

Abstract
Avian H7N9 influenza viruses are group 2 influenza A viruses that have been identified as the etiologic agent for a current major outbreak that began in China in 2013 and may pose a pandemic threat. Here, we examined the human H7-reactive antibody response in 75 recipients of a monovalent inactivated A/Shanghai/02/2013 H7N9 vaccine. After 2 doses of vaccine, the majority of donors had memory B cells that secreted IgGs specific for H7 HA, with dominant responses against single HA subtypes, although frequencies of H7-reactive B cells ranged widely between donors. We isolated 12 naturally occurring mAbs with low half-maximal effective concentrations for binding, 5 of which possessed neutralizing and HA-inhibiting activities. The 5 neutralizing mAbs exhibited narrow breadth of reactivity with influenza H7 strains. Epitope-mapping studies using neutralization escape mutant analysis, deuterium exchange mass spectrometry, and x-ray crystallography revealed that these neutralizing mAbs bind near the receptor-binding pocket on HA. All 5 neutralizing mAbs possessed low numbers of somatic mutations, suggesting the clones arose from naive B cells. The most potent mAb, H7.167, was tested as a prophylactic treatment in a mouse intranasal virus challenge study, and systemic administration of the mAb markedly reduced viral lung titers.
Introduction
Influenza type A viruses comprise a phylogenetically and antigenically diverse group of viruses that infect both human and animal populations. Influenza is a common cause of yearly epidemics and, less frequently, global pandemics in humans. The viral surface envelope protein HA is the principal target of neutralizing antibodies. HA has a conserved membrane-proximal stem region and a globular head region with a shallow receptor-binding pocket. The globular head region surrounding the receptor-binding pocket tolerates extensive sequence and structural variation, which allows constant antigenic drift of the viruses. The sequence and structure of the receptor-binding pocket determine the affinity for diverse sialic acid cellular receptors in birds and mammals, with a wide host range.
There are currently 18 known subtypes of influenza type A HA, which can be classified broadly into 2 groups according to their amino acid sequences and structural features (designated groups 1 and 2). Pandemics occurred in 1957, 1968, and 1981, caused by H2N2, H3N2, and H1N1 viruses, respectively. Seasonal H3 viruses have circulated in humans since 1968, and seasonal H1 viruses circulated in humans from the late 1970s until 2009, when a new H1 virus pandemic occurred. Influenza type A viruses from additional subtypes circulate in avian or animal populations, and several of these have caused sporadic outbreaks in humans, typically involving direct bird- or animal-to-human contact. Zoonotic influenza infections can be quite severe due to a lack of preexisting immunity in humans as well as properties intrinsic to some influenza viruses that can make them more pathogenic than circulating human viruses.
Since March 2013, novel H7N9 avian influenza viruses have caused a major outbreak in humans in China. As of March 2015, there have been 571 laboratory-confirmed human cases of H7N9, many of which were characterized by severe clinical course, including 212 deaths (1). In January 2015, health officials in British Columbia reported the first 2 cases of human infection in North America with avian influenza A (H7N9) virus in 2 travelers following a trip to Hong Kong and mainland China (1).
Although there appears to be a strong association of infection with direct exposure to live poultry markets, there are still many unknown features of H7N9 virus biology, including the natural animal reservoir, the main exposure source for humans, and the prevalence of the virus in bird or animal populations. Viral phylogenetic and genome-sequence analysis studies suggest that the H7N9 viruses infecting humans have a complex genetic background derived from avian influenza viruses, possibly from as many as 4 wild and domestic bird species (2–5). Sustained human-to-human transmission of H7N9 viruses has not been documented, although the WHO has identified 17 family clusters of infection involving 2 or more people to date (1).
H7N9 viruses are influenza A viruses classified in group 2, such as human H3N2 viruses that currently circulate in humans. However, H7N9 and H3N2 viruses are members of different viral clades; therefore, most of the human population is likely immunologically naive to H7 subtype viruses. While there is no evidence of sustained human-to-human transmission of H7N9 viruses, laboratory studies suggest that mutation of 2 to 3 amino acids within the receptor-binding pocket of HA is sufficient for increasing the affinity of avian viruses for binding to the mammalian type receptor (6). Recent studies in ferrets showed that circulating H7N9 viruses already have acquired the potential to spread by respiratory droplet transmission (7, 8), which may be a risk factor for establishing human-to-human transmission. These data suggest that H7N9 viruses could pose a human pandemic threat with relatively few mutations in HA. Therefore, it is necessary to test candidate H7N9 vaccines and to determine the molecular basis for human neutralizing antibody responses against H7N9 viruses. Results from a recent phase II double-blind clinical trial with 980 enrolled adults suggested that 2 doses of an experimental monovalent inactivated H7N9 vaccine are necessary to induce neutralizing antibody titers (9). Administration of vaccine with AS03 adjuvant induced higher antibody titers than administration of unadjuvanted or MF59-adjuvanted vaccine (9).
We studied human B cell responses to H7 HA in 75 individuals who participated in a clinical trial of an experimental monovalent vaccine candidate containing inactivated A/Shanghai/02/2013 (H7N9) antigens. Here, we assayed the frequency of H7 HA–specific EBV-transformed B cells before vaccination and 3 weeks after a second dose of vaccine. We also generated a panel of 20 human hybridomas secreting H7-specific mAbs from 12 different donors. The mAbs were characterized in detail, and 12 of the 20 were found to bind to H7 HA with half-maximal effective concentrations (EC50) of less than 0.5 μg/ml. Five of the mAbs possessed H7-specific neutralizing and hemagglutination inhibition (HAI) activities. These studies confirm that, while most donors were immunologically naive to H7 HA antigens prior to immunization, most were able to produce B cells secreting H7-specific IgGs after experimental vaccination with 2 doses of a conventional subunit H7 vaccine candidate. Sequence analysis of the antibody variable gene sequences encoding H7-reactive mAbs revealed that both primary H7-specific and H3-crossreactive memory B cell responses occurred following vaccination with H7 antigens.
Results
Memory B cell frequencies against diverse influenza HA antigens.
Peripheral blood mononuclear cells (PBMCs) were collected from donors who participated in a multicenter trial conducted by the NIH Vaccine Treatment and Evaluation Unit (VTEU) (DMID 13-0033). The study was a phase II human clinical trial with monovalent inactivated influenza A/Shanghai/02/2013, testing 10 different prime and boost regimens, with doses ranging from 3.75 to 45 μg of HA protein either without adjuvant or with MF59 or AS03 adjuvant. A total of 75 donors participated in this substudy, all of whom were recruited at Vanderbilt University.
We transformed cryopreserved PBMCs from day 0 (day of vaccination) or day 42 (21 days after the second dose of vaccine) with EBV. The number of cell clusters forming lymphoblastoid cell lines (LCLs) was determined, and supernatants were tested by ELISA for secretion of human IgG that bound to recombinant A/Shanghai/02/2013 H7 HA. Any well for which the binding of antibodies in supernatant was greater than 2 SDs above background was deemed positive, and the percentage of H7-reactive EBV-transformed B cells was calculated as the number of positive wells divided by the total number of LCLs in all wells, multiplied by 100. The limit of detection for frequency of H7-reactive B cells was 0.033%. On the day of the first vaccination, 27 of 75 donors had LCLs secreting IgG that bound to H7 HA, although the frequencies were low, with a median percentage below the limit of detection and a mean of 0.047% (Figure 1). The highest frequencies of H7-reactive B cells prior to vaccination were seen in 2 donors, who showed frequencies of 0.33% and 0.75%. At day 42, 61 of 75 donors (81%) had LCLs secreting IgG that bound H7 HA at frequencies greater than the limit of detection, with a range of 0% to 2.6% (Figure 1); the median frequency was 0.18%, and the mean was 0.48%. The H7-reactive LCL frequencies differed significantly before and after vaccination (P < 0.0001). Notably, the donor with the highest prevaccination H7-reactive B cell frequency had 0.75% H7-reactive B cells before vaccination, but only 0.66% after vaccination.
