

Abstract
Background
Hypoxia-tolerant human glioma cells reduce oxygen consumption rate in response to oxygen deficit, a defense mechanism that contributes to survival under moderately hypoxic conditions. In contrast, hypoxia-sensitive cells lack this ability. As it has been previously shown that hypoxia-tolerant (M006x, M006xLo, M059K) and -sensitive (M010b) glioma cells express differences in mitochondrial function, we investigated whether mitochondrial DNA-encoded mutations are associated with differences in the initial response to oxygen deficit.
Results
The mitochondrial genome was sequenced and 23 mtDNA alterations were identified, one of which was an unreported mutation (T-C transition in base pair 14634) in the hypoxia-sensitive cell line, M010b, that resulted in a single amino acid change in the gene encoding the ND6 subunit of NADH:ubiquinone oxidoreductase (Complex I). The T14634C mutation did not abrogate ND6 protein expression, however, M010b cells were more resistant to rotenone, an agent used to screen for Complex I mutations, and adriamycin, an agent activated by redox cycling. The specific function of mtDNA-encoded, membrane-embedded Complex I ND subunits is not known at present. Current models suggest that the transmembrane arm of Complex I may serve as a conformationally driven proton channel. As cellular respiration is regulated, in part, by proton flux, we used homology-based modeling and computational molecular biology to predict the 3D structure of the wild type and mutated ND6 proteins. These models predict that the T14634C mutation alters the structure and orientation of the trans-membrane helices of the ND6 protein.
Conclusion
Complex I ND subunits are mutational hot spots in tumor mtDNA. Genetic changes that alter Complex I structure and function may alter a cell's ability to respond to oxygen deficit and consolidate hypoxia rescue mechanisms, and may contribute to resistance to chemotherapeutic agents that require redox cycling for activation.
Background
Human tumors frequently contain hypoxic cells that are more resistant to the killing effects of ionizing radiation and certain chemotherapeutic agents, are genetically unstable and metastasize frequently [1]. Further, hypoxic tumor sub-volumes are a negative prognostic indicator for local-regional control and progression-free survival in human tumors such as squamous cell carcinoma of the head and neck and uterine cervix [2,3]. Thus, hypoxic tumor cells are a source of local treatment failure and disease progression.
The mechanisms by which normal cells, tissues and organisms tolerate hypoxic stress are of great interest in comparative physiology and provide paradigms that can be tested in tumor model systems. While our current understanding of hypoxic stress defense mechanisms is incomplete, some general principles have emerged. Hypoxia-tolerant vertebrate species utilize a repertoire of molecular and biochemical strategies to adapt to and survive periods of oxygen limitation. The first of these is a metabolic or "defense" phase that is initiated within minutes of sensing an oxygen deficit, followed by an "adaptation" phase that proceeds over hours or days [4]. The metabolic changes that occur during the "defense" phase result in reversible suppression of oxygen consumption, significant down-regulation of ATP-consuming processes, and membrane channel arrest, which collectively result in conditions in which ATP supply and demand remain closely coupled. During the ensuing "adaptation" phase, a limited number of genes that encode for proteins whose function is to ensure survival under oxygen limiting conditions are selectively expressed. Transcriptional activation of genes such as VEGF and GLUT1 by hypoxia-inducible factor (HIF1) is the best understood example of the defense mechanisms utilized during this adaptation phase [5]. Thus, successful adaptation to hypoxia requires both metabolic changes that control bioenergetics and transcriptional activation of selected hypoxia rescue genes.
The presence of necrosis, and by inference, hypoxia, is a pathognomonic feature of human GBM tumors [6]. GBM model systems have been developed to investigate the mechanisms by which these tumor cells adapt to and survive under hypoxic conditions. A cardinal feature of hypoxia-tolerant GBM cells is their ability to significantly reduce their rate of oxygen consumption and preserve clonogenic potential when incubated under moderately hypoxic conditions [7]. In contrast, hypoxia-sensitive GBM cells maintain a steady oxygen consumption rate under oxygen limiting conditions, yet show a reduced clonogenic survival. In this regard, hypoxia-tolerant cells show several characteristics common to hypoxia-tolerant vertebrates as they have an effective oxygen sensing mechanism and the ability to consolidate hypoxia rescue mechanisms. Interestingly, hypoxia-tolerant and -sensitive GBM cells also display notable differences in mitochondrial function as exemplified by the ability of hypoxia-tolerant cells to stabilize mitochondria membrane potential and preserve intracellular ATP under hypoxic conditions and their relatively greater sensitivity to mitochondrial poisons [8]. These results are consistent with the hypothesis that control of respiration and energy production are essential hypoxia adaptation mechanisms in human tumors.
In eukaryotic cells, mitochondria are the site of energy production and ATP synthesis by oxidative phosphorylation (OXPHOS). The OXPHOS system consists of multiple carriers and molecules collectively referred to as the electron transport chain (ETC), including electron acceptors, coenzyme Q, cytochrome c, and five multiprotein complexes designated as Complexes I – V [9]. Oxidation of fuel molecules produces NADH and FADH2, and electrons generated from NADH/FADH2 are transferred between Complexes I – IV to molecular oxygen, the final electron acceptor. The transfer of electrons releases energy that is stored in the form of a proton gradient across the mitochondrial membrane, and this energy is used to produce ATP. Individual cells contain many mitochondria (e.g., ~1500 in a liver cell), and each mitochondrion has hundreds to thousands of DNA molecules. mtDNA is a circular, intronless molecule ~16.6 kb in size that encodes 13 polypeptide components of the ETC, 22 tRNAs and 2 rRNAs [10]. mtDNA has a higher mutation rate than nuclear DNA (nDNA) as the mtDNA molecule is not protected by histones, is exposed to reactive oxygen species generated during oxidative phosphorylation, and is replicated by DNA pol-γ that copies with low fidelity due to the absence of a proof-reading function [11]. Deletions or mutations in mtDNA underlie several complex human disorders [e.g., MELAS Syndrome, Leber's hereditary optic neuropathy (LHON)], the pathophysiology of which includes alterations in mitochondrial bioenergetics [10]. Among patients with mtDNA-associated syndromes, Complex I deficiency is also the most common disorder [12], and of the several documented mtDNA Complex I mutations, most are associated with LHON or LHON-associated syndromes [13,14].
Recently, human tumors of a variety of histopathological types, including GBM [11], have been reported to harbor mtDNA mutations [15,16]. In human tumor mtDNA, both the non-coding D-loop region and Complex I are mutational hotspots [15,17], with mutations that result in coding changes most frequently observed in Complex I [18,19]. Most human tumors show homoplasmic mutations, i.e., the same mutations are present in all mitochondria in all cells of the tumor [20]. It is well established that certain nuclear DNA mutations provide a selective advantage or are associated with tumor progression, thus, it is reasonable to postulate that similar principles may apply to mutations in mtDNA. At the present time, there is virtually no information about which, if any, mtDNA mutations are of functional importance in human tumors. Nonetheless, mtDNA mutations hold the promise of being useful clinical markers in the diagnosis and clinical management of cancer patients.
As previous results indicated that differences in mitochondrial function exist among hypoxia-tolerant and -sensitive human tumor cells, we investigated whether alterations to the mtDNA underlie differences in the metabolic response to hypoxia. We sequenced the entire mitochondrial genome in four GBM cell lines and identified several known polymorphisms and an unreported transition mutation (T14634C) in the hypoxia-sensitive cell line, M010b. The T14634C mutation results in a single amino acid (M64V) change in the gene encoding the ND6 subunit of Complex I. This mutation does not alter ND6 protein expression or mtDNA mass in M010b cells. However, Complex I function is altered as M010b cells are significantly more resistant to both rotenone [8], an agent used to screen Complex I mutations [21], and adriamycin, a chemotherapeutic drug activated by Complex I redox cycling [22]. Homology-based modelling and computational molecular biology was used to predict the 3D structures of the wild type and mutated ND6 proteins and interactions of their helices and amino acids. The T14634C mutation alters the structure and orientation of the trans-membrane helices of the ND6 subunit. The finding that mitochondrial membrane potential, which is a measure of total proton flux in the ETC, is dysregulated in M010b cells under hypoxic conditions [8] is consistent with this hypothesis and the modelling predictions. Thus, this mutation provides a link to the defective respiratory control in M010b cells in response to hypoxia as models suggest that mtDNA-encoded, membrane-embedded Complex I ND subunits function in proton translocation, and cellular respiration is regulated, in part, by proton flux. Hypoxia-sensitive M010b cells do not inhibit respiration in response hypoxic challenge. As this characteristic makes them vulnerable hypoxic stress, determination of the underlying mechanism may lead to the development of strategies that could be used to induce this condition in hypoxia-tolerant human tumor cells.
