

Abstract
Background
The importance of various inflammatory cytokines in maintaining tumor cell growth and viability is well established. Increased expression of the proinflammatory cytokine macrophage migration inhibitory factor (MIF) has previously been associated with various types of adenocarcinoma.
Methods
MIF IHC was used to localize MIF in human bladder tissue. ELISA and Western blot analysis determined the synthesis and secretion of MIF by human bladder transitional cell carcinoma cells. The effects of MIF inhibitors (high molecular weight hyaluronate (HA), anti-MIF antibody or MIF anti-sense) on cell growth and cytokine expression were analyzed.
Results
Human bladder cancer cells (HT-1376) secrete detectable amounts of MIF protein. Treatment with HA, anti-MIF antibody and MIF anti-sense reduced HT-1376 cell proliferation, MIF protein secretion, MIF gene expression and secreted inflammatory cytokines. Our evidence suggests MIF interacts with the invariant chain, CD74 and the major cell surface receptor for HA, CD44.
Conclusions
This study is the first to report MIF expression in the human bladder and these findings support a role for MIF in tumor cell proliferation. Since MIF participates in the inflammatory response and bladder cancer is associated with chronic inflammatory conditions, these new findings suggest that neutralizing bladder tumor MIF may serve as a novel therapeutic treatment for bladder carcinoma.
Background
According to 2003 estimates, urinary bladder cancer will be diagnosed in 57,400 Americans and will result in 12,500 deaths [1]. Of these new cases, 80 to 90% will originally present as tumors of the epithelium or submucosa, with the majority being transitional cell carcinomas [2,3]. Transurethral resection of bladder tumor remains the initial line of defense in treatment of superficial bladder cancer. However, this treatment is hardly adequate as the recurrence rate in treated patients approaches 50 to 70% and 5 to 40% of recurrent cancers progress [2,4]. In an attempt to curb the reoccurrence rate, a variety of immunotherapies and chemotherapies have been devised, with the most common being intravesical bacillus Calmette-Guerin [4]. The high rate of mortality associated with invasive urinary bladder cancer and the high incidence of reoccurrence after treatment demonstrate the need for a better understanding of bladder cancer and new therapeutic agents for treatment.
Chronic inflammation is an established risk factor for the development of bladder cancer [5]. Recently, studies by this lab localized a proinflammatory cytokine, macrophage migration inhibitory factor (MIF) in the urothelium of experimental rats [6]. Chemical or lipopolysaccharide-induced cystitis was found to induce increases in the protein levels and mRNA expression of MIF in nervous system structures innervating the bladder suggesting a role for MIF in bladder inflammation [7,8]. From these findings, we hypothesized that MIF may function similarly in the human bladder.
MIF is a ubiquitously expressed protein that is able to manifest itself as a cytokine, hormone, or enzyme [9]. Consequently, it maintains a key regulatory role in inflammation and both specific and nonspecific immunity. As a proinflammatory cytokine, MIF counter-regulates the effects of glucocorticoids and stimulates the secretion of certain other cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β [10], thus assuming a role in the pathogenesis of inflammatory, immune diseases and cancer including septic shock [11], rheumatoid arthritis [12], Crohn's disease [13], and lung [14], breast [15], and prostate [16,17] cancers.
In addition to its roles in inflammation and immunity, MIF is suggested to be involved in tumor cell growth and differentiation [18]. It has been reported that MIF mRNA is over-expressed in both prostatic [16,17] and breast [15] tumors. MIF has also been associated with the growth of lymphoma cells, melanoma cells, and colon cancer cells [18]. Treatment with anti-MIF immunoglobulin therapy has been shown to possess anti-tumor activity [19].
Although MIF is associated with cancer angiogenesis, progression and metastasis the exact mechanism of this cytokine's action is unknown, as a receptor has only recently been identified as the cell surface form of the invariant chain (CD74) [20]. CD74 regulates loading of exogenous derived peptides onto major histocompatibility class II heterodimers, but a small portion of the total cell CD74 content is expressed on cell surfaces [21]. Activation of cell surface CD74 requires interaction with CD44, a major adhesion molecule expressed in most cell types that has a strong affinity for hyaluronan (HA) [22,23]. CD44's extracellular domain is cleaved by membrane bound matrix metalloproteinases [24] and the resulting soluble CD44 reported as the most dominant form of CD44 expressed in tumors [25].
Currently, no information exists on the expression of MIF in the human bladder or its association with bladder cancer. In an effort to identify new therapies for the treatment of bladder cancer, the aims of the present study include documentation of MIF synthesis and secretion by human bladder cancer epithelial cells. An additional aim of this study is determination of the effects of high molecular weight HA (a linear glucosaminoglycan) known to inhibit MIF [26], as well as anti-MIF antibody and MIF anti-sense oligonucleotides on in vitro bladder cancer cell growth and cytokine expression. Finally this study ascertains whether MIF associates with CD74 or CD44 in bladder cancer cells.
Methods
MIF in human bladder
Tissue microarray slides were obtained from the Cooperative Human Tissue Network (CHTN, block 2002N1A) of the National Cancer Institute, The National Institutes of Health, Bethesda, MD and used for both immunohistochemistry (IHC) and in situ hybridization to document MIF location in human bladder tissue. MIF IHC was performed as described previously [27]. Control slides were not treated with primary antiserum. MIF biotinylated probes for in situ were prepared by reverse transcription, followed by amplification of a 254 base pair (bp) MIF fragment [17]. T7 RNA polymerase promoters were ligated to the PCR product (Ambion, Austin, TX) and single stranded RNA oligonucleotide probes produced by in vitro T7 transcription (Promega, Madison, WI) with incorporation of biotinylated-dUTP. Tissue arrays were hybridized overnight with 100 ng probe, washed at a final stringency of 0.1 × SSC at 60°C for 30 min and probes detected with avidin-peroxidase (ISH-B1, Sigma, St. Louis, MO). Negative controls included hybridization with sense-biotinylated probes and hybridization without probe addition. A poly d(T) biotinylated probe was used as a positive control. Slides were not counterstained.
Cell culture
HT-1376 cells (ATCC, Manassas, VA) were plated at 1.0 × 105 cells per well overnight in DMEM supplemented with 5% FBS, then rendered quiescent by overnight incubation in serum free medium. Media was exchanged for that with no supplement or one of the following: sodium hyaluronate (0–500 μg/ml; Healon®, Pharmacia & Upjohn, Kalamazoo, MI), monoclonal anti-human MIF antibody (0–500 μg/ml; MAB289, R&D Systems, Minneapolis, MN) or anti-sense MIF mRNA (0–500 ng/ well). Controls for antibody treatment included addition of non-specific mouse monoclonal antibody (Sigma) and for anti-sense treatment addition of MIF sense mRNA at the appropriate concentrations.