Figure 1. Frequency of influenza antigen–specific B cells in H7N9 vaccine recipients.

PBMCs from day 0 (day of vaccination) or day 42 (3 weeks after second vaccination) from 75 donors were transformed with EBV. LCLs were enumerated, and supernatants were assayed for the presence of IgG that bound A/Shanghai/02/2013 H7 HA. The percentage of H7-specific B cells of total transformable B cells was calculated by the number of wells with ODs greater than 2 SDs above background divided by the total number of LCLs times 100. Lines represent the means, and error bars represent SEM. P value was calculated using Wilcoxon test.
Five representative donors were chosen to screen for LCL IgG reactivity against group 1 and group 2 HAs. Donor 185 and 190 samples were chosen because they had B cell reactivity frequencies against A/Shanghai/02/2013 H7 HA greater than 1 SD above the mean. Donor 198 and 233 samples were examined because they had B cell reactivity frequencies against A/Shanghai/02/2013 H7 HA within 1 SD of the mean. A donor 208 sample was examined because this specimen exhibited a B cell H7 reactivity frequency below the mean, near the median value. The total number of wells with detectable reactivity against any HA ranged from 78 to 186, with most wells displaying reactivities against only 1 HA subtype (Figure 2). Wells with reactivity against 3 or 4 HAs occurred, but were rare, with the exception of donor 190, where 20 wells had IgG that bound H7, H3, and H1 HAs. The most common heterosubtypic binding pattern was H7-H3 cross reactivity. For three donors, 185, 190, and 233, their most frequent B cell reactivity was to H3 HAs. The other 2 donors, 198 and 208, had their most frequent B cell reactivity to H1 HAs. Thus, each of the tested donors exhibited either H1-reactive or H3-reactive dominant responses, despite their recent H7 vaccinations.
Figure 2. Circos plots and summary table representing number of B cell lines with reactivity for specified HAs in 5 donors.

Circos plots from 5 representative donors and a table summary of the results are shown. PBMCs from donors 185, 190, 198, 208, and 233 from day 42 were immortalized with EBV. LCL supernatants were screened by ELISA for the presence of IgGs that bound A/Shanghai/02/2013 or A/New York/55/2004 H7 HAs (H7), A/Hong Kong/1/1968 or A/Victoria/361/2011 H3 HAs (H3), A/California/2009 or A/Texas/36/1991 H1 HAs (H1), or A/turkey/Wisconsin/1/1966 or A/Hong Kong/1073/1999 H9 HA (H9). Each circle has 384 positions, with each representing 1 well of a 384-well plate. Each ring represents results from ELISA using 1 HA protein. The height of the bar represents OD405. Presence of IgG was defined as an OD of greater than 2 SDs above background. Black, gold, aqua or coral bars indicate that a well contained IgG that bound to 1, 2, 3, or 4 HA proteins, respectively. The table represents a summary of data from all 5 Circos plots. The numbers of wells that were reactive against 1 (black), 2 (gold), 3 (aqua), or all 4 (coral) HAs were counted and are indicated in the top portion of the table. The bottom portion of the table represents a tally of all wells with reactivity to any HA and shows the total number of positive wells out of 384 wells tested.
Human hybridoma–derived mAb binding to A/Shanghai/02/2013 H7 HA.
A representative selection of the LCLs described in Figure 1 was chosen for fusion, cell cloning, and cell culture expansion to generate human hybridoma cell lines secreting mAbs. Twenty hybridoma lines were generated from twelve donors, and then cells were cloned from each line by single-cell flow cytometric sorting, and mAbs from each clone were purified. Of the 20 mAbs, 11 had EC50 values for binding in ELISA to HA that were below 1 μg/ml (Table 1). mAbs H7.57, H7.169, and H7.134 all exhibited EC50 values in the ng/ml range. Each of the mAbs with low EC50 values was tested for HAI activity against Eurasian lineage H7N9 viruses, a Eurasian-lineage H7N3 avian virus, and a North American lineage H7N3 avian virus. The lowest concentration of mAbs tested was 2 μg/ml. Of 12 mAbs, 5 had HAI activity at concentrations lower than 2 μg/ml. All 5 mAbs with HAI activity were active against the A/Shanghai/02/2013 H7N9 vaccine strain, with H7.167 exhibiting particularly potent inhibiting activity with a half-maximal inhibitory concentration (IC50) of 31 ng/ml (Table 1). All 5 mAbs with HAI activity also had inhibiting activity against Eurasian-lineage H7N9 and H7N3 viruses and the North American–lineage H7N3 virus. Each of the mAbs also was tested for HAI activity against A/California/7/2009 (H1N1) and A/Victoria/361/2011 (H3N2) viruses, but activity was not detected for these viruses (data not shown).
Table 1. Binding, HAI, and microneutralization titers for 11 H7-reactive human mAbs with low EC50 values for binding to H7 HA protein.

Each of the mAbs also was tested in viral neutralization assays against the A/Shanghai/02/2013 H7N9 strain that was the antigenic basis for the vaccine. Each of the mAbs with HAI activity also exhibited neutralizing activity against the parental strain of virus, with H7.167 again being the most potent mAb (Table 1). One mAb, designated H7.96, exhibited potent neutralizing activity for A/Sydney/5/1997 (H3N2), even though it did not have HAI activity against any H7 viruses tested, suggesting it likely was derived from a memory B cell induced by prior H3 exposure.
Breadth of binding of H7-reactive mAbs with diverse influenza type A group 1 or group 2 HA molecules.
We tested the breadth of binding to diverse HA molecules by ELISA for the 11 H7-reactive mAbs with low EC50 values. ELISA plates were coated with 1 of 17 different recombinant influenza type A HA proteins from group 1 or 2 viruses. Each of the mAbs was diluted to 1 μg/ml for the binding assay, and the optical density at 405 nm (OD405) is shown in Figure 3, with darker orange representing a higher OD405 (i.e., greater magnitude of binding). The 5 mAbs with HAI activity (H7.169, H7.137, H7.57, H7.167, and H7.5) are indicated in bold and bound only to H7 HAs. Of the 5 HAI mAbs, 4 bound to 2 of 3 H7 HA proteins tested, whereas H7.5 bound to all 3 H7 HAs tested. Three mAbs with low EC50 values, H7.92, H7.3, and H7.141, which did not exhibit HAI activity, also bound only to H7 HAs (Figure 3). Two mAbs with low EC50 values, but that lacked HAI activity, H7.134 and H7.105, bound to most of the group 2 HAs tested, with the exception of A/Victoria/3/1975 H3 HA. One H7-reactive mAb, H7.96, bound to HAs from both group 1 and group 2 viruses.
Figure 3. Binding breadth of H7-reactive mAbs.

Binding of each of the 11 H7-reactive mAbs with low EC50 values or the mAb FI6v3 control IgG was tested by ELISA against a panel of recombinant HA molecules at a concentration of 1 μg/ml. Numbers show a mean of OD405 of 4 different wells. Cells are shaded in orange according to OD reading, with higher readings shown as darker. The 5 mAbs previously found to have HAI activity are indicated in bold font.
Epitope mapping of 5 mAbs with HAI activity.