Results
Mutation Analysis
The sequence of the mitochondrial genome (13 coding genes plus the D-loop regulatory region) of hypoxia-tolerant (M006x, M006xLo, M059K) and -sensitive (M010b) GBM cells was determined using automated analysis and primers previously published [16]. Sequence alterations that differ from the Cambridge sequence were checked against databases of known sequence polymorphisms available on the MITOMAP website [23]. A total of 23 different sequence changes were identified, some of which were found in more than one cell line, for a total of 44 alterations (Table 1). Of these, 39 were base pair changes, and five were insertions of a single nucleotide. Of the 23 sequence alterations, 19 were previously reported polymorphisms, 1 was an unreported polymorphism that does not result in an amino acid change, 2 were previously unreported alterations in the non-coding D-loop, and 1 was an unreported mutation. This mutation in M010b cells was a T-C transition in base pair 14634 that results in a single amino acid change (met → val) in the gene encoding the ND6 subunit of NADH:ubiquinone oxidoreductase (OXPHOS Complex I).
Table 1.
mtDNA Sequence Analysis of Human GBM Cells. The mtDNA of hypoxia-tolerant (M006x, M006xLo, M059K) and hypoxia-sensitive (M010b) GBM cells was sequenced. Sequences that differ from the Cambridge sequence were checked against databases of known sequence polymorphisms available on the MITOMAP website [23]. A previously unreported mutation (T14634C) in the ND6 subunit of M010b cells was discovered. (insert C = a single "C" inserted into the D-loop poly-C tract; n.c. = no amino acid change; poly = polymorphism).
| Mutation | M006x | M006xLo | M059K | M010b | Region | Type | aa Change | Previously Reported |
| 64 | C-T | C-T | D-loop | transition | n.c. | Yes | ||
| 152 | T-C | D-loop | transition | H strand replication origin | Yes | |||
| 195 | T-C | D-loop | transition | n.c. | Yes | |||
| 263 | A-G | A-G | A-G | A-G | D-loop | transition | n.c. | Yes |
| 303–309 | insert C | insert C | D-loop | insertion | n.c. | Yes | ||
| 311–315 | insert C | insert C | insert C | D-loop | insertion | n.c. | Yes | |
| 320 | C – T | D-loop | transition | replication primer (n.c.) | No | |||
| 343 | C – G | C – G | D-loop | transversion | n.c. | No | ||
| 750 | A – G | A – G | 12S rRNA | transition | 12S rRNA | Yes | ||
| 4769 | A-G | A-G | A-G | ND2 | transition | met-met | Yes | |
| 8860 | A-G | A-G | A-G | A-G | ATPase6 | transition | thr-ala | Yes |
| 8919 | A-G | ATPase6 | transition | glu-glu | No | |||
| 11719 | G-A | ND4 | transition | gly-gly | Yes | |||
| 14470 | T-C | ND6 | transition | gly-gly | Yes | |||
| 14634 | T-C | ND6 | transition | met-val | No | |||
| 14798 | T-C | Cyt. B | transition | phe-leu | Yes | |||
| 15326 | A-G | A-G | A-G | A-G | Cyt. B | transition | thr-ala | Yes |
| 16129 | G-A | D-loop | transition | 7S rRNA (n.c.) | Yes | |||
| 16189 | T-C | T-C | D-loop | transition | n.c. | Yes | ||
| 16220 | A-C | D-loop | transition | n.c. | Yes | |||
| 16264 | C-T | D-loop | transition | n.c. | Yes | |||
| 16316 | A-G | D-loop | transition | n.c. | Yes | |||
| 16519 | T-C | T-C | T-C | T-C | D-loop | transition | n.c. | Yes |
mtDNA Copy Number
We used a PCR-based assay to test whether the ND6 mutation in M010b cells resulted in relatively increased mitochondrial mass [24]. The relative copy number of mtDNA in GBM cells lines and normal human fibroblasts was determined by generating standard curves to compare the amount of product amplified from a region of the mtDNA cytochrome b gene to that of an internal standard corresponding to products amplified from a segment of the nuclear DNA β-actin gene in the same DNA isolation (Fig. 1A). Results indicated that normal human fibroblasts have ~1000-fold greater number of mtDNA copies as compared to the diploid nuclear genome. The relative mtDNA content of GBM cells was also greater than that of their respective nuclear genomes, with ~20- to ~250-fold greater numbers of mtDNA copies. Of interest, the relative mtDNA copy number of M010b cells carrying the ND6 mutation was not significantly greater than that of other GBM cell lines with wild type ND6.
Figure 1.

mtDNA copy number and proteins in M010b cells. A, mtDNA copy number in GBM cell lines (M010b, M059K, M006x, M006xLo) and a normal fibroblast cell strain (GM38) by quantitative PCR. Graphical analyses of cytochrome b (■) and β-actin (▼) amplified from serially diluted template DNA. Standard curves were derived from three or more independent PCR experiments. Error bars show mean ± s.d. B, 35S-labeled mitochondrial translation products in M010b cells. The ND6 protein (indicated by the arrow) was identified on the basis of its relative position within the electrophoretic migration pattern of the 13 mtDNA-encoded proteins [25].
mtDNA Translation Products
As ND6 mutations have been reported previously to result in a near absence of the protein [21], we tested whether M010b cells with the T14634C ND6 mutation expressed ND6 protein. Mitochondrial translation products were labeled in vivo and separated using high-resolution polyacrylamide gel electrophoresis [25]. Identification of individual proteins according to their gel migrations pattern is facilitated by the fact that mtDNA has only 13 protein-coding genes. Analysis of newly synthesized mitochondrial proteins showed that the ND6 protein is expressed in M010b cells (Fig. 1B), suggesting that this mutation does not result in a truncated or unstable polypeptide.
Complex I Activity
A flow cytometric assay [26] was used to compare overall Complex I activity of normal human fibroblasts (GM38) and hypoxia-tolerant (M006x, M006xLo, M059K) and -sensitive (M010b) GBM cells. In this assay, cells with severe Complex I deficiency will show a significant reduction in the relative ratio of dihydroethidium (DE) fluorescence after treatment with rotenone when compared to normal controls. A one-way ANOVA test (Prism GraphPad data analysis software) was used to compare the results of these five groups of cells. The results shown in Fig. 2 indicate that there are no significant differences in overall Complex I activity when M10b cells are compared to normal fibroblasts or GBM cells lacking ND mutations (p < 0.051; R2 = 0.28).
Figure 2.

Complex I Activity. A flow cytometric assay of overall Complex I activity in normal human fibroblasts (GM38) and hypoxia-tolerant (M006x, M006xLo, M059K) and -sensitive (M010b) GBM cells. Changes in the relative ratio of dihydroethidium fluorescence after treatment of cells with rotenone are shown. Data shown for five or more separate experiments for each cell line. Error bars show mean ± s.d.
Cell Growth and Cytotoxicity
Cells with severe defects in oxidative phosphorylation fail to grow in vitro when glucose in the medium is replaced by galactose [27]. We used this screening system to test whether the ND6 mutation in M010b cells was associated with a significant respiratory chain defect. The growth of M010b cells in media containing glucose or galactose as substrates was analyzed using the nonlinear regression curve fit function for exponential growth provided by Prism GraphPad data analysis software (Fig. 3). The curve fit function predicted a doubling time of 5.9 and 9.0 days for glucose and galactose cultures, respectively. An F test was used to compare the curves and test whether they were different. The calculated p value was 0.012 from which we conclude that the glucose and galactose curves differ overall.
Figure 3.
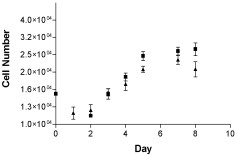
M010b growth assay. Cell growth assay for M010b cells grown for 1–8 days in DMEM/medium containing either 10 mM glucose (▲) or 5 mM galactose (■) as substrates. Data are from three independent experiments. Error bars show mean ± s.d.
We next tested whether the ND6 mutation was associated with altered sensitivity to adriamycin, a commonly used chemotherapeutic agent that requires redox activation for its cytotoxic effect. A MTT assay was used to compare the cytotoxic effect of adriamycin on M010b cells vs. GBM cells that show wild type ND6 (Fig. 4A). The data were analyzed using the paired t-test function of the StatView data analysis software program. M010b cells were significantly (p < 0.01) more resistant than M006x, M006xLo and M059K cells to the cytotoxic effects of adriamycin over the dose range of 0.5 – 50 μM.