Anti-sense MIF was prepared by reverse transcription of total RNA collected from the HT-1376 cells, followed by amplification of a 254 base pair (bp) MIF fragment (nucleotides 67–321). T7 RNA polymerase promoters were ligated to the PCR product (Lig-N-Scribe, Ambion, Austin, TX) and single stranded RNA oligonucleotide probes were produced from this template by in vitro transcription (Promega, Madison, WI) were then alkaline treated and neutralized to produce small random fragments. Transfection of the cells was carried out using LipofectAMINE PLUS Reagent (Invitrogen, Carlsbad, CA). Anti-sense (or sense) RNA was mixed with 10 μl of serum-free DMEM and 1 μl of PLUS Reagent followed by 0.5 μl of LipofectAMINE Reagent then diluted with 10 μl of serum-free DMEM. 50 μl of serum-free DMEM/F12 was added to each well, followed by 20 μl of transfection mix. Plates incubated at 37°C and 5% CO2 for 3 h. Following incubation, 70 μl of 1% serum DMEM was added to the wells.
Proliferation was assessed using a colorimetric assay (MTS, Promega). Upon media removal from the wells, 100 μl of DMEM containing 20 μl MTS was added. Cells were incubated for 30 min, followed by measurement of the absorbance of the formazon product at 490 nm. For quantification, relative numbers of cells per well were determined using a generated standard curve. The exact cell counts were determined by direct counting and two-fold dilutions of cells in 100 μl volumes of 105 to 102 cells per well were attached for 8 h, then incubated with MTS reagent for 30 min. The growth curve to estimate cell counts was produced by plotting the OD 490 nm of the 30 min MTS incubation versus the exact cell number. Data are expressed total cell numbers and are the treatment group means (n = 6) from three separate experiments.
Protein analysis
Following 24 h exposure to the different treatments, culture media was collected and immediately frozen at -80°C and cells lysed by the direct addition of 100 μl RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS and 50 mM Tris, pH 7.5) with protease inhibitors (Sigma).
Cell lysate samples were tested for caspase-3 activity using reaction buffer (20 mM HEPES pH 7.4 containing 100 mM NaCl, 10 mM DTT, 0.1% CHAPS, and 10% sucrose) and 4 mM DEVD-pNA substrate (Sigma) followed by incubation in the dark at 37°C for 2 h. Absorbance levels were read at 405 nm.
Protein concentrations were determined by modified Bradford assay (BioRad, Hercules, CA). Proteins (30 μg/ml total) were separated by polyacrylamide gel electrophoresis [7] and transferred to PVDF for Western blot analysis. CD44 and CD74 protein bands were visualized using specific antibodies (Santa Cruz, Ann Arbor, MI). The resultant protein bands were quantified by digital imaging [7]. Data are expressed as integrated density values (IDV, band area times relative intensity) and are the treatment group means of Western blot determinations performed from three separate experiments.
Secreted and intracellular levels of MIF were quantified using a sandwich ELISA [17]. Data are expressed as pg/105 cells and are the treatment group means from three separate experiments.
An array for secreted human cytokine proteins (Human Cytokine Array V, RayBio, Norcross, GA) was used to determine relative changes in secreted cytokines. Membranes with immobilized antibodies were incubated for 14 h with either 1 ml of the control media (untreated, non-specific IgG treated or sense- treated) or 1 ml of the experimental media pooled from 12 wells, followed by biotin-conjugated antibodies, then horseradish-peroxidase (HRP)-conjugated streptavidin (0.2 μg/ml) (DuoSet, R&D Systems) and detected by chemiluminescence (ECL Plus, Amersham, Piscataway, NJ). Integrated density values were calculated for each spot (area times relative intensity). Then positive control signals on each membrane were used to normalize cytokine signal intensities using SigmaScan Pro image-analysis software (Jandel Scientific Software, San Raphael, CA). A two-fold change in spot intensity from treated culture medium compared to control culture medium was considered significant. For each treatment, the array analysis was performed twice using conditioned medium from two separate experiments. The mean treatment control data are expressed as a mean protein ratio ± the standard error, which were determined by dividing the normalized integrated density values of the cytokine spot from all of the control cultures by the integrated density value of the positive control (HRP) spot. The effect of treatment on cytokine secretion was determined by dividing the cytokine spot normalized integrated density value from the treatment culture by the cytokine spot normalized integrated density value from the control culture. Data are expressed as +, greater than 2 fold increase; -, greater than 2 fold decrease; (-) decreased beyond detection.
Gene expression analysis
Total RNA was obtained from cells by the direct addition of TRIzol Reagent (Invitrogen). 1 μg of extracted total RNA was reverse transcribed (Promega) and MIF amplified by PCR using as described [27]. IL – 1β or TNF-α were amplified as described [28]. Control conditions and quantification of resultant PCR bands intensities were as described [8]. Data are expressed as a ratio of target PCR band area intensity divided by 18 S rRNA internal standard band area intensity.
Immunoprecipitation
The association of MIF with CD74 and CD44 in HT-1376 cells was ascertained by immunoprecipitation with either anti-human CD74 or CD44 (3 μg/ml, Santa Cruz) using Protein G agarose beads per the manufacturer's protocol (Kirkegaard & Perry Laboratories, Gaithersburg, MD), followed by detection on Western blots. All incubations were performed at 4°C with continuous rocking. Cleared cell lysates were incubated overnight with antibody and equilibrated Protein G agarose beads (Kirkegaard & Perry Laboratories, Gaithersburg, MD). Controls included incubation of pre-cleared samples with nonspecific polyclonal IgG (Sigma) and samples without addition of antibody in the immunoprecipitation reaction.
Statistical analysis
Statistical analysis was performed using Prism statistical analysis software version 4 (GraphPad Software). For ELISA and MTS assays linear regression with interpolation was used to determine either antigen concentration or cell number, respectively. The results of all other experiments were analyzed using unpaired two-tailed t test or analysis of variance (ANOVA). Data are reported as the mean values ± standard error of the means (SEM). Statistical significance was determined at values of p < 0.05.
Results
MIF localization in normal human bladder
MIF protein is readily detected within the normal human urothelium (Fig. 1A). MIF protein was primarily localized throughout the urothelial cytoplasm with some evidence of perinuclear localization (Fig. 1A). In addition, there was intense apical staining in the surface urothelial cells (Fig. 1A, arrows). MIF in situ established that the urothelium was the source of the detected MIF protein as MIF mRNA was detected in the urothelial cytoplasm (Fig. 1B). The in situ signal was variable, but detectable through the full thickness of the urothelium.
Figure 1.
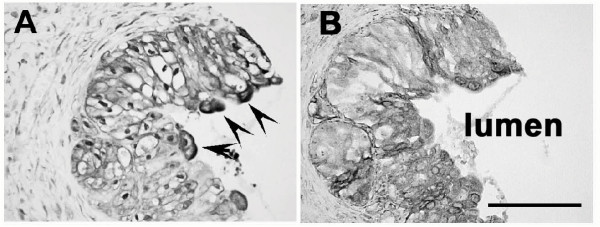
Localization of MIF in human bladder – A) MIF IHC – MIF protein is localized to the urothelial cells with mostly cytoplasmic staining and some evidence of perinuclear localization. The apical surface of the urothelial cells exhibits intense immunostaining (arrows). B) MIF in situ hybridization – MIF mRNA is localized primarily to the urothelial cytoplasm.