We used competition-binding studies to determine how many major antigenic sites were recognized by mAbs with HAI activity in this panel. We used a real-time biosensor to determine whether 2 antibodies could bind HA antigen successively. Briefly, His-tagged HA antigen was loaded onto Ni-NTA tips on an instrument to measure biolayer interferometry; then tips were dipped into 2 different mAbs in succession. If the first antibody in suspension blocked subsequent binding of antigen by a second antibody in suspension to antigen on the tip, these mAbs were considered to be part of the same competition-binding group. In Figure 4A, the primary mAb applied is listed in the left column and the second antibody applied is listed along the top row. The maximum signal from binding of the primary (uncompeted) antibody to antigen-coated tips was determined. The signal from binding of the same antibody applied as a secondary (competed) antibody was determined and calculated as a percentage of uncompeted binding. Numbers in the boxes in Figure 4A indicate percentage of uncompeted binding. A primary antibody was defined as fully blocking if the secondary mAb bound with less than 10% of the maximum uncompeted signal, intermediate in blocking if the signal was between 10% and 49% of the maximum uncompeted signal, and nonblocking if the signal was 50% or greater than the maximum uncompeted signal. mAbs with HAI activity are listed in bold. The 5 mAbs with HAI activity fell into 2 overlapping competition-binding groups, as indicated by the orange and blue boxes (Figure 4A). One additional clone, the mAb H7.92, which did not exhibit HAI activity, fell into a competition-binding group with mAbs that did possess HAI activity, suggesting it binds near the receptor-binding pocket, but not in a manner that inhibits virus-receptor interactions. Two other mAbs, H7.3 and H7.105, did not exhibit HAI activity at concentrations below 2 μg/ml. These mAbs fell into their own competition-binding groups, suggesting they bind HA in a region not adjacent to the receptor-binding pocket.
Figure 4. Epitope mapping of H7-reactive mAbs.

(A) Competition-binding assays were used to bin mAbs into groups that bound the same antigenic regions. Using biolayer interferometry, His-tagged trimeric A/Shanghai/2/2013 H7 HA0 protein was immobilized on Ni-NTA biosensor tips. Two different IgGs then were loaded successively to determine whether the first mAb blocked binding of the second mAb to HA. The first mAb added is listed in the vertical column, and the second mAb added is listed in the horizontal row. The maximal signal of each mAb alone was calculated. The signal of each second mAb was calculated in comparison with that of the mAb alone. mAbs listed in bold font are the mAbs with HAI activity. mAbs in the same competition-binding groups are highlighted in orange and blue boxes. (B) Regions of mAb contact are mapped onto the surface of A/Shanghai/02/2013 H7 HA, as determined by sequence analysis of antibody escape mutant viruses (magenta) or DXMS experiments (blue). The side view is shown for H7.169, H7.137, H7.57, and H7.167. The top view is shown for H7.5. Escape mutant virus HA altered residues are listed below the mAb name in magenta.
We mapped the footprint of the 5 HAI mAbs on HA using 2 different experimental methods: deuterium exchange mass spectrometry (DXMS) and the generation and analysis of antibody neutralization escape mutant viruses. Recombinant A/Shanghai/02/2013 H7 head domain protein was labeled with deuterated water in the presence or absence of H7.169, H7.137, H7.57, H7.167, or H7.5 antibodies. H7 head domain protein was digested with pepsin, and deuterium labeling of the resulting peptides was measured by mass spectrometry (MS). A decrease in deuterium binding suggested residues in an HA1 peptide that were in contact with a mAb. The peptides that exhibited decreased deuterium labeling when incubated with mAbs are indicated in bold (Figure 4B).
Antibody neutralization escape mutant viruses were generated for mAbs H7.169, H7.137, H7.167, and H7.5, and the variant residues in HA of those mutants are shown in magenta on the surface of H7 HA (Figure 4B). Passaging virus in the presence of H7.169 or H7.137 resulted in S143L or A149D mutations (H3 numbering), respectively, in the classic HA antigen 130-loop. mAb H7.167, the most potent mAb with HAI activity, selected for a variant with 2 mutations N157D and N158BD, which are in the HA antigen 150-loop. mAb H7.5 selected a virus with a mutation of A250T, which is not in a classic antigenic loop, but this mutation is predicted to add a glycosylation site to the HA. The location of each of these antibody escape virus mutations was consistent with the epitope-mapping findings in the DXMS and competition-binding data (Figure 4B).
H7.167 binds a highly conserved epitope adjacent to the receptor-binding site of H7 HA.
To investigate the structural basis of H7 neutralization by H7.167, we determined the crystal structure of H7.167 Fab in complex with the H7N9 A/Shanghai/02/2013 (Sh2/H7) HA at 4.65 Å resolution, with good refinement statistics at this resolution, in part due to high-quality molecular replacement models (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI85317DS1). The asymmetric unit consists of an HA protomer liganded by 1 Fab, which bound adjacent to the receptor-binding site (RBS) (Figure 5, A and B). Despite the low resolution, the main chains of each component, especially the interface between HA and Fab, were observed clearly and could be traced in the electron density (Supplemental Figure 1). On average, a total of 1,138 Å2 was buried at the interface (551 Å2 on HA and 587 Å2 on the Fab), where the heavy and light chains contributed to 53% and 47% of the Fab buried surface area, respectively. These findings are consistent with the observation that H7.167 exhibits potent HAI activity (Table 1). Moreover, the variable domain of the heavy chain showed obvious clashes when human receptor analog LSTc was docked into the RBS of H7 HA (Figure 5B). H7.167 targets a highly conserved epitope across H7 HAs using all of the complementarity-determining regions (CDRs) except LCDR2 (Figure 5C and Figure 6B) through a series of hydrogen bonds and van der Waals interactions. At this resolution, the heavy chain appears to bind to numerous residues, including R131 and N157 to N158B close to the 150-loop, 130-loop, and 190-helix, which constitute the RBS, while the light chain appears to bind additional residues, including F161, S187, K200, and N248, which are distant from the RBS (Figure 6A). The contacting residues in H7 HA, including the insertion of D158A and N158B in the 150-loop, differ remarkably from the corresponding residues in H1/H2/H3/H5 HAs (Supplemental Table 2). Moreover, the 150-loop has a different conformation in H7 HA compared with other subtype HAs (Supplemental Figure 2), where N157 and N158B are key residues responsible for the neutralization activity of the antibody (Supplemental Figure 3). These observations are consistent with the finding that H7.167 inhibitory activity is specific to the H7 subtype.
Figure 5. The antibody H7.167 binds a conserved epitope adjacent to the RBS of H7 HA.
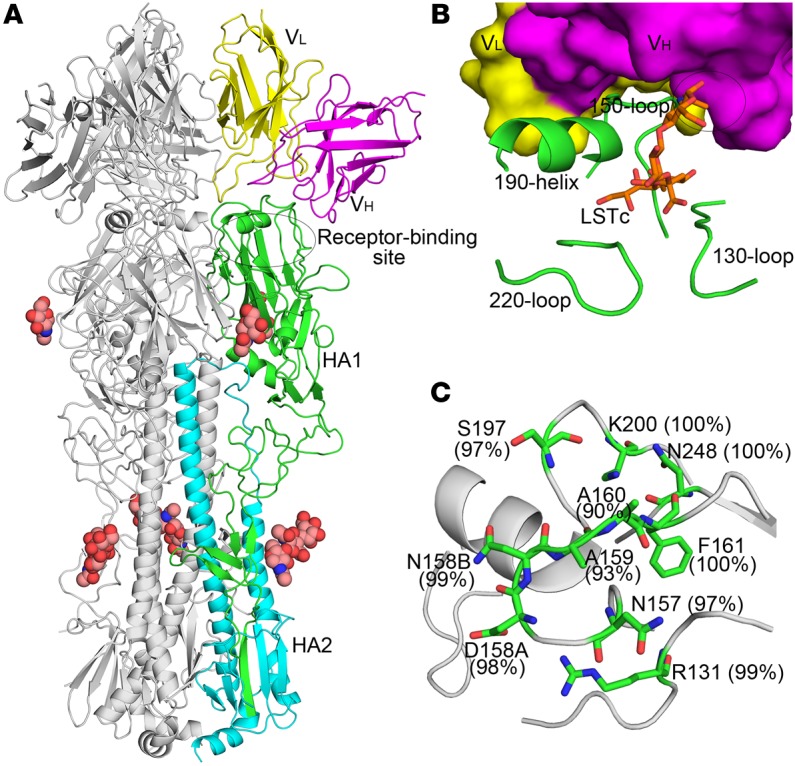
(A) Crystal structure of H7N9 (A/Shanghai/2/2013, abbreviated as Sh2) HA in complex with H7.167 Fab. One HA/Fab protomer of the trimeric complex is colored with HA1 in green, HA2 in cyan, the Fab heavy chain in magenta, and the Fab light chain in yellow. Only the Fv of the Fab is shown. The other 2 protomers are colored gray. Glycans are shown as spheres (carbon in light pink, oxygen in red, and nitrogen in blue). The constant domain of the Fab cannot be modeled due to disorder that arises from flexibility about the Fab elbow region. (B) Close-up view of the interaction between H7.167 and Sh2/H7 HA. The coloring is similar to that shown in A, but H7.167 is shown in a surface representation. The human receptor analog LSTc (orange sticks) was docked into the RBS of the complex by its superimposition with human receptor analog from human H7 HA–LSTc complex (PDB ID: 4BSE). The RBS elements (130-loop, 150-loop, 190-helix, and 220-loop) are also labeled. The variable domain of the heavy chain (VH) shows an obvious clash (labeled as an ellipse) with 1 glycan of LSTc. (C) Sequence conservation of the H7.167 footprint across H7 viruses. HA1 is shown in gray, and the epitope residues are shown as green sticks. The weighted percentage of identity of each residue with the H7 consensus sequence is indicated.
Figure 6. Interaction of H7.167 with Sh2/H7 illustrating the neutralizing epitope.

(A) Surface presentation of H7.167 footprint (green) in Sh2/H7 HA (gray) highlighting antibody residues contacting HA. The relative location of RBS is labeled. (B) The electrostatic potential surface of HA is illustrated (red, negative, –5.4 kT; blue, positive, +5.4 kT; white, neutral) in the same orientation as in A, with the H7.167 contacting residues in the 5 CDRs that interact with HA represented as sticks and color coded by CDR. H7.167 binds HA with both chains, using 5 of the 6 CDRs (LCDR2 makes no contacts) of the heavy and light chains in recognition (magenta and yellow, respectively).
In vivo efficacy of H7-specific antibodies.
H7.167 or a similarly prepared irrelevant human mAb (BDBV280, which is specific for Bundibugyo virus glycoprotein) were injected i.p. into mice 1 day prior to i.n. challenge with A/Shanghai/2/2017 H7-PR8 reassorted virus. Animals were administered 5 mg/kg or 1.65 mg/kg of mAb. Each experimental group had 11 mice. Three mice from each group were euthanized at 3 or 5 days after virus inoculation, and lung viral titers were assayed. Five mice from each group were weighed daily. Mice injected with the irrelevant mAb had 3.6 × 106 or 8.9 × 105 PFU/g lung tissue at 3 or 5 days after inoculation, respectively (Figure 7A). At 3 or 5 days after inoculation, mice who had received 1.65 mg/kg of mAb H7.167 had 4.4 × 103 or 3.2 × 103 PFU/g lung tissue respectively, an approximately 1,000-fold (3.0 or 2.4 log10 PFU/g) reduction in virus replication in lung tissue (Figure 7A). Mice who received 5 mg/kg of mAb H7.167 1 day prior to virus inoculation had an approximately 10,000-fold reduction of replication (mean of 510 or 560 PFU/g lung tissue at 3 or 5 days after inoculation, respectively). Only mice that were injected with 5 mg/kg BDBV280 lost weight during the experiment (Figure 7B).
Figure 7. Viral lung titers and weight from mice treated with H7.167.

Mice were injected with an irrelevant mAb, BDBV280, or H7.167 at 1.65 mg/kg or 5 mg/kg i.p. 1 day prior to viral inoculation. Mice were anesthetized and inoculated with 10,000 PFU H7-PR8 reassortant virus i.n. and were euthanized on day 3 (n = 3) or 5 (n = 3) to measure the presence of virus in lung tissue (A) or weighed every day for 2 weeks (n = 5) (B). P value was calculated using an unpaired t test with equal SD.
Antibody variable genes encoding H7N9-reactive mAbs are unique and comparatively unmutated.
We determined the antibody isotype and the nucleotide sequence of the antibody variable gene regions of each of the H7-reactive mAbs. All antibodies were of the IgG1 subclass (data not shown) and were encoded by distinct antibody genes (Table 2). There were no duplicated HCDR3 sequences, indicating the antibodies represented independently derived clones. The mAbs with HAI activity are indicated in bold. All mAbs with HAI activity exhibited very low mutation frequencies in their antibody variable gene sequences. A direct comparison of the mutation frequency in variable genes of H7-reactive mAbs with or without HAI activity showed a significant difference (Figure 8A). The somatic mutations identified included both silent and coding mutations. We examined the number of amino acid mutations in both heavy and light chains by comparing the sequences to (a) inferred germline genes of the H7-specific mAbs, (b) H7-reactive mAbs with heterosubtypic binding profiles, (c) previously published H5-specific or H1-specific mAbs, or (d) broadly neutralizing influenza mAbs. The H7-specific mAbs were significantly less mutated than H1-specific, broadly neutralizing, or H7-reactive mAbs with heterosubtypic binding profiles (Figure 8B).
Table 2. Antibody variable genes encoding 11 H7-reactive human mAbs.

Figure 8. Mutation frequency of influenza mAbs.

(A) Antibody variable gene sequences were determined and analyzed using the IMGT analysis tool. The percentage of identity of antibody heavy-chain variable genes as compared with inferred germline genes is shown for H7-specific mAbs with or without HAI activity. Nucleotide mutation frequencies are shown and represent both coding and noncoding mutations. Lines represent the means, and error bars represent SEM. P values were calculated using the Mann-Whitney U test. (B) The total number of amino acid mutations in heavy and light chains as compared with inferred germline genes, as determined by IMGT analysis. Genes encoding H7-specific or H7 mAbs with heterosubtypic binding activity from this manuscript were compared with sequences of previously published H5-specific, H1-specific, or broadly neutralizing influenza mAbs. The lines represent the means, and error bars represent SEM. P values were calculated using the Mann-Whitney U test.
Discussion
Here, we report the isolation of naturally occurring human mAbs induced by H7 influenza immunization and their mechanism of action. A number of these antibodies exhibited remarkable neutralizing potency, with neutralizing activity as low as an IC50 of 16 ng/ml (H7.167). Such antibodies have promise as prophylactic or therapeutic biological drugs for management of H7 virus exposure or infection. Moreover, the identification of the molecular and structural features of the epitopes recognized by these mAbs reveals critical aspects of the structural determinants of neutralization for H7 viruses.
The magnitude of human B cell responses to experimental immunization with novel avian influenza HA–based vaccine candidates is low, and the associated serum antibody responses are of poor quality. The molecular and cellular basis for the poor immunogenicity of avian influenza virus vaccines in humans is poorly understood. In general, it has proven remarkably difficult to induce high-quality neutralizing antibody responses to avian-origin influenza HA vaccines in humans without large protein doses or use of new adjuvants (10–12). It has not been clear whether or not these HA molecules possess some antigenic features associated with avian host adaptation that render them particularly lacking in immunogenicity for the human immune system. Atomic resolution structures of representative H5, H7, and H9 HA molecules have been determined (6, 12, 13) and reveal minor differences in the surface structures of the HAs, but no apparent major structural features that account for the markedly low immunogenicity of these HA antigens in humans. The work reported here reveals that humans vaccinated with a conventionally prepared H7 subunit vaccine are capable of generating B cells encoding antibodies with low EC50 values for binding and potent neutralizing activity against H7 influenza viruses. Using diverse epitope-mapping techniques, including sequence analysis of antibody neutralization escape mutant viruses, hydrogen-DXMS, HA-binding studies, and x-ray crystallography, we determined the molecular basis of recognition of H7 HA in these donors. Members of the mAb panel described here recognized diverse features of the HA, including the major antigenic 130- and 150-loops within the globular head as well as an epitope on the HA head domain that is located behind the 190 helix. In particular, insertion of 158A and 158B residues imparts a unique conformation to the 150-loop in H7 HA and is likely associated with the particular specificity of H7.167.
Sequence analysis of the antibody variable gene sequences was revealing. Each of the mAbs isolated was distinct. For the most part, the frequency of somatic mutations in the variable region sequences was very low compared with that of sequences of influenza-specific human mAbs that we have isolated previously to influenza viruses. The number of mutations in the H7-reactive clones reported here ranged from 2 to 23, with an average of 10. In contrast, we found higher numbers of mutations in influenza HA–specific human mAbs in studies of seasonal H1 or even H5 vaccination (14–18), with average numbers of antibody-variable gene mutations of 18 or 12, respectively. Even the most potent neutralizing clones described here, with neutralizing activity of less than 20 ng/ml, possessed low numbers of somatic mutations. Likely, this genetic profile stems from the fact that the H7-specific response is a primary immune response due to the lack of prior exposure to this novel HA molecule. Interestingly, we did identify mAbs that were H7-reactive, but not H7-specific, as they had a cross-reactive heterosubtypic binding profile (such as the H7.134, H7.105, and H7.96 clones that bound to both H3 and H7 HA molecules). The antibody variable gene sequences of these clones possessed larger numbers of somatic mutations, suggesting they arose from memory B cells induced by prior H3 or other seasonal HA exposure that were restimulated by exposure to the H7 HA in a crossreactive manner. The mAb designated H7.96, which bound to both group 1 and 2 HA proteins, was particularly broad in its recognition pattern of diverse influenza A HA subtypes. This mAb neutralized at least 1 strain of H3N2 virus, but it did not neutralize H7N9 virus. The data here on antigenic relatedness of H3 and H7 HA epitopes are consistent with a recent report from others on 3 mAbs isolated from individuals with seasonal influenza immunity (but not H7 exposure) that bind and neutralize both H3 and H7 viruses (19). Consistent with those data, we show that some individuals mount H3-H7 crossreactive responses after experimental vaccination with a monovalent subunit A/Shanghai/02/2013 H7N9 vaccine candidate.
Although these mAbs are the first reported naturally occurring human mAbs induced by H7 influenza immunization, there is a previous report of H7-specific murine mAbs and one of engineered human mAbs that recognize H7 HA. Five murine mAbs reactive with A/Shanghai/02/2013 H7 HA were reported, 3 of which exhibited HAI activity and recognized a single antigenic site (designated site A in H3 influenza viruses as mapped by loss of binding to an HA variant, R131G) (20). We found that 3 human mAbs with HAI activity described here (H7.57, H7.137, and H7.169) also bound to this antigenic site, since passage of virus in the presence of mAbs H7.169 or H7.137 generated antibody neutralization escape mutant viruses with HA polymorphisms S132L or A138D, respectively, which are adjacent to position 131. These 3 human mAbs, which bind to the equivalent of antigenic site A in H3 nomenclature, exhibited very potent HAI activity (with IC50 values in the 0.1 to 1 μg/ml range). This antigenic site appears to be a dominant neutralizing determinant in at least 2 species. Another recent report describes isolation of human mAbs from randomly combined heavy-chain/light-chain phage display antibody libraries constructed from the RNA of blood cells of A/Shanghai/02/2013 H7N9 survivors (21). All mAbs from those experiments had a single HCDR3 sequence, exhibited loss of binding to HA variants with V186G and L226Q changes in the receptor-binding domain, and displayed neutralizing activity in a range similar to that of some of the vaccine-induced mAbs described here.
Potent neutralizing antibody responses to influenza antigens typically require help from activated CD4+ T cells following immunization, especially those T cells with markers associated with T follicular helper cell function (22). Some informatics analysis of predicted T cell epitopes suggested T cell help for H7N9 antibody responses may be limiting (23). Experimental studies of human T cell reactivities from subjects with prior exposure to seasonal influenza H1 and H3, but not H7, suggest that over half of such individuals have CD4+ T cells that respond to peptides from regions in H1 or H3 HA that are conserved in H7, especially in the genetically conserved HA2 domain (24). It is of interest that the most potent H7-specific neutralizing mAbs isolated here did not require accumulation of large numbers of somatic mutations, for which repetitive encounter with antigen and robust T cell help in germinal centers is generally required.
Taken together, these data suggest that when humans are exposed to novel avian H7 HA antigens, 2 principal components of the B cell repertoire are engaged. First, a primary B cell response occurs that consists of naive B cells that specify antibodies with H7-specific reactivity patterns and the antibody variable genes encoding these antibodies possess low numbers of somatic mutations that arise during the primary response. It is reassuring to find that the H7 HA antigen is not necessarily immunologically silent in humans, nor are humans incapable of generating B cells encoding high-potency neutralizing antibodies to H7 viruses. Despite the relatively low number of somatic mutations in the H7-specific mAbs described here, some of these clones are very potent inhibitors of replication. For example, the H7.167 clone has neutralizing activity as potent as that of almost any influenza-specific mAb ever reported. Clearly, there is not a genetic deficiency in humans for mounting responses to H7 viruses, but the fact that the first vaccination with H7 antigen is mostly a primary response constitutes a challenge for more robust responses.
Methods
Human sample acquisition.
The samples used in this study were from subjects enrolled in a phase II randomized, doubled-blinded, controlled study in healthy adults to assess safety, reactogenicity, and immunogenicity of a monovalent influenza A/H7N9 virus vaccine at different dosages with or without AS03 or MF59 adjuvant (DMID 13-0033; clinicaltrials.gov NCT01942265). The study was conducted at 5 NIH-funded VTEUs, but all samples used in this report were collected at the Vanderbilt University VTEU site. Vaccine was provided by Sanofi Pasteur, and AS03 adjuvant (manufactured by GlaxoSmithKline) and MF59 adjuvant (manufactured by Novartis) were sourced from the US national pre-pandemic influenza vaccine stockpile. Medically stable adults between 19 and 64 years of age were eligible to participate. Vaccine was administered at day 0 and day 21. Ten different vaccine regimens were tested. Monovalent influenza A/H7N9 virus vaccine was delivered in 2 doses administered at different dosages (3.75, 7.5, or 15 μg of HA/0.5 ml dose) given without adjuvant or with AS03 or MF59 adjuvant (manufactured by Novartis Vaccines and Diagnostics). A detailed description of the vaccine regimens was described previously (9). Blood was collected at day 0 and day 42, and PBMCs were isolated from a buffy coat using Histopaque 1077 (Sigma-Aldrich) and cryopreserved at a concentration of 1 × 107 cells/ml.
Generation of human hybridomas and purification of mAbs.
Hybridomas were generated using previously reported methods (25). Briefly, PBMCs from day 42 were transformed with EBV in the presence of Chk2 inhibitor (Sigma-Aldrich, catalog C3742), cyclosporin A (Sigma-Aldrich), and CpG10103. The CpG10103 was synthesized as an oligonucleotide, TCGTCGTTTTTCGGTCGTTTT, containing phosphorothioate bonds (Invitrogen). The EBV-transformed cells were plated in 384-well microtiter plates and grown for 10 days, then screened by ELISA for binding to recombinant soluble trimeric H7 HA based on the sequence of the A/Shanghai/02/2013 strain. Cells from wells with supernatants containing antibodies that reacted with H7 HA in ELISA were collected, expanded by incubating in 96-well plates with irradiated human PBMC feeder layers, and then fused with HMMA2.5 myeloma cells using a BTX ECM 2001 electrofusion device. Hybridomas generated by this process were screened by ELISA, and those secreting antibodies binding to H7 HA were subcloned by sorting a single live cell per well by flow cytometry. mAbs secreted into the supernatants of cloned hybridoma cells were purified by affinity purification with HiTrap Protein G Columns (GE) and a peristaltic pump, as directed by the manufacturer. The 50-kDa centrifugal filters were used for buffer exchange and concentration of purified antibodies, as directed by the manufacturer (Millipore). Concentrations of purified antibodies in DPBS were calculated with a NanoDrop 2000 spectrophotometer (Thermo Fisher).
B cell frequencies.
PBMCs collected at both day 0 and day 42 were isolated from blood in a subset of subjects by density gradient centrifugation. Cells were aliquoted into cryovials, cryopreserved, and stored in liquid nitrogen until testing. Previously cryopreserved PBMC samples were thawed and transformed as above. The EBV-transformed cells were plated in 384-well microtiter plates and grown for 10 days, then screened by ELISA for binding to each of the several recombinant HA proteins (described below). The minimal frequency of HA-reactive B cells was estimated based on the number of wells with HA-reactive supernatants compared with the total number of LCL colonies in the transformation plates (calculation: HA – reactive B cell frequency = [number of wells with HA-reactive supernatants] ÷ [number of LCL colonies in the plate] × 100). Data plots for multiple antigens were generated using Circos software (26).
HA expression and purification.
DNA copies of the genes encoding the HA proteins of A/Shanghai/02/2013 H7, A/New York/107/2003 H7, A/Netherlands/219 H7, A/Shearwater/Western Australia/2576/1979 H15, A/Victoria/261/2011 H3, A/New York/55/2004 H3, A/Panama/2007/1999 H3, A/mallard duck/Astrakhan/263/1982 H14, A/duck/Czechoslovakia/1956 H4, A/turkey/Wisconsin/1/1966 H9, A/Texas/36/1991 H1, A/Solomon Islands/03/2006, A/Vietnam/1203/2004 H5, A/Japan/305/1957 H2, A/Singapore/1/1957 H2, and A/black-headed gull/Sweden/1999 H16 were sequence optimized for human cell expression and synthesized without the transmembrane domain or the multi-basic cleavage site (when applicable) and with the addition of DNA sequences encoding a C-terminal GCN4 trimerization domain and a 6× histidine epitope tag. The synthesized cDNAs were subcloned into the pcDNA3.1+ mammalian cell expression vector (Invitrogen). Protein was expressed by transient transfection of 293F cells (Invitrogen) according to the manufacturer’s instructions, with the exception that polyethyleneimine was used as the transfection reagent as previously described (27). Cells were grown for 7 days, then harvested by centrifugation at 2,500 g. Supernatant was passed through a 0.45-μm membrane. The clarified supernatant was applied to a HiTrap TALON Crude Column (GE Healthcare), as directed by the manufacturer, with a peristaltic pump. Purified proteins were concentrated and buffer exchanged twice with DPBS using an Amicon Ultra centrifugal concentrator with a 50-kDa cutoff membrane (Millipore).
Antibody gene cloning and sequence analysis.
Antibody heavy- and light-chain variable genes were cloned by molecular means from each of the HA-reactive hybridoma lines that had been cloned biologically by flow cytometry. Briefly, RNA was extracted using the RNeasy Kit (QIAGEN) and reverse-transcriptase PCR (RT-PCR) amplification of antibody gene cDNAs was performed using primer sets described in Supplemental Table 3. The sequence of the antibody cDNAs was determined by automated Sanger sequence analysis. Sequence analysis of antibody variable gene sequences in the cDNAs was performed using the international ImMunoGeneTics information system (IMGT) (28).
EC50 and breadth binding analyses.
HA proteins were coated onto immunoassay 384-well plates (Nunc) in DPBS at 1 μg/ml overnight; then antigen was removed and plates were blocked with 0.8% nonfat dry milk and 2% goat serum in PBS plus 0.1% Tween-20 for 30 minutes. Antibodies were applied in triplicate to the plates at a concentration range of 10 μg/ml to 50 pg/ml for EC50 analysis and 1 μg/ml for breadth of binding analysis in block using 3-fold serial dilutions. The presence of antibodies bound to HA was determined using anti-human IgG alkaline phosphatase conjugate (Meridian Life Science Inc.) and p-nitrophenol phosphate substrate tablets (Sigma-Aldrich), with OD405 after 60 minutes. A nonlinear regression analysis was performed on the resulting curves using Prism software version 5 (GraphPad) to calculate EC50 values.
Biosensor competition-binding assays.
Antibody competition-binding groups were defined using biosensor assays on an Octet Red instrument (ForteBio). A/Shanghai/02/2013 H7 HA protein was loaded onto Ni-NTA tips (ForteBio) at a concentration of 10 μg/ml. Antigen-loaded tips were dipped successively into 2 different IgGs at a concentration of 50 μg/ml. If binding of the primary antibody blocked the binding of the secondary antibody, it was defined as a competitor. If binding of the primary antibody did not block the binding of the secondary antibody, it was not defined as a competitor. Blocking was defined as reduction of the signal by 90%, as compared with uncompeted binding.
HAI and virus neutralization assays.
HAI assays were performed as described elsewhere (29). Working stocks for each influenza virus strain were prepared by diluting each virus stock to a final HA titer of 8 HA units/50 μl. Each mAb was diluted initially to 2 μg/ml in PBS; then 2-fold dilutions of the indicated mAb were mixed with 25 μl of the working stock of each influenza virus strain (each well contained a final volume of 50 μl). The mAb virus samples then were incubated at room temperature for 30 minutes to allow virus neutralization. To each well, 50 μl of a 0.5% suspension of turkey red blood cells was then added. The assay was incubated on ice until hemagglutination occurred.
Virus neutralization assays (VNA) were performed as described elsewhere (30). Briefly, 2-fold dilutions (50 μl) of the indicated mAbs were mixed with 200 PFU of influenza virus. The mAb virus samples were incubated at room temperature for 60 minutes to allow for virus neutralization. The serum-virus samples then were transferred to MDCK cells cultured in 96-well flat-bottom plates. Following virus absorption for 60 minutes, the serum-virus inocula were removed, and the MDCK cells were cultured for 4 days in Opti-MEM I medium supplemented with 1 μg/ml of tosylsulfonyl phenylalanyl chloromethyl ketone– trypsin (TPCK-trypsin) (Sigma-Aldrich). Virus production was determined by HA assay.
Generation of mAb escape mutant viruses.
Recombinant PR8 influenza virus expressing the H7 HA and N9 NA of A/Shanghai/2/2013 (PR8 Shanghai/2 H7N9) was produced by reverse genetics and virus stocks prepared in specific pathogen–free embryonated chicken eggs (Charles River Laboratories). To map the binding site on A/Shanghai/2/2013 H7 HA for each indicated mAb, 1 × 106 PFU of PR8 Shanghai/2 H7N9 were incubated with 50 ng of the indicated mAb. The mAb/virus sample then was inoculated into 8-day-old embryonated chicken eggs, and virus production was assessed by HA assay. This process was repeated 3 times, and then plaque assays were conducted with MDCK cells to plaque isolate clones of potential antibody escape mutant viruses. cDNA of the viral genomic segment (vRNA) encoding the H7 HA then was prepared by reverse transcription with sequence-specific primers, and the HA gene sequence analyzed by Sanger sequencing (Macrogen Corp.).
Peptide fragmentation and DXMS.
To maximize peptide probe coverage, the optimized quench condition was determined prior to deuteration studies (31, 32). In short, HA head domain was diluted with H2O buffer (8.3 mM Tris, 150 mM NaCl, in H2O, pH 7.15) at 0°C and then quenched with 0.8% formic acid (v/v) containing various concentration of GuHCl and Tris(2-carboxyethyl)phosphine (TCEP). The samples then were frozen on dry ice and stored at –80°C until they were transferred to the cryogenic autosampler. 2.2 M GuHCl and 100 mM TCEP in 0.8% formic acid gave an optimal peptide coverage map.
The samples later were thawed automatically on ice and then immediately passed over an AL-20–pepsin column (16 μl bed volume, 30 mg/ml porcine pepsin; Sigma-Aldrich). The resulting peptides were collected on a C18 trap and separated using a C18 reversed phase column (Vydac) running a linear gradient of 0.046% (v/v) trifluoroacetic acid, 6.4% (v/v) acetonitrile to 0.03% (v/v) trifluoroacetic acid, and 38.4% (v/v) acetonitrile over 30 minutes, with column effluent directed into an Orbitrap Elite mass spectrometer (Thermo-Fisher Scientific). Data were acquired in both data-dependent MS/MS mode and MS1 profile mode. Proteome Discoverer software (Thermo Finnigan Inc.) was used to identify the sequence of the peptide ions. DXMS Explorer (Sierra Analytics Inc.) was used for the analysis of the mass spectra as described previously (33). H7 mAb–bound HAs were prepared by mixing H7 mAbs with monomeric H7 HA head domain at a 1:1.3 stoichiometric ratio. The mixtures were incubated at 25°C for 30 minutes. All functionally deuterated samples, with the exception of the equilibrium-deuterated control, and buffers were prechilled on ice and prepared in the cold room.
Functional deuterium-hydrogen exchange reaction was initiated by diluting free HA or antibody-bound HA stock solution with D2O buffer (8.3 mM Tris, 150 mM NaCl, in D2O, pDREAD 7.15) at a 1:3 v/v ratio. At 10, 100, and 1,000 seconds, quench was added to the respective samples, and then samples were frozen at –80°C. In addition, nondeuterated samples and equilibrium-deuterated back-exchange control samples were prepared as previously described (31, 32, 34). The centroids of the isotopic envelopes of nondeuterated, functionally deuterated, and fully deuterated peptides were measured using DXMS Explorer and then converted to corresponding deuteration levels with corrections for back exchange (35).
Preparation of H7 HA–H7.167 Fab complex.
Recombinant soluble H7N9 (A/Shanghai/02/2013) HA was produced using a baculovirus expression system, as described previously (36). The expressed uncleaved HA protein (HA0) was purified via a His-tag affinity purification, dialyzed against 20 mM Tris-HCl pH 8.0, 100 mM NaCl, and then cleaved by trypsin (New England BioLabs) to produce uniformly cleaved (HA1/HA2) protein and to remove the trimerization domain and His6 tag. The digested protein was purified further by gel filtration chromatography using a Superdex-200 column (Pharmacia). The HA protein eluted as a trimer and was concentrated to approximately 10 mg/ml.
H7.167 Fab fragment was obtained by specific cleavage of H7.167 IgG using immobilized papain (Pierce, catalog 20341). The Fabs were separated from the Fc fragment by protein A chromatography and then buffer exchanged to 50 mM NaAc, 50 mM NaCl, pH 5.5. The identity of the fragments was confirmed by Western blotting using a rabbit anti-human IgG κ-chain, goat anti-rabbit/HRP antibody combination. The Fabs were subjected to cation exchange chromatography (after dilution in 50 mM sodium acetate buffer, pH 5.0, and separation over a gradient of 0–1 M NaCl) and gel filtration. Fab concentration was measured by optical absorption at 280 nm.
The purified H7 HA and H7.167 Fab were mixed in a molar ratio of approximately 1:1.2 and incubated overnight at 4°C to allow complex formation. Saturated complexes then were purified from unbound Fab by gel filtration and were concentrated to approximately 8 mg/ml in 10 mM Tris, pH 8.0, and 150 mM NaCl.
Crystallization and x-ray structure determination and refinement.
H7 HA–H7.167 Fab complex crystals were grown using the automated Rigaku CrystalMation robotic system at the Joint Center for Structural Genomics (JCSG) by sitting drop vapor diffusion. Crystals grew at 20°C by mixing concentrated complex (~8 mg/ml) with 10% (w/v) polyethylene glycol (PEG) 8000, 0.1 M HEPES, pH 7.5, and 8% (v/v) ethylene glycol. Crystals were cryoprotected in mother liquor supplemented with 10% (w/v) ethylene glycol, flash cooled, and stored in liquid nitrogen until data collection.
The crystals diffracted to 4.65 Å resolution at beamline 12-2 at the Stanford Synchrotron Radiation Lightsource (SSRL) and the diffraction data were indexed in space group I213 (Supplemental Table 1) with a Vm of 6.2 Å3/Da and 83% solvent content. The structure was determined by molecular replacement with Phaser (37) using the H7N9 (A/Shanghai/02/2013) HA monomer (PDB ID: 4N5J) as the initial search model. The variable domain of the heavy chain from 1 Fab (PDB ID: 3NH7) and the variable domain of the light chain from another Fab (PDB ID: 3QOT), which share high sequence similarity (with 97% and 83% identities for the light- and heavy-chain variable domains, respectively), were then used as search models. One HA and the variable domains corresponding to 1 Fab were found in the asymmetric unit. Finally, molecular replacement using the constant domain of the Fab (PDB ID: 3QOT) as a search model did not give solutions because of disorder due to the flexible elbow angle between the variable and constant domains, as found in a number of Fab structures (38–40). The model was rebuilt iteratively using Coot (41) and refined in Phenix (42). Refinement parameters included rigid body refinement (for HA1 and HA2 in the HA molecule and the variable domains of the heavy chain and light chains of the Fab molecule) and restrained refinement, including TLS refinement (as for the rigid body refinement above).
Structural analyses.
Hydrogen bonds and van der Waals contacts were calculated using HBPLUS and CONTACSYM, respectively (43, 44). Surface area upon Fab binding was calculated using MS. MacPyMOL (DeLano Scientific) was used to render figures. Kabat numbering was applied to the coordinate files using the AbNum server (45). The final coordinates were validated using the JCSG quality control server (v2.8), which includes MolProbity (46).
Sequence analysis of the antibody epitopes.
The full-length and nonredundant influenza A HA sequences were downloaded from the Influenza Virus Resource at the National Center for Biotechnology Information (NCBI) database (47). At the time of download (December 31, 2015), the data set included 1,070 H7 HA sequences (including viruses form human, avian, and other hosts). The sequences were aligned using MUSCLE with default parameters (48). The sequence conservation of the H7.167 epitope is reported in Figure 5C.
Mouse passive immunization and influenza virus challenge.
Eight-week-old female BALB/c mice were purchased from The Jackson Laboratory. Mice were passively immunized with the indicated mAbs by i.p. injection 1 day prior (day –1) to challenge infection. On the day of inoculation (day 0), mice were anesthetized by i.p. injection of a ketamine/xylazine cocktail. Prior to inoculation with virus, the passively immunized mice were bled and serum samples collected to determine HI titers for the H7.167 mAbs. The anesthetized mice were inoculated i.n. with 10,000 PFU of PR8 Shanghai/2 H7N9 influenza virus per mouse. Inoculated mice (n = 5) were weighed daily to assess morbidity from influenza virus infection, as assessed by body weight changes. On days 3 and 5 after inoculation, a subgroup of mice (n = 3) was euthanized humanely to determine pulmonary virus titers. Pooled mouse sera were treated with receptor-destroying enzyme (RDE) (Vibrio cholera filtrate; Sigma-Aldrich) essentially as described elsewhere (30) and then assessed for HI titer as described above.
Statistics.
In neutralization assays, IC50 values were calculated after log transformation of antibody concentrations using a 3-parameter nonlinear fit analysis of antibody log10 concentration versus response with R2 values greater than 0.85. IC50 values with nonoverlapping 95% CIs were identified as differing significantly. In ELISA assays, EC50 values were calculated after log transformation of antibody concentrations using sigmoidal dose-response nonlinear fit analysis with R2 values of greater than 0.85. The EC50 values that retained nonoverlapping 95% CIs were identified as differing significantly. To analyze ELISA data obtained for testing complex orientation, a 1-way ANOVA statistical analysis was performed to compare the sets of variants with EC50 values of less than 1 μg/ml to variants with no detectable binding at 10 μg/ml. All statistics were analyzed using Prism software version 5 (GraphPad). A P value of less than 0.05 was considered significant.
Study approval.
PBMCs were collected in the Vanderbilt Clinical Research Center after informed consent from otherwise healthy subjects with a prior history of experimental H7N9 subunit vaccination, as described in Methods. The protocol and consent forms were approved prior to study by the Vanderbilt University Institutional Review Board Committee. The animal protocol covering the H7N9 influenza virus challenge infections of passively immunized mice was reviewed and approved by the Institutional Animal Care and Use Committee at ISMMS.
Protein data accession number.
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank (PDB 5F45 for A/Shanghai/02/2013 [H7N9] HA in complex with H7.167 Fab).
Author contributions
NJT, HZ, SB, SL, IAW, RAA, and JEC designed the research study and experiments. NJT, HZ, SB, GS, NK, RML, RGB, YY, SG, AB, SMY, MTR, DL, SL, AGS, and RAA conducted experiments and acquired and analyzed data. CBC and KME conducted the parental vaccine study and acquired PBMCs. NJT and JEC wrote the manuscript. All authors read and commented on the manuscript.
Supplementary Material
Acknowledgments
The authors thank F. Smith-House (Vanderbilt) and Sarah Reiss and Rachel G. Blinick (ISMMS) for technical support; Deborah Hunter, Julie Anderson, Mary Jones, Wendi McDonald, Sara Anderson, Shanda Phillips, and Belinda Gayle Johnson for clinical support during human sample acquisition; and Jill Janssen of the Vanderbilt Clinical Trials Center for regulatory support. We thank X. Dai and M. Elsliger for crystallographic and computational support; Henry Tien of the Robotics Core at the Joint Center for Structural Genomics for automated crystal screening; and the staff at the SSRL beamlines 12-2. The SSRL is a Directorate of Stanford Linear Accelerator Center National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the NIH, the National Institute of General Medical Sciences (NIGMS) (including P41GM103393), and the National Center for Research Resources (NCRR) (P41RR001209). The original clinical trial of the H7 vaccine (DMID 13-0033) was supported by the NIH and performed in the Vanderbilt Vaccine and Treatment Evaluation Unit. This work was supported by NIH grant R01 AI106002, National Institute of Allergy and Infectious Diseases (NIAID) contracts HHSN272200900047C and HHSN2720100007C (to J.E. Crowe Jr.) and HHSN27220800007C (to K.M. Edwards), Vanderbilt NIH Clinical and Translational Science Award (CTSA) grant UL1 RR024975, Department of Defense (DoD) grant HDTRA1-10-1-0067, by NIH grants P01AI097092 (to A. García-Sastre and R.A. Albrecht) and R56 AI117675 (to I.A. Wilson), and by the Center of Research in Influenza Pathogenesis (CRIP), an NIAID-funded Center of Excellence in Influenza Research and Surveillance (CEIRS) (contract HHSN272201400008C, to A. García-Sastre and R.A. Albrecht), and NIH grants R01 GM020501 and R01 AI101436 (to S. Li). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Heng Zhang’s present address is: Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, China.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(4):1482–1494. doi:10.1172/JCI85317.
Contributor Information
Heng Zhang, Email: zhangh@scripps.edu.
Sandhya Bangaru, Email: sandhya.bangaru@Vanderbilt.Edu.
Gopal Sapparapu, Email: gopal.sapparapu@Vanderbilt.Edu.
Nurgun Kose, Email: nurgun.kose@Vanderbilt.Edu.
Yingchun Yu, Email: yuantou8@gmail.com.
Stephen Graham, Email: sdgraham118@gmail.com.
Andre Branchizio, Email: andre.j.branchizio@Vanderbilt.Edu.
David Lee, Email: David.Lee@med.wmich.edu.
Sheng Li, Email: s4li@ucsd.edu.
References
- 1.World Health Organization WHO RISK ASSESSMENT of Human infections with avian influenza A(H7N9) virus. [February 23, 2015]; [February 1, 2016];WHO website. http://www.who.int/influenza/human_animal_interface/influenza_h7n9/RiskAssessment_H7N9_23Feb20115.pdf
- 2.Gao R, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368(20):1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 3.Liu D, et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet. 2013;381(9881):1926–1932. doi: 10.1016/S0140-6736(13)60938-1. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, et al. Towards a better understanding of the novel avian-origin H7N9 influenza A virus in China. Sci Rep. 2013;3: doi: 10.1038/srep02318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kageyama T, et al. Genetic analysis of novel avian A(H7N9) influenza viruses isolated from patients in China, February to April 2013. Euro Surveill. 2013;18(15): [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens J, et al. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312(5772):404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, et al. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science. 2013;341(6144):410–414. doi: 10.1126/science.1240532. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, et al. Infectivity, transmission, and pathology of human-isolated H7N9 influenza virus in ferrets and pigs. Science. 2013;341(6142):183–186. doi: 10.1126/science.1239844. [DOI] [PubMed] [Google Scholar]
- 9.Jackson LA, et al. Effect of varying doses of a monovalent H7N9 influenza vaccine with and without AS03 and MF59 adjuvants on immune response: a randomized clinical trial. JAMA. 2015;314(3):237–246. doi: 10.1001/jama.2015.7916. [DOI] [PubMed] [Google Scholar]
- 10.Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M. Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N Engl J Med. 2006;354(13):1343–1351. doi: 10.1056/NEJMoa055778. [DOI] [PubMed] [Google Scholar]
- 11.Bart SA, et al. A cell culture-derived MF59-adjuvanted pandemic A/H7N9 vaccine is immunogenic in adults. Sci Transl Med. 2014;6(234): doi: 10.1126/scitranslmed.3008761. [DOI] [PubMed] [Google Scholar]
- 12.Ha Y, Stevens DJ, Skehel JJ, Wiley DC. H5 avian and H9 swine influenza virus haemagglutinin structures: possible origin of influenza subtypes. EMBO J. 2002;21(5):865–875. doi: 10.1093/emboj/21.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong X, et al. Receptor binