:4783–4796. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor TJ, Knipe DM. 2004. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol 78:5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohni KN, Livingston CM, Cortez D, Weller SK. 2010. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J Virol 84:12152–12164. doi: 10.1128/JVI.01643-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohni KN, Mastrocola AS, Bai P, Weller SK, Heinen CD. 2011. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J Virol 85:12241–12253. doi: 10.1128/JVI.05487-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liptak LM, Uprichard SL, Knipe DM. 1996. Functional order of assembly of herpes simplex virus DNA replication proteins into prereplicative site structures. J Virol 70:1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everett RD, Sourvinos G, Leiper C, Clements JB, Orr A. 2004. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J Virol 78:1903–1917. doi: 10.1128/JVI.78.4.1903-1917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukonis CJ, Burkham J, Weller SK. 1997. Herpes simplex virus type 1 prereplicative sites are a heterogeneous population: only a subset are likely to be precursors to replication compartments. J Virol 71:4771–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carrington-Lawrence SD, Weller SK. 2003. Recruitment of polymerase to herpes simplex virus type 1 replication foci in cells expressing mutant primase (UL52) proteins. J Virol 77:4237–4247. doi: 10.1128/JVI.77.7.4237-4247.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor TJ, McNamee EE, Day C, Knipe DM. 2003. Herpes simplex virus replication compartments can form by coalescence of smaller compartments. Virology 309:232–247. doi: 10.1016/S0042-6822(03)00107-7. [DOI] [PubMed] [Google Scholar]
- 18.Chang L, Godinez WJ, Kim I-H, Tektonidis M, de Lanerolle P, Eils R, Rohr K, Knipe DM. 2011. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc Natl Acad Sci U S A 108:E136–E144. doi: 10.1073/pnas.1103411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bush M, Yager DR, Gao M, Weisshart K, Marcy AI, Coen DM, Knipe DM. 1991. Correct intranuclear localization of herpes simplex virus DNA polymerase requires the viral ICP8 DNA-binding protein. J Virol 65:1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNamee EE, Taylor TJ, Knipe DM. 2000. A dominant-negative herpesvirus protein inhibits intranuclear targeting of viral proteins: effects on DNA replication and late gene expression. J Virol 74:10122–10131. doi: 10.1128/JVI.74.21.10122-10131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uprichard SL, Knipe DM. 2003. Conformational changes in the herpes simplex virus ICP8 DNA-binding protein coincident with assembly in viral replication structures. J Virol 77:7467–7476. doi: 10.1128/JVI.77.13.7467-7476.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makhov AM, Griffith JD. 2006. Visualization of the annealing of complementary single-stranded DNA catalyzed by the herpes simplex virus type 1 ICP8 SSB/recombinase. J Mol Biol 355:911–922. doi: 10.1016/j.jmb.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 23.Tolun G, Makhov AM, Ludtke SJ, Griffith JD. 2013. Details of ssDNA annealing revealed by an HSV-1 ICP8-ssDNA binary complex. Nucleic Acids Res 41:5927–5937. doi: 10.1093/nar/gkt266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Donnell ME, Elias P, Funnell BE, Lehman IR. 1987. Interaction between the DNA polymerase and single-stranded DNA-binding protein (infected cell protein 8) of herpes simplex virus 1. J Biol Chem 262:4260–4266. [PubMed] [Google Scholar]
- 25.Makhov AM, Sen A, Yu X, Simon MN, Griffith JD, Egelman EH. 2009. The bipolar filaments formed by herpes simplex virus type 1 SSB/recombination protein (ICP8) suggest a mechanism for DNA annealing. J Mol Biol 386:273–279. doi: 10.1016/j.jmb.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mumtsidu E, Makhov AM, Konarev PV, Svergun DI, Griffith JD, Tucker PA. 2008. Structural features of the single-stranded DNA-binding protein of Epstein-Barr virus. J Struct Biol 161:172–187. doi: 10.1016/j.jsb.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 27.Mapelli M, Mühleisen M, Persico G, van Der Zandt H, Tucker PA. 2000. The 60-residue C-terminal region of the single-stranded DNA binding protein of herpes simplex virus type 1 is required for cooperative DNA binding. J Virol 74:8812–8822. doi: 10.1128/JVI.74.19.8812-8822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mapelli M, Panjikar S, Tucker PA. 2005. The crystal structure of the herpes simplex virus 1 ssDNA-binding protein suggests the structural basis for flexible, cooperative single-stranded DNA binding. J Biol Chem 280:2990–2997. doi: 10.1074/jbc.M406780200. [DOI] [PubMed] [Google Scholar]
- 29.Gao M, Knipe DM. 1989. Genetic evidence for multiple nuclear functions of the herpes simplex virus ICP8 DNA-binding protein. J Virol 63:5258–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK. 2012. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog 8:e1002862. doi: 10.1371/journal.ppat.1002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grady LM, Bai P, Weller SK. 2014. HSV-1 protein expression using recombinant baculoviruses. Methods Mol Biol 1144:293–304. doi: 10.1007/978-1-4939-0428-0_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reuven NB, Staire AE, Myers RS, Weller SK. 2003. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J Virol 77:7425–7433. doi: 10.1128/JVI.77.13.7425-7433.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Carrington-Lawrence SD, Bai P, Weller SK. 2005. Mutations in the putative zinc-binding motif of UL52 demonstrate a complex interdependence between the UL5 and UL52 subunits of the human herpes simplex virus type 1 helicase/primase complex. J Virol 79:9088–9096. doi: 10.1128/JVI.79.14.9088-9096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Livingston CM, DeLuca NA, Wilkinson DE, Weller SK. 2008. Oligomerization of ICP4 and rearrangement of heat shock proteins may be important for herpes simplex virus type 1 prereplicative site formation. J Virol 82:6324–6336. doi: 10.1128/JVI.00455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shelton LS, Albright AG, Ruyechan WT, Jenkins FJ. 1994. Retention of the herpes simplex virus type 1 (HSV-1) UL37 protein on single-stranded DNA columns requires the HSV-1 ICP8 protein. J Virol 68:521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith S, Reuven N, Mohni KN, Schumacher AJ, Weller SK. 2014. Structure of the herpes simplex virus 1 genome: manipulation of nicks and gaps can abrogate infectivity and alter the cellular DNA damage response. J Virol 88:10146–10156. doi: 10.1128/JVI.01723-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heilbronn R, zur Hausen H. 1989. A subset of herpes simplex virus replication genes induces DNA amplification within the host cell genome. J Virol 63:3683–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makhov AM, Taylor DW, Griffith JD. 2004. Two-dimensional crystallization of herpes simplex virus type 1 single-stranded DNA-binding protein, ICP8, on a lipid monolayer. Biochim Biophys Acta 1701:101–108. doi: 10.1016/j.bbapap.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Ozgur S, Damania B, Griffith J. 2011. The Kaposi's sarcoma-associated herpesvirus ORF6 DNA binding protein forms long DNA-free helical protein filaments. J Struct Biol 174:37–43. doi: 10.1016/j.jsb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozgur S, Griffith J. 2014. Interaction of Kaposi's sarcoma-associated herpesvirus ORF6 protein with single-stranded DNA. J Virol 88:8687–8695. doi: 10.1128/JVI.03652-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor TJ, Knipe DM. 2003. C-terminal region of herpes simplex virus ICP8 protein needed for intranuclear localization. Virology 309:219–231. doi: 10.1016/S0042-6822(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 42.Gao M, Knipe DM. 1992. Distal protein sequences can affect the function of a nuclear localization signal. Mol Cell Biol 12:1330–1339. doi: 10.1128/MCB.12.3.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao M, Knipe DM. 1993. Intragenic complementation of herpes simplex virus ICP8 DNA-binding protein mutants. J Virol 67:876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukonis CJ, Weller SK. 1997. Formation of herpes simplex virus type 1 replication compartments by transfection: requirements and localization to nuclear domain 10. J Virol 71:2390–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamatake RK, Bifano M, Hurlburt WW, Tenney DJ. 1997. A functional interaction of ICP8, the herpes simplex virus single-stranded DNA-binding protein, and the helicase-primase complex that is dependent on the presence of the UL8 subunit. J Gen Virol 78(Part 4):857–865. doi: 10.1099/0022-1317-78-4-857. [DOI] [PubMed] [Google Scholar]
- 46.Falkenberg M, Elias P, Lehman IR. 1998. The herpes simplex virus type 1 helicase-primase. Analysis of helicase activity. J Biol Chem 273:32154–32157. [DOI] [PubMed] [Google Scholar]
- 47.Van Bortle K, Corces VG. 2012. Nuclear organization and genome function. Annu Rev Cell Dev Biol 28:163–187. doi: 10.1146/annurev-cellbio-101011-155824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA. 2009. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 49.Brangwynne CP. 2013. Phase transitions and size scaling of membrane-less organelles. J Cell Biol 203:875–881. doi: 10.1083/jcb.201308087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hyman AA, Weber CA, Jülicher F. 2014. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- 51.Lee CF, Brangwynne CP, Gharakhani J, Hyman AA, Jülicher F. 2013. Spatial organization of the cell cytoplasm by position-dependent phase separation. Phys Rev Lett 111:088101. doi: 10.1103/PhysRevLett.111.088101. [DOI] [PubMed] [Google Scholar]