Treatment effects on cell proliferation
Hyaluronan, anti-MIF or MIF antisense reduced HT-1376 cell proliferation in a dose-dependent manner (Fig 2A). The first dose that resulted in a statistically significant decrease in cell proliferation was determined the minimum effective concentration to be used in subsequent experiments. Minimum effective concentrations of all treatments (hyaluronan, 100 μg/ml; monoclonal anti-human MIF antibody, 50 μg/ml; anti-sense MIF mRNA, 200 ng/ml); resulted in a small but significant reduction in cell numbers 24 h post-treatment and 48 h post-treatment when compared to untreated control cells (Fig. 2B, p < 0.01). This reduction in total cell numbers was not attributable to apoptosis induction as no capsase-3 activity was detected in cell lysates from HA, anti-MIF antibody or anti-sense treated cell cultures (data not shown). There was no significant reduction in cell proliferation in non-specific IgG (anti-human IgG1 antibody, 50 μg/ml) or MIF-sense (sense MIF mRNA, 200 ng/ml) treated cells indicating that the reduction in cell numbers was not due to either non-specific antibody effects or the transfection protocol, respectively (data not shown). HT-1376 cells secrete large amounts of hyaluronidase and various proteases [29], the effects of the various treatments on MIF expression and cytokine production were examined at 24 h so as to limit potential confounds associated with hyaluronan degradation and antibody proteolysis.
Figure 2.
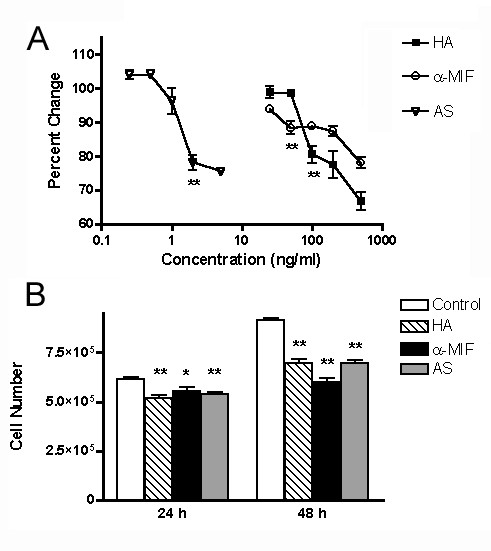
Effect of treatments on HT-1376 cell proliferation. A) Dose response curve – Effect of varying concentrations of HA, anti-MIF antibody and MIF anti-sense on HT-1376 cell growth over 24 h period. Data are expressed as percentage of control for each condition. Control conditions: HA, DMEM plus 1% BSA, anti-MIF antibody; non-specific mouse IgG1 at 50 μg/ml; antisense, MIF sense RNA at 2 ng/ml. The first dose to produce a statistically significant decrease in cell proliferation is listed for each treatment (* p < 0.05; ** p <0.01) and was used as the minimal effective dose for subsequent experiments. B) Minimal effective dose – Effect of minimal effective doses of HA, anti-MIF antibody and MIF anti-sense on HT-1376 cells following 24 and 48 h treatment. All treatments resulted in a decrease cell proliferation at both time periods, with a greater effects observed after 48 hours. Data are expressed as cell number as measured by MTS assay (* – p < 0.05, ** – p < 0.01)
Treatment effects on MIF production by bladder cancer cells
HT-1376 cells constitutively secrete MIF at concentrations comparable to those determined in prostate cancer cells [27]. The secreted MIF amounts significantly declined with all treatments with the greatest decrease seen with anti-MIF treatment (Fig. 3A; p < 0.001).
Figure 3.
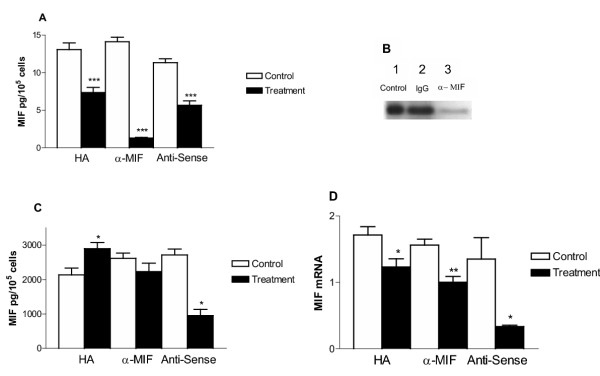
Effect of treatments on MIF secretion, intracellular content and gene expression. A. MIF secretion – Conditioned medium following 24 h growth was assayed for MIF content by ELISA. Data are expressed as culture medium MIF concentration in pg per 105 cells. All treatments decreased MIF in the culture media. B. Western blot of anti-MIF treated conditioned medium following 24 h treatment- 12 kDa MIF band is present in all samples. Lane 1, DMEM 1% BSA; Lane 2, non-specific mouse IgG1; Lane 3, anti-MIF antibody treatment. C. Intracellular MIF content – Changes in intracellular MIF 24 h post treatment. Data are expressed as cleared cell lysate MIF concentration in pg per 105 cells. Note that HA treatment increased, while MIF antisense treatment decrease the concentration of MIF in the cell lysates. Anti-MIF treatment did not produce a significant effect. D. MIF gene expression -MIF mRNA content was quantified in cells following 24 h and the indicated treatment. Data are expressed as a relative intensity ratio. PCR band intensity was determined from the formula, total intensity = area × average intensity. The relative intensity ratio is determined from the total intensity of gene specific PCR product band divided by the 18 S rRNA band intensity (internal standard). All treatments resulted in a decrease in MIF mRNA with the greatest effects seen following MIF anti-sense treatment. (* – p < 0.05, ** – p < 0.01, *** – p < 0.001).
It could be argued that anti-MIF treatment does not reduce MIF secretion, but rather the resulting interaction between secreted MIF and the exogenously added antibody results in an inability to detect MIF in the culture medium by ELISA. We performed Western blot analysis of culture medium, since under reducing conditions MIF protein should dissociate from anti-MIF IgG. Western blot analysis revealed a single 12 kDa band corresponding to the known molecular weight of MIF [30]. These results confirmed that the anti-MIF treatment inhibited MIF secretion, as Western blot analysis detected little MIF protein in cell culture medium after anti-MIF treatment (Fig. 3B, lane 3).
Concomitant with the decreased secreted MIF were changes in the intracellular MIF content (Fig. 3C). HA treatment resulted in a small but significant increase in the intracellular MIF content (p < 0.05), while anti-MIF treatment had no effect on intracellular MIF content and MIF antisense treatment resulted in a decrease in total MIF content, as would be expected (p < 0.05). MIF mRNA amounts decreased significantly with all treatments when compared to control cultures (Fig. 3D).
Treatment effects on TNF-α and IL-1β expression
MIF blockade has been shown to affect the production of various proinflammatory cytokines [10]. RT-PCR analysis was performed to determine whether the HA, anti-MIF antibody and MIF-antisense altered the expression of TNF-α and IL-1β mRNA in HT-1376 cells (Fig. 4). HA had no effect on TNF-α mRNA, while anti-MIF antibody and MIF anti-sense significantly decreased TNF- mRNA amounts when compared to untreated cells (Fig. 4A). IL-1β expression, on the other hand, was not affected by any treatment (Fig. 4B).
mRNA amounts when compared to untreated cells (Fig. 4A). IL-1β expression, on the other hand, was not affected by any treatment (Fig. 4B).
Figure 4.
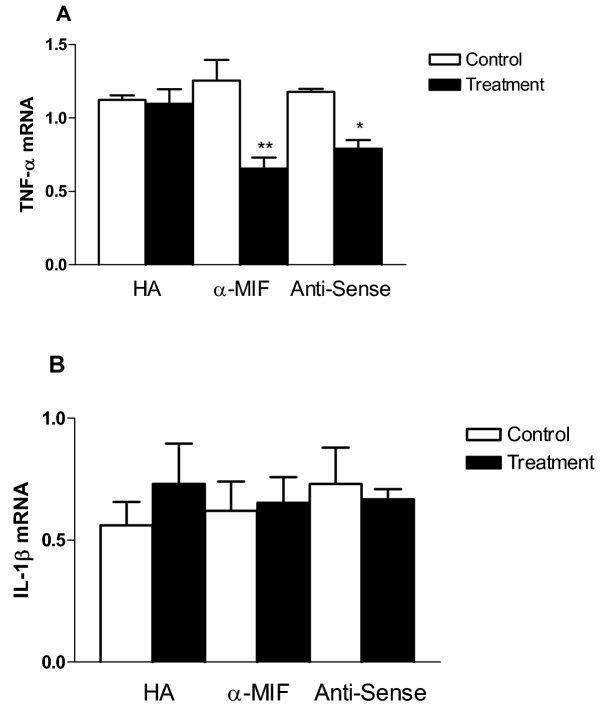
Effect of MIF Inhibition on TNF-α and IL-1β gene expression – A. TNF-α expression – TNF-α mRNA content was quantified in cells following 24 h with the indicated treatment. Anti-MIF and MIF antisense treatments decreased the levels of TNF-alpha mRNA. HA treatment, however, was not effective. B. IL-1β expression – IL-1β mRNA content was quantified in cells following 24 h with the indicated treatment. No significant difference in IL-1β expression was seen with any of the treatments. In both A and B the data is expressed as a relative intensity ratio. PCR band intensity was determined from the formula, total intensity = area × average intensity. The relative intensity ratio is determined from the total intensity of gene specific PCR product band divided by the 18 S rRNA band intensity (internal standard), (* – p < 0.05, ** – p < 0.01).
Treatment effects on secreted cytokine
Treatments effects on the amounts of proinflammatory cytokines detected in culture medium was studied using protein arrays. The conditioned medium from all HT-1376 controls (untreated, non-specific IgG and sense) contained 65 out of the possible 79 cytokines on the array. There was no difference in the protein ratios of the 65 secreted cytokines in the different controls (Table 1). Secretion of 18 cytokines was downregulated by all three treatments (>2-fold, Table 1). Significant among the cytokines downregulated by all three treatments were the CXC chemokines CXCL5 and CXCL1, previously shown by others as induced by MIF resulting in increased small lung cell tumor cell angiogenic activity (Table 1A, [31]). Nine of the 18 cytokines downregulated by all three treatments were CC chemokines (Table 1).
Table 1.
Secreted cytokine array. Secreted proinflammatory cytokines down-regulated by HA, anti-MIF antibody and MIF anti-sense. Integrated density values (IDV, average intensity times total area) were determined for each individual cytokine dot. Individual cytokine dot intensities were normalized to HRP-positive controls on each membrane. Mean control data for each cytokine spot are expressed as protein ratios (cytokine specific band integrated density value divided by HRP-positive control integrated density value). Treatment data are expressed as +, greater than 2 fold increase in secretion compared with control cytokine spot; -, greater than 2 fold decrease in secretion compared with control cytokine spot; (-), decreased beyond detection.
| Cytokine Family | |||||
| Control (Mean Protein Ratio) | HA | α-MIF | AS | ||
| Chemokine CC | |||||
| CCL2 | 1.27 ± 0.04 | - | (-) | ||
| CCL4 | 0.30 ± 0.02 | - | (-) | (-) | |
| CCL5 | 0.18 ± 0.01 | - | (-) | (-) | |
| CCL11 | 0.11 ± 0.01 | (-) | (-) | (-) | |
| CCL15 | 0.41 ± 0.04 | (-) | (-) | - | |
| CCL17 | 0.12 ± 0.01 | - | (-) | (-) | |
| CCL18 | 1.97 ± 0.07 | - | (-) | - | |
| CCL20 | 0.69 ± 0.04 | (-) | - | ||
| CCL22 | 0.19 ± 0.02 | - | (-) | - | |
| CCL23 | 0.12 ± 0.01 | (-) | (-) | (-) | |
| CCL24 | 0.14 ± 0.01 | - | (-) | - | |
| CCL26 | 0.20 ± 0.01 | (-) | (-) | (-) | |
| CXC Chemokine | |||||
| CXCL1 | 0.16 ± 0.02 | - | - | (-) | |
| CXCL2 | 0.15 ± 0.02 | - | - | (-) | |
| CXCL5 | 0.19 ± 0.02 | - | (-) | (-) | |
| CXCL6 | 0.26 ± 0.03 | - | - | - | |
| CXCL7 | 1.56 ± 0.03 | - | (-) | - | |
| CXCL8 | 0.32 ± 0.02 | - | - | ||
| CXCL9 | 0.16 ± 0.01 | - | (-) | - | |
| CXCl10 | 1.60 ± 0.17 | - | - | - | |
| CXCL12 | 0.15 ± 0.01 | - | (-) | - | |
| CXCL13 | 0.34 ± 0.02 | - | (-) | - | |
| CXC3C Chemokine | |||||
| CX3CL1 | 0.19 ± 0.01 | - | (-) | ||
| Cytokines | |||||
| Leptin | 0.19 ± 0.02 | - | (-) | (-) | |
| LIF | 3.20 ± 0.04 | - | |||
| MIF | 0.64 ± 0.04 | (-) | (-) | (-) | |
| OSM | 0.20 ± 0.02 | - | (-) | (-) | |
| Interferon | |||||
| INF-γ | 0.17 ± 0.02 | - | (-) | (-) | |
| Neurotropins | |||||
| BDNF | 1.10 ± 0.05 | - | - | ||
| GDNF | 1.30 ± 0.07 | - | |||
| NT-3 | 2.00 ± 0.11 | - | (-) | ||
| NT-4 | 0.79 ± 0.04 | - | (-) | - | |
| Enzymes | TIMP-1 | 2.54 ± 0.11 | - | ||
| TIMP-2 | 5.29 ± 0.29 | - | + | ||
| Growth Factors | |||||
| Ang-1 | 0.20 ± 0.02 | (-) | (-) | ||
| EGF | 0.17 ± 0.03 | - | (-) | - | |
| FGF-4 | 0.24 ± 0.03 | - | (-) | (-) | |
| FGF-6 | 0.18 ± 0.01 | - | (-) | - | |
| FGF-9 | 0.81 ± 0.05 | - | |||
| HGF | 0.12 ± 0.02 | - | (-) | (-) | |
| MCSF | 0.15 ± 0.02 | - | (-) | (-) | |
| PGIF | 0.64 ± 0.05 | - | (-) | - | |
| Inhibin | 0.17 ± 0.02 | - | (-) | (-) | |
| TGF β-2 | 3.20 ± 0.03 | - | |||
| TGF β-3 | 0.77 ± 0.02 | - | (-) | (-) | |
| Hormone-like protein | |||||
| IGF-1 | 0.14 ± 0.01 | (-) | (-) | (-) | |
| IGFBP-1 | 2.40 ± 0.10 | - | |||
| IGFBP-2 | 1.40 ± 0.06 | - | - | ||
| IGFBP-3 | 2.50 ± 0.12 | - | (-) | ||
| IGFBP-4 | 0.11 ± 0.01 | - | (-) | - | |
| TP-3 | 0.13 ± 0.01 | - | (-) | (-) | |
| Interleukins | |||||
| IL-1α | 0.20 ± 0.01 | - | (-) | (-) | |
| IL-1β | 0.14 ± 0.02 | - | (-) | (-) | |
| IL-2 | 0.14 ± 0.04 | - | (-) | (-) | |
| IL-12 | 0.16 ± 0.01 | - | (-) | (-) | |
| IL-15 | 0.10 ± 0.09 | - | (-) | (-) | |
| IL-16 | 0.27 ± 0.04 | - | (-) | - | |
| PDGF Superfamily | |||||
| PDGF-B | 0.17 ± 0.02 | - | (-) | (-) | |
| P1GF | 0.59 ± 0.04 | - | (-) | - | |
| VEGF | 3.20 ± 0.48 | - | + | - | |
| TNF Superfamily | |||||
| TNFSF14 | 0.19 ± 0.01 | - | - | (-) | |
| TNFSF2 | 0.13 ± 0.02 | (-) | (-) | - | |
| TNFRSF11B | 4.40 ± 0.04 | - | |||
| CD Ligands | |||||
| Fit-3 | 0.07 ± 0.01 | - | - | (-) | |
HA decreased the amounts of 27 of the secreted cytokines with a complete loss of detectable TNF-α and MIF with no effect on the secreted amounts of IL-1β. Anti-MIF antibody decreased the amounts of 55 of the secreted cytokines with complete loss of TNF-α, IL-1β and MIF. While, MIF anti-sense treatment resulted in a decrease in 61 of the secreted cytokines and reduction of secreted TNF-α (4-fold) with complete loss of detectable secreted MIF and IL-1β.
The secreted cytokine array identified two cytokines not previously identified as secreted by bladder cancer cells (as determined by PubMED search); MDC (macrophage derived chemokine, CCL22) and fractalkine (CX3CL1).
Treatment effects on CD 74 (MIF receptor) and CD 44 (HA receptor)
Based upon recent experiments by other groups [20], the interaction of MIF with CD74 and CD44 are key to MIF signal transduction. Immunoprecipitation assays were performed to ascertain whether MIF interacts with CD74 or CD44 in HT-1376 cells. MIF was detected by Western blotting of protein complexes precipitated from HT-1376 cell lysates by CD74 antibody (Fig. 5A, lane 1) and CD44 antibody (Fig. 5A, lane 2). The MIF amounts associated with CD74 were much less than that associated with CD44. This likely reflects the relative abundance of these two proteins in HT-1376 cells, which is demonstrated in the Western blots of untreated cultures (Fig. 5C and 5E, lane 1). Interaction of CD74 with CD44 was demonstrated by immunoprecipitation with CD44 antibody with detection of CD74 on Western blots (Fig. 5A, lane 5). Antigen specific bands were absent in Western blots using non-specific antibody or with no antibody in the immunoprecipitation (Fig. 5A, lane 3, 4). Confirmation of the production of CD74 by HT-1376 cells was accomplished by Western blots (Fig. 5C, lane 1). In all instances, treatment resulted in a significant increase in CD74 protein (Fig. 5C, lanes 2 – 4 respectively).
Figure 5.
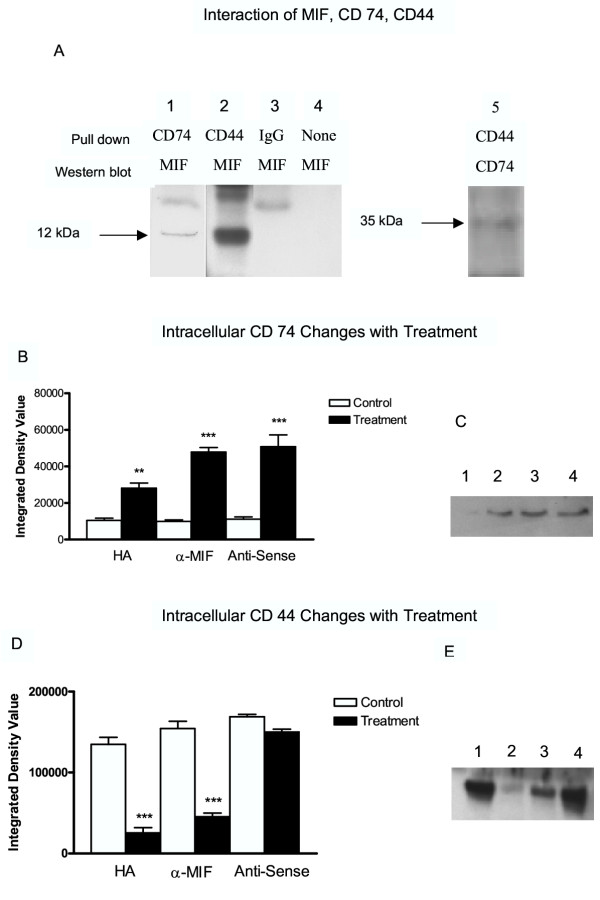
Effect of treatment on CD74 and CD44 proteins A. MIF pull down experiment – Cell lysates from 24 h control cultures were immunoprecipitated with CD74 antibody (lane 1) or CD44 antibody (lane 2,5) and the protein G binding protein complexes separated by polyacrylamide gel electrophoresis, followed by Western blotting using polyclonal anti-MIF or CD74 antibody. Lane 1, CD74 antibody; lane 2, CD44 antibody; lane 3, non-specific antibody; lane 4, no antibody added; lane 5, CD44 antibody. B. CD 74 Western blot analysis – Proteins in cell lysates from 24 h cultures treated with indicated reagents were separated by polyacrylamide gel electrophoresis, followed by Western blotting using polyclonal anti-CD74 antibody. Bands were quantified by digital imaging and are expressed as integrated density values (band area × relative intensity) and are presented as the mean ± SEM of three separate experiments of quadruplicate cultures. All treatments increased the levels of CD74 in the cell lysates. C. CD 74 Western blot – Proteins in cell lysates from 24 h cultures treated with indicated reagents were separated by polyacrylamide gel electrophoresis, followed by Western blotting using polyclonal anti-CD74 antibody. Blot is representative of those from three separate experiments. D. CD 44 Western blot analysis – Proteins in cell lysates from 24 h cultures treated with indicated reagents were separated by polyacrylamide gel electrophoresis, followed by Western blotting using polyclonal anti-CD44 antibody. Bands were quantified by digital imaging and are expressed as integrated density values (band area × relative intensity) and are presented as the mean ± SEM of three separate experiments of quadruplicate cultures. HA and anti-MIF treatments significantly decreased the levels of CD44 in cell lysates. MIF antisense had no effect. E. CD 44 Western blot – Proteins in cell lysates from 24 h cultures treated with indicated reagents were separated by polyacrylamide gel electrophoresis, followed by Western blotting using polyclonal anti-CD44 antibody. Blot is representative of those from three separate experiments.(** – p < 0.01, *** – p < 0.001).
CD44 production by HT-1376 cells was confirmed by Western blot analysis (Fig. 5E, lane 1). The effect of treatments on CD44 protein concentrations was determined by Western blot analysis (Fig. 5D,5E). A major band at approximately 60 kDa was identified, which corresponds to the known size of soluble CD44 [24]. Soluble CD44 amounts were significantly decreased by HA and anti-MIF antibody treatment (p < 0.001, Fig. 5E, lane 2 and 3 respectively), while the CD44 concentrations were not affected by MIF anti-sense treatments (Fig. 5D lane 4).
Discussion
MIF is a unique proinflammatory cytokine, found preformed in various tissues, which plays a critical role in immune response and inflammation [10]. Over the past several years evidence has accumulated supporting a role for MIF in various disease pathologies. In particular, significant experimental data have emerged linking MIF to the development and aggressiveness of adenocarcinoma [32]. Increased MIF expression has been associated with a wide variety of tumor cell types including breast [33], prostate [16,17], lung [34], colon [35], liver [36], and glioblastoma [37]. Prior studies in this laboratory determined that MIF is located in the rat bladder [6]. Here we report for the first time on the presence of MIF in the human urothelium and MIF synthesis and secretion by HT-1376 human transitional cell carcinoma cells.
HA treatment significantly decreased secreted MIF amounts. In this instance, the interference with MIF secretion is evidenced by the significant increase in intracellular MIF content along with the attenuation of MIF gene expression. Anti-MIF antibody treatment resulted in the maximum suppression of MIF secretion (90.1%). Conversely, MIF anti-sense treatment decreased both the intracellular MIF content and the amount of secreted MIF, suggesting that MIF anti-sense treatment interfered with MIF synthesis.
Endogenous MIF secretion may activate autocrine and/or paracrine regulatory loops that enhance or sustain cancer cell growth [38]. While, the precise molecular mechanism behind MIF's function in tumorigenesis and metastasis is unknown numerous studies have determined that reduction in tumor MIF results in reduced cell proliferation [39], apoptosis induction [40], and reduction in the secretion of other growth factors and cytokines [41]. In accordance with previous findings, all the treatments used in the present study were found to have a suppressive effect on HT-1376 cell proliferation.
MIF is suggested to function as a regulator of pro-inflammatory gene and cytokine expression, and thus, alterations in MIF activity are expected to affect the expression and secretion of other proinflammatory molecules and cytokines [41]. The role of MIF in stimulating the production of additional cytokines, including TNF-α, IL-1β, among others has been previously documented [41,42]. MIF's exerts an upstream effect on TNF-α production [43]. Interestingly, the present study identified MIF blockade as a key means to reduce the production of various proinflammatory and growth promoting cytokines and growth factors by bladder tumor cells. All examined treatments significantly altered the secretion of a wide range of cytokines. In particular anti-MIF antibody and MIF anti-sense treatments both abolished the secretion of TNF-α and IL-1β. The decreased gene expression of TNF-α and the altered cytokine secretion patterns lend further evidence for MIF's role as a modulator of cytokine production.
The observed reduction in cytokine production by MIF blockade raises further questions about the physiological relationship between MIF and other cytokines and the possible importance of MIF interaction with HT-1376 cells. Recently CD74 was established as a MIF cell surface receptor [20]. CD74, also known as the invariant chain acts as a chaperone for major histocompatibility class (MHC) II proteins [44]. CD74 plays a critical role in this process by influencing both the expression and peptide loading of MHC II molecules for antigen presentation. Previously other investigators determined that greater than constitutive amounts of recombinant MIF downregulated concentrations of CD74's intracellular ligand, MHC II [45]. In the present studies we establish that HT-1376 cells produce the MIF receptor and that its expression is upregulated when its extracellular ligand (MIF) concentration is reduced. These findings support the hypothesis that MIF modulates CD74 and MHC II expression.
Recently it has been established that activated CD74 acts as a signaling molecule by nuclear activation of NF-κβ [46]. However, the intracellular portion of CD74 does not contain domains predicted to activate downstream signaling molecules. Of note, studies have suggested that CD74's interaction with CD44 results in cellular activation [22,23]. HA binds the pleiomorphic cell surface molecule CD44, which functions in cell proliferation and adhesion [47]. Recent studies indicate high molecular weight HA binding to CD44 hinders its cleavage, thus interrupting CD44 induced angiogenesis and tumor promotion [48]. Membrane associated metalloproteinases cleave the extracellular domain of CD44 thus providing changes in cell adhesion to the extracellular matrix, which results in increased tumor cell migration [49]. In noncancerous cells the intracellular domain of intact CD44 binds to merlin resulting in a growth inhibitory signal [50]. It is suggested that CD44 functions in MIF signal transduction in some cells via horizontal recruitment of the protein into the MIF-CD74 complex [20]. From the present study it appears that although high molecular weight HA, which interferes with CD44 function, reduces MIF secretion and expression, this is still not sufficient to completely abrogate cell proliferation and cytokine secretion.
We developed a tentative model of MIF- CD44- CD74 interaction. Based upon our current model CD74 is the rate-limiting member of MIF signal transduction complex as it is found in rather low concentrations in HT-1376 cells when compared to CD44 or MIF. In HT-1376 cells large quantities of MIF are constitutively synthesized and secreted, which interact with soluble CD44. The soluble CD44-MIF complex interacts with CD74 to induce activation of cytokine production and cell proliferation presumably through ERK1/2 [51]. Treatment of HT-1376 cells with high molecular weight HA blocks cleavage of CD44 resulting in decline in cell proliferation and cytokine production. Anti-MIF antibody prevents interaction of MIF with soluble CD44 again blocking MIF signal transduction. MIF anti-sense treatment results in inadequate MIF amounts to interact with soluble CD44, which also results in reduced cell proliferation and cytokine production.
Conclusions
In summary, the present study localized MIF within human bladder tissue and identifies MIF synthesis and secretion by human transitional cell carcinoma cells. The results reported here, point towards MIF inhibition as a directed therapeutic intervention in bladder cancer. Intravesical instillation of HA, anti-MIF antibodies, and MIF anti-sense presents as new potential therapies for bladder cancer treatment. Pharmacological interference of the hypothetical MIF-CD44-CD74 complex may offer a new approach to treating bladder cancer.
Competing interests
None declared.
Author's contributions
KLMS participated in the design of the study, ELISA, PCR experiments, Western blotting, statistical analyses and performed in situ hybridizations. ECL carried out all cell culture and proliferation studies. PLV participated in the design of the study, immunohistochemical analysis and dose response studies. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgments
Veterans Administration Merit Review Awards (PLV; KLMS) and the Bay Pines Foundation (KLMS) supported this work. Gary A. Smith Jr. and Michael Bellino provided excellent technical assistance. We thank Dr. Irving Nadelhaft for the use of his microscope and digital camera system.
Contributor Information
Katherine L Meyer-Siegler, Email: Katherine.Siegler@med.va.gov.
Erica C Leifheit, Email: leifheec@eckerd.edu.
Pedro L Vera, Email: pvera@hsc.usf.edu.
References
- Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- Small EJ, Halabi S, Dalbagni G, Pruthi R, Phillips G, Edelman M, Bajorin D, Cancer and Leukemia Group B Overview of bladder cancer trials in the cancer and leukemia group B. Cancer. 2003;97:2090–2098. doi: 10.1002/cncr.11299. [DOI] [PubMed] [Google Scholar]
- Piazza GA, Thompson WJ, Pamukeu R, Alila HW, Whitehead CM, Liu L, Fetter JR, Gresh WE, Jr, Klein-Szanto AJ, Farnell DR, Eto I, Grubbs CJ. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 2001;61:3961–3968. [PubMed] [Google Scholar]
- Herr HW, Badalament RA, Amato DA, Laudone VP, Fair WR, Whitmore WF., Jr Superficial bladder cancer treated with bacillus Calmette-Guerin: a multivariate analysis of factors affecting tumor progression. J Urol. 1989;141:22–29. doi: 10.1016/s0022-5347(17)40575-1. [DOI] [PubMed] [Google Scholar]
- La Vecchia C, Negri E, D'Avanzo B, Savoldelli R, Francesci S. Genital and urinary tract disease and bladder cancer. Cancer Res. 1991;51:629–631. [PubMed] [Google Scholar]
- Vera PL, Meyer-Siegler KL. Anatomical location of macrophage migration inhibitory factor in urogenital tissues, peripheral ganglia and lumbosacral spinal cord of the rat. BMC Neuroscience. 2003;4:17. doi: 10.1186/1471-2202-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera PL, Ordorica RC, Meyer-Siegler KL. Hydrochloric acid-induced changes in macrophage migration inhibitory factor in the bladder, peripheral and central nervous system of the rat. J Urol. 2003;170:623–627. doi: 10.1097/01.ju.0000066001.10343.d5. [DOI] [PubMed] [Google Scholar]
- Meyer-Siegler KL, Ordorica RC, Vera PL. Macrophage migration inhibitory factor is upregulated in an endotoxin-induced model of bladder inflammation in rats. J Interferon and Cytokine Res. 2004;24:55–63. doi: 10.1089/107999004772719918. [DOI] [PubMed] [Google Scholar]
- Swope MD, Lolis E. Macrophage migration inhibitory factor: cytokine, hormone or enzyme? Rev Physiol Biochem Pharmacol. 1999;139:1–32. doi: 10.1007/BFb0033647. [DOI] [PubMed] [Google Scholar]
- Baugh JA, Bucala R. Macrophage migration inhibitory factor. Crit Care Med. 2002;30:S27–S35. doi: 10.1097/00003246-200201001-00004. [DOI] [PubMed] [Google Scholar]
- Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- Leech M, Metz C, Hall P, Hutchinson P, Gianis K, Smith M, Weedon H, Holdsworth SR, Bucala R, Morand EF. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis & Rheumatism. 1999;42:1601–1608. doi: 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- de Jong YP, Abadia-Molina AC, Satoskar AR, Clarke K, Rietdijk ST, Faubion WA, Mizoguchi E, Metz CN, Alsahli M, ten Hove T, Keates AC, Lubetsky JB, Farrell RJ, Michetti P, van Deventer SJ, Lolis E, David JR, Bhan AK, Terhorst C, Sahli MA. Development of chronic colitis is dependent on the cytokine MIF. Nat Immunol. 2001;2:1061–1066. doi: 10.1038/ni720. [DOI] [PubMed] [Google Scholar]
- Kamimura A, Kamachi M, Nishihira J, Ogura S, Isobe H, Dosaka-Akita H, Ogata A, Shindoh M, Ohbuchi T, Kawakami Y. Intracellular distribution of macrophage migration inhibitory factor predicts the prognosis of patients with adenocarcinoma of the lung. Cancer. 2000;89:334–341. doi: 10.1002/1097-0142(20000715)89:2<334::AID-CNCR18>3.3.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Bando H, Matsumoto G, Bando M, Muta M, Ogawa T, Funata N, Nishihira J, Koike M, Toi M. Expression of macrophage migration inhibitory factor in human breast cancer: association with nodal spread. Jpn J Cancer Res. 2002;93:389–396. doi: 10.1111/j.1349-7006.2002.tb01269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Siegler K, Hudson PB. Enhanced expression of macrophage migration inhibitory factor in prostatic adenocarcinoma metastases. Urology. 1996;48:448–452. doi: 10.1016/S0090-4295(96)00207-5. [DOI] [PubMed] [Google Scholar]
- Meyer-Siegler KL, Bellino MA, Tannenbaum M. Macrophage migration inhibitory factor evaluation compared with prostate specific antigen as a biomarker in patients with prostate carcinoma. Cancer. 2002;94:1449–1456. doi: 10.1002/cncr.10354. [DOI] [PubMed] [Google Scholar]
- Nishihira J, Ishibashi T, Fukushima T, Sun B, Sato Y, Todo S. Macrophage migration inhibitory factor (MIF): Its potential role in tumor growth and tumor-associated angiogenesis. Ann NY Acad Sci. 2003;995:171–182. doi: 10.1111/j.1749-6632.2003.tb03220.x. [DOI] [PubMed] [Google Scholar]
- Chesney J, Metz C, Bacher M, Peng T, Meinhardt A, Bucala R. An essential role for macrophage migration inhibitory factor in angiogenesis and the growth of a murine lymphoma. Molecul Med. 1999;5:181–191. [PMC free article] [PubMed] [Google Scholar]
- Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF Signal Transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraight C, vanEndert P, Moller P, Lipp J, Ling N, MacLennan CKN, Moldenhauer G. Human major histocompatibility class II invariant chain is expressed on the cell surface. J Biol Chem. 1990;265:5787–5792. [PubMed] [Google Scholar]
- Naujokas MF, Morin M, Anderson MS, Peterson M, Miller J. The chondroitin sulfate form of invariant chain can enhance stimulation of T cell responses through interaction with CD44. Cell. 1993;74:257–268. doi: 10.1016/0092-8674(93)90417-O. [DOI] [PubMed] [Google Scholar]
- Naujokas MF, Arneson LS, Fineschi B, Peterson ME, Sitterding S, Hammond AT, Reilly C, Lo D, Miller J. Potent effects of low levels of MHC class II-associated invariant chain on CD4+ T cell development. Immunity. 1995;3:359–372. doi: 10.1016/1074-7613(95)90120-5. [DOI] [PubMed] [Google Scholar]
- Cichy J, Bals R, Potempa J, Mani A, Pure E. Proteinase-mediated release of epithelial cell associated CD44. J Biol Chem. 2002;277:44440–44447. doi: 10.1074/jbc.M207437200. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Tsuiki H, Kenyon LC, Godwin AK, Emlet DR, Holgado-Madruga M, Lanham IS, Joynes CJ, Vo KT, Guha A, Matsumoto M, Ushio Y, Saya H, Wong AJ. Proteolytic cleavage of the CD44 adhesion molecule in multiple human tumors. Am J Pathology. 2002;160:441–447. doi: 10.1016/S0002-9440(10)64863-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Gaag R, Broersma L, Koornneef L. The influence of high molecular weight sodium hyaluronate (Healon) on the production of migration inhibitory factor. Curr Eye Res. 1987;6:1433–1440. doi: 10.3109/02713688709044507. [DOI] [PubMed] [Google Scholar]
- Meyer-Siegler K, Fattor RA, Hudson PB. Expression of macrophage migration inhibitory factor in the human prostate. Diag Mol Path. 1998;7:44–50. doi: 10.1097/00019606-199802000-00008. [DOI] [PubMed] [Google Scholar]
- Seydel KB, Ellen LI, Swanson PE, Stanely SL., Jr Human intestinal epithelial cells produce proinflammatory cytokines in response to infection in a SCID mouse-human intestinal xenograft model of amebiasis. Infect Immun. 1997;65:1631–1639. doi: 10.1128/iai.65.5.1631-1639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokeshwar VB, Young MJ, Goudarzi G, Iida N, Yudin AI, Cherr GN, Selzer MG. Identification of bladder tumor-derived hyaluronidase: Its similarity to HYAL1. Cancer Res. 1999;59:4464–4470. [PubMed] [Google Scholar]
- Meyer-Siegler K. Cox-2 specific inhibitor, NS-398, increases macrophage migration inhibitory factor expression and induced neuroendocrine differentiation in C4-2b prostate cancer cells. Molecular Medicine. 2001;7:850–860. [PMC free article] [PubMed] [Google Scholar]
- Mitchell RA, Bucala R. Tumor growth-promoting properties of macrophage migration inhibitory factor (MIF) Semin Cancer Biol. 2000;10:359–66. doi: 10.1006/scbi.2000.0328. [DOI] [PubMed] [Google Scholar]
- Bini L, Magi B, Marzocchi B, Arcuri F, Tripodi S, Cintorino M, Sanchez JC, Frutiger S, Hughes G, Pallini V, Hochstrasser DF, Tosi P. Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis. 1997;18:2832–2841. doi: 10.1002/elps.1150181519. [DOI] [PubMed] [Google Scholar]
- White ES, Flaherty KR, Carskadon S, Brant A, Iannettoni MD, Yee J, Orringer MB, Arenberg DA. Macrophage migration inhibitory factor and CXC chemokine expression in non-small cell lung cancer: role in angiogenesis and prognosis. Clin Cancer Res. 2003;9:853–860. [PubMed] [Google Scholar]
- Takahashi N, Nishihira J, Sato Y, Kondo M, Ogawa H, Ohshima T, Une Y, Todo S. Involvement of macrophage migration inhibitory factor (MIF) in the mechanism of tumor cell growth. Mol Med. 1998;4:707–714. [PMC free article] [PubMed] [Google Scholar]
- Akbar SM, Abe M, Murakami H, Tanimoto K, Kumagi T, Yamashita Y, Michitaka K, Horiike N, Onji M. Macrophage migration inhibitory factor in hepatocellular carcinoma and liver cirrhosis; relevance to pathogenesis. Cancer Lett. 2001;171:125–132. doi: 10.1016/S0304-3835(01)00606-1. [DOI] [PubMed] [Google Scholar]
- Ren Y, Tsui HT, Poon RT, Ng IO, Li Z, Chen Y, Jiang G, Lau C, Yu WC, Bacher M, Fan ST. Macrophage migration inhibitory factor: Roles in regulating tumor cell migration and expression of angiogenic factors in hepatocellular carcinoma. Int J Cancer. 2003;107:22–29. doi: 10.1002/ijc.11287. [DOI] [PubMed] [Google Scholar]
- Bacher M, Schrader J, Thompson N, Kuschela K, Gemsa D, Waeber G, Schlegel T. Up-regulation of macrophage migration inhibitory factor gene and protein expression in glial tumor cells during hypoxic and hypoglycemic stress indicates a critical role for angiogenesis in glioblastoma multiforme. Am J Pathol. 2003;162:11–17. doi: 10.1016/S0002-9440(10)63793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Kasuya K, Nishihira J, Magami Y, Tsuchida A, Aoki T, Koyanagi Y. Suppression of tumor growth through introduction of an antisense plasmid of macrophage migration inhibitory factor. Int J Mol Med. 2002;10:579–583. [PubMed] [Google Scholar]
- Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawara T, Nishihira J, Takeda H, Hige S, Kato M, Sugiyama T, Iwanaga T, Nakamura H, Mizue Y, Asaka M. Amelioration of dextran sulfate sodium-induced colitis by anti-macrophage migration inhibitory factor antibody in mice. Gastroenterology. 2002;123:256–70. doi: 10.1053/gast.2002.34236. [DOI] [PubMed] [Google Scholar]
- Lolis E, Bucala R. Macrophage migration inhibitory factor. Expert Opin Ther Targets. 2003;7:153–164. doi: 10.1517/eott.7.2.153.23778. [DOI] [PubMed] [Google Scholar]
- Murakami H, Akbar SM, Matsui H, Horiike N, Onji M. Macrophage migration inhibitory factor activates antigen-presenting dendritic cells and induces inflammatory cytokines in ulcerative colitis. Clin Exp Immunol. 2002;128:504–510. doi: 10.1046/j.1365-2249.2002.01838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matza D, Kerem A, Shachar I. Invariant chain, a chain of command. Trends in Immunology. 2003;24:264–268. doi: 10.1016/S1471-4906(03)00073-5. [DOI] [PubMed] [Google Scholar]
- Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavitsky AB, Xianli J. In vitro and in vivo regulation by macrophage migration inhibitory factor (MIF) of expression of MHC-II, costimulatory, adhesion, receptor, and cytokine molecules. Cellular Immunol. 2002;217:95–104. doi: 10.1016/S0008-8749(02)00516-6. [DOI] [PubMed] [Google Scholar]
- Matza D, Wolstein O, Dikstein R, Shachar I. Invariant chain induces B cell maturation by activating TAFII105-NF-κβ dependent transcription program. J Biol Chem. 2001;276:27203–27206. doi: 10.1074/jbc.M104684200. [DOI] [PubMed] [Google Scholar]
- Cichy J, Pure E. The liberation of CD44. J Cell Biol. 2003;161:839–843. doi: 10.1083/jcb.200302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara KN, Murai T, Nishinakamura H, Kawashima H, Saya H, Miyasaka M. Hyaluronan oligosaccharides induce CD44 cleavage and promote cell migration in CD44-expressing tumor cells. J Biol Chem. 2003;278:32259–32265. doi: 10.1074/jbc.M300347200. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Kawano Y, Murakami D, Sasayama T, Araki N, Miki T, Wong AJ, Saya H. Proteolytic release of CD44 intracellular domain and its role in CD44 signaling pathway. J Cell Biol. 2001;155:755–762. doi: 10.1083/jcb.200108159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, Gutmann DH, Ponta H, Herrlich P. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001;15:968–980. doi: 10.1101/gad.189601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]