Figure 4.

Adriamycin and TTFA cytotoxicity. Effects of adriamycin or TTFA on GBM cell survival. Serial dilutions of A., adriamycin, or B, TTFA were added to GBM cells growing in multi-well plates. Cell viability was determined 96 h later using a MTT assay. Error bars show mean ± s.d. from three or more independent experiments. When not visible, errors were smaller than the symbol. ▲, M010b; ■, M006x; ●; M006xLo; ◆, M059K.
Thenoyltrifluoroacetone (TTFA) is a mitochondrial Complex II inhibitor [28]. We reasoned that if the ND6 mutation in M010b cells resulted in compensatory up-regulation of Complex II activity, then M010b would be more sensitive to the cytotoxic effects of TTFA than GBM cells with wild type ND6. However, no significant difference in sensitivity to TTFA was observed among the GBM cell lines (Fig 4B), suggesting that M010b cells were not more reliant on electron transfer through Complex II.
HIF-1α Protein
The hypoxia-inducible transcription factor, HIF-1, is a heterodimer of HIF-1α and HIF-1β subunits. The HIF-1β subunit is constitutively expressed whereas the HIF-1α protein is rapidly degraded under normoxic conditions but is stable in hypoxia. This property suggests that HIF-1 may function as an oxygen sensor in coordinating transcriptional responses to hypoxic stress. We therefore tested whether the hypoxia-sensitive phenotype of M010b cells was, in part, a consequence of failure to stabilize HIF-1α under hypoxic conditions. Western blot analysis showed that HIF-1α protein stabilization in response to hypoxia is not affected by the ND6 mutation (Fig. 5). Under aerobic conditions, the HIF-1α protein is near absent in both hypoxia-tolerant (M006x; ND6 wild type) and -sensitive (M010b; ND6 mutant) cells. However, when cells were maintained under physiologically relevant hypoxic conditions (0.6% O2), HIF-1α protein was stabilized as indicated by detection of an band on western blot probed with an antibody to HIF-1α. As expected, re-oxygenation of previously hypoxic cells by transfer to atmospheric oxygen concentrations (~18% O2) for 1 h prior to protein harvest resulted in reduced HIF-1α protein levels as compared to that of fully hypoxic cells that were not re-oxygenated.
Figure 5.

HIF-1α protein expression. Western blot analysis of HIF-1α protein expression in hypoxia-tolerant (M006x) or hypoxia-sensitive (M010b) cells. Lanes 1,4, cells incubated (24 h) then lysed under ambient aerobic conditions (18% O2); Lanes 2,5, cells incubated in hypoxia (0.6% O2 × 24 h) followed by 1 h re-oxygenation prior to lysis; Lanes 3,6, cells incubated in hypoxia (0.6% O2 × 24 h) then lysed in an atmosphere of 1% O2.
Homology-Based Modeling of ND6 Protein
Homology-based modelling and computational chemistry were used to obtain a homology-based model of human ND6 and its mutations. (Additional file 1 provides details of methodology used to model the probable interactions and structure of key residues involved in the maintenance of the 3D structure of the human ND6 protein and its mutations. References to the GROMACS 3.0 modeling package are provided.) Prediction of the secondary structure of the human ND6 protein indicated six possible hydrophobic domains (putative hydrophobic TMHs) that connected to each other by five loops (two external loops and three internal loops) (Fig. 6A). This is consistent with models reported by others [29]. The first transmembrane helix, TMH I is the shortest helix and TMH V is the longest one. The first internal loop, IL I, (the longest extramembrane random coil) is divided into two anti-parallel beta-sheet structures by a short random coil. The interaction of ND6 with a small protein B18 (NADH-ubiqinone oxidoreductase B18 subunit) [30] may explain the short length of TMH I and special structure of IL I.
Figure 6.
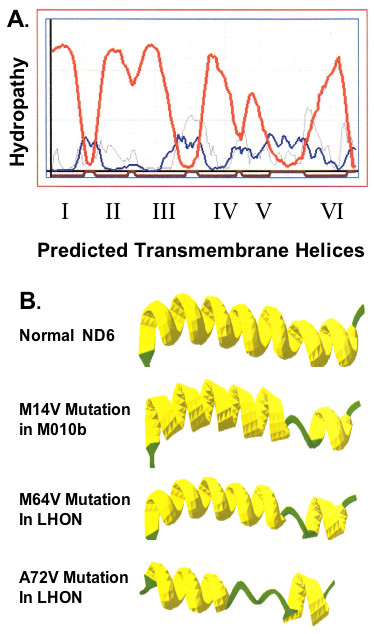
ND6 protein secondary structure. Predicted secondary structure for human ND6 protein. A, Hydropathy plot for the human ND6 protein. Red line: transmembrane helix preference; Blue line: beta-turn preference; Gray line: modified hydrophobic moment index; Dark red line: predicted transmembrane helix. B, The secondary structures predicted for the complete ND6 proteins. The models predict six hydrophobic domains (putative hydrophobic transmembrane helices) that are connected to each other by five loops (two external loops and three internal loops). The secondary structure of the normal protein is shown for reference; changes in secondary structure predicted to occur as a result of the mutation M14V in M010b cells; the M64V mutation in LHON and the A72V mutation in LHON are illustrated.
The secondary structures predicted for the complete normal and three mutated ND6 proteins are shown in Fig. 6B. (Additional file 2 provides the secondary structure predicted for the normal human ND6 protein. The positions of mutated amino acids are indicated. Additional file 3 shows helical wheel projection maps of the six transmembrane helices of the human ND6 protein. Each amino acid's position in the helix is indicated.)
To validate our computational approach, the same parameters were used to predict the structural consequences of two LHON mutations: 1) the M64V ND6 mutation that is associated with degree of visual loss, and 2) the A72V mutation that is more severe, leading to early onset blindness in all affected individuals. The multiple alignment results showed that the most important differences between family members occurred in TMH V and EL II. This conclusion is based on calculations of 1) the 3D-model of the human ND6 protein and phylogenetic relations between its close sequence homologues; 2) the residues' variety at each position of the multiple sequence alignment; and 3) the normalized conservation scores (NCS) or averaged conservation index, as shown in Additional file 4. (Additional file 4 also summarizes the hydrogen bonding pattern that directly affect the 3D structure of TMH V.) Based on the 3D-model of human ND6 and phylogenetic relations between its close sequence homologues, the residue's variety at each position of the multiple sequence alignment, and also the normalized conservation scores or the averaged conservation index, TMH V and the ELII have the least average conservation scores. Calculations of the non-covalent amino acid interactions and the interactions patterns of the 3D structures of the normal protein and its mutants showed that these two regions, as well as TMH I, BT I and TMH IV, likely have functionally important roles in the maintenance of secondary and tertiary structure of the protein and thus may directly influence protein function.
The refined 3D structures corresponding to the wild type ND6 protein, the M14V mutation in M010b cell, and the M64V and A72V mutations in LHON patients, are shown in Fig. 7. Superimposition of the normal ND6 protein and its pathogenic mutations showed disorientation and conformational changes in TMH V. The root mean square values of the superimposed 3D structures were calculated in order to quantitate these changes. For the mutated ND6 proteins, these calculations showed significant changes in the accessible surface area of the proteins as compared to normal. (Additional file 5 shows root mean square values of superimposed 3D structures of normal and mutated ND6 proteins.) Interestingly, these changes were greatest for the A72V mutation that is associated with the most clinically severe LHON phenotype.
Figure 7.
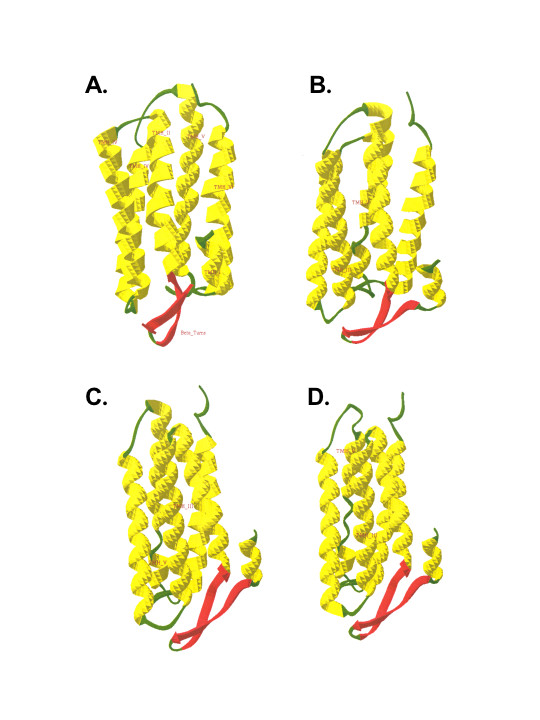
Homology-based modeling of ND6 proteins. Homology-based modeling of normal and mutated ND6 3D protein structures. By comparison to the predicted 3D structure of the normal ND6 protein, these models predict that the M14V mutation in M010b cells, the M64V mutation in LHON, and the A72V mutation in LHON would significantly disrupt the orientation of the entire ND6 protein within the mitochondrial membrane, and would alter the interactions of the individual helices of each protein.
Discussion
Tumor cells that survive moderately hypoxic microenvironments are a source of treatment resistance and tumor progression. Mitochondrial control of energy production is an important component of hypoxia defense mechanisms as the ability to match ATP supply and demand under oxygen limiting conditions is a general feature of hypoxia tolerant organisms [4]. Consistent with this, previous work has demonstrated that hypoxia-sensitive GBM cells lack the ability to stabilize mitochondria membrane potential and preserve intracellular ATP under hypoxic conditions [8]. Dysregulation of bioenergentics is also a hallmark of multi-system syndromes such as LHON or MELAS in which mtDNA is mutated. Based on this evidence, we wished to determine whether mtDNA mutations underlie the differential sensitivity to hypoxia previously observed in GBM cells. We sequenced the mitochondrial genome of four human GBM cells and identified a mutation (T14634C) in the ND6 subunit of NADH:ubiquinone oxidoreductase (Complex I) in the hypoxia-sensitive cell line, M010b. Search of databases available on the MitoMap website [23] indicate that this mutation that has not been previously reported.
Complex I is a large (~1 million Da) multimeric complex comprised of at least 43 subunits, 7 of which (ND1-6, and 4L) are encoded by the mtDNA. Complex I proteins assemble into an L-shaped structure with the hydrophobic ND1–ND6 sub-units spanning the inner mitochondrial membrane and the nuclear encoded proteins extending into the matrix space [31]. The transfer of electrons from NADH to ubiquinone is catalyzed by Complex I, and the free energy released by this process is used to pump protons across the inner mitochondrial membrane to the cytoplasm, thus establishing an electrochemical gradient across the membrane. The function of the individual Complex I subunits is largely unknown, however, the mtDNA-encoded, membrane-embedded subunits are postulated to be involved in proton translocation [21,29]. The specific roles and exact locations of the ND subunits within the membrane domain of Complex I are incompletely understood, however, biochemical studies suggest that the ND6 subunit is located at or near the quinone redox site within the hydrophobic transmembrane arm [29,32]. We previously used specific inhibitors of mitochondrial Complexes I, III and IV, together with electron transport chain uncouplers, to assess the activity of respiratory chain complexes in cells with (M010b) and without (M006x) the T14634C mutation. These results have been published [8] and showed that M010b cells are relatively resistant to rotenone, an agent used to screen for Complex I mutations, but are relatively sensitive to antimycin A, an inhibitor that binds to the ubiquinone reduction site in Complex III, and myxothiazol, an inhibitor of ubiquinol oxidation. Together, these results are of particular interest because they demonstrate that the ND6 mutation in M010b cells is associated with a common phenotype previously described for three primary ND6 mutations in LHON. In the case of the pathogenic LHON mutations, cells samples did not show overall differences in Complex I activity but did show Complex I defects that were detected as changes in sensitivity to specific effectors of Complex I quinone reactivity such as myxothiazol [29,33,34]. Although the function of Complex I ND subunits still remains unclear [35], together these findings suggest that the ND6 protein contributes to the quinone-binding site of Complex I [33]. They also highlight the fact that ND6 Complex I mutations are subtle and difficult to detect solely by monitoring overall differences in Complex I activity.
In rodent cells, ND6 mutations can result in absence of the cognate protein and failure to assemble the Complex I membrane arm [14,21]. In contrast, the 14484 ND6 mutation in a LHON patient is associated with expression of a mutated protein [33]. In the case of the T14634C mutation reported here, analysis of mtDNA translation products indicated that ND6 protein is present in M010b cells. We therefore assessed possible effects of this mutation on Complex I function by comparing the growth rate of M010b cells maintained in medium containing either glucose or galactose as carbon sources. This growth assay is a technique commonly employed to screen fibroblasts or lymphoblasts obtained from patients suspected to harbor respiratory chain defects [27]. When galactose replaces glucose in cell culture medium, cellular ATP is generated by mitochondrial oxidation of pyruvate. Under these conditions, cells with severe defects in mitochondrial metabolism die within the first day of growth in galactose-containing medium, whereas cells with mild defects become detached and die within 3–4 days [27]. In the case of M010b cells, the glucose vs. galactose growth curves do differ overall, however, the curves do not begin to diverge until day 5. This would suggest that the ND6 mutation confers a "subtle" Complex I defect. In non-malignant tissues, mutations in mtDNA-encoded Complex I ND subunits are known to confer a broad range of phenotyes [13]. In this regard, the phenotype associated with the T14634C mutation in M010b cells (mutated protein, subtle Complex I defect) is more similar to that observed in a LHON patient with a 14488 ND6 mutation [33] than it is to that observed in mutagenized rodent cells.
To further test the functional significance of the ND6 mutation, we treated GBM cells with a drug that targets Complex I. Adriamycin, a member of the anthracycline class of anticancer drugs, is activated in mitochondria by Complex I redox cycling between the quinone and the semiquinone, which in the presence of oxygen, generates ROS [22]. Tumor cells that lack Complex I activity as a consequence of mtDNA deletions are resistant to adriamycin [36]. In this study, we tested a range of adriamycin concentrations that included the peak plasma concentration (~2 μM) that that can be achieved after iv infusion [37]. M010b cells were significantly more resistant to adriamycin over a broad dose range (0.5 – 10 μM) when compared to other GBM cells with wild type mtDNA Complex I genes. Thus, Complex I mutations that frequently occur in human tumors have the potential to contribute to resistance to chemotherapeutic agents that require redox cycling for their activation.
Both Complex I and Complex II can transfer electrons to coenzyme Q, which then shuttles them to Complex III, ultimately resulting in the production of ATP. To test whether the ND6 mutation in Complex I resulted in a compensatory up-regulation of Complex II activity, we treated GBM cells with the Complex II inhibitor, TTFA. M010b cells were not differentially sensitive to TTFA, suggesting that the ND6 mutation does not elicit a greater reliance on Complex II activity for energy production. Similarly, Barrientos and Moraes [38] demonstrated that rotenone-induced Complex I deficiency did not increase Complex II protein expression in human osteosarcoma cells.
Mouse cells carrying ND6 mutations that result in complete absence of the protein show increased mtDNA copy number, a phenomenon believed to reflect compensatory mtDNA amplification [21]. Therefore, we used a PCR-based method to compare relative mtDNA copy number in M010b cells, three GBM cells that lack this mutation, and normal human fibroblasts. Normal fibroblasts show ~1000-fold greater mtDNA copy number as compared to nuclear DNA, a finding consistent with the fact that mammalian cells contain of the order of thousands of mtDNA molecules [24]. No evidence of increased mtDNA was observed in M010b cells as compared to other GBM cell lines. It is not possible to calculate total mtDNA copy number for the GBM cell lines as they are aneuploid and therefore may contain more than 2 copies of the nuclear β-actin gene that was used as an internal standard. Nonetheless, the finding the relative mtDNA copy number of M010b cells is similar to that of the other GBM cells is consistent with our other data that indicates the T14634C ND6 mutation confers a 'subtle' phenotype.
Analysis of Complex I function has been largely focused on electron transport activity, however, both bacterial and eukaryotic Complex I transmembrane subunits are suggested to function as a conserved, conformation-driven proton pump [39]. While details of the mechanisms regulating proton translocation remain controversial [40], metabolic control analyses indicate that cellular respiration is regulated, in part, by proton flux [41]. Recently, it has been shown that oxygen conformance (i.e., the ability to decrease oxygen consumption rate under hypoxic conditions) in hypoxia-tolerant cardiomyocytes is controlled, in part, by regulation of the rate of proton leak [42]. Hypoxia-tolerant GBM cells also show an oxygen conforming phenotype, whereas M010b cells do not [7]. Together, these finding suggested that changes in proton translocation arising from structural alterations in ND subunit proteins might modify the control of cellular respiration in response to hypoxic challenge. We used homology-based modeling and computational molecular biology to predict the 3D structure of the wild type and mutated ND6 proteins. Our calculations showed ND6 mutations confer potentially significant changes in the accessible surface area of the proteins and orientation of individual amino acids, the consequences of which are reflected in a secondary structure perturbation from helix to coil. Thus, the structural changes predicted to occur in the ND6 subunit as a result of the T14634C mutation may alter its proton conductance properties, and may ultimately contribute to the defective respiration control observed in M010b cells. However, verification of this hypothesis awaits a high resolution crystal structure of Complex I, or its individual ND subunits.
There is considerable interest in identifying the 'oxygen sensor' both in normal tissues systems as well as tumors. HIF-1 family of transcription factors up-regulates the expression of a repertoire of hypoxia rescue genes whose products permit cell survival under hypoxic condition [5]. Under aerobic conditions, the HIF-1α subunit is targeted for proteasomal degradation by the dioxygenase, prolyl-4-hydroxylase. In the absence of oxygen, prolyl-4-hydroxylase is inactive, the HIF-1α subunit is stabilized and subsequently activated by post-translational phosphorylation events [43]. HIF-1α protein is also stabilized in response to hormones, toxic metals and growth factors [44-46]. Although it has been postulated that hypoxia-independent stabilization of HIF-1α is mediated by production of ROS, not all studies support a role for ROS in this process [47]. As mtDNA mutations can alter ROS production, we tested whether M010b cells would show HIF-1α stabilization at low oxygen concentrations. The hypoxic sensitivity of M010b cells is manifest as a significant reduction in clonogenic cell survival under physiologically relevant hypoxic conditions [7]. Absence of HIF-1α stabilization, and subsequent failure to transcriptionally activate the expression of hypoxia rescue genes, would be consistent with this phenotype. However, our results showed that incubation of M010b cells in atmospheres of reduced oxygen tensions similar to those that can occur in human tumors resulted in stabilization of HIF-1α protein. This is consistent with previous findings that expression the HIF-1 target gene, VEGF, is up-regulated in M010b cells in response to hypoxia [7]. Together, these results suggest that HIF-1α stabilization is a necessary but not sufficient condition to ensure tumor cell survival in hypoxia. The model of hypoxia defense mechanisms formalized by Hochachka and colleagues [4] postulated that successful hypoxia adaptation requires both the ability to activate metabolic changes that link ATP demand to ATP supply and to up-regulate a repertoire of hypoxia defense genes, many of which are controlled at the transcriptional level by HIF-1. Although M010b cells can stabilize HIF-1α protein and trans-activate target genes under hypoxic conditions, we postulate that they remain sensitive to hypoxia due to an inability to modulate respiration and conserve intracellular ATP stores [8].
While it is attractive to suggest a genetic link between the Complex I ND6 T14634C mutation and the hypoxic sensitivity of M010b cell, it is inherently difficulty to assign a single phenotype to a given mtDNA mutation as study of mitochondrial diseases has shown that the same mtDNA mutation can result in variable phenotypic expression among different individuals or pedigrees [48]. As well, different mtDNA mutations can be associated with similar phenotypes [10]. For example, LHON is associated with mutations in ND1, ND4 and ND6 mtDNA genes [49]. We postulate that a similar principle may apply in the case of human tumors. Complex I is a mutational hotspot in human tumor mtDNA. Thus, the T14634C mutation reported here, or other ND subunit mutations the effect changes in protein structure, might be associated with or modulate tumor cell response to hypoxic stress.
Conclusions
We have identified a mutation (T14634C) in mtDNA-encoded Complex I ND6 subunit that is associated with a hypoxia-sensitive phenotype in the human glioma cell line, M010b. M010b cells show mild Complex I deficiency and resistance to adriamycin. Homology-based modelling predicts that this mutation disrupts the conformation of the ND6 protein. As Complex I mtNDA mutations are common among human tumors, further analysis of their role in tumor response to hypoxia would be of value.
Methods
Cell lines
The origin of the GBM cell lines used in this study has been previously described [50,51]. The M010b, M006 and M059K cell lines were derived directly from GBM tumor resections. The M006x cell line was derived from a M006 tumor xenograft. The M006xLo cell line was derived by incubating M006x cells at 0.6% oxygen for two weeks. GM38 cells are a normal human fibroblast strain. All cell lines were cultured in DMEM/F12 media supplemented with 10% fetal calf serum and 1% L-glutamine.
Muatation analysis
Total DNA was isolated from M006x, M006xLo, M059K and M010b cells using a standard phenol/chloroform extraction procedure. The mtDNA was amplified using seven different 1–3 kb overlapping fragments to eliminate the possibility of amplifying nuclear mitochondrial pseudogenes [16]. The 13 mitochondrial coding genes plus the D-loop regulatory region were sequenced using primers previously published [16] and an ABI Prism Big Dye II Terminator mix. The sequence of the GBM cell lines was compared to the normal "Cambridge sequence" and deviations from normal were analyzed using the MitoAnalyzer computer program provided on the Mitomap website [23]. Genetic alterations identified by sequencing were checked against databases of previously reported polymorphisms maintained on the Mitomap website. These were MITOMAP: MtDNA Coding Region Sequence Polymorphisms and MITOMAP: MtDNA Control Region (D-Loop) Sequence Polymorphisms. Complete references for all previously reported polymorphisms are provided by direct links from the databases. Mutations not previously reported were re-sequenced using different PCR fragments and sequencing primers [52] to confirm their authenticity.
Mitochondrial Translation Products
Mitochondrial protein synthesis was analyzed according to Chomyn [25]. Briefly, M010b cells (6 × 105) were seeded onto 10 cm plates, incubated for five days to 80–90% confluence, washed with methionine/cysteine-free DMEM and then incubated for 15 min in the same medium containing emetine (100 μg/ml), a cytoplasmic translation inhibitor. 35S-methionine/cysteine (7.15 mCi; >1000 Ci/mmol; Amersham) was then added (0.2 mCi/ml) and the cells incubated for an additional 2 h. The labelled cells were then washed, scraped off and lysed in 1% RIPA buffer (1% NP40, 0.5% deoxycholate, 0.1% SDS). Protein samples (10 μg) were electrophoresed through an SDS-polyacrylamide gel (10–20% gradient). The gel was then fixed, dried, and the electrophoretic pattern of mitochondrial proteins visualized using autoradiography film. The ND6 subunit was identified by its relative position within the migration pattern of the 13 mtDNA encoded proteins.
mtDNA Copy Number
mtDNA copy number was measured by a PCR-based assay that uses the mitochondrial cytochrome b gene as a marker of the entire mtDNA genome [24]. DNA was harvested from confluent cultures of M059K, M006x, M006xLo, M010b, and GM38 cells using the DNeasy tissue kit (Qiagen). The DNA was quantified by spectrophotometry. Serial dilutions (2-2–2-16) were made for each DNA sample, starting with 25 ng of DNA. A region of the mitochondrion-encoded cytochrome b gene was amplified from the serially diluted template DNA using primers F41 and R29 as described by Polyak et al. [16]. β-actin was amplified as a control, using the primers BA-67 and BA-68 as described by Horikoshi et al. [53]. PCR conditions were as follows: 1X PCR buffer (Amersham Biosciences), 0.34 mM MgCl2, 0.1 μM forward primer, 0.1 μM reverse primer, 0.4 mM dNTPs, and 0.02 units/μL Taq polymerase (Amersham Biosciences). The following cycling conditions were used: 95°C for 5 min, followed by 35 cycles of: 50 sec at 95°C, 50 sec at 57°C, 50 sec at 72°C, with a final extension of 5 min at 72°C. Electrophoresis of PCR products was performed on 1% agarose gels; PCR products were then stained with ethidium bromide. Quantitation was performed using the Typhoon 9400 imager and ImageQuant software (Amersham Pharmacia). Standard curves derived from three or more experiments were analyzed using the linear regression curve fit function provided by Prism GraphPad data analysis software.
Complex I Activity
A flow cytometric assay was used to screen for defects in overall Complex I activity [26]. Briefly, cells were grown DMEM/F12 culture medium supplemented with 10% fetal calf serum until plates reached ~90% confluency. Cells were then washed with PBS and placed in serum-free medium for 24 h. Cells were then washed with PBS, trypsinized, counted and divided into aliquots of 106 cells. Rotenone (20 μM) was then added for 30 min to inhibit respiratory chain activity. Control cells were incubated in PBS alone. Control and rotenone-treated cells were then washed, resuspended in PBS containing dihyroethidium (DE) (Molecular Probes, 10 μM final concentration), and incubated at room temperature for 15 min. Cells were then immediately analyzed by flow cytometry using a FACSort flow cytometer. At least 10,000 gated cells were counted and the mean FL3 fluoresence peak was calculated. Relative fluorescence ratios were calculated by dividing the mean level of FL3 fluoresence of DE plus rotenone treated cells by that of cells treated with DE alone. Results for each cell line were based on the mean ± s.d. of five or more separate experiments.
Cell Growth Assays
A technique described by Robinson [27] was used to screen M010b cells for respiratory chain defects. Briefly, cells (1.5 × 104/well) were seeded into six well tissue culture plates in DMEM medium containing either 10 mM glucose or 5 mM galactose as substrates. At daily intervals (day 1–8), replicate plates were trypsinized and cell counts determined using an electronic particle counter (Coulter). Growth curves determined for each cell line were based on the mean ± s.d. of three replicate experiments.
Cytotoxicity Assays
Cells were seeded in 96 well plates (2 × 104 cells/well) and allowed to adhere for 24 h. Serial dilutions of adriamycin (0.5 – 50 μM) or TTFA (25 – 5000 μM) were then added, cells returned to the incubator for 96 h after which the spent media was replaced with 200 μl of fresh media containing MTT (500 μg/ml) [54]. The cells were incubated for an additional 4 h, then lysed with 40% formalin/20% SDS, and viability was determined using a plate reader set to record absorbance at 600 – 660 nm.
HIF-1α Protein Expression
Preparation of whole cell lysates and Western blotting analysis were as previously described [55]. For this study, cells were seeded onto 60 mm glass plates (2 × 105/plate) in complete medium and incubated under normoxic conditions (18% O2; 5% CO2) in a standard tissue culture incubator, or in specially designed leak-proof aluminum chambers [56] that maintained an atmosphere of 0.6% O2; 5% CO2; balance N2 at 37°C. Aerobic cells were lysed 24 h later under ambient conditions. To maintain hypoxic conditions, cylinders containing the hypoxic cells were transferred to a specially designed glove box (PlasLabs) in which an atmosphere of 1% O2 was maintained. The cylinders were then opened, the plates removed and cells lysed while in an oxygen-reduced atmosphere. To simulate re-oxygenation, a second set of cylinders containing hypoxic cells was opened under ambient conditions and then placed in a standard incubator for 1 h prior to lysis under ambient conditions. HIF-1α protein expression was detected by chemiluminescence using a HIF-1α primary antibody (Novus Biologicals), followed by a horseradish peroxidase-conjugated secondary antibody.
Homology-Based Modeling of ND6
The refined three-dimensional structures of bacteriorhodopsin, as a template for modeling studies of membrane-bound proteins, were obtained from the Brookhaven Protein Databank [PDB Entry 1M0L [57] resolution of 1.47 Å and 1C3W [58], resolution of 1.55 Å]. The primary structure of the human NADH: ubiquinone oxidoreductase (Complex I) [59,60] and its mutations [61-67], were obtained from the Swiss-Prot databank (NU6M_HUMAN, Entry P03923).
The multiple alignment between NU6M family, extracted from the PSI-BLAST [68] output, was performed with the CLUSTAL W [69] method (with default parameters) as implemented in the ConSurf (surface-mapping of phylogenetic information using "Maximum Likelihood" paradigm) [70], and also with the Lipman-Pearson method [71], by using the Dayhoff Similarity Table [72] as implemented in the MULTALIN [73] software. We used several 'Gap Penalties' in the latter method to obtain an optimal alignment, and finally the comparisons were refined manually. In overall view, the alignments were generated on the basis of residue identity, homology parameters and conservation patterns [74,75].
Because of the location of the ND6 protein and its special topology, the prediction of the secondary structure and hydropathy plot of human ND6 was performed with several methods and scales [58,70,71,74-82], and finally with manual refinement with attention to overall topology and alignment result.
The MODELLER [83] software was used in order to obtain a homology-based model of human ND6 and its mutations from the bacteriorhodopsin structure by changing the side chains of those residues whose predicted secondary structures were similar to those of the corresponding residues of bacteriorhodopsin. The handling and visualization of the 3D molecular structure and the construction of missing H-atoms were accomplished with molecular modeling Swiss-PdbViewer [84] software. Details of the procedures used for molecular dynamics and molecular mechanics simulations [85,86] are provided in the supplementary material.
We studied the ND6 protein and eight pathogenic mutations in two forms: 1) only TMHs of the protein and 2) a complete model of the protein: N-terminal and C-Terminal, loops and TMHs. We constructed models for the following forms of ND6: 1) normal ND6 protein; 2) possible primary mutation (M14V) in M010b cells; 3) possible rare primary mutation (I26M) in LHON [61]; 4) possible rare primary mutation (G36S) in LHON [67]; 5) possible rare primary mutation (Y59C) in LHON [65]; 6) possible rare primary mutation (M64I) in LHON [64,66]; 7) primary mutation (M64V) in LHON characterized by visual loss in both eyes, from which ~50% of those affected recover visual function [64,66]; 8) primary rare and more severe mutation (A72V) in LHON characterized by no recovery from blindness [62]; and 9) possible primary mutation: (Y165C) [63]. From this framework and with attention to rapid structure generation and high-volume protein preparation in structural proteomics studies, we used HOME_MODELLER and HOME_OPTIMIZER, two simple and user-friendly Perl multi-optional command line scripts on the basis of MODELLER [83] and GROMACS [86] packages, prepared to accept primary sequence of a protein, as an input, and automatically produce fully refined and minimized 3D model(s) of protein, as an output.
Statistics
Statistical analyses were performed using StatView for the Mac (Abacus Concepts Inc.) or Prism GraphPad data analysis software.
Abbreviations
The abbreviations used are: GBM, glioblastoma multiforme; mtDNA, mitochondrial DNA; OXPHOS, oxidative phosphorylation; LHON, Leber's hereditary optic neuropathy; DMEM, Dulbecco's modified Eagle media; SDS, sodium dodecyl sulphate; DE, dihydroethidium; PBS, phosphate buffered saline; TTFA, thenoyltrifluoroacetone; MTT, methylthiazoletetrazolium; HIF, hypoxia-inducible factor; EL, external loop; IL, internal loop; TMH, transmembrane helix; BT, beta turn; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor.
Authors' contributions
CD carried out the mtDNA sequencing, the ND6 protein analysis, and wrote first draft of the manuscript. BH-N performed the computational molecular biology and homology-based modelling and contributed to writing of manuscript. EY the analyzed Complex I defect and performed cytotoxicity assays. NS determined the mtDNA mass. MP contributed to data analysis and manuscript writing. JA-T conceived of the study, participated in its design and coordination, and wrote the final manuscript. All authors read and approved the final manuscript.
Supplementary Material
Molecular Dynamics and Molecular Modeling
Secondary Structure Predictions
Helical Wheel Projection Maps of the Human ND6 Protein
3D Structure of the Human ND6 Protein
Root Mean Square Values of Superimposed 3D Structures
Acknowledgments
Acknowledgements
We thank Ivy Ma for excellent technical assistance. This work was supported by an award from the Canadian Institutes of Health Research (CIHR). EY was supported by an award from the Alberta Heritage Foundation for Medical Research. NS is supported by the Strategic Training Program in Translational Cancer Research, a partnership between CIHR, the Alberta Cancer Board and the National Cancer Institute of Canada.
Contributor Information
Carrie DeHaan, Email: cldehaan@unimelb.edu.au.
Bahram Habibi-Nazhad, Email: bhabibinazhad@yahoo.com.
Elizabeth Yan, Email: esparney@hotmail.com.
Nicole Salloum, Email: salloum@ualberta.ca.
Matthew Parliament, Email: matthewp@cancerboard.ab.ca.
Joan Allalunis-Turner, Email: joan.turner@ualberta.ca.
References
- Dachs GU, Chaplin DJ. Microenvironmental control of gene expression: implications for tumor angiogenesis, progression, and metastasis. Semin Radiat Oncol. 1998;8:208–216. doi: 10.1016/s1053-4296(98)80046-5. [DOI] [PubMed] [Google Scholar]
- Fyles A, Milosevic M, Hedley D, Pintilie M, Levin W, Manchul L, Hill RP. Tumor hypoxia has independent predictor impact only in patients with node-negative cervix cancer. J Clin Oncol. 2002;20:680–687. doi: 10.1200/JCO.20.3.680. [DOI] [PubMed] [Google Scholar]
- Nordsmark M, Overgaard J. A confirmatory prognostic study on oxygenation status and loco-regional control in advanced head and neck squamous cell carcinoma treated by radiation therapy. Radiother Oncol. 2000;57:39–43. doi: 10.1016/S0167-8140(00)00223-1. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/S0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- Wilden J, Moore I. Brain Oncology: Biology, Diagnosis and Therapy. Dordrecht, Martinus Nijhoff; 1987. Histological factors in the prognosis of malignant glioma; pp. 243–247. [Google Scholar]
- Allalunis-Turner MJ, Franko AJ, Parliament MB. Modulation of oxygen consumption rate and vascular endothelial growth factor mRNA expression in human malignant glioma cells by hypoxia. Br J Cancer. 1999;80:104–109. doi: 10.1038/sj.bjc.6690328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte ML, Parliament M, Franko A, Allalunis-Turner J. Variation in mitochondrial function in hypoxia-sensitive and hypoxia- tolerant human glioma cells. Br J Cancer. 2002;86:619–624. doi: 10.1038/sj.bjc.6600087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Kirches E, Krause G, Warich-Kirches M, Weis S, Schneider T, Meyer-Puttlitz B, Mawrin C, Dietzmann K. High frequency of mitochondrial DNA mutations in glioblastoma multiforme identified by direct sequence comparison to blood samples. Int J Cancer. 2001;93:534–538. doi: 10.1002/ijc.1375. [DOI] [PubMed] [Google Scholar]
- van den Heuvel L, Smeitink J. The oxidative phosphorylation (OXPHOS) system: nuclear genes and human genetic diseases. Bioessays. 2001;23:518–525. doi: 10.1002/bies.1071. [DOI] [PubMed] [Google Scholar]
- Schon EA. Mitochondrial genetics and disease. Trends Biochem Sci. 2000;25:555–560. doi: 10.1016/S0968-0004(00)01688-1. [DOI] [PubMed] [Google Scholar]
- Chomyn A. Mitochondrial genetic control of assembly and function of complex I in mammalian cells. J Bioenerg Biomembr. 2001;33:251–257. doi: 10.1023/A:1010791204961. [DOI] [PubMed] [Google Scholar]
- Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287:2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- Parrella P, Xiao Y, Fliss M, Sanchez-Cespedes M, Mazzarelli P, Rinaldi M, Nicol T, Gabrielson E, Cuomo C, Cohen D, Pandit S, Spencer M, Rabitti C, Fazio VM, Sidransky D. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res. 2001;61:7623–7626. [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Parrella P, Nomoto S, Cohen D, Xiao Y, Esteller M, Jeronimo C, Jordan RC, Nicol T, Koch WM, Schoenberg M, Mazzarelli P, Fazio VM, Sidransky D. Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res. 2001;61:7015–7019. [PubMed] [Google Scholar]
- Yeh JJ, Lunetta KL, van Orsouw NJ, Moore F. D. = Jr, Mutter GL, Vijg J, Dahia PL, Eng C. Somatic mitochondrial DNA (mtDNA) mutations in papillary thyroid carcinomas and differential mtDNA sequence variants in cases with thyroid tumours. Oncogene. 2000;19:2060–2066. doi: 10.1038/sj.onc.1203537. [DOI] [PubMed] [Google Scholar]
- Mondica-Napolitano JS, Singh KK. Mitochondria as targets for detection and treatment of cancer. Expert Reviews in Molecular Medicine. http://www-ermm.cbcu.cam.ac.uk/02004453h.htm [DOI] [PubMed]
- Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261:3060–3067. [PubMed] [Google Scholar]
- MitoMap http://www.mitomap.org
- Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31:e61. doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996;264:197–211. doi: 10.1016/S0076-6879(96)64020-8. [DOI] [PubMed] [Google Scholar]
- Setterfield K, WIlliams AJ, Donald J, Thorburn DR, Kirby DM, Trounce IA, Chriostodolou J. Flow cytometry in the study of mitochodrial respiratory chain disorders. Mitochondrion. 2002;1:437–445. doi: 10.1016/S1567-7249(02)00008-9. [DOI] [PubMed] [Google Scholar]
- Robinson BH. Use of fibroblast and lymphoblast cultures for detection of respiratory chain defects. Methods Enzymol. 1996;264:454–464. doi: 10.1016/S0076-6879(96)64041-5. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Fariss MW. Thenoyltrifluoroacetone, a potent inhibitor of carboxylesterase activity. Biochem Pharmacol. 2002;63:751–754. doi: 10.1016/S0006-2952(01)00871-1. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Brown DT, Andrews RM, Singh-Kler R, Riordan-Eva P, Lindley J, Applegarth DA, Turnbull DM, Howell N. The mitochondrial ND6 gene is a hot spot for mutations that cause Leber's hereditary optic neuropathy. Brain. 2001;124:209–218. doi: 10.1093/brain/124.1.209. [DOI] [PubMed] [Google Scholar]
- Robinson BH. Human complex I deficiency: clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochim Biophys Acta. 1998;1364:271–286. doi: 10.1016/S0005-2728(98)00033-4. [DOI] [PubMed] [Google Scholar]
- Hofhaus G, Weiss H, Leonard K. Electron microscopic analysis of the peripheral and membrane parts of mitochondrial NADH dehydrogenase (complex I) J Mol Biol. 1991;221:1027–1043. doi: 10.1016/0022-2836(91)80190-6. [DOI] [PubMed] [Google Scholar]
- Cardol P, Matagne RF, Remacle C. Impact of mutations affecting ND mitochondria-encoded subunits on the activity and assembly of complex I in Chlamydomonas. Implication for the structural organization of the enzyme. J Mol Biol. 2002;319:1211–1221. doi: 10.1016/S0022-2836(02)00407-2. [DOI] [PubMed] [Google Scholar]
- Carelli V, Ghelli A, Bucchi L, Montagna P, De Negri A, Leuzzi V, Carducci C, Lenaz G, Lugaresi E, Degli Esposti M. Biochemical features of mtDNA 14484 (ND6/M64V) point mutation associated with Leber's hereditary optic neuropathy. Ann Neurol. 1999;45:320–328. doi: 10.1002/1531-8249(199903)45:3<320::AID-ANA7>3.3.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Jun AS, Trounce IA, Brown MD, Shoffner JM, Wallace DC. Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystonia. Mol Cell Biol. 1996;16:771–777. doi: 10.1128/mcb.16.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Hu P, Park JS, Deng JH, Song X, Chomyn A, Yagi T, Attardi G. Genetic and functional analysis of mitochondrial DNA-encoded complex I genes. Ann N Y Acad Sci. 2004;1011:272–283. doi: 10.1196/annals.1293.026. [DOI] [PubMed] [Google Scholar]
- Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene. 1999;18:6641–6646. doi: 10.1038/sj.onc.1203056. [DOI] [PubMed] [Google Scholar]
- Ozols RF, Willson JK, Weltz MD, Grotzinger KR, Myers CE, Young RC. Inhibition of human ovarian cancer colony formation by adriamycin and its major metabolites. Cancer Res. 1980;40:4109–4112. [PubMed] [Google Scholar]
- Barrientos A, Moraes CT. Simultaneous transfer of mitochondrial DNA and single chromosomes in somatic cells: a novel approach for the study of defects in nuclear-mitochondrial communication. Hum Mol Genet. 1998;7:1801–1808. doi: 10.1093/hmg/7.11.1801. [DOI] [PubMed] [Google Scholar]
- Friedrich T. Complex I: a chimaera of a redox and conformation-driven proton pump? J Bioenerg Biomembr. 2001;33:169–177. doi: 10.1023/A:1010722717257. [DOI] [PubMed] [Google Scholar]
- Ludwig B, Bender E, Arnold S, Huttemann M, Lee I, Kadenbach B. Cytochrome C oxidase and the regulation of oxidative phosphorylation. Chembiochem. 2001;2:392–403. doi: 10.1002/1439-7633(20010601)2:6<392::AID-CBIC392>3.3.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Ainscow EK, Brand MD. Top-down control analysis of ATP turnover, glycolysis and oxidative phosphorylation in rat hepatocytes. Eur J Biochem. 1999;263:671–685. doi: 10.1046/j.1432-1327.1999.00534.x. [DOI] [PubMed] [Google Scholar]
- Casey TM, Pakay JL, Guppy M, Arthur PG. Hypoxia causes downregulation of protein and RNA synthesis in noncontracting Mammalian cardiomyocytes. Circ Res. 2002;90:777–783. doi: 10.1161/01.RES.0000015592.95986.03. [DOI] [PubMed] [Google Scholar]
- Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- Page EL, Robitaille GA, Pouyssegur J, Richard DE. Induction of hypoxia-inducible factor-1alpha by transcriptional and translational mechanisms. J Biol Chem. 2002;277:48403–48409. doi: 10.1074/jbc.M209114200. [DOI] [PubMed] [Google Scholar]
- Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem. 2000;275:26765–26771. doi: 10.1074/jbc.M003325200. [DOI] [PubMed] [Google Scholar]
- Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, Shi X, Jiang BH. Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J Biol Chem. 2002;277:31963–31971. doi: 10.1074/jbc.M200082200. [DOI] [PubMed] [Google Scholar]
- Salnikow K, Su W, Blagosklonny MV, Costa M. Carcinogenic metals induce hypoxia-inducible factor-stimulated transcription by reactive oxygen species-independent mechanism. Cancer Res. 2000;60:3375–3378. [PubMed] [Google Scholar]
- Grossman LI, Shoubridge EA. Mitochondrial genetics and human disease. Bioessays. 1996;18:983–991. doi: 10.1002/bies.950181208. [DOI] [PubMed] [Google Scholar]
- Valentino ML, Avoni P, Barboni P, Pallotti F, Rengo C, Torroni A, Bellan M, Baruzzi A, Carelli V. Mitochondrial DNA nucleotide changes C14482G and C14482A in the ND6 gene are pathogenic for Leber's hereditary optic neuropathy. Ann Neurol. 2002;51:774–778. doi: 10.1002/ana.10193. [DOI] [PubMed] [Google Scholar]
- Allalunis-Turner MJ, Barron GM, Day R. S. = 3d, Dobler K, Urtasun RC. Heterogeneity in response to treatment with buthionine sulfoximine or interferon in human malignant glioma cells. International Journal of Radiation Oncology, Biology, Physics. 1992;22:765–768. doi: 10.1016/0360-3016(92)90520-r. [DOI] [PubMed] [Google Scholar]
- Parliament MB, Franko AJ, Allalunis-Turner MJ, Mielke BW, Santos CL, Wolokoff BG, Mercer JR. Anomalous patterns of nitroimidazole binding adjacent to necrosis in human glioma xenografts: possible role of decreased oxygen consumption. British Journal of Cancer. 1997;75:311–318. doi: 10.1038/bjc.1997.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Taylor GA, Durham SE, Turnbull DM. The determination of complete human mitochondrial DNA sequences in single cells: implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res. 2001;29:E74–4. doi: 10.1093/nar/29.15.e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi T, Danenberg KD, Stadlbauer TH, Volkenandt M, Shea LC, Aigner K, Gustavsson B, Leichman L, Frosing R, Ray M, et al. Quantitation of thymidylate synthase, dihydrofolate reductase, and DT-diaphorase gene expression in human tumors using the polymerase chain reaction. Cancer Res. 1992;52:108–116. [PubMed] [Google Scholar]
- Campling BG, Pym J, Baker HM, Cole SP, Lam YM. Chemosensitivity testing of small cell lung cancer using the MTT assay. Br J Cancer. 1991;63:75–83. doi: 10.1038/bjc.1991.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania A, Urban S, Yan E, Hao C, Barron G, Allalunis-Turner J. Expression and localization of cyclin-dependent kinase 5 in apoptotic human glioma cells. Neuro-oncol. 2001;3:89–98. doi: 10.1215/S1522851700000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CJ, Howell RL, Biaglow JE. Ascorbate anion potentiates cytotoxicity of nitro-aromatic compounds under hypoxic and anoxic conditions. Br J Cancer. 1979;39:321–329. doi: 10.1038/bjc.1979.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert B, Cupp-Vickery J, Hornak V, Smith S, Lanyi J. Crystallographic structure of the K intermediate of bacteriorhodopsin: conservation of free energy after photoisomerization of the retinal. J Mol Biol. 2002;321:715–726. doi: 10.1016/S0022-2836(02)00681-2. [DOI] [PubMed] [Google Scholar]
- Juretic D, Jkeroncic A, Zucic D. Sequence analysis of membrane proteins with web server SPLIT. Croatica Chemica Acta. 1999;72:975–997. [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N. Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs. Proc Natl Acad Sci U S A. 1995;92:532–536. doi: 10.1073/pnas.92.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries DD, Went LN, Bruyn GW, Scholte HR, Hofstra RM, Bolhuis PA, van Oost BA. Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia. Am J Hum Genet. 1996;58:703–711. [PMC free article] [PubMed] [Google Scholar]
- Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci U S A. 1994;91:6206–6210. doi: 10.1073/pnas.91.13.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzuki S, Noer AS, Lertrit P, Thyagarajan D, Kapsa R, Utthanaphol P, Byrne E. Normal variants of human mitochondrial DNA and translation products: the building of a reference data base. Hum Genet. 1991;88:139–145. doi: 10.1007/BF00206061. [DOI] [PubMed] [Google Scholar]
- Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun. 1992;187:1551–1557. doi: 10.1016/0006-291x(92)90479-5. [DOI] [PubMed] [Google Scholar]
- Leo-Kottler B, Christ-Adler M, Baumann B, Zrenner E, Wissinger B. Leber's hereditary optic neuropathy: clinical and molecular genetic results obtained in a family with a new point mutation at nucleotide position 14498 in the ND 6 gene. Ger J Ophthalmol. 1996;5:233–240. [PubMed] [Google Scholar]
- Sudoyo H, Sitepu M, Malik S, Poesponegoro HD, Marzuki S. Leber's hereditary optic neuropathy in Indonesia: two families with the mtDNA 11778G>A and 14484T>C mutations. Hum Mutat. 1998;Suppl 1:S271–4. doi: 10.1002/humu.1380110186. [DOI] [PubMed] [Google Scholar]
- Besch D, Leo-Kottler B, Zrenner E, Wissinger B. Leber's hereditary optic neuropathy: clinical and molecular genetic findings in a patient with a new mutation in the ND6 gene. Graefes Arch Clin Exp Ophthalmol. 1999;237:745–752. doi: 10.1007/s004170050307. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos P, Rao JK. Prediction of protein structure. Methods Enzymol. 1986;130:185–207. doi: 10.1016/0076-6879(86)30012-0. [DOI] [PubMed] [Google Scholar]
- Lipman DJ, Pearson WR. Rapid and sensitive protein similarity searches. Science. 1985;227:1435–1441. doi: 10.1126/science.2983426. [DOI] [PubMed] [Google Scholar]
- Dayhoff HO, Schwartz RM, Orcutt BC. Atlas of Protein Sequence and Structure. Vol. 5. Washington DC, National Biomedical Research Foundation; A model of evolutionary change in proteins; pp. 345–352. [Google Scholar]
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Hulme EC, Birdsall MJM, Buckley MJ. Annual Review of Pharmacology and Toxicology. Vol. 30. Palo Alto, CA, Annual Reviews Inc.; 1990. Muscarinic receptor subtypes; pp. 633–673. [DOI] [PubMed] [Google Scholar]
- Eisenberg D, Schwartz E, Komarony M, Wall R. Normalized consensus hydrophobicity scale. Journal of Molecular Biology. 1984;179:125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- Roseman MA. Hydrophilicity of polar amino acid side-chains is markedly reduced by flanking peptide bonds. J Mol Biol. 1988;200:513–522. doi: 10.1016/0022-2836(88)90540-2. [DOI] [PubMed] [Google Scholar]
- Miyazawa S, Jernigan RL : Estimation of effective inter-residue contact energies from protein crystal structures: quasi-chemical approximation. . Macromolecules. 1985;18:34–552. [Google Scholar]
- Rost B, Fariselli P, Casadio R. Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci. 1996;5:1704–1718. doi: 10.1002/pro.5560050824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B, Sander C. Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins. 1994;19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C. Conservation and prediction of solvent accessibility in protein families. Proteins. 1994;20:216–226. doi: 10.1002/prot.340200303. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Journal of Molecular Modeling. 2001;7:306–317. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Molecular Dynamics and Molecular Modeling
Secondary Structure Predictions
Helical Wheel Projection Maps of the Human ND6 Protein
3D Structure of the Human ND6 Protein
Root Mean Square Values of Superimposed 3D Structures