

Abstract
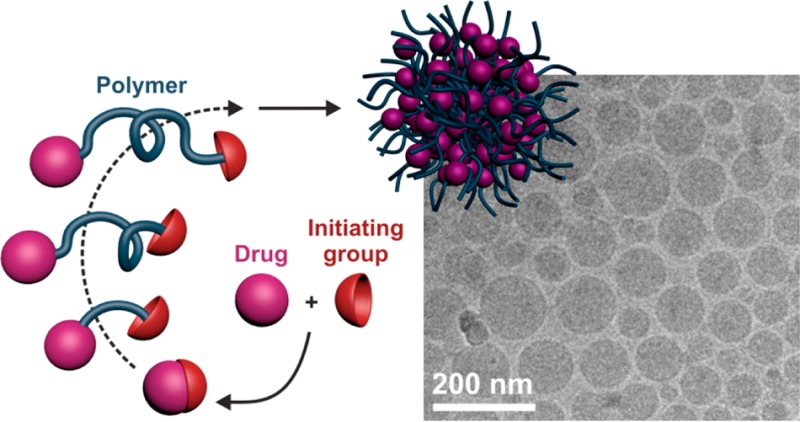
In the field of nanomedicine, the global trend over the past few years has been toward the design of highly sophisticated drug delivery systems with active targeting and/or imaging capabilities, as well as responsiveness to various stimuli to increase their therapeutic efficacy. However, providing sophistication generally increases complexity that could be detrimental in regards to potential pharmaceutical development. An emerging concept to design efficient yet simple drug delivery systems, termed the “drug-initiated” method, consists of growing short polymer chains from drugs in a controlled fashion to yield well-defined drug–polymer prodrugs. These materials are obtained in a reduced amount of synthetic steps and can be self-assembled into polymer prodrug nanoparticles, be incorporated into lipid nanocarriers or be used as water-soluble polymer prodrugs. This Perspective article will capture the recent achievements from the “drug-initiated” method and highlight the great biomedical potential of these materials.
Introduction
Nanoscaled systems for drug delivery,1 such as liposomes,2 micelles,3 polymer nanoparticles4 or polymersomes,5 have received remarkable attention and are in the process of becoming a well-established technology to serve as efficient therapeutic tools against serious diseases including cancer, infectious or neurodegenerative disorders;6−8 the evidence being the increasing number of systems under clinical trials.9 Precisely delivering drugs to diseased areas in the body is crucial in view of drugs’ nonspecific cell and tissue biodistribution and of the rapid metabolization and excretion from the body of some of them.10 The vast majority of drug delivery systems rely on the physical encapsulation of drugs into nanoparticulate assemblies during the formulation process. This concept has been extensively proven over the past few decades and several drug-loaded nanocarriers have even reached the market.9
Even though drug-loaded liposomal formulations (notably, Myocet and Caelyx/Doxil)2,9 are in the lead in terms of bench-to-bedside translation compared to synthetic polymer nanoparticles (as some of them are still under clinical trials), nanocarriers based on polymers have attracted considerable interest owing to the great flexibility and robustness offered by polymer synthesis methods, the broad diversity of polymers that can be produced (in terms of nature, properties and compositions) and their relative ease of functionalization.4 The considerable development of macromolecular engineering11−13 has indeed offered new opportunities in the design of highly sophisticated polymer nanoparticles by making them able to (i) target selectively diseased tissues by active targeting mechanisms through their surface functionalization with biologically active ligands; (ii) serve as diagnostic tools after encapsulation or coupling of various imaging agents and (iii) perform a spatiotemporal release of their content under the action of many different endogenous or exogenous stimuli (e.g., pH, redox status, temperature, magnetic field, light, etc.).4,14−18 These advanced features, however, have increased the complexity of drug delivery systems. This could be detrimental in regards to their potential pharmaceutical development for which high yield, purity and reproducibility, as well as easy scale-up and low costs are needed.19
Drug-loaded polymer nanocarriers are typically obtained by drug encapsulation during the self-assembly of preformed polymers in aqueous solution. Consequently, they exhibit several important drawbacks that may hamper their further translation to the clinical setting and therefore to the market: (i) the “burst release”,20 which consists in the abrupt release postadministration of a large fraction of adsorbed drug can induce significant toxicity; (ii) the difficulty to encapsulate drugs that are poorly miscible to the polymer matrix and (iii) the poor drug loading (typically a few percent) that requires a high concentration of nanocarrier to obtain a therapeutic effect and can also generate toxicity.
These strong limitations have, however, been partially tackled by taking benefit from the prodrug21 concept by developing polymer prodrug nanocarriers.22 A prodrug is formed by the conjugation between a drug and a promoiety; that is a functional group used to improve its physicochemical, biopharmaceutical or pharmacokinetic properties (a prodrug is usually inactive until the linkage between the drug and the promoeity gets cleaved). Similarly to lipid prodrug nanocarriers,23 which rely on the covalent conjugation of drugs to lipidic building blocks, polymer prodrug nanocarriers are obtained by drug conjugation to a polymer scaffold. In that case, the “burst release” is suppressed as the drug release is governed by cleavage (e.g., hydrolytic, enzymatic, reductive, etc.) of the drug from the polymer. Drug loading and encapsulation of poorly soluble drugs are also improved. Nevertheless, development of polymer prodrug nanocarriers often requires complex synthetic routes caused by a series of further synthetic steps beyond that of the polymer itself, generally including protection, deprotection, coupling and purification steps. Efficient yet simple synthetic strategies for producing polymer prodrug delivery systems are therefore highly desirable.
An emerging approach, termed the “drug-initiated” method (Figure 1), consists of preparing polymer prodrugs by growing a single polymer chain from a drug in a controlled fashion. It has met some success in addressing all the above-mentioned requirements and drawbacks, and can therefore be considered as a valuable strategy to prepare easily efficient drug delivery systems. This Perspective article will capture the recent achievements deriving from this approach and highlight the great biomedical potential of these materials.
Figure 1.
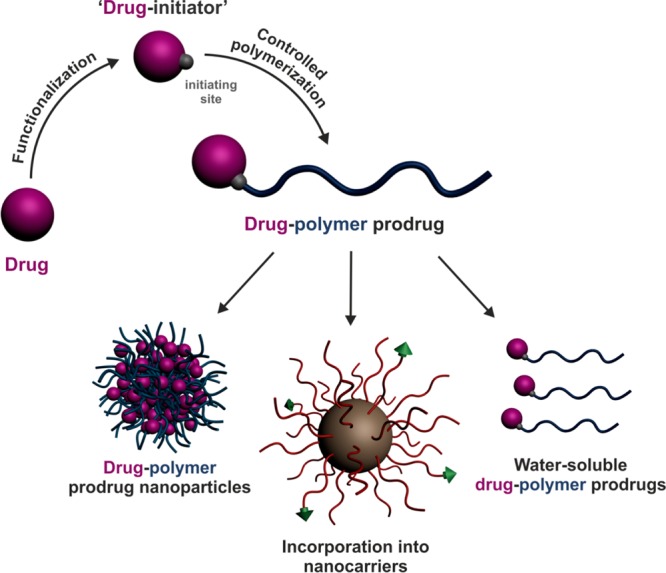
Design of polymer prodrugs by the “drug-initiated” method and their use in drug delivery.
General Considerations
With the “drug-initiated” method, a drug is used to initiate the controlled polymerization of a monomer to yield a drug–polymer prodrug. The direct consequence of this methodology relies on the structural homogeneity of the resulting materials in terms of molar mass distribution and chain-end functionalization. Indeed, if the polymerization is well-controlled (i.e., rapid and quantitative initiation as well as negligible termination/transfer reactions), all polymer chains have nearly the same molar mass and are end-functionalized with one drug molecule. On a more practical point, the only purification step required after the polymerization simply consists in the removal of unreacted monomer. The simplicity of both the synthetic approach and the final material structure is also anticipated to give high batch-to-batch reproducibility and easy scale-up. This could represent crucial advantages compared to traditional drug-delivery systems in regards to their potential pharmaceutical development. A direct analogy can actually be made between the “drug-initiated” method and the “grafting from” approach (also often termed “surface-initiated”),24−26 which generally consists of growing polymer chains from bulky substrates (e.g., surfaces, nanoparticles, proteins, etc.). When compared to the opposite strategy (termed “grafting to”), involving the coupling of preformed α-functional polymers to a substrate, the “grafting from” approach also leads to (i) higher conjugation efficacies due to a lower steric hindrance nearby the conjugation site and (ii) facilitated purification as only unreacted monomer has to be removed.
Another specificity of this approach is the expected quantitative loading efficacy (LE), as each drug molecule should initiate a polymer chain growth and be retained at the chain end. Also, given that the drug loading (DL) represents the mass fraction of the drug in the polymer prodrug (according to DL = MWdrug/Mn,polymer prodrug, where MWdrug is the molecular weight of the drug and Mn,polymer prodrug is the number-average molar mass of the polymer prodrug), it can be easily fine-tuned simply by adjusting the polymer chain length: the lower the Mn, the higher the DL (Figure 2). This enables great flexibility toward the design of drug delivery systems with tunable drug contents, up to high values.
Figure 2.
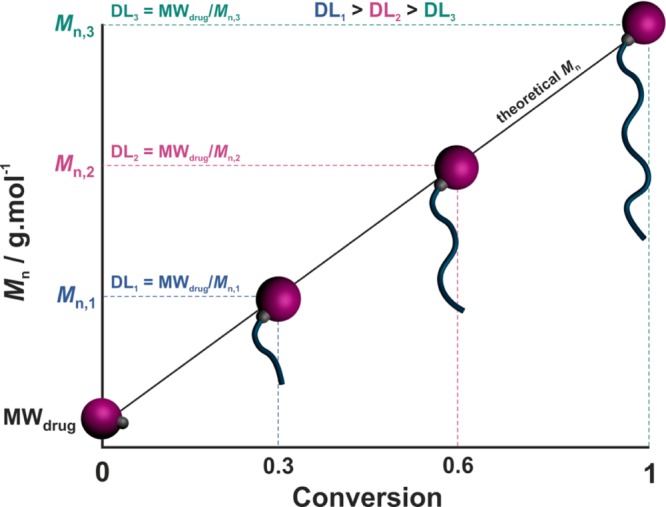
Schematic representation of the evolution of the number-average molar mass (Mn) and the drug loading (DL, according to DL = MWdrug/Mn with MWdrug = molecular weight of the drug) of the drug–polymer prodrug with monomer conversion during the “drug-initiated” synthesis of a polymer prodrug.
Not only is this construction method virtually applicable to any kind of drugs (providing they either inherently possess a suitable initiation site or they can be functionalized to introduce an initiating moiety), which facilitates the loading of poorly soluble ones, but also to many different polymers depending on the polymerization method used. Therefore, it has great potential for the design of many different drug polymer prodrugs with therapeutic activities against various diseases.
Depending on the nature of the drug and of the polymer promoiety (that is the polymer linked to the drug), as well as the formulation process used, “drug-initiated” polymer prodrugs can form polymer prodrug nanoparticles, water-soluble polymer prodrugs, or can be encapsulated into nanocarriers (Figure 1). After a brief description of the polymerization techniques used to build these materials, the synthesis and biomedical applications of the different “drug-initiated” polymer prodrugs reported in the literature are discussed.
Polymerization Techniques
Two important classes of controlled polymerization techniques have been successfully employed to prepare well-defined polymer prodrugs from the “drug-initiated” method: ring-opening polymerization (ROP)11 and reversible-deactivation radical polymerization (RDRP).12,13
In practice, appropriate ROP and RDRP conditions allow for controlled growth of the polymer chain, with linear increase of the number-average molar mass (Mn) vs monomer conversion (Figure 2) and low dispersity (Đ = Mw/Mn, with Mw the weight-average molar mass).27 The number-average degree of polymerization (DPn) of the final polymer is therefore predictable and is equal to the monomer-to-initiator molar ratio (times the monomer conversion if the polymerization is stopped prior to completion). The polymer also exhibits high chain-end fidelity; that is quantitative α- and ω-functionalization.
Ring Opening Polymerization (ROP)
ROP is the polymerization technique of choice to prepare well-defined biodegradable polyesters.28 Representative polyesters are polylactide (PLA), polyglycolide (PGA), poly(δ-valerolactone) (PVL), poly(ε-caprolactone) (PCL) and poly(trimethylene carbonate) (PTMC), which are obtained from ROP of lactide (LA), glycolide (G), δ-valerolactone (δVL), ε-caprolactone (εCL) and trimethylene carbonate (TMC), respectively (Figure 3). Importantly, PLA, PGA and their copolymers (poly(lactide-co-glycolide), PLGA) have gained Food and Drug Administration (FDA) approval for use in humans as a result of their biocompatibility and biodegradability.
Figure 3.
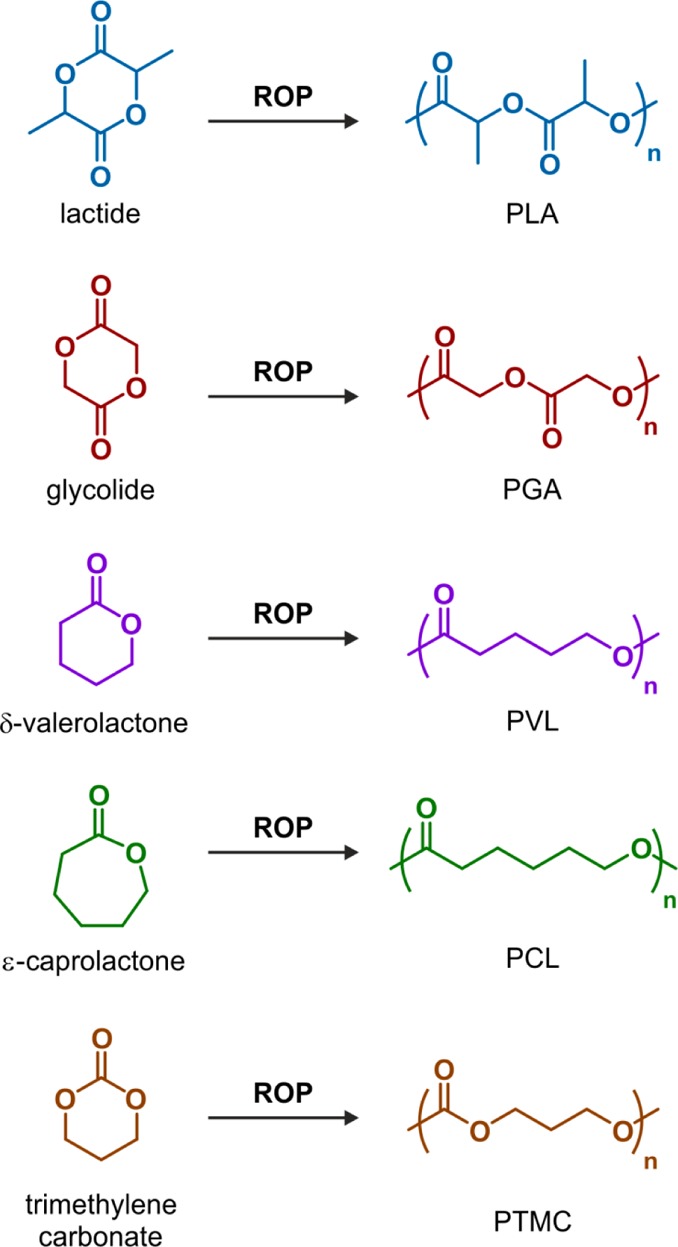
Structures of lactide (LA), glycolide (GA), δ-valerolactone (VL), ε-caprolactone (CL), trimethylene carbonate (TMC), and their corresponding polymers (PLA, PGA, PVL, PCL and PTMC, respectively) obtained by ring-opening polymerization (ROP).
ROP can be performed following different mechanisms: (i) coordination–insertion polymerization; (ii) ionic (anionic or cationic) polymerization and (iii) nucleophilic polymerization. Metal alkoxides (MORs) are well-known initiators for the ROP of cyclic esters via a coordination–insertion mechanism (Figure 4).11,29 They are usually prepared in situ by mixing hydroxyl-containing compounds (R–OH) with appropriately designed active metal complexes (LM, with L = ligand and M = metal). The resulting MOR initiates and controls the ROP, hence leading to quantitative insertion of the hydroxyl-containing compound into the polyester chain-end. Even though the coordination–insertion mechanism is still the most popular method, metal-free nucleophilic polymerizations mediated by organocatalysts has gained increasing interest as more robust, economical and environmentally friendly alternatives.30
Figure 4.
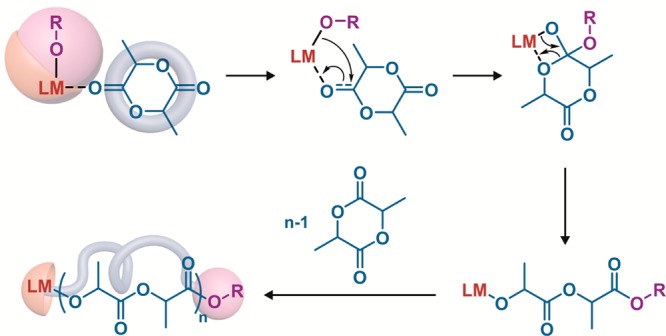
Use of metal alkoxides (MORs) as initiators for the synthesis of polylactide (PLA) by ring-opening polymerization (ROP) via a coordination–insertion mechanism. M = metal; L = ligand; OR = hydroxyl-containing molecule.
Reversible-Deactivation Radical Polymerization (RDRP)
RDRP techniques have emerged as simple routes to prepare well-defined vinyl polymers with high degree of structural uniformity, comparable to those obtained by ionic and coordination–insertion polymerizations.12,31 However, because RDRP is based on a radical mechanism, milder reaction conditions can be applied (e.g., no stringent purification of the reactants and extensive drying procedures) and greater versatility in terms of macromolecular architectures and functionalization are usually witnessed. Among the different RDRP techniques developed so far, nitroxide-mediated polymerization (NMP),32−34 atom transfer radical polymerization (ATRP)35−39 and reversible addition–fragmentation chain transfer (RAFT),40−43 represent the three most representative ones.
RDRP techniques are based on a reversible deactivation mechanism between active (macro)radicals (that can propagate) and dormant species (that cannot propagate) to minimize irreversible transfer and termination reactions.27 NMP is based on a reversible termination reaction between a growing (macro)radical and a free nitroxide to form a (macro)alkoxyamine (Figure 5a).32 This equilibrium between active and dormant species presents the advantage of being a thermal process where no catalyst nor bimolecular exchange is required. NMP is usually initiated by a preformed alkoxyamine;44 that is a two-in-one molecule that cleaves at elevated temperature to release an initiating radical and a nitroxide.32 ATRP is also based on a reversible termination reaction during which reversible activation of halide species by a transition-metal complex (e.g., copper, ruthenium, iron or nickel) takes place, usually with nitrogen-donor ligands, MtXn/L, via a redox process involving a ±1 change in the formal oxidation state of the metal (Figure 5a).36,37,39,45 RAFT polymerization is controlled by a reversible transfer reaction between a growing (macro)radical and a dormant (macro)RAFT agent (Figure 5b).40−42 The RAFT group is typically a thiocarbonylthio group such as dithioester, trithiocarbonate, xanthate or dithiocarbamate. Conversely to NMP and ATRP, the RAFT equilibrium between active and dormant species requires conventional radical initiation and is established after the addition of the growing radical Pi• onto the dormant species Pj, producing an intermediate radical followed by its fragmentation. It then leads to the growing radical Pj• and the dormant species Pi.
Figure 5.

Reversible-deactivation radical polymerization (RDRP) based (a) on a reversible termination mechanism or (b) on a reversible transfer mechanism. M = monomer. NMP = nitroxide-mediated polymerization; ATRP = atom-transfer radical polymerization; RAFT = reversible addition–fragmentation chain transfer polymerization.
Even though the carbon–carbon backbone of vinyl polymers is not readily degradable compared to that of polyesters, many different strategies have been developed to insert discrete or multiple labile functions into vinyl structures to confer degradability.46−48
Polymer Prodrug Nanoparticles
The most important class of drug-loaded polymer nanocarriers is undoubtedly polymer nanoparticles.4 The drug-initiated method was therefore logically used for the design of polymer prodrugs that were further processed into polymer prodrug nanoparticles by an emulsification method. Nanoprecipitation represents the most used emulsification method for preparing polymer nanoparticles from preformed polymers.49 The polymer is usually solubilized into a water-miscible organic solvent (e.g., acetone, DMSO, THF, etc.) followed by addition of the resulting polymer solution into water (a nonsolvent of the polymer).50 Formation of nanoparticles is instantaneous and removal of the organic solvent is subsequently performed under reduced pressure to obtain an aqueous suspension of polymer nanoparticles.
Drug–Polyester Prodrug Nanoparticles
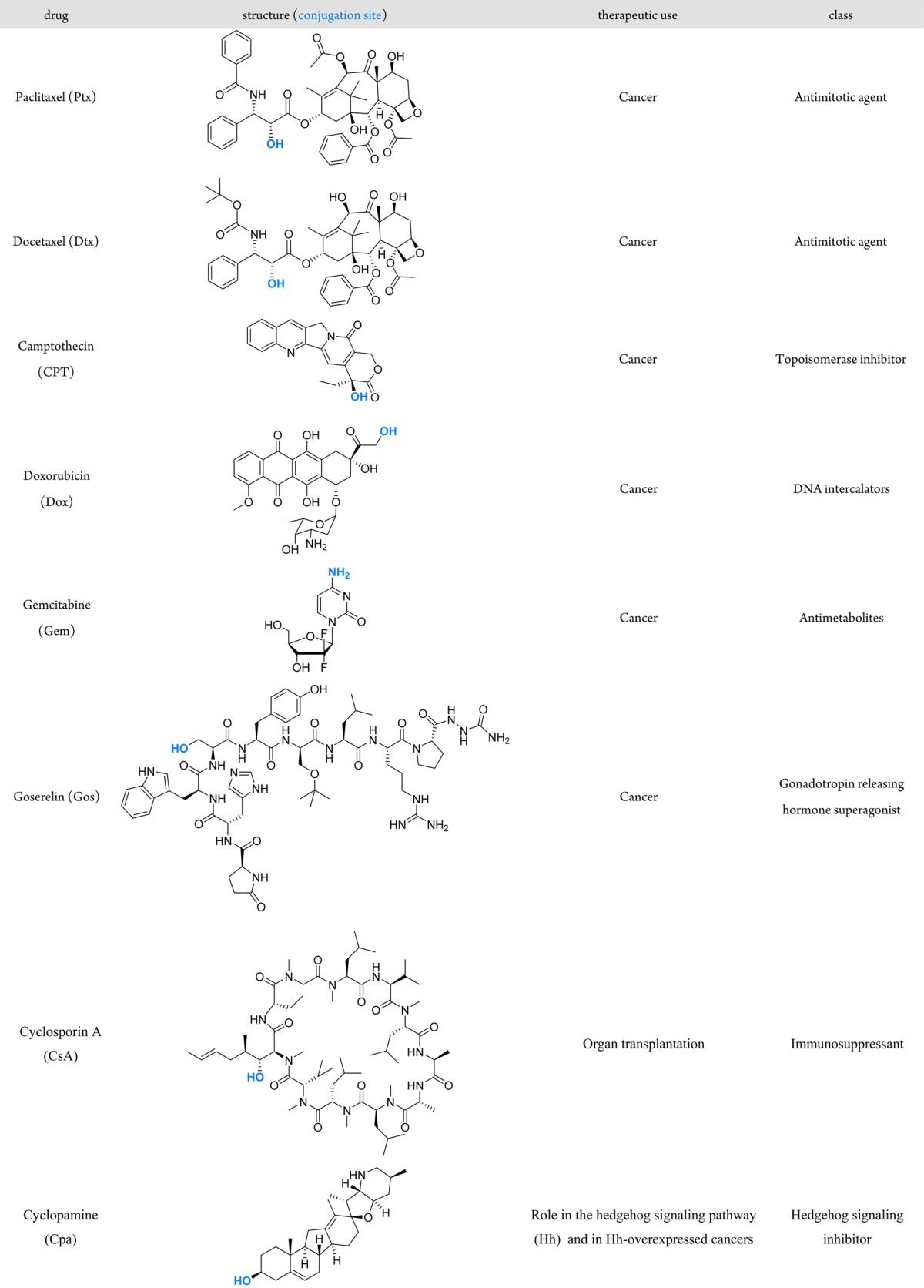
Given the initiation step of ROP by the coordination–insertion mechanism,11 hydroxyl-containing drugs can be used as initiators to prepare drug–polyester prodrugs after conversion into MORs. This was demonstrated with five well-established anticancer drugs (paclitaxel (Ptx),51−54 docetaxel (Dtx),52−55 camptothecin (CPT),52,54−56 doxorubicin (Dox)54,55 and goserelin (Gos)55), an immunosuppressive agent (cyclosporin A (CsA))57 and a drug involved in the hedgehog signaling pathway (Hh) as a potential candidate for Hh-overexpressed cancers (cyclopamine (Cpa).55 The structure of these drugs, their conjugation sites, as well as their therapeutic use and class are indicated in Table 1.
Table 1. Structure of Drugs Used To Synthesize Drug−Polymer Prodrug Nanoparticles by the “Drug-Initiated” Method.
To achieve polyester prodrugs from these hydroxyl-containing drugs, (BDI-X)MN(TMS)2 (BDI = 2-((2,6-dialkylphenyl)amino)-4-((2,6-dialkylphenyl)imino)-2-pentene, X = R1R2R3, M = Zn or Mg, TMS = trimethylsilyl) was used as a catalyst to convert them into efficient MORs (Figure 6).52,53,55 This catalyst was initially developed by Coates and co-workers for the ROP of LA.58 When the hydroxyl-containing drug is mixed equimolarly with the catalyst, a (BDI-X)M–drug complex is formed in situ (note that the structure was uncharacterized but tentatively illustrated as a monomeric M–drug complex). It can then initiate and mediate the ROP at room temperature leading to one drug molecule at the polymer chain-end.
Figure 6.

Structure of (BDI-X)MN(TMS)2 (BDI = 2-((2,6-dialkylphenyl)amino)-4-((2,6-dialkylphenyl)imino)-2-pentene, X = R1R2R3, M = Zn or Mg, TMS = trimethylsilyl) catalyst to convert hydroxyl containing compounds into metal alkoxides (MORs) for subsequent ring-opening polymerization (ROP) of lactide.
Not only was this catalyst able to form efficient drug-based MORs from drugs containing one hydroxyl group (i.e., CPT,56 CsA57 and Cpa55), but its bulky structure also enabled site-specific LA initiation for drugs bearing more than one hydroxyl groups via discrimination between their different steric environments. For instance, among the three available hydroxyl groups of Ptx, a Ptx–M complex was solely formed through the 2′-OH group in agreement with their respective steric hindrance (2′-OH < 1-OH < 7-OH).52 Dox, which also possesses three hydroxyl groups (4′-OH, 9-OH, 14-OH), was selectively functionalized through its 14-OH group, which is most sterically accessible.55 The same high regioselectivity through the 2′-OH group was also observed with Dtx, which has four hydroxyl groups (2′-OH, 1-OH, 7-OH and 10-OH) with different steric environments.53
It has been shown that subtle structural variations of the catalyst had a dramatic impact on the polymerization outcome. For instance, (BDI-II)MgN(TMS)2 led to high site-specific control but rather broad dispersity during Ptx-mediated ROP of LA (Đ = 1.47 for Ptx-PLA200).52 Conversely, the zinc analogue, (BDI-II)ZnN(TMS)2, gave lower dispersity (Đ = 1.02) without being at the expense of the chemoselectivity as the 2′-OH group was still exclusively functionalized by the PLA chain.52 A similar trend was also observed with Dox (Figure 7). The ROP mediated by (BDI-II)MgN(TMS)2 led to a dispersity of 1.5 whereas (BDI-II)ZnN(TMS)2 led to a better control (Đ ∼ 1.2).55 This is in line with earlier results showing that Zn-based alkoxides gave slightly slower but better controlled ROP than their Mg counterparts.58 Other structural variations were focused on the substituents of the N-aryl groups and at the β-position of the BDI ligand (R1, R2 and R3, Figure 6).53,55 Under similar Ptx-initiated ROP conditions, a progressive increase of the bulkiness the N-aryl substituents (BDI-EE < BDI-EI < BDI-II) led to better control of LA polymerization with final dispersities gradually decreasing from 1.30 to 1.02.53 However, substituting the hydrogen atom by a nitrile moiety at the β-position of the BDI (BDI-IICN, Figure 6) had marginal effect on the polymerization. With CPT, the use of BDI-EI was found to be the overall best catalyst for the ROP of LA in terms of control and CPT incorporation whereas BDI-II led to higher dispersity.56
Figure 7.
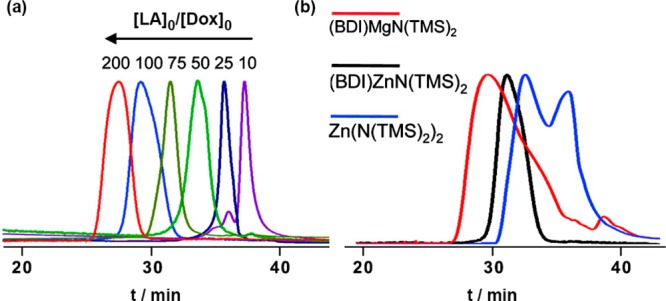
(a) SEC (UV detection) analysis of Dox-PLA synthesized by Dox/(BDI-II)ZnN(TMS)2-mediated ROP of LA at [LA]0/[Dox]0 = 10, 25, 50, 75, 100 and 200. (b) SEC (UV detection) analysis of Dox-PLA synthesized by Dox/(BDI-II)ZnN(TMS)2-, Dox/(BDI-II)MgN(TMS)2- and Dox/Zn(N(TMS)2)2-mediated ROP of LA at a [LA]0/[Dox]0 = 100. Adapted with permissions from ref (55).
Several lines of evidence supported that formation of drug-based MORs and subsequent polymerization did not affect the structure of the drug. For instance, HPLC and MS analyses showed no degradation of Ptx and Dox when reacted with (BDI-II)ZnN(TMS)2, and model initiation steps using succinic anhydride demonstrated that the drug was unaffected by the formation of the mono adduct.52,53 Likewise, this was shown for CPT, whose lactone ring preservation is essential for the antitumor activity.56 Hydrolysis of Ptx-PLA, CPT-PLA, Dox-PLA and Gos-PLA also released intact parent drugs that excluded their potential degradation during the prodrug synthesis.52,55
ROP of LA was investigated with all drugs from Table 1. The main characteristics of the different drug–polyester prodrug nanoparticles that have been synthesized, together with their biological evaluations are reported in Table 2. In general,52,53,55,56 targeted DPn were in the 10–200 range (which corresponds to Mn ∼ 1.4–30 kg·mol–1), and the right choice of catalyst led to good control and agreement between theoretical Mn and experimental ones, low dispersities, as well as >95% drug incorporation efficacy (except for Gos were it was >81%55). Not only different drugs have been used as ROP initiators, but also different monomers including CL,53 VL,53 TMC53 and phenyl O-carboxyanhydride (Phe-OCA)54 have been successfully controlled, which demonstrated a high level of versatility as it enabled materials with different properties to be synthesized. Interestingly, whereas (BD-II)ZnN(TMS)2 was an appropriate catalyst for CL, VL and TMC (note that a polymerization temperature of 50 °C was required for TMC),53 (BDI-EI)ZnN(TMS)2 gave the best control for the ROP of Phe-OCA.54
Table 2. Synthesis, Macromolecular Characteristics and Biological Evaluation of Drug–Polyester Prodrug Nanoparticles.
| drug | catalyst | polymer (Mn, kg·mol–1)a | drug loading (wt %)b | biological evaluation(s) | ref |
|---|---|---|---|---|---|
| paclitaxel (Ptx) | (BDI-II)ZnN(TMS)2 | PLA (6.9–27.2)c | 3.0–10.9c | in vitro (targeting) | (51) |
| PLA (2.2–14.4)d | 5.6–28.3d | in vitro (cytotoxicity) | (52) | ||
| PVL (14.2–30.4)c | 2.7–5.7c | (53) | |||
| PCL (19.4)c | 4.2c | (53) | |||
| PTMC (13.8)c | 5.8c | (53) | |||
| (BDI-EI)ZnN(TMS)2 | P(Phe-OCA) (3.7–14.8)d | 5.4–18.7c | (54) | ||
| camptothecin (CPT) | (BDI-EI)ZnN(TMS)2 | PLA (1.4–14.2)d | 2.4–19.5d | in vitro (cytotoxicity) | (56) |
| (BDI-II)ZnN(TMS)2 | PLA (1.4)d | 19.5d | in vitro (cytotoxicity) | (52) | |
| PLA (3.6–14.4)d | 2.4–8.8d | (55) | |||
| (BDI-EI)ZnN(TMS)2 | P(Phe-OCA) (4.0–15.1)c | 2.3–8.1c | in vitro (cytotoxicity), in vivo (biodistribution, pharmacokinetics, toxicity) | (54) | |
| docetaxel (Dtx) | (BDI-II)ZnN(TMS)2 | PLA (1.4)d | 35.9d | in vitro (cytotoxicity) | (52) |
| PLA (1.4–3.6)d | 18.3–35.9d | (55) | |||
| PCL (10.4)c | 7.2c | (53) | |||
| (BDI-EI)ZnN(TMS)2 | P(Phe-OCA) (3.7)d | 16.1c | (54) | ||
| doxorubicin (Dox) | (BDI-II)ZnN(TMS)2 | PLA (1.7–18.3)c | 3.6–27.4d | in vitro (cytotoxicity) | (55) |
| (BDI-EI)ZnN(TMS)2 | P(Phe-OCA) (3.7)d | 10.3c | (54) | ||
| cyclosporin (CsA) | (BDI-II)ZnN(TMS)2 | PLA (15.7)c | 7.1c | in vitro (suppression of T cell proliferation and production of inflammatory cytokines); in vivo (targeting lymph nodes, suppression of T cell proliferation) | (57) |
| cyclopamine (Cpa) | (BDI-II)ZnN(TMS)2 | PLA (7.2–14.4)d | 2.8–5.4d | (55) | |
| goserelin (Gos) | (BDI-II)ZnN(TMS)2 | PLA (1.4–14.4)d | 8.1–46.8d | (55) |
Mn of the polymer alone (i.e., without drug).
Drug loading of the polymer prodrug conjugate alone (that is before poststabilization, if any).
Determined experimentally.
Calculated from [monomer]0/[initiator]0. PLA = polylactide, PVL = poly(δ-valerolactone), PCL = poly(ε-caprolactone), PTMC = poly(trimethylene carbonate), P(Phe-OCA) = phenyl O-carboxyanhydride.
As the drug loading can be finely tuned by varying the [M]0/[I]0 ratio (where I = initiator), high drug contents can be easily achieved by targeting low Mn. For instance, the maximum drug loadings achieved for the tested drugs with PLA as the polymer promoiety were Ptx (28.3 wt %),52 CPT (19.5 wt %),52 Dtx (35.9 wt %),55 Dox (27.4 wt %),55 CsA (7.1 wt %),57 Cpa (5.4 wt %)55 and Gos (46.8 wt %).55 These values are much greater than those usually observed for traditional drug-loaded nanoparticles by means of physical encapsulation, which are usually ca. 3–5 wt %.
Nanoprecipitation of drug–polyester conjugates in water gave narrowly dispersed polymer prodrug nanoparticles (Figure 8a). Their average diameters ranged from 50 to 240 nm depending on the nature of the polymer prodrug and the nanoprecipitation conditions. Increasing the polymer concentration in the organic solution led to higher average diameters whereas switching from acetone to DMF as the organic solvent had the opposite effect (leading to ca. 20–30 nm difference).51,52,55,57 In general, sub-100 nm nanoparticles were obtained at low concentrations; typically below 0.5 mg·mL–1.
Figure 8.

Formation of drug–polyester prodrug nanoparticles by nanoprecipitation and poststabilization by poly(ethylene glycol)-b-polylactide (PEG-b-PLA) amphiphilic diblock copolymer.
Whereas good colloidal stability was obtained in water, rapid aggregation in PBS occurred,51,52 presumably because of salt-induced screening of repulsive forces and absence of stabilizing groups. Reducing the polymer chain length to reach high drug loadings also decreased the polymer/polymer interaction and therefore led to less stable nanoparticles. This may limit their use, as colloidal stability in biological media is crucial to safely perform in vitro and in vivo experiments. However, this limitation was alleviated by (i) the use of Phe-OCA in place of LA to increase hydrophobic interactions between polyester chains (Figure 9)54 and (ii) stabilizing drug–PLA nanoparticles by PEG-based macromolecular surfactants such as PEG-b-PLA or PEG-b-PLA-b-PEG, by either sequential or conanoprecipitation (Figure 8b).51,52,54,57 Note that best stabilizing effects were obtained when Phe-OCA and PEG-based surfactants were used concomitantly.54
Figure 9.
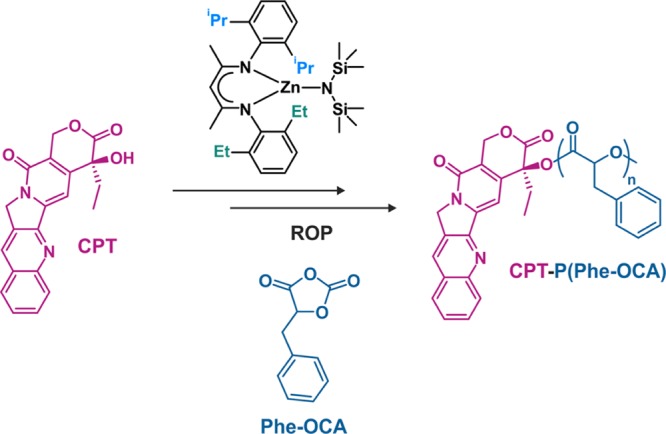
Synthesis of CPT-poly(phenyl O-carboxyanhydride) (CPT-P(Phe-OCA)) from phenyl O-carboxyanhydride (P(Phe-OCA)) and (BDI-EI)ZnN(TMS)2.
Switching from PLA to P(Phe-OCA) improved the colloidal stability of PEGylated CPT-based prodrug nanoparticles in PBS over a 30 min time interval and in human serum buffer up to 5 days.54 More stable diameters upon dilution as well as a higher CMC were also measured for PEG-b-P(Phe-OCA) compared to PEG-b-PLA nanoparticles (Figure 10). However, P(Phe-OCA) may not have the same degradation and toxicological profiles as PLA. Besides, although PEGylation led to much slower clearance from the blood (as shown with Cu64-labeled PEG-b-PLA/P(Phe-OCA) nanoparticles),54 it significantly decreased the drug loading and complicated the formulation process, which makes the “drug-initiated” method less advantageous and bring it back closer to regular drug-loaded polymer nanoparticles. Yet, the presence on the surface of PEG chains provided a mean to perform active targeting of extracellular prostate-specific membrane antigen (PSMA) via their carbodiimide-assisted coupling to an A10 aptamer, as shown from model Cy5-PLA/PLA-b-PEG-COOH nanoparticles.51
Figure 10.
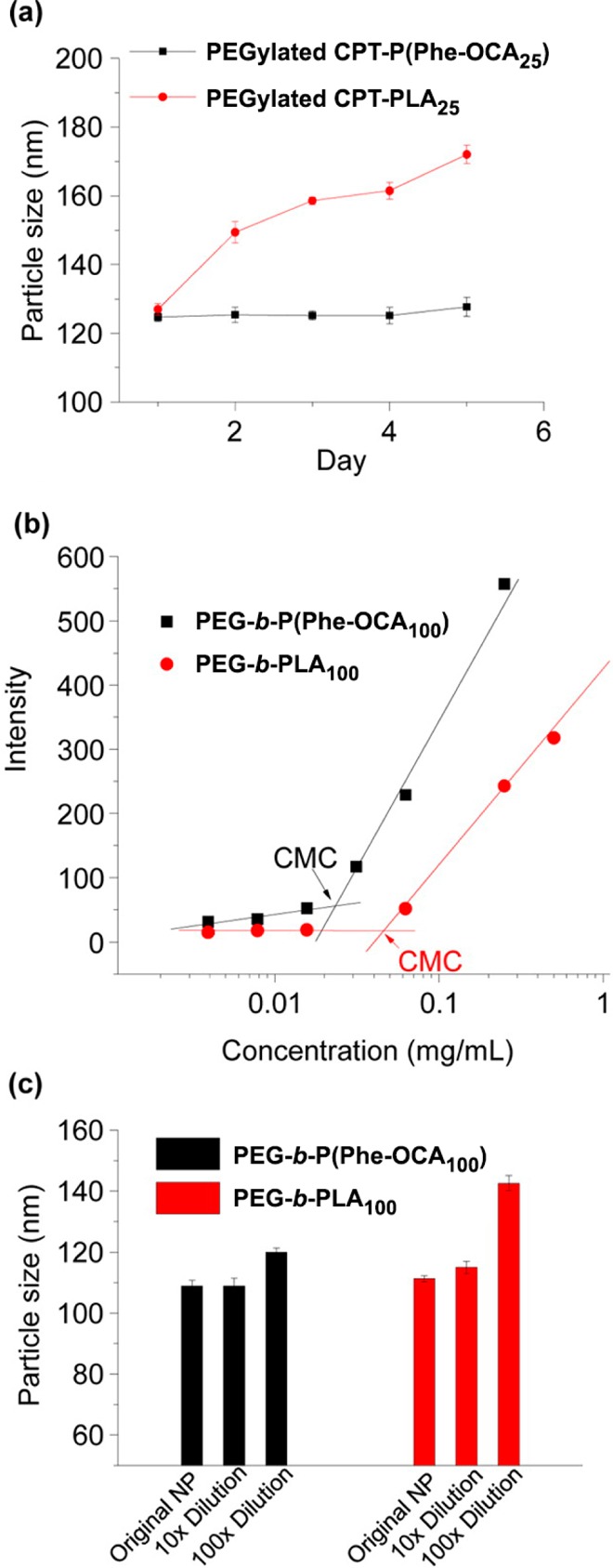
(a) Stability of PEGylated CPT-P(Phe-OCA25) and PEGylated CPT-PLA25 nanoparticles in human serum buffer. (b) Intensity of Nile Red versus concentration of PEG5k-P(Phe-OCA100) and PEG5k-PLA100, and CMC determination (CMC = 2.2 × 10–2 mg·mL–1 and 4.5 × 10–2 mg·mL–1, respectively). (c) Particle size variation of PEGylated P(Phe-OCA100) and PEGylated PLA100 nanoparticles with or without dilution determined by DLS. Adapted with permissions from ref (54).
Investigations on lyophilization of nanoparticle suspensions for long-term storage are often neglected, but it is an important parameter for potential clinical translation. Among all tested cryoprotectants, bovine serum albumin (BSA) was shown to provide the best protection to Ptx-PLA/PEG-b-PLA-b-PEG nanoparticles during lyophilization.51 After resuspension, only a moderate increase of the diameter (from 89 to 106 nm) and a still low particle size distribution were obtained. CPT-P(Phe-OCA) nanoparticles gave the same trend using human serum albumin (HAS) as cryoprotectant. However, no long-term stability measurement was performed.
Sustained drug release (i.e., absence of burst release) from these prodrug nanoparticles was obtained in PBS,52,54−57 as opposed to PLA nanoparticles with physically encapsulated drugs giving ∼80% of drug release within 24 h.52,55 It was also shown that the higher the polymer chain length, the lower the drug release.52,54,55 This is likely due to (i) a lower access of ester groups to the aqueous environment and (ii) a lower diffusion of the drug out of the nanoparticles due to increased polymer chain entanglement.
Biological evaluation of anticancer polyester prodrug nanoparticles based on Ptx,52 CPT,56 Dtx52 and Dox55 was exclusively investigated in vitro, by performing cytotoxicity experiments (MTT assay) on different cancer cell lines. Although nanoparticles exhibited significant anticancer activities, the half-maximal inhibitory concentrations (IC50) of all polyester prodrug nanoparticles were systematically higher than the free parent drugs, irrespectively of the nature of the drug and of the polymer chain length. This was expected for a prodrug, as the drug must be cleaved from the polymer promoiety before being active whereas a free drug is immediately active. Interestingly, the shorter the polymer chain length, the lower the IC50. The lowest IC50 values were reached for a targeted DPn of 10 and were 389 nM (CPT-PLA),56 970 nM (Dox-PLA),55 111 nM (Ptx-PLA)52 and 180 nM (Dtx-PLA)52 whereas IC50 values for the free parent drugs were 95 nM (CPT), 507 nM (Dox), 87 nM (Ptx) and 10 nM (Dtx). PEGylated CsA-PLA nanoparticles were shown to suppress T-cell proliferation and production of inflammatory cytokines in vitro in a comparable manner to free CsA.57 To deliver selectively PEG-b-PLA/Cy5-PLA/CsA-PLA nanoparticles in vivo to the lymph nodes, the main loci for T-cell activation, they were first internalized into dendritic cells (DCs) followed by injection of the resulting nanoparticle-loaded DC in mice. They were able to migrate to the lymph nodes leading to a significant reduction of T-cell priming without systemic release.
Drug–Polyvinyl Prodrug Nanoparticles
Drugs do not naturally possess functionalities suitable to mediate RDRP. Therefore, to grow vinyl polymer chains from drugs in a controlled fashion, the drug has to be first derivatized by a functional group allowing implementation of RDRP.
NMP and RAFT have been used to prepare polymer prodrug nanoparticles by the “drug-initiated” method59 (note that water-soluble polymer prodrugs were prepared by ATRP and RAFT following a similar approach,60,61 see section titled Water-Soluble Polymer Prodrugs). Many different vinyl monomers with various functionalities can be polymerized by radical polymerization, which makes it the most versatile polymerization technique. Unfortunately, the choice of monomers of potential interest for biomedical applications is very limited, which is the main consequence of the nondegradability and nonmetabolization of vinyl polymers,46 as well as the applications they are intended to be used for. A notable exception is poly(oligo(ethylene glycol) methyl ether methacrylate) (POEGMA),62 which has been extensively used for protein PEGylation as alternative to traditional linear PEG.63,64 In this particular case, degradation is not a prerequisite, as water-soluble polymers such as POEGMA of moderate molar masses can be excreted through renal filtration (a commonly accepted renal excretion limit is ∼40–60 kDa).65 For hydrophobic vinyl polymers, however, because they are not readily excretable, an interesting strategy is to design biocompatible polymers or polymers with strong structural analogies with biocompatible materials. This approach was illustrated by the use two different vinyl polymers containing isoprenoid units and gemcitabine (Gem, Table 1) as a drug, a nucleoside analogue with demonstrated activity against a broad range of solid tumors.66 Beyond its strong anticancer activity, the advantage of using Gem also relied on its hydrophilicity, likely conferring a certain degree of amphiphilicity to the resulting polymer prodrug and thus promoting nanoparticle stabilization. Also, the coupling strategy aimed at protecting Gem from rapid deamination by deoxycytidine deaminase, leading to greater in vivo anticancer activity than free Gem.67
To perform RAFT from Gem, the 3′,5′-hydroxyl-protected drug was first derivatized with 4-cyano-4-[(dodecylsulfanylthiocarbonyl)-sulfanyl]pentanoic acid through its 4-N position to give the corresponding Gem-RAFT agent (Figure 11a).68 It was then used to control the polymerization of squalenyl-methacrylate (SqMA), a monomer based on squalene (Sq), which is a lipidic precursor in cholesterol biosynthesis that is widely distributed in nature.69 Sq also served as a building block for the synthesis of molecular prodrugs, which self-assembled in aqueous solution to form supramolecular nanostructures.70−73 After deprotection, each Gem-PSqMA polymer prodrug was therefore composed of a Gem chain-end and a methacrylate backbone with pending copies of squalene. By adjusting the polymerization conditions, a small library of well-defined Gem-PSqMA was obtained with Mn ranging from 4.4 to 11.3 kg·mol–1 (which corresponds to DPn = 7–14) and low dispersities (1.18–1.28). Nanoprecipitation led to highly stable and narrowly dispersed Gem-PSqMA nanoparticles of average diameters in the 120–156 nm range (Figure 11b,c) with no noticeable influence of the Mn. A noticeable feature of these nanoparticles were the strongly negative surface charges (ca. −60 mV), as shown by ζ-potential measurements, which represents a decisive criterion for their colloidal stability. Depending on the PSqMA chain length, DL varied from 2.5 to 7.2 wt %.
Figure 11.

(a) Synthetic pathway for gemcitabine–poly(squalenyl methacrylate) (Gem-PSqMA) prodrug by reversible addition–fragmentation chain transfer (RAFT) polymerization. (b) DLS data giving the average diameter in intensity. (c) Cryo-TEM of Gem-PSqMA10 nanoparticles. The RAFT moiety was omitted on the schematic representation of the nanoparticle for clarity. Adapted with permissions from ref (68).
To confer stealth properties, conanoprecipitation of Gem-PSqMA with Sq-PEG was successfully attempted68 and led to PEGylated Gem-PSqMA nanoparticles as shown by XPS and complement activation assay. The latter resulted in observing protein C3 fragmentation into C3b, which has a central role in triggering the immune system response against foreign bodies.74 Cytotoxicity assays showed significant anticancer activities on various cancer cell lines (L1210 WT, MiaPaCa-2, A549, CCRF-CEM and P388S) with very low IC50 values (20–180 nM depending on the cell line) and no marked influence of the polymer chain length. Similarly to drug–polyester nanoparticles, IC50 of all Gem-PSqMA nanoparticles were higher than that of free Gem due to their prodrug nature. Importantly, Gem-PSqMA14 nanoparticles exhibited significant anticancer activity in vivo against human pancreatic (MiaPaCa-2) carcinoma xenograft model in mice (8 i.v. injections, Gem equivalent dose of 3.4 mg·kg–1 per injection).75 Gem-PSqMA14 nanoparticles gave an important tumor growth inhibition of ∼75% conversely to control experiments (i.e., untreated mice, nonfunctionalized PSqMA nanoparticles and free Gem at the same dose than Gem-PSqMA14 nanoparticles), for which tumor progression was rapid (Figure 12a,c). No weight loss was observed with prodrug nanoparticles, supporting absence of toxicity to the mice (Figure 12b). Also, immunohistochemical analysis of tumor biopsies was performed and revealed an important reduction in normal vasculature, together with significant antiproliferative and apoptotic effects.75
Figure 12.
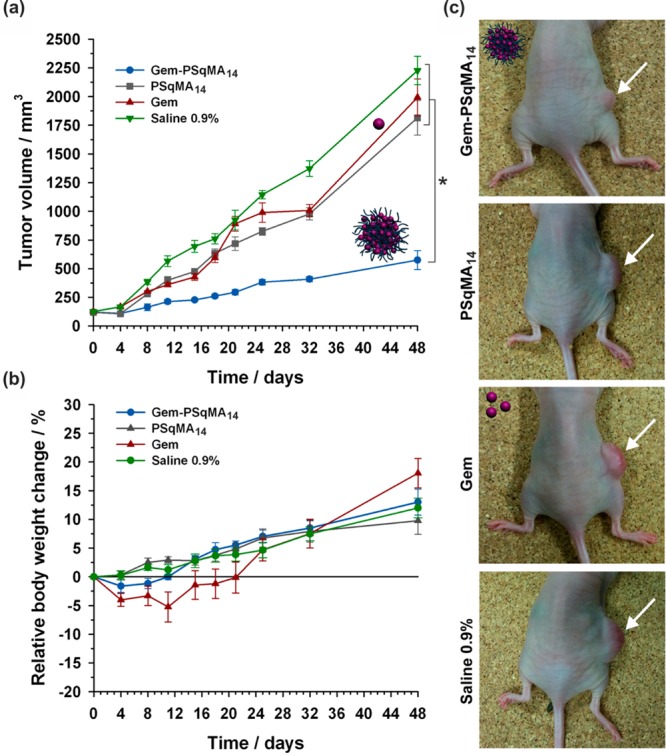
Evolutions of (a) tumor volume and (b) relative body weight change with time following intravenous injection (on days 0, 4, 8, 11, 15, 18, 21 and 25) of Gem (green −▼–, 3.4 mg·kg–1), Gem-PSqMA14 nanoparticles (blue −●–, 3.4 mg·kg–1 Gem-equivalent dose), control (gray −■–, saline 0.9%) and PSqMA14 nanoparticles (red −▲–, same dose of polymer as Gem-PSqMA14). (c) Pictures showing the position of the implanted tumor on representative mouse at day 32 for all treatments. Adapted with permissions from ref (75).
Gem was also efficiently derivatized through its 4-N position by a secondary alkoxyamine based on the SG1 nitroxide using PyBOP as a coupling agent (Figure 13a).76 In this case, protection of Gem hydroxyl groups was not required and short polyisoprene (PI) chains of controlled molar masses were directly grown from the Gem–alkoxyamine by NMP77,78 to give well-defined Gem-PI polymer prodrugs. PI was selected for its interesting properties such as chemical and enzymatic degradability, as well as its biocompatibility and its structural similarity with natural polyisoprenoids such as squalene, vitamin E, retinol, etc. By varying the polymerization time, a small library of Gem-PI was prepared with low Mn (0.84–2.51 kg·mol–1, which corresponds to DPn ∼ 4–28) and narrow molar mass distributions (Đ = 1.28–1.40). Controlled polymerization of a low molecular weight monomer like isoprene enabled low Mn to be obtained and thus high DLs to be reached; from 10.5 to 31.2 wt %. Nanoparticles of Gem-PI with average diameters of 130–160 nm (Figure 13b), narrow particle size distributions and strongly negative surface charges (from −66 to −77 mV), were obtained by nanoprecipitation and exhibited remarkable colloidal stability over several weeks.
Figure 13.

(a) Synthesis of gemcitabine–polyisoprene (Gem-PI) prodrug by nitroxide-mediated polymerization (NMP). The nitroxide moiety was omitted on the schematic representation of the nanoparticle for clarity. (b) Cryogenic transmission electron microscopy of Gem-PI28 nanoparticles. Adapted with permissions from ref (76).
Cytotoxicity assays were performed on different cancer cell lines and confirmed the anticancer activity of Gem-PI nanoparticles with IC50 values in the nM range.76 It was noted that for the Mn tested, the higher the Mn, the lower the IC50 values. Surprisingly, the opposite trend was observed with polyester prodrug nanoparticles (see section “Drug–Polyester Prodrug Nanoparticles”), for which the shortest PLA chain length gave the highest cytotoxicity. This discrepancy may arise from several parameters beyond the fact of using different drugs: (i) the polymers are different in terms of structure and degradability, which may drastically alter the colloidal disassembly and the drug release; (ii) the surface hydrophobicity of the nanoparticles is likely to be different and this may have a role in opsonin adsorption leading to different rates of endocytosis and (iii) the drug positioning inside the nanoparticles may also be different (e.g., at the surface, deep into the core, etc.), which could impact their cleavage from the polymer. The therapeutic efficacy of Gem-PI nanoparticles was also demonstrated in vivo on human pancreatic (MiaPaCa-2) tumor bearing mice.76 Only four injections were performed, and the equivalent Gem dose was increased up to 7 mg·kg–1 per injection. Gem-PI nanoparticles of two different Mn, 1.2 kg·mol–1 (Gem-PI9) and 2.5 kg·mol–1 (Gem-PI28), were administered and showed remarkable anticancer activity compared to control experiments (Figure 14a). The tumor growth reduction was even more pronounced with nanoparticles from Gem-PI28, giving a tumor growth inhibition of 72%. This was in good agreement with in vitro assays and tended to show a good correlation between in vitro and in vivo experiments. In addition, Gem-PI treated mice maintained a rather constant body weight conversely to mice treated with free Gem (∼10% weight loss), hence supporting both the efficient anticancer activity of Gem-PI nanoparticles and the disappearance of Gem-related toxic effects (Figure 14b).
Figure 14.
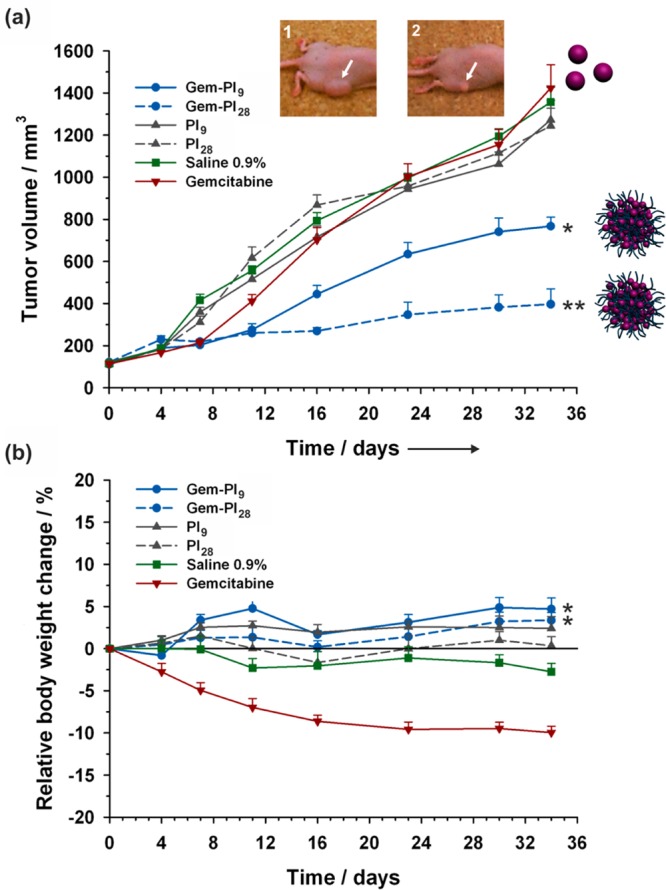
Evolutions of (a) tumor volume and (b) relative body weight change with time following intravenous injection (on days 0, 4, 8 and 12) of Gem (red −▼–, 7 mg·kg–1), Gem-PI nanoparticles [blue −●– (Gem-PI9) and blue --●-- (Gem-PI28), 7 mg·kg–1 Gem-equivalent dose], control (green −■–, saline 0.9%) and PI nanoparticles [gray −▲–, (PI9) and gray --▲--, (PI28), same dose of polymer as Gem-PI]. White arrows point to the position of the implanted tumor on representative mouse at end point for Gem-treated group (inset 1) and Gem-PI28-treated group (inset 2). Adapted with permissions from ref (76).
Incorporation into Nanocarriers
Another interesting use of drug–polymer prodrugs obtained by the “drug-initiated” method concerns their incorporation into nanocarriers and the benefit that can be taken from this approach compared to encapsulation of small drug molecules. By combining the advantages of a macromolecular prodrugs with those of sophisticated nanocarriers featuring stealth and targeting abilities, spatiotemporal controlled drug delivery can be achieve. For instance, ∼60 nm hybrid nanoparticles composed of a Ptx-PLA25 core (synthesized by Ptx-initiated LA from (BDI-II)ZnN(TMS)2, DL = 19.2 wt %), surrounded by a lipid monolayer composed of sybean lecithin and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), as well as a hydrophilic shell of anchored PEG-DSPE were prepared by self-assembly and nanoprecipitation (Figure 15a). They were further surface-functionalized by a functional vascular targeting peptide (KLWVLPK) from DSPE-PEG-maleimide using thiol/maleimide conjugation. Ptx release was achieved over a period of 10–12 days in vitro, and nanoparticles inhibited human aortic smooth muscle cell proliferation in vitro (Figure 15b). Model nanoparticles (with Alexa Fluor 647-PLGA as substitute for Ptx-PLA25) showed greater in vivo vascular retention during percutaneous angioplasty compared to nontargeted nanoparticles.
Figure 15.
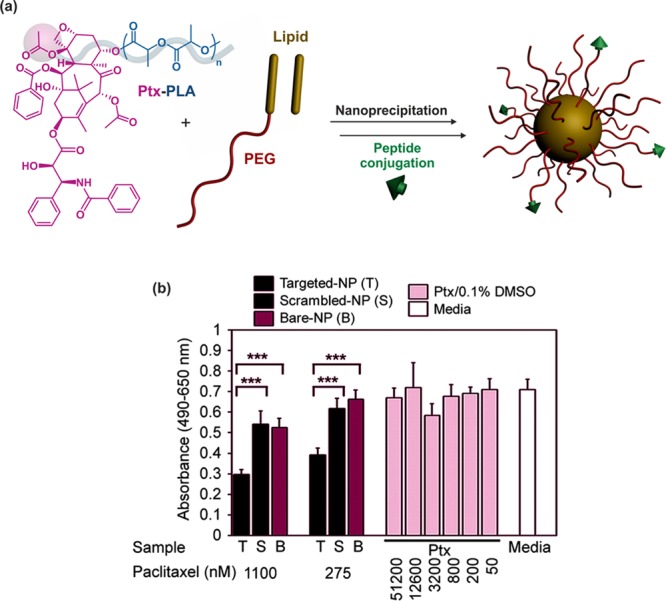
(a) Structure of paclitaxel–polylactide (Ptx-PLA) and formation of KLWVLPK peptide-functionalized lipid–polymer hybrid nanoparticles composed of a Ptx-PLA core and a PEGylated lipidic shell. (b) Human aortic smooth muscle cell (haSMC) cytotoxicity study showing the targeted drug release from KLWVLPK peptide-functionalized hybrid nanoparticles. Adapted with permissions from ref (79).
The second benefit of incorporating macromolecular prodrug into nanocarriers relies on the possibility to incorporate concurrently different drug–polymer prodrugs and accurately control the drug ratio to achieve efficient multidrug delivery systems. This was illustrated by the design of hybrid nanoparticles with a core of Dox-PLA66/CPT-PLA67 (DL = 5.4 and 3.5 wt %, respectively) of variable ratio (1:1, 3:1 and 1:3), surrounded by a lipid bilayer to which was anchored PEG chains to confer stabilizing and stealth properties (Figure 16). It was shown that a single population of nanoparticles containing both Dox and CPT was obtained (and not two separate populations of each type of nanoparticles). Preliminary in vitro results on MB-435 breast cancer cells (by MTT assays) tended to confirm the benefit of the dual drug-loaded nanoparticles compared to a physical mixture of individual nanoparticles.
Figure 16.

Formation of dual polymer prodrug-loaded PEGylated lipid–polymer hybrid nanoparticles from concomitant encapsulation of CPT-PLA and Dox-PLA polymer prodrugs.
Water-Soluble Polymer Prodrugs
The “drug-initiated” method is applicable to many different monomers. If the monomer is hydrophilic, the resulting drug–polymer prodrug is likely to be highly soluble in water, whatever the nature of the drug. This has been shown with the synthesis of poly(methacryloyloxyethyl phosphorylcholine) (PMPC)60 and poly(hydroxylpropyl methacrylamide) (PHPMA).61 Although PMPC reduces plasma protein adsorption and is considered as a biomembrane-mimetic polymer,80 PHPMA is nonimmunogenic as well as nontoxic, and has been mainly used as drug carriers via multiple side chain conjugation.81
CPT was derivatized at the 20-OH position with 2-bromopropionyl bromide or 2-bromoisobutyryl bromide to yield the corresponding CPT-ATRP initiators with an ester linkage between CPT and the initiating part.60 Well-defined CPT-terminated PMPC (Đ ∼ 1.21–1.40, Mn = 6.5–17 kg·mol–1) were obtained using Cu(I)Br/bipy as the catalyst in DMSO/MeOH at room temperature with no marked difference between the two initiators. A glycine-linked initiator was also prepared but led to lower initiation efficiency, slightly higher dispersity and longer reaction times, in agreement with literature on amide-based ATRP initiators. As expected, CPT-PMPC prodrugs were highly water-soluble and essentially molecularly dissolved with poor local solvation of the drug but no significant aggregation. Biological evaluation of these polymer prodrugs was, however, not reported.
7-Ethyl-10-hydroxycamptothecin (SN-38) is a topoisomerase inhibitor deriving from CPT and is 100–1000 times more active than its water-soluble prodrug counterpart CPT-11.82 SN-38 is, however, much less water-soluble than CPT-11. Its use as an initiator for RDRP of hydrophilic monomers to increase its water-solubility and therefore its therapeutic index has been attempted. Protected SN-38 on the 10-OH position (termed 10-Boc-SN-38) was derivatized throught its 20-OH position with 4-cyano-4-[(do-decylsulfanylthiocarbonyl)sulfanyl]pentanoic acid as a RAFT agent and then used to mediate the polymerization of HPMA under AIBN initiation (Figure 17a).61 Water-soluble 10-Boc-SN-38-PHPMA prodrugs of tunable chain length were obtained with dispersities below 1.3 (for Mn = 6.7–25.2 kg·mol–1) and drug loadings ranging from 1.6 to 5.9 wt %. Cell viability experiments by MTS assay on X63-Ag8 and HT-29 cell line monocultures demonstrated good retention of the anticancer activity of the parent SN-38 with concentration-dependent cytotoxicity and induction of apoptosis. Also, the lower the Mn, the lower the IC50. Expectedly, given the prodrug nature of SN-38-PHPMA, higher IC50 than that of the free drug were always observed. However, the anticancer activity of SN-38-PHPMA showed no improvement compared to that of the PEG-SN-38 prodrug. Interestingly, MTS assay was also performed on a coculture of both cancer (X63-Ag8) and nonmalignant (L929) cell lines, intending to mimic the in vivo situation where cancer cells are in close proximity to healthy ones (Figure 17b). The cocultivation assays showed higher toxicity toward cancer cells compared to healthy ones, demonstrating the selectivity of the treatment.
Figure 17.

(a) Synthesis of 10-Boc-SN-38-poly(hydroxylpropyl methacrylamide) (10-Boc-SN-38-PHPMA) polymer prodrug by RAFT polymerization. (b) Apoptosis induction in a coculture of X63-Ag8 and L929 cells from SN-38 or SN-38-PHPMA polymer prodrugs after 48 h of incubation. The number of apoptotic/necrotic L929 cells (left) and apoptotic/necrotic X63-Ag8 cells (right) was determined using annexin-V (Ann.-V) and propidium iodide (PI) staining, and flow cytometry. Adapted with permissions from ref (61).
Conclusion and perspectives
Although the drug-initiated synthesis of polymer prodrugs for application in nanomedicine has been developed only very recently, a great deal of work has already been done and relevant data have been collected providing insight into the potential of this new approach. In particular, its simplicity is a strong asset compared to traditional drug delivery system as polymer prodrug delivery systems are achieved in a reduced number of synthetic steps with facilitated purification procedures. Its applicability to various drugs and polymers, as well as the fine-tuning of the drug loading and the possibility to use such materials in different ways (e.g., polymer prodrug nanoparticles, incorporation into nanocarriers and water-soluble polymer prodrugs) confer the methodology with exceptional flexibility and versatility, which are crucial features in drug delivery. Finally, biological evaluations of the different drug delivery systems have shown very promising results both in vitro and in vivo, which gave some credibility to this method. Even though the therapeutic strategy is somewhat different, growing polymer chains from drugs also appears much easier than doing so from proteins or peptides, for obvious steric, structural complexity and fragility reasons of natural macromolecules. Promising results have nevertheless been reported from different RDRP-derived polymer–peptide/proteins systems,83−87 but most of them used model proteins. Their successful application to real pathological situations would therefore be a strong achievement and strengthen the “drug-initiated/grafting from” toolbox for biomedical applications.
So far, the “drug-initiated” synthesis of polymer prodrugs has been achieved by ROP and RDRP techniques. Growing polyester chains from drugs by ROP obviously ensures that the resulting materials are fully degradable, which is of paramount importance when biomedical applications are envisioned. However, the flexibility offered by RDRP techniques (in terms of polymer nature, composition and functionalities) as well as the many possibilities to insert discrete or multiple degradable groups in vinyl backbones46 make them credible candidates in nanomedicine. This is true especially as drug-initiated synthesized polyvinyl prodrugs are yet the sole systems with demonstrated in vivo anticancer activity. In the context of “bench to bedside” translation, in vivo demonstration of the efficacy of nanomedicines is indeed essential and a necessary step toward preclinical studies and potential clinical trials, before it gets a chance to reach the market.
To explore better the capabilities of the drug-initiated synthesis of polymer prodrugs, further development should be directed toward broadening the range of drugs and polymer promoieties. Changing polymers’ nature and composition, and conferring them with additional features such as stimuli responsiveness could give access to both new nanoobjects with unprecedented properties. In particular, having the possibility to fine-tune the drug release kinetics and achieve a wide range of kinetic regimes would be an attractive feature to adapt to different pathological situations. From the standpoint of the process, combining the drug-initiated method and the formation of nanoparticles into a single process would also be of considerable interest in the field. Knowing that nanoparticle concentrations obtained from emulsification of preformed polymers are generally moderate, this would enable both high drug loadings to be reached and highly concentrated nanoparticle suspensions to be obtained in one-step. In this context, the polymerization-induced self-assembly (PISA),88−92 which relies on the formation of a wide range of polymer nanoparticle morphologies during the polymerization process, seems very promising to achieve this goal.
However, all these improvements should be made as simple as possible to avoid falling back into traditional nanoparticle weaknesses. Also, the majority of achievements have been obtained in the field of cancer therapy and it would be beneficial to apply this strategy to other pathologies such as infectious and antiparasitic diseases, as well as neurosciences.
Given the simplicity of the “drug-initiated” method is postulated to be a key parameter for the construction of efficient drug delivery systems, I would like to end this Perspective article by quoting Antoine de Saint-Exupery the same way George Whitesides did at the end of his brilliant and so inspiring TED2010 talk entitled “Toward a science of simplicity”: “Il semble que la perfection soit atteinte non quand il n’y a plus rien à ajouter, mais quand il n’y a plus rien à retrancher.”, “It seems that perfection is attained not when there is nothing more to add, but when there is nothing more to take away.” Antoine de Saint-Exupery. Terre des Hommes, Chapitre III, L’avion.
Acknowledgments
Some results described in this Perspective article were obtained thanks to the European Research Council under the European Community’s Seventh Framework Programme FP7/2007-2013 (Grant Agreement No. 249835) and the Ph.D. training program in France of the University of Science and Technology of Hanoi. The author also thanks the CNRS and University Paris-Sud for financial support.
Biography
Julien Nicolas completed his Ph.D. in 2005 under the supervision of Prof. Bernadette Charleux at the University Pierre and Marie Curie in Paris (France), where he studied nitroxide-mediated polymerization. He then joined the group of Prof. David M. Haddleton at the University of Warwick (UK), as a postdoctoral researcher (Marie Curie Intra-European Fellowship) in the field of polymer–protein bioconjugates. Since 2007, he is a CNRS researcher at Institut Galien Paris-Sud (Univ. Paris-Sud) in Châtenay-Malabry (France), where his current research activities lie in advanced macromolecular synthesis and in the design of innovative polymer-based nanomedicines. He is (co)author of more than 70 peer review articles in international journals, 5 patents and 13 book chapters. See http://julnicolas.free.fr/ for further details, or follow him on Twitter: @julnicolas.
The author declares no competing financial interest.
Notes
† This Perspective is part of the Up-and-Coming series.
References
- Farokhzad O. C.; Langer R. Impact of Nanotechnology on Drug Delivery. ACS Nano 2009, 3, 16–20. 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- Allen T. M.; Cullis P. R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Delivery Rev. 2013, 65, 36–48. 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- Bae Y.; Kataoka K. Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv. Drug Delivery Rev. 2009, 61, 768–784. 10.1016/j.addr.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Nicolas J.; Mura S.; Brambilla D.; Mackiewicz N.; Couvreur P. Design and Functionalization Strategies for Biodegradable/Biocompatible Polymer-Based Nanoparticles Applied in Targeted Drug Delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. 10.1039/C2CS35265F. [DOI] [PubMed] [Google Scholar]
- De Oliveira H.; Thevenot J.; Lecommandoux S. Smart polymersomes for therapy and diagnosis: fast progress toward multifunctional biomimetic nanomedicines. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology 2012, 4, 525–546. 10.1002/wnan.1183. [DOI] [PubMed] [Google Scholar]
- Vauthier C.; Dubernet C.; Fattal E.; Pinto-Alphandary H.; Couvreur P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv. Drug Delivery Rev. 2003, 55, 519–548. 10.1016/S0169-409X(03)00041-3. [DOI] [PubMed] [Google Scholar]
- Brambilla D.; Le Droumaguet B.; Nicolas J.; Hashemi S. H.; Wu L.-P.; Moghimi S. M.; Couvreur P.; Andrieux K. Nanotechnologies for Alzheimer’s disease: therapy, diagnosis and safety issues. Nanomedicine NBM 2011, 7, 521–540. 10.1016/j.nano.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Erb S. E. Nanotechnology in drug delivery. Drug Delivery Syst. 2009, 24, 63–70. [Google Scholar]
- Wang R.; Billone P. S.; Mullett W. M. Nanomedicine in Action: An Overview of Cancer Nanomedicine on the Market and in Clinical Trials. J. Nanomater. 2013, 2013, 1–12. 10.1155/2013/629681. [DOI] [Google Scholar]
- Langer R. Drug delivery and targeting. Nature 1998, 392, 5–10. [PubMed] [Google Scholar]
- Dechy-Cabaret O.; Martin-Vaca B.; Bourissou D. Controlled Ring-Opening Polymerization of Lactide and Glycolide. Chem. Rev. 2004, 104, 6147–6176. 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K. Macromolecular engineering: From rational design through precise macromolecular synthesis and processing to targeted macroscopic material properties. Prog. Polym. Sci. 2005, 30, 858–875. 10.1016/j.progpolymsci.2005.06.004. [DOI] [Google Scholar]
- Braunecker W. A.; Matyjaszewski K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. 10.1016/j.progpolymsci.2006.11.002. [DOI] [Google Scholar]
- Sapsford K. E.; Algar W. R.; Berti L.; Gemmill K. B.; Casey B. J.; Oh E.; Stewart M. H.; Medintz I. L. Functionalizing Nanoparticles with Biological Molecules: Developing Chemistries that Facilitate Nanotechnology. Chem. Rev. 2013, 113, 1904–2074. 10.1021/cr300143v. [DOI] [PubMed] [Google Scholar]
- Mura S.; Nicolas J.; Couvreur P. Stimuli-Responsive Nanocarriers for Drug Delivery Systems. Nat. Mater. 2013, 12, 991–1003. 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- Elsabahy M.; Wooley K. L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012, 41, 2545–2561. 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilkoti A.; Dreher M. R.; Meyer D. E.; Raucher D. Targeted drug delivery by thermally responsive polymers. Adv. Drug Delivery Rev. 2002, 54, 613–630. 10.1016/S0169-409X(02)00041-8. [DOI] [PubMed] [Google Scholar]
- Torchilin V. P. Multifunctional nanocarriers. Adv. Drug Delivery Rev. 2012, 64, 302–315. 10.1016/j.addr.2012.09.031. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; Al Zaki A.; Hui J. Z.; Muzykantov V. R.; Tsourkas A. Multifunctional Nanoparticles: Cost Versus Benefit of Adding Targeting and Imaging Capabilities. Science 2012, 338, 903–910. 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Brazel C. S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Controlled Release 2001, 73, 121–136. 10.1016/S0168-3659(01)00248-6. [DOI] [PubMed] [Google Scholar]
- Rautio J.; Kumpulainen H.; Heimbach T.; Oliyai R.; Oh D.; Jarvinen T.; Savolainen J. Prodrugs: design and clinical applications. Nat. Rev. Drug Discovery 2008, 7, 255–270. 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- Delplace V.; Couvreur P.; Nicolas J. Recent Trends in the Design of Anticancer Polymer Prodrug Nanocarriers. Polym. Chem. 2014, 5, 1529–1544. 10.1039/C3PY01384G. [DOI] [Google Scholar]
- Mura S.; Bui D. T.; Couvreur P.; Nicolas J. Lipid prodrug nanocarriers in cancer therapy. J. Controlled Release 2015, 208, 25–41. 10.1016/j.jconrel.2015.01.021. [DOI] [PubMed] [Google Scholar]
- Edmondson S.; Osborne V. L.; Huck W. T. S. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 2004, 33, 14–22. 10.1039/b210143m. [DOI] [PubMed] [Google Scholar]
- Barbey R.; Lavanant L.; Paripovic D.; Schuwer N.; Sugnaux C.; Tugulu S.; Klok H. A. Polymer Brushes via Surface-Initiated Controlled Radical Polymerization: Synthesis, Characterization, Properties, and Applications. Chem. Rev. 2009, 109, 5437–5527. 10.1021/cr900045a. [DOI] [PubMed] [Google Scholar]
- Sumerlin B. S. Proteins as Initiators of Controlled Radical Polymerization: Grafting-from via ATRP and RAFT. ACS Macro Lett. 2011, 1, 141–145. 10.1021/mz200176g. [DOI] [PubMed] [Google Scholar]
- Goto A.; Fukuda T. Kinetics of Living Radical Polymerization. Prog. Polym. Sci. 2004, 29, 329–385. 10.1016/j.progpolymsci.2004.01.002. [DOI] [Google Scholar]
- Albertsson A.-C.; Varma I. K. Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications. Biomacromolecules 2003, 4, 1466–1486. 10.1021/bm034247a. [DOI] [PubMed] [Google Scholar]
- Thomas C. M. Stereocontrolled ring-opening polymerization of cyclic esters: synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. 10.1039/B810065A. [DOI] [PubMed] [Google Scholar]
- Kamber N. E.; Jeong W.; Waymouth R. M.; Pratt R. C.; Lohmeijer B. G. G.; Hedrick J. L. Organocatalytic Ring-Opening Polymerization. Chem. Rev. 2007, 107, 5813–5840. 10.1021/cr068415b. [DOI] [PubMed] [Google Scholar]
- Qiu J.; Charleux B.; Matyjaszewski K. Controlled/living radical polymerization in aqueous media: homogeneous and heterogeneous systems. Prog. Polym. Sci. 2001, 26, 2083–2134. 10.1016/S0079-6700(01)00033-8. [DOI] [Google Scholar]
- Nicolas J.; Guillaneuf Y.; Lefay C.; Bertin D.; Gigmes D.; Charleux B. Nitroxide-Mediated Polymerization. Prog. Polym. Sci. 2013, 38, 63–235. 10.1016/j.progpolymsci.2012.06.002. [DOI] [Google Scholar]
- Hawker C. J.; Bosman A. W.; Harth E. New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerizations. Chem. Rev. 2001, 101, 3661–3688. 10.1021/cr990119u. [DOI] [PubMed] [Google Scholar]
- Grubbs R. B. Nitroxide-Mediated Radical Polymerization: Limitations and Versatility. Polym. Rev. 2011, 51, 104–137. 10.1080/15583724.2011.566405. [DOI] [Google Scholar]
- Matyjaszewski K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. 10.1021/ma3001719. [DOI] [Google Scholar]
- Matyjaszewski K.; Xia J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- Kamigaito M.; Ando T.; Sawamoto M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101, 3689–3745. 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- Limer A. J.; Haddleton D. M. Transition metal mediated living radical polymerisation. Prog. React. Kinet. Mech. 2004, 29, 187–241. 10.3184/007967404777726223. [DOI] [Google Scholar]
- Ouchi M.; Terashima T.; Sawamoto M. Transition Metal-Catalyzed Living Radical Polymerization: Toward Perfection in Catalysis and Precision Polymer Synthesis. Chem. Rev. 2009, 109, 4963–5050. 10.1021/cr900234b. [DOI] [PubMed] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379–410. 10.1071/CH05072. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process-A First Update. Aust. J. Chem. 2006, 59, 669–692. 10.1071/CH06250. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process - A Second Update. Aust. J. Chem. 2009, 62, 1402–1472. 10.1071/CH09311. [DOI] [Google Scholar]
- Perrier S.; Takolpuckdee P. Macromolecular design via reversible addition-fragmentation chain transfer (RAFT)/xanthates (MADIX) polymerization. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 5347–5393. 10.1002/pola.20986. [DOI] [Google Scholar]
- Hawker C. J.; Barclay G. C.; Orellana A.; Dao J.; Devonport W. Initiating systems for nitroxide-mediated living free radical polymerizations: synthesis and evaluation. Macromolecules 1996, 29, 5245–5254. 10.1021/ma951905d. [DOI] [Google Scholar]
- Tsarevsky N. V.; Matyjaszewski K. ″Green″ Atom Transfer Radical Polymerization: From Process Design to Preparation of Well-Defined Environmentally Friendly Polymeric Materials. Chem. Rev. 2007, 107, 2270–2299. 10.1021/cr050947p. [DOI] [PubMed] [Google Scholar]
- Delplace V.; Nicolas J. Degradable vinyl polymers for biomedical applications. Nat. Chem. 2015, 7, 771–784. 10.1038/nchem.2343. [DOI] [PubMed] [Google Scholar]
- Rikkou M. D.; Patrickios C. S. Polymers prepared using cleavable initiators: Synthesis, characterization and degradation. Prog. Polym. Sci. 2011, 36, 1079–1097. 10.1016/j.progpolymsci.2011.01.004. [DOI] [Google Scholar]
- Agarwal S. Chemistry, chances and limitations of the radical ring-opening polymerization of cyclic ketene acetals for the synthesis of degradable polyesters. Polym. Chem. 2010, 1, 953–964. 10.1039/c0py00040j. [DOI] [Google Scholar]
- Thioune O.; Fessi H.; Devissaguet J. P.; Puisieux F. Preparation of pseudolatex by nanoprecipitation: Influence of the solvent nature on intrinsic viscosity and interaction constant. Int. J. Pharm. 1997, 146, 233–238. 10.1016/S0378-5173(96)04830-2. [DOI] [Google Scholar]
- Beck-Broichsitter M.; Nicolas J.; Couvreur P. Solvent selection causes remarkable shifts of the ″Ouzo region″ for poly(lactide-co-glycolide) nanoparticles prepared by nanoprecipitation. Nanoscale 2015, 7, 9215–9221. 10.1039/C5NR01695A. [DOI] [PubMed] [Google Scholar]
- Tong R.; Yala L.; Fan T. M.; Cheng J. The formulation of aptamer-coated paclitaxel–polylactide nanoconjugates and their targeting to cancer cells. Biomaterials 2010, 31, 3043–3053. 10.1016/j.biomaterials.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong R.; Cheng J. Paclitaxel-Initiated, Controlled Polymerization of Lactide for the Formulation of Polymeric Nanoparticulate Delivery Vehicles. Angew. Chem., Int. Ed. 2008, 47, 4830–4834. 10.1002/anie.200800491. [DOI] [PubMed] [Google Scholar]
- Tong R.; Cheng J. Drug-Initiated, Controlled Ring-Opening Polymerization for the Synthesis of Polymer–Drug Conjugates. Macromolecules 2012, 45, 2225–2232. 10.1021/ma202581d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q.; Tong R.; Xu Y.; Baek K.; Dobrucki L. W.; Fan T. M.; Cheng J. Drug-Initiated Ring-Opening Polymerization of O-Carboxyanhydrides for the Preparation of Anticancer Drug–Poly(O-carboxyanhydride) Nanoconjugates. Biomacromolecules 2013, 14, 920–929. 10.1021/bm301999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong R.; Cheng J. Ring-Opening Polymerization-Mediated Controlled Formulation of Polylactide–Drug Nanoparticles. J. Am. Chem. Soc. 2009, 131, 4744–4754. 10.1021/ja8084675. [DOI] [PubMed] [Google Scholar]
- Tong R.; Cheng J. Controlled Synthesis of Camptothecin–Polylactide Conjugates and Nanoconjugates. Bioconjugate Chem. 2009, 21, 111–121. 10.1021/bc900356g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi J.; Tang L.; Moore R.; Tong R.; El Haddad N.; Akiyoshi T.; Mfarrej B.; Yang S.; Jurewicz M.; Ichimura T.; Lindeman N.; Cheng J.; Abdi R. Polylactide-cyclosporin A nanoparticles for targeted immunosuppression. FASEB J. 2010, 24, 3927–3938. 10.1096/fj.10-154690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain B. M.; Cheng M.; Moore D. R.; Ovitt T. M.; Lobkovsky E. B.; Coates G. W. Polymerization of Lactide with Zinc and Magnesium β-Diiminate Complexes: Stereocontrol and Mechanism. J. Am. Chem. Soc. 2001, 123, 3229–3238. 10.1021/ja003851f. [DOI] [PubMed] [Google Scholar]
- Harrisson S.; Maksimenko A.; Bui D. T.; Desmaële D.; Couvreur P.; Nicolas J., The Drug-Initiated Method: A Convenient Approach for the Synthesis of Efficient Polymer Prodrug Nanoparticles. In Controlled Radical Polymerization: Materials; American Chemical Society: 2015; Vol. 1188, pp 257–272. [Google Scholar]
- Chen X.; McRae S.; Parelkar S.; Emrick T. Polymeric Phosphorylcholine–Camptothecin Conjugates Prepared by Controlled Free Radical Polymerization and Click Chemistry. Bioconjugate Chem. 2009, 20, 2331–2341. 10.1021/bc900339x. [DOI] [PubMed] [Google Scholar]
- Williams C. C.; Thang S. H.; Hantke T.; Vogel U.; Seeberger P. H.; Tsanaktsidis J.; Lepenies B. RAFT-Derived Polymer–Drug Conjugates: Poly(hydroxypropyl methacrylamide) (HPMA)–7-Ethyl-10-hydroxycamptothecin (SN-38) Conjugates. ChemMedChem 2012, 7, 281–291. 10.1002/cmdc.201100456. [DOI] [PubMed] [Google Scholar]
- Lutz J.-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 3459–3470. 10.1002/pola.22706. [DOI] [Google Scholar]
- Nicolas J.; Mantovani G.; Haddleton D. M. Living radical polymerization as a tool for the synthesis of polymer-protein/peptide bioconjugates. Macromol. Rapid Commun. 2007, 28, 1083–1111. 10.1002/marc.200700112. [DOI] [Google Scholar]
- Le Droumaguet B.; Nicolas J. Recent advances in the design of bioconjugates from controlled/living radical polymerization. Polym. Chem. 2010, 1, 563–598. 10.1039/b9py00363k. [DOI] [Google Scholar]
- Pasut G.; Veronese F. M. Polymer–drug conjugation, recent achievements and general strategies. Prog. Polym. Sci. 2007, 32, 933–961. 10.1016/j.progpolymsci.2007.05.008. [DOI] [Google Scholar]
- Hertel L. W.; Boder G. B.; Kroin J. S.; Rinzel S. M.; Poore G. A.; Todd G. C.; Grindey G. B. Evaluation of the Antitumor Activity of Gemcitabine (2′,2′-Difluoro-2′-deoxycytidine). Cancer Res. 1990, 50, 4417–4422. [PubMed] [Google Scholar]
- Heinemann V.; Xu Y.-Z.; Chubb S.; Sen A.; Hertel L. W.; Grindey G. B.; Plunkett W. Cellular Elimination of 2′,2′-Difluorodeoxycytidine 5′-Triphosphate: A Mechanism of Self-Potentiation. Cancer Res. 1992, 52, 533–539. [PubMed] [Google Scholar]
- Bui D. T.; Maksimenko A.; Desmaele D.; Harrisson S.; Vauthier C.; Couvreur P.; Nicolas J. Polymer prodrug nanoparticles based on naturally occurring isoprenoid for anticancer therapy. Biomacromolecules 2013, 14, 2837–2847. 10.1021/bm400657g. [DOI] [PubMed] [Google Scholar]
- Abe I.; Rohmer M.; Prestwich G. D. Enzymatic cyclization of squalene and oxidosqualene to sterols and triterpenes. Chem. Rev. 1993, 93, 2189–2206. 10.1021/cr00022a009. [DOI] [Google Scholar]
- Bui D. T.; Nicolas J.; Maksimenko A.; Desmaele D.; Couvreur P. Multifunctional squalene-based prodrug nanoparticles for targeted cancer therapy. Chem. Commun. 2014, 50, 5336–5338. 10.1039/C3CC47427E. [DOI] [PubMed] [Google Scholar]
- Couvreur P.; Reddy L. H.; Mangenot S.; Poupaert J. H.; Desmaele D.; Lepetre-Mouelhi S.; Pili B.; Bourgaux C.; Amenitsch H.; Ollivon M. Discovery of new hexagonal supramolecular nanostructures formed by squalenoylation of an anticancer nucleoside analogue. Small 2008, 4, 247–253. 10.1002/smll.200700731. [DOI] [PubMed] [Google Scholar]
- Couvreur P.; Stella B.; Reddy L. H.; Hillaireau H.; Dubernet C.; Desmaële D.; Lepêtre-Mouelhi S.; Rocco F.; Dereuddre-Bosquet N.; Clayette P.; Rosilio V.; Marsaud V.; Renoir J.-M.; Cattel L. Squalenoyl Nanomedicines as Potential Therapeutics. Nano Lett. 2006, 6, 2544–2548. 10.1021/nl061942q. [DOI] [PubMed] [Google Scholar]
- Arias J. L.; Reddy L. H.; Othman M.; Gillet B.; Desmaële D.; Zouhiri F.; Dosio F.; Gref R.; Couvreur P. Squalene Based Nanocomposites: A New Platform for the Design of Multifunctional Pharmaceutical Theragnostics. ACS Nano 2011, 5, 1513–1521. 10.1021/nn1034197. [DOI] [PubMed] [Google Scholar]
- Kazatchkine M. D.; Carreno M. P. Activation of the complement system at the interface between blood and artificial surfaces. Biomaterials 1988, 9, 30–35. 10.1016/0142-9612(88)90066-X. [DOI] [PubMed] [Google Scholar]
- Maksimenko A.; Bui D. T.; Desmaële D.; Couvreur P.; Nicolas J. Significant Tumor Growth Inhibition from Naturally Occurring Lipid-Containing Polymer Prodrug Nanoparticles Obtained by the Drug-Initiated Method. Chem. Mater. 2014, 26, 3606–3609. 10.1021/cm501500k. [DOI] [Google Scholar]
- Harrisson S.; Nicolas J.; Maksimenko A.; Bui D. T.; Mougin J.; Couvreur P. Nanoparticles with In Vivo Anticancer Activity from Polymer Prodrug Amphiphiles Prepared by Living Radical Polymerization. Angew. Chem., Int. Ed. 2013, 52, 1678–1682. 10.1002/anie.201207297. [DOI] [PubMed] [Google Scholar]
- Harrisson S.; Couvreur P.; Nicolas J. SG1 Nitroxide-Mediated Polymerization of Isoprene: Alkoxyamine Structure/Control Relationship and α,ω-Chain-End Functionalization. Macromolecules 2011, 44, 9230–9238. 10.1021/ma202078q. [DOI] [Google Scholar]
- Harrisson S.; Couvreur P.; Nicolas J. Use of Solvent Effects to Improve Control over Nitroxide-Mediated Polymerization of Isoprene. Macromol. Rapid Commun. 2012, 33, 805–810. 10.1002/marc.201100866. [DOI] [PubMed] [Google Scholar]
- Chan J. M.; Zhang L.; Tong R.; Ghosh D.; Gao W.; Liao G.; Yuet K. P.; Gray D.; Rhee J.-W.; Cheng J.; Golomb G.; Libby P.; Langer R.; Farokhzad O. C. Spatiotemporal controlled delivery of nanoparticles to injured vasculature. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2213–2218. 10.1073/pnas.0914585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi N.; Williams D. F. Preparation of non-thrombogenic materials using 2-methacryloyloxyethyl phosphorylcholine. Biomaterials 2003, 24, 2431–2435. 10.1016/S0142-9612(03)00113-3. [DOI] [PubMed] [Google Scholar]
- Kopeček J.; Kopečková P. HPMA copolymers: Origins, early developments, present, and future. Adv. Drug Delivery Rev. 2010, 62, 122–149. 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawato Y.; Aonuma M.; Hirota Y.; Kuga H.; Sato K. Intracellular Roles of SN-38, a Metabolite of the Camptothecin Derivative CPT-11, in the Antitumor Effect of CPT-11. Cancer Res. 1991, 51, 4187–4191. [PubMed] [Google Scholar]
- Nicolas J.; San Miguel V.; Mantovani G.; Haddleton D. M. Fluorescently tagged polymer bioconjugates from protein derived macroinitiators. Chem. Commun. 2006, 4697–4699. 10.1039/b609935a. [DOI] [PubMed] [Google Scholar]
- Averick S.; Simakova A.; Park S.; Konkolewicz D.; Magenau A. J. D.; Mehl R. A.; Matyjaszewski K. ATRP under Biologically Relevant Conditions: Grafting from a Protein. ACS Macro Lett. 2011, 1, 6–10. 10.1021/mz200020c. [DOI] [PubMed] [Google Scholar]
- Heredia K. L.; Bontempo D.; Ly T.; Byers J. T.; Halstenberg S.; Maynard H. D. In Situ Preparation of Protein-″Smart″ Polymer Conjugates with Retention of Bioactivity. J. Am. Chem. Soc. 2005, 127, 16955–16960. 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]
- Boyer C.; Bulmus V.; Liu J.; Davis T. P.; Stenzel M. H.; Barner-Kowollik C. Well-defined protein-polymer conjugates via in situ RAFT polymerization. J. Am. Chem. Soc. 2007, 129, 7145–54. 10.1021/ja070956a. [DOI] [PubMed] [Google Scholar]
- De P.; Li M.; Gondi S. R.; Sumerlin B. S. Temperature-regulated activity of responsive polymer-protein conjugates prepared by grafting-from via RAFT polymerization. J. Am. Chem. Soc. 2008, 130, 11288–11289. 10.1021/ja804495v. [DOI] [PubMed] [Google Scholar]
- Delaittre G.; Nicolas J.; Lefay C.; Save M.; Charleux B. Surfactant-free synthesis of amphiphilic diblock copolymer nanoparticles via nitroxide-mediated emulsion polymerization. Chem. Commun. 2005, 614–616. 10.1039/b415959d. [DOI] [PubMed] [Google Scholar]
- Delaittre G.; Nicolas J.; Lefay C.; Save M.; Charleux B. Aqueous suspension of amphiphilic diblock copolymer nanoparticles prepared in situ from a water-soluble poly(sodium acrylate) alkoxyamine macroinitiator. Soft Matter 2006, 2, 223–231. 10.1039/b515267d. [DOI] [PubMed] [Google Scholar]
- Charleux B.; Delaittre G.; Rieger J.; D’Agosto F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45, 6753–6765. 10.1021/ma300713f. [DOI] [Google Scholar]
- Warren N. J.; Armes S. P. Polymerization-Induced Self-Assembly of Block Copolymer Nano-objects via RAFT Aqueous Dispersion Polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. 10.1021/ja502843f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groison E.; Brusseau S.; D’Agosto F.; Magnet S.; Inoubli R.; Couvreur L.; Charleux B. Well-Defined Amphiphilic Block Copolymer Nanoobjects via Nitroxide-Mediated Emulsion Polymerization. ACS Macro Lett. 2011, 1, 47–51. 10.1021/mz200035b. [DOI] [PubMed] [Google Scholar]