

ABSTRACT
Herpesviruses infect cells using the conserved core fusion machinery composed of glycoprotein B (gB) and gH/gL. The gH/gL complex plays an essential but still poorly characterized role in membrane fusion and cell tropism. Our previous studies demonstrated that the conserved disulfide bond (DB) C278/C335 in domain II (D-II) of Epstein-Barr virus (EBV) gH has an epithelial cell-specific function, whereas the interface of D-II/D-III is involved in formation of the B cell entry complex by binding to gp42. To extend these studies, we compared gH of the alphaherpesvirus pseudorabies virus (PrV) with gH of the gammaherpesvirus EBV to identify functionally equivalent regions critical for gH function during entry. We identified several conserved amino acids surrounding the conserved DB that connects three central helices of D-III of PrV and EBV gH. The present study verified that the conserved DB and several contacting amino acids in D-III modulate cell surface expression and thereby contribute to gH function. In line with this finding, we found that DB C404/C439 and T401 are important for cell-to-cell spread and efficient entry of PrV. This parallel comparison between PrV and EBV gH function brings new insights into how gH structure impacts fusion function during herpesvirus entry.
IMPORTANCE The alphaherpesvirus PrV is known for its neuroinvasion, whereas the gammaherpesvirus EBV is associated with cancer of epithelial and B cell origin. Despite low amino acid conservation, PrV gH and EBV gH show strikingly similar structures. Interestingly, both PrV gH and EBV gH contain a structural motif composed of a DB and supporting amino acids which is highly conserved within the Herpesviridae. Our study verified that PrV gH uses a minimal motif with the DB as the core, whereas the DB of EBV gH forms extensive connections through hydrogen bonds to surrounding amino acids, ensuring the cell surface expression of gH/gL. Our study verifies that the comparative analysis of distantly related herpesviruses, such as PrV and EBV, allows the identification of common gH functions. In addition, we provide an understanding of how functional domains can evolve over time, resulting in subtle differences in domain structure and function.
INTRODUCTION
A variety of enveloped viruses enter host cells by using a core fusion machinery to facilitate the initial receptor-binding step, followed by fusion of the host cell membrane with the virion envelope. Most enveloped viruses use one or two surface glycoproteins to bind to receptor and mediate fusion. In contrast, the herpesvirus core fusion machinery consists of the heterodimeric complex gH/gL and the fusogen gB, with an additional receptor-binding protein such as gD in the alphaherpesvirus pseudorabies virus (PrV) or gp42 in the gammaherpesvirus Epstein-Barr virus (EBV) (1). For infectious entry, PrV requires gB and gH/gL as well as gD, which binds to specific host cell receptors such as nectin-1 and -2 and HveD (2), whereas EBV requires gB, gH/gL, and gp42 for B cell entry, with gp42 binding the receptor HLA class II (3). In contrast, for infection of epithelial cells, EBV uses gH/gL to bind to integrin receptors so that only gH/gL and gB are required for entry into these cells (3).
Although functions of the herpesvirus receptor-binding proteins and the fusogen gB are understood to some extent, the role of the gH/gL complex remains enigmatic. Recently, the crystal structures of herpes simplex virus 2 (HSV-2) and EBV gH/gL as well as PrV gH (4–6) provided a structural basis for gaining more insight into the role of gH during fusion. The gH homologs can be divided into four domains (Fig. 1A and B) demonstrating strikingly similar structures despite low amino acid conservation (4, 6). The gL-interacting domain I (D-I) is composed of the N-terminal residues of gH interacting with gL and is connected via the linker helix with D-II, forming a large groove in EBV gH/gL. The flexibility of the large groove is necessary for gH/gL function during EBV-mediated fusion (7). In support of this finding, it was shown that binding of the epithelial cell receptor integrin results in a conformational change of the large groove (8). Moreover, a recent study confirmed that flexibility between the structural features of D-II and the interface of D-II and D-III is necessary for function of PrV gH during fusion (9).
FIG 1.

Side-by-side comparison of EBV gH/gL and PrV gH. The EBV gH/gL (PDB code 3PHF) (A) and PrV gH (PDB code 2XQY) (B) structures (4, 6) are shown as ribbon diagrams. The four domains (D) are indicated in different colors. Previously identified structural aspects, such as the bifunctional KGD motif (red) (24), DBs (orange spheres), and syntaxin-like bundle (SLB) formed by three helices, are shown. The structural views of D-III of EBV gH/gL (C) and PrV gH (D), including the conserved DB (orange spheres) and conserved surrounding amino acids (blue sticks), are shown as ribbon diagrams. (E) Amino acid alignment of the conserved DB (orange stars) of a partial D-III (corresponding to amino acids 404 to 421 and 448 to 485 of EBV gH) is shown with the correlated secondary structures, such as alpha-helices and β-sheets (red) for PrV (top) and EBV (bottom) gH.
Since the amino acid conservation between EBV and PrV gH is only modest, in contrast to the high degree of conservation of gB, we hypothesized that stretches of conserved amino acids and/or motifs would reflect fundamental commonalities in gH function during herpesvirus entry. We chose PrV and EBV gH since they not only are among the best-studied herpesviruses regarding fusion with host cells but also exhibit strikingly similar gH structures (Fig. 1A and B). In addition, they differ in their host ranges. While PrV has one of the broadest host ranges among the Herpesviridae, EBV infection is restricted to human or nonhuman primate cells (3, 10).
The DB of D-III is highly conserved and the only DB of gH which is not exposed at the surface of the protein, suggesting an internal stabilizing function. Therefore, we hypothesized that this DB is essential for maintenance of the conformation of gH/gL and, in consequence, gH/gL cell surface expression. Moreover, the requirement for flexibility of the large groove and the D-II/D-III interface (7, 9) might suggest that the DB is also important for the integrity of D-III and virus entry. To test these hypotheses, we mutated the conserved DB (Fig. 1C and D) by amino acid substitution. Interestingly, we found that the conserved DB of D-III contributes to cell surface expression of gH/gL and thereby gH function during fusion.
MATERIALS AND METHODS
Cell culture and antibodies.
Cells were grown in cell culture medium supplemented with 10% Corning CellGro Fetal Bovine Serum Premium, Mediatech Inc. (VWR). CHO-K1 Chinese hamster ovary cells (a gift from Jeffrey D. Esko), CHO cells expressing nectin-1 (CHO-nectin-1) (2), and human gastric adenocarcinoma cells (AGS) were passaged in Ham's F-12 medium (Corning cellgro), whereas RK13 rabbit kidney epithelial cells (ATCC CCL-37), PK13 pig kidney epithelial cells (ATCC CRL-6489), and ST swine testis fibroblasts (ATCC CRL-1746) were grown in Dulbecco's modified Eagle's medium (DMEM) (Corning CellGro). For the EBV fusion assay, human B lymphoblasts (Daudi-T7-29) and human embryonic kidney 293-T14 cells (HEK-293-T14), which stably express T7 RNA polymerase (11, 12), were grown in RPMI 1640 medium and DMEM (Corning CellGro). All cells were kindly provided by Nanette Susmarski.
We used mouse monoclonal antibody (MAb) E1D1 (13) and a rabbit polyclonal antiserum (14), both specific for EBV gH/gL, as well as a rabbit polyclonal antiserum against PrV gH (15) and a mouse monoclonal antibody (6C5) to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Abcam).
Plasmids and mutagenesis.
The site-directed mutagenesis of PrV and EBV gH was done by using mutagenic oligonucleotide primers specific for pcDNA-PrV-gH (16) or pSG5-EBV-gH and using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's protocol. Correct mutagenesis was verified by sequencing (Northwestern Genomics Core Facility).
CELISA.
For cell enzyme-linked immunosorbent assay (CELISA), CHO-K1 cells were transiently transfected with 0.5 μg of plasmid DNA of EBV or PrV gL and either wild-type (wt) or mutant gH using Lipofectamine 2000 (Invitrogen). After 16 h, transfected cells were detached, counted by a Countess cell counter (Invitrogen), and plated (0.5 × 105) into a 96-well plate. After 24 h, the transfected CHO cells were incubated with specific antibodies against EBV gH (E1D1, 1:500; CL40, 1:1,000; CL59, 1:1,000; and HL800, 1:500) or PrV gH (1:1,500). CELISA was further performed as described previously (17).
Fusion assay.
For EBV fusion, CHO-K1 cells were transiently transfected with plasmids expressing a luciferase reporter containing a T7 promoter (0.8 μg) and EBV gL (0.5 μg) and gB (0.8 μg), either wt or mutant gH (0.5 μg), and, for B cell fusion, gp42 (0.5 μg) using Lipofectamine 2000 (Invitrogen) (18). For PrV fusion, CHO-K1 effector cells were transfected with plasmids expressing a T7 RNA polymerase (0.8 μg), PrV gL (0.5 μg), gB (0.8 μg), gD (0.5 μg), and either wt or mutant gH (0.5 μg). At the same time, the target cells, such as RK13, ST, PK13, and CHO-nectin-1 cells, were transfected with the luciferase reporter plasmid containing a T7 promoter (2 μg). After 4 h (PrV) or 16 h (EBV), transfected cells were detached and counted. Effector cells were then mixed in a ratio of 1:1 (0.5 × 106) with target cells, such as HEK-293-T14 and Daudi-T7-29 cells for EBV fusion and RK13, ST, PK13, and CHO-nectin-1 cells for PrV fusion. After 16 h (PrV) or 24 h (EBV), the luciferase activity was analyzed on a PerkinElmer Wallac Victor2 multilabel plate reader.
Immunoprecipitation, SDS-PAGE, and Western blot analysis of PrV and EBV gH proteins.
CHO-K1 cells were transfected and detached as described above (see “CELISA”). For immunoprecipitation, equal amounts of transfected cells (1 × 107) were lysed with 1% Triton X-100 lysis buffer (20 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1% Triton X-100) plus 1% protease inhibitor cocktail set I (Calbiochem), incubated with desired antibodies against EBV gH and gH/gL overnight, and treated with protein A/G plus agarose (Santa Cruz Biotechnology) for 1.5 h. For sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), samples were resuspended in phosphate-buffered saline (PBS) and 4× SDS loading buffer (60 mM Tris-HCl [pH 6.8], 4% SDS, 25% glycerol, 0.01% bromophenol blue, 10% 2-mercaptoethanol). After being heated for 5 min at 100°C, the samples were separated on a 4 to 20% polyacrylamide gel and electrotransferred onto nitrocellulose membranes (Bio-Rad). The protein bands on the membranes were visualized by using primary rabbit polyclonal anti-EBV-gH/gL (1:100) in 1% bovine serum albumin–Tris-buffered saline–Tween (BSA-TBST), anti-PrV-gH (1:30,000) and -gL (1:10,000) in TBST, mouse monoclonal anti-GAPDH (1:5,000), and an IRDye800-conjugated anti-rabbit or anti-mouse secondary antibody (Li-Cor Biosciences, Lincoln, NE) and then detected on an Odyssey Fc Western blotting imager using Image Studio, version 2.0 (Li-Cor Biosciences).
In vitro entry studies.
PrV gH recombinants were generated as described previously (19). For plaque size analysis, RK13 cells were infected with 100 PFU per well under plaque assay conditions and fixed with 3% paraformaldehyde after 2 days. For each virus, 30 plaques were measured with a Nikon Eclipse Ti-S fluorescence microscope using Nikon NIS-Elements imaging software (Nikon Instruments Inc.). The values were calculated relative to the plaque size of wt PrV strain Ka, which was set at 100%. For penetration kinetics, RK13 cells were infected with 150 PFU per well with either wt or mutant virus on ice for 1 h. After medium exchange, cells were incubated for 0, 5, 10, 20, or 40 min at 37°C, and then either extracellular virus was inactivated by low-pH treatment or cells were washed with PBS only as a 100% penetration control. After 2 days, cells were fixed and stained with crystal violet, and plaques were counted.
Graphical analysis.
The structural views of EBV gH/gL (PDB code 3PHF) (6) and PrV gH (PDB code 2XQY) (4) were generated using the PyMOL molecular graphics system, version 1.3 (Schrödinger, LLC).
Sequence alignment of gH.
The gH gene sequences (GenBank accession numbers are in parentheses) of the following were compared: HSV-1 (AAG17895.1); HSV-2 (CAB06746.1); varicella-zoster virus (VZV) (ABF22268.1); PrV (CAA41678.1); human herpesvirus 5 (HHV-5) (CAA00301.1); HHV-6 (CAA58382.1); HHV-7 (AAB64293.1); macacine herpesvirus 3 (McHV-3) (AAP50630.1); McHV-4 (AAK95464.1); murid herpesviruses 1 (AAA20190.1), 2 (NP_064175.1), and 4 (NP_044860.1); caviid herpesvirus 2 (P87730.2); suid herpesviruses 2 (YP_008492985.1), 3 (AAM22122.1), 4 (AAO12364.1), and 5 (AAO12326.1); equid herpesviruses 2 (NP_042618.1) and 5 (ACY71880.1); Kaposi's sarcoma-associated herpesvirus (KSHV) (ADB08188.1); saimiriine herpesvirus 2 (P16492.1); callitrichine herpesvirus 3 (CalHV-3) (AAK38222.1); and EBV (P03231.1). Figure 1E shows a summary of the alignment with representative viruses (HSV-1 and -2; VZV; PrV; HHV-5, -6, and -7; KSHV; EBV; CalHV-3; and McHV-4). The amino acid sequences of gH were aligned using T-Coffee Expresso (http://tcoffee.crg.cat/apps/tcoffee/do:expresso) (20), including the PDB files of HSV-2, and EBV gH/gL as well as PrV gH (PDB codes 2XQY, 3M1C, and 3PHF) (4–6). The alignment was modified using Jalview (http://www.jalview.org/) (21).
RESULTS
The DB in D-III of PrV and EBV gH is surrounded by a group of conserved amino acids.
The crystal structures of HSV-2 and EBV gH/gL as well as PrV gH identified the DB as a structural feature of D-III (4–6). Since this DB connects three central helices of D-III and is the only buried DB of gH, we hypothesized that it might function as a stabilizing structural feature for this domain and thereby be an important determinant of gH/gL expression. To analyze if additional amino acids supported the role of this DB, we performed an amino acid sequence alignment using T-Coffee Expresso, which uses the relevant structures of gH in determining amino acid alignments (Fig. 1E). We used gH sequences from alpha-, beta- and gammaherpesviruses. Based on the alignment, a number of additional conserved amino acids were identified. Interestingly, these amino acids are located around the conserved DB in EBV and PrV gH (Fig. 1C and D). Focusing on the location and interactions of these amino acids in the crystal structures, we identified hydrogen bonds between the DB and the framing amino acids (Fig. 2A and B). Based on the crystal structure of PrV gH, we also identified hydrogen bonds between cysteine 404 (C404) and arginine 444 (R444), which contacts serine 442 (S442). Additionally, S442 forms hydrogen bonds with C439, and alanine 440 (A440) contacts leucine 436 (L436) (Fig. 2A). In contrast, the crystal structure of EBV gH/gL revealed more extensive connections to surrounding amino acids, including aromatic residues, with DB C454/C478 forming the core of the interactions (Fig. 2B). Besides the contact of C454 and R483, which is also present in PrV gH, C454 makes a bond with threonine 451 (T451). R483 also contacts S481, as seen for PrV, and additionally binds to D485. S481 establishes hydrogen bonds with C478, as seen for PrV gH, but in the case of EBV it also binds proline 477 (P477) and the preceding S476 contacts tyrosine 479 (Y479) by hydrogen bonding. Therefore, we included the aromatic residues of EBV gH and the corresponding amino acids of PrV gH in this study. To analyze the significance of these amino acids (Fig. 2A and B), we performed a site-specific mutagenesis screen by altering the cysteines of the DB to serine and the surrounding conserved amino acids to alanine. In addition, we changed the aromatic residues found in EBV gH to corresponding amino acids of PrV gH and vice versa.
FIG 2.

The DB and contacting amino acids approve the cell surface expression of PrV gH and EBV gH/gL. The arrangements of the conserved amino acids surrounding the conserved DB's connecting three central helices of D-III of PrV (PDB code 2XQY) (A) and EBV gH (PDB code 3PHF) (B) are shown as ribbon diagrams, and the surface expression-required amino acids are highlighted in red. The amino acids (sticks) are colored by element (C and H in gray, N in blue, O in red, and S in orange), and the hydrogen bonds are indicated as black dashes. Surface expression of wt and mutant gH of PrV (C) and EBV (D) is also shown. CELISA was performed with either the monoclonal conformation-specific antibodies against EBV gH (CL40 and CL59) and gH/gL (E1D1) or polyclonal antisera against PrV and EBV gH (HL800). Mean values and standard deviations from three independent experiments are shown.
The conserved DB of D-III is important for cell surface expression of gH/gL.
To test if the DB and contacting amino acids are required for cell surface expression of gH/gL, we performed a cell-based ELISA using a polyclonal antiserum recognizing PrV gH. For EBV, we used conformation-specific monoclonal antibodies against gH (CL40 and CL59) and gH/gL (E1D1) as well as a polyclonal antiserum detecting gH (HL800). E1D1 recognizes a complex epitope coformed by residues of gL and the N terminus of gH, whereas CL59 is directed against C-terminal residues 501 to 628 of gH (8, 18, 22, 23). For PrV gH, cell surface expression of the T401A, P438A, and C404S/C439S mutants was strongly decreased, whereas most of the mutants showed wt-like expression (Fig. 2C). Similar to the PrV mutants, the corresponding EBV T451A, P477A, and C454S/C478S mutants lacked cell surface expression (Fig. 2D). Moreover, the EBV S476A, R483A, and D485A mutants as well as the aromatic-residue F475A/L and Y479A mutants exhibited a strong decrease in cell surface expression. The substitution by another aromatic residue (F475Y and Y479F) did not cause a defect in EBV gH/gL expression. We also investigated the mutant proteins using reducing SDS-PAGE conditions followed by Western blot analysis. All the gH mutants migrated similarly to the wt proteins (Fig. 3A and B), indicating that all mutants were expressed but were not all exposed at the cell surface. Interestingly, there was a dependence on an increased number of conserved amino acids for EBV gH/gL compared with PrV gH with regard to cell surface expression.
FIG 3.

The defect in cell surface expression of the DB and specific amino acid mutants strongly correlates with a loss in fusion function of PrV and EBV gH. Western blot analysis of wt and mutant gH was performed by using polyclonal antibodies against PrV gH (A) and EBV gH/gL (B) as well as anti-GAPDH as a loading control. The fusion activities of wt and mutant gH of PrV (C) and EBV (D) are also shown. For the virus-free luciferase-based fusion assay, CHO-KI cells were transfected with the fusion-required glycoproteins of either PrV (gD, gB, and gH/gL) or EBV (gp42, gB, and gH/gL). Averages with standard deviations from three independent experiments are shown.
The conserved residues of D-III are not involved in cell-type-specific gH fusion function.
The structural features of D-II and D-III are necessary for the binding of EBV gH/gL to either the epithelial cell receptor or gp42 (14, 24–26). Interestingly, residues involved in the binding of gp42, forming the EBV B cell entry complex (26), are in close proximity to the conserved DB and its contacting amino acids. To test if the mutated amino acids are involved in cell-specific function of gH, we used a virus-free cell-based fusion assay with luciferase as a reporter. Since it is known that epithelial and B cells are the primary targets of EBV, we used stably T7 polymerase-expressing HEK-293 and Daudi B cells. For the PrV mutants, we used CHO cells, expressing the gD receptor nectin-1, and two porcine cell lines (ST and PK13) mimicking host conditions as well as the rabbit kidney cell line RK13, which is frequently used for the propagation of PrV. PrV gH T401A, C404S/C439S, and P438A mutants lacked fusion activity correlating with a defect in cell surface expression (Fig. 3C). Moreover, mutant R444A showed a strong defect in fusion activity despite wt-like cell surface expression. Correlating with the results for PrV gH, EBV gH T451A, C454S/C478S, F475A/L, S476A, P477A, Y479A, R483A, and D485A mutants showed a strong decrease in fusion activity (Fig. 3D). To summarize, the loss of correct cell surface expression strongly correlates with a cell-type-independent defect in fusion activity for both EBV and PrV gH, with only the PrV R444A mutant showing cell surface expression and a reduction in fusion activity.
A conserved tyrosine is important for PrV and EBV gH function during fusion despite different effects on cell surface expression.
Based on the sequence alignment, we identified a conserved tyrosine, Y363 in PrV gH and Y404 in EBV gH, that forms conserved hydrogen bonds and interactions with adjacent amino acids in the same helix and with an amino acid of a D-IV β-sheet extending into the interface between D-III and D-IV (Fig. 4A and B). Y363 of PrV gH forms hydrogen bonds with V359 and V367, which are located in the same helix (α-7), and interacts to L589 located at the tip of sheet β-11 (Fig. 4A). Similarly, Y404 of EBV gH forms hydrogen bonds with helix residues H407 and L400/L401, as well as Y633, located in sheet 4β-8 (Fig. 4B). The hydrogen bonding between the conserved tyrosine of PrV and EBV gH to a corresponding amino acid in D-IV could be involved in stabilizing the interface between D-III and D-IV. To test if these amino acids and their observed hydrogen bonding are important for cell surface expression of gH/gL and/or fusion, we mutated the relevant amino acids and analyzed the mutant proteins for cell surface expression and fusion activity. Cell surface localization of the PrV gH V359A and Y363A mutants was like that of the wt, but the L589Y mutant exhibited a very modest decrease, to about 70% of wt levels (Fig. 4C). In contrast, the L400A/L401A and Y404A mutants were not detectable at the cell surface, whereas most of the EBV gH mutant proteins revealed wt-like cell surface expression, including the Y633A mutant (Fig. 4D). Furthermore, we tested the gH mutants by SDS-PAGE followed by Western blotting. PrV (Fig. 4E) and EBV (Fig. 4F) mutants migrated similarly to the wt. Most of them showed wt-like fusion activity, which correlated with wt-like cell surface expression (Fig. 4G and H). In contrast, PrV gH V359A and Y363A mutants had a significant decrease in fusion activity despite wt-like cell surface localization (Fig. 4G). The corresponding EBV gH mutants, the L400A/L401A and Y404 mutants, showed a similar defect in fusion activity (Fig. 4H) but also had a strong decrease in cell surface expression. Taken together, these results showed that the conserved tyrosine (Y363 and Y404) and the hydrogen-bound residues V359 of PrV gH and L400/L401 of EBV gH are important for gH function during fusion, whereas the residues of EBV gH are also important for cell surface localization. Moreover, the interacting amino acids within D-IV (PrV L589 and EBV Y663) are not important for either localization or fusion activity.
FIG 4.

The conserved tyrosine and a contacting amino acid are important for function of gH during fusion. The conserved tyrosine and contacting amino acids of PrV (PDB code 2XQY) (A) and EBV gH (PDB code 3PHF) (B) are shown as sticks and labeled by element as well as the involved secondary structures are displayed in ribbon diagrams. The surface expression-required amino acids are highlighted in red. Shown are cell surface expression and fusion activities of wt and mutant gH of PrV (C and G) and EBV (D and H). Mean values and standard deviations from three independent experiments are shown. Western blot analysis of wt and mutant gH was performed by using polyclonal antibodies against PrV gH (E) and EBV gH/gL (F) as well as anti-GAPDH.
EBV mutants defective in cell surface expression are still recognized by conformation-specific antibodies.
Our gH mutants can be divided into three categories. Category I mutants (PrV gH V367A, L436A/F, S437A, A440F/Y, S442A, D446A, and L589A/Y mutants and EBV gH H407A, F475Y, Y479F, S481A, and Y633A/L/F mutants) were characterized by wt-like expression, cell surface localization, and fusion. Category II mutants (PrV gH V359A, Y363A, and R444A mutants) were expressed and located to the cell surface but showed a significant defect in fusion. Category III mutants (PrV gH T401A, C404S/C439S, and P438A mutants and EBV gH L400A/L401A, Y404A, T451A, C454S/C478S, F475A/L, S476A, P477A, Y479A, R483A, and D485A mutants) were expressed but did not localize to the cell surface, correlating with a defect in fusion activity. To further investigate the surface localization defect of the EBV category III mutants, we used the conformation-specific monoclonal antibodies against gH (CL40 and CL59) and gH/gL (E1D1) for precipitation from whole-cell lysates. The selected, representative category III mutants, such as the Y404A, C454S/C478S, and R483A mutants, were recognized by CL59, CL40, and E1D1, which recognizes an epitope coformed by the N termini of gH and gL (Fig. 5). The precipitation by the conformation-specific antibody E1D1 indicates the interaction of gH and gL, despite a loss of cell surface localization. The corresponding PrV gH mutants were precipitated with the available polyclonal gH and gL antisera, indicating wt-like interaction of gH and gL (data not shown).
FIG 5.
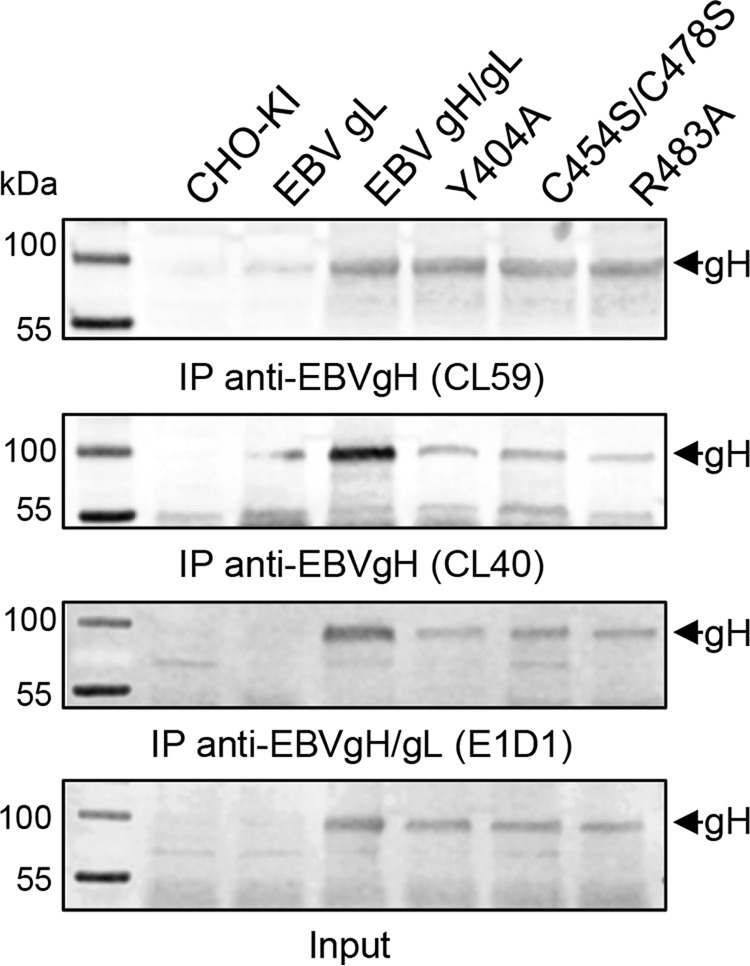
Immunoprecipitation (IP) of EBV gH using monoclonal conformation-specific antibodies. Immunoprecipitation analysis for wt and selected mutant gH proteins was performed by using monoclonal conformation-specific antibodies against EBV gH (CL40 and CL59) and gH/gL (E1D1). The input is shown. Western blot analyses were done with polyclonal EBV gH/gL-specific antiserum. IP experiments were repeated at least twice.
The conserved DB of D-III is required for efficient PrV entry.
To further investigate the impact of the DB and supporting amino acids for the role of gH during entry, we generated virus recombinants using BAC mutant pPrV-ΔgHABF and transfer plasmid pcDNA-gHKDE (9) carrying the selected amino acid changes. Infectious virus progenies were characterized by Western blotting, demonstrating wt-like gH expression, but the PrV DB mutant lacked the slower-migrating mature form of gH (data not shown). PrV expressing gH mutated in the DB was replication competent but showed a 10-fold decrease in titers (data not shown). Plaque assays revealed that the PrV gHC404S/C439S strain formed only small foci and the PrV gHT401A strain also exhibited a severe defect in cell-to-cell spread, whereas the spread defect was modest in the Y363A and R444A virus mutants (Fig. 6A and B), despite their apparent defects in the cell-based fusion assay. To investigate if the DB is important for gH not only during cell-to-cell spread but also during entry of free virions, we analyzed penetration kinetics of PrV recombinants expressing mutated gH. As expected, the PrV gHC404S/C439S strain showed a strong delay in penetration, whereas the delay in penetration of the T401A virus mutant was less significant (Fig. 6C). Taken together, the results show that DB C404/C439 and the contacting amino acid T401 are important for efficient cell entry and cell-to-cell spread.
FIG 6.
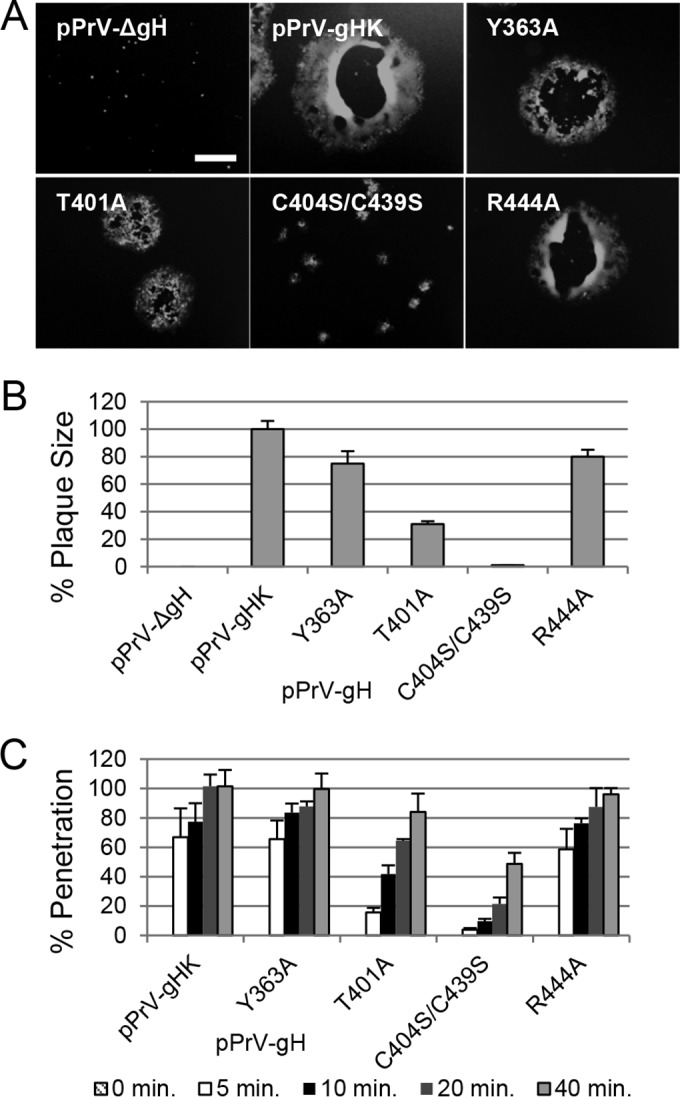
The conserved DB is important for the role of gH during PrV entry. (A) Shown are images of the plaques. Scale bar represents 400 μm. (B) For analysis of the plaque size, 30 plaques were measured in three experiments, with wild-type PrV-Ka set as 100%. (C) For penetration kinetics, RK13 cells were infected on ice for 1 h and incubated for the indicated times at 37°C. Then extracellular virus was inactivated with citric acid. Averages with standard deviations from three independent experiments are shown.
DISCUSSION
Herpesvirus gH/gL is thought to be triggered by the receptor-binding signal to activate gB for initiation of the fusion process (27). It is also an important determinant of the cell tropism for some herpesviruses, such as EBV (6, 14, 24, 28, 29) and human cytomegalovirus (HCMV) (30–32). Therefore, it is likely that gH has evolved to execute a conserved core function in the transmission of cell receptor-binding signals, but gH has also developed species-dependent adaptations for gH functions important for cell tropism. To further analyze the role of gH, we studied the alpha- and gammaherpesviruses PrV and EBV based on available structure data. We focused on the region comprising the DB in D-III of gH, which is highly conserved among the Herpesviridae and is buried inside the domain connecting three central helices (4, 6). Besides the DB, we identified additional amino acids in D-III that are conserved among the gH homologs. Our data demonstrate that the conserved DB acts as a core of an extensive interaction network by forming hydrogen bonds with surrounding amino acids that connect the three central helices of D-III. Mutation of this conserved motif identified a region that is important for cell surface localization of both PrV and EBV gH/gL. This surface localization defect in gH with mutations of the DB and specific surrounding amino acids correlates strongly with a reduction in fusion activity. The DB was also essential for efficient entry and direct cell-to-cell spread of PrV. Mutation of the conserved tyrosine (Y404 and Y363) identified a specific role for surface localization of EBV gH/gL but not for PrV gH/gL. However, the tyrosine (Y363 and Y404) and the contacting amino acid (V359 and L400/L401) are important for fusion activity of both PrV and EBV. Interestingly, mutation of the conserved Y363 within the virus induced only a slight decrease in cell-to-cell spread and a delay in penetration of PrV.
The extensive network of the DB and surrounding amino acids of D-III of EBV gH is mediated by contacting aromatic residues. A specific feature for EBV compared to PrV is the shorter, only one-turn helix 3α-7 beginning with the highly conserved P477 followed by C478 and a gammaherpesvirus-specific aromatic residue, Y479, and ending with L480. An additional aromatic residue (F475) is conserved in most gH homologs but not in PrV gH. F475 is located in the loop preceding helix 3α-7 and together with Y479 forms an extensive network of hydrogen bonds with DB C454/C478 as the core within D-III of EBV. Replacement with other aromatic residues did not result in a defect in cell surface localization and fusion function. Interestingly, the network of contacting amino acids with DB C404/C439 of PrV gH as the core is more restricted than with EBV gH based on the crystal structures. In support of this finding, P438 causes a kink within helix α-11 of PrV gH (19). Here, we showed that the conserved motif important for cell surface localization of PrV gH is restricted to T401, C404/C439, and P438. Subtle structural differences between the conserved motifs of D-III of gH may explain why 4 amino acids are relevant for cell surface expression of PrV gH, whereas a more extensive network is required for EBV gH.
PrV gH is known to be surprisingly tolerant to a variety of mutations (9, 19, 33). Therefore, it is not unexpected that the highly conserved DB is not essential for PrV replication, although it causes a loss of cell-to-cell spread ability and a significant defect in penetration. In contrast, this conserved DB in D-III is essential for replication of other alphaherpesviruses, such as VZV and HSV (34, 35).
Interestingly, attenuated PrV strain Bartha (PrV-Ba), which is widely used for vaccination against PrV infection in pigs, expresses a gH which contains a P438S substitution as well as other mutations (36). However, several studies demonstrated that the lack of virulence of PrV-Ba is due to a large deletion encompassing the gI, gE, pUS9, and pUS2 genes as well as the amino acid substitutions in pUL21. Thus, at least in this virus background, P438S does not appear to affect PrV virulence (37–39). Correlating with a previous study (19), PrV recombinants expressing gHC404S/C439S or T401A are replication competent, despite a delay in penetration and loss of cell-to-cell spread ability.
The conserved amino acids Y404, T451, R483, and D485 of EBV gH and Y363, T401, and R444 of PrV gH (Fig. 1C and D) are oriented toward D-IV, which might explain their importance for fusion. In contrast, D446 of PrV gH is directed outwards from the protein (Fig. 1D), explaining why this amino acid is involved in neither surface localization nor fusion activity. Taken together, the results show that residues of D-III reaching into the interface of D-IV are important for gH function during fusion of both PrV and EBV. Our data are supported by a previous mutagenesis study showing that changes of the residues L529/P530, which participate in the D-III/D-IV-interface, resulted in disruption of fusion activity of EBV (18).
Our comparative analysis verifies that the DB is an important structural determinant of D-III required for proper cell surface localization of gH/gL and thereby for its function during gB-mediated fusion. Our study brings insights into how this common motif is adapted to the specific needs of gH during PrV and EBV infection.
ACKNOWLEDGMENTS
We thank Lindsey Hutt-Fletcher for kindly providing monoclonal antibodies and the members of the Jardetzky and Longnecker laboratories for help and advice, especially Nanette Susmarski, Jia Chen, and Sarah Connolly for technical expertise and helpful discussions.
REFERENCES
- 1.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 3.Longnecker R, Kieff E, Cohen J. 2013. Epstein-Barr virus, 6th ed Lippincott Wilkins and Williams, Philadelphia, PA. [Google Scholar]
- 4.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Jardetzky TS, Longnecker R. 2013. The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. J Virol 87:3620–3627. doi: 10.1128/JVI.03245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chesnokova LS, Hutt-Fletcher LM. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J Virol 85:13214–13223. doi: 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Böhm S, Eckroth E, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 12 November 2014. Structure-based functional analyses of domains II and III of pseudorabies virus glycoprotein H. J Virol doi: 10.1128/JVI.02765-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mettenleiter TC. 2008. Pseudorabies virus, p 341–351. In Mahy BWJ, van Regenmortel MHV (ed), Encyclopedia of virology, 3rd ed, vol 1 Elsevier Ltd, Oxford, United Kingdom. [Google Scholar]
- 11.Omerović J, Lev L, Longnecker R. 2005. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with epithelial and B cells. J Virol 79:12408–12415. doi: 10.1128/JVI.79.19.12408-12415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silva AL, Omerovic J, Jardetzky TS, Longnecker R. 2004. Mutational analyses of Epstein-Barr virus glycoprotein 42 reveal functional domains not involved in receptor binding but required for membrane fusion. J Virol 78:5946–5956. doi: 10.1128/JVI.78.11.5946-5956.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oba DE, Hutt-Fletcher LM. 1988. Induction of antibodies to the Epstein-Barr virus glycoprotein gp85 with a synthetic peptide corresponding to a sequence in the BXLF2 open reading frame. J Virol 62:1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Möhl BS, Sathiyamoorthy K, Jardetzky TS, Longnecker R. 17 September 2014. The conserved disulfide bond within domain II (D-II) of Epstein-Barr virus (EBV) gH has divergent roles in membrane fusion with epithelial cells and B cells. J Virol doi: 10.1128/JVI.02272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klupp BG, Mettenleiter TC. 1999. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J Virol 73:3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McShane MP, Longnecker R. 2005. Analysis of fusion using a virus-free cell fusion assay. Methods Mol Biol 292:187–196. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Borza CM, Hutt-Fletcher LM. 2005. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J Virol 79:10923–10930. doi: 10.1128/JVI.79.17.10923-10930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schröter C, Klupp BG, Fuchs W, Gerhard M, Backovic M, Rey FA, Mettenleiter TC. 3 September 2014. The highly conserved proline at position 438 in pseudorabies virus gH is important for regulation of membrane fusion. J Virol doi: 10.1128/JVI.01204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 21.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Turk SM, Hutt-Fletcher LM. 1995. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J Virol 69:3987–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol 74:6324–6332. doi: 10.1128/JVI.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Rowe CL, Jardetzky TS, Longnecker R. 2012. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio 3(1):e00290-11. doi: 10.1128/mBio.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A 106:20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Möhl BS, Chen J, Jiang W, Mellins ED, Longnecker R, Zhou ZH, Jardetzky TS. 2014. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog 10:e1004309. doi: 10.1371/journal.ppat.1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stampfer SD, Heldwein EE. 26 October 2012. Stuck in the middle: structural insights into the role of the gH/gL heterodimer in herpesvirus entry. Curr Opin Virol doi: 10.1016/j.coviro.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8:594–599. doi: 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- 29.Hutt-Fletcher LM, Chesnokova LS. 2010. Integrins as triggers of Epstein-Barr virus fusion and epithelial cell infection. Virulence 1:395–398. doi: 10.4161/viru.1.5.12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adler B. 2015. A viral pilot for HCMV navigation? Viruses 7:3857–3862. doi: 10.3390/v7072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Revello MG, Gerna G. 2010. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev Med Virol 20:136–155. doi: 10.1002/rmv.645. [DOI] [PubMed] [Google Scholar]
- 32.Zhou M, Lanchy JM, Ryckman BJ. 2015. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol 89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs W, Backovic M, Klupp BG, Rey FA, Mettenleiter TC. 2012. Structure-based mutational analysis of the highly conserved domain IV of glycoprotein H of pseudorabies virus. J Virol 86:8002–8013. doi: 10.1128/JVI.00690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cairns TM, Landsburg DJ, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology 332:550–562. doi: 10.1016/j.virol.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 35.Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci U S A 108:18412–18417. doi: 10.1073/pnas.1111333108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog 7:e1002282. doi: 10.1371/journal.ppat.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Porat T, DeMarchi J, Pendrys J, Veach RA, Kaplan AS. 1986. Proteins specified by the short unique region of the genome of pseudorabies virus play a role in the release of virions from certain cells. J Virol 57:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klupp BG, Lomniczi B, Visser N, Fuchs W, Mettenleiter TC. 1995. Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212:466–473. doi: 10.1006/viro.1995.1504. [DOI] [PubMed] [Google Scholar]
- 39.Lomniczi B, Watanabe S, Ben-Porat T, Kaplan AS. 1987. Genome location and identification of functions defective in the Bartha vaccine strain of pseudorabies virus. J Virol 61:796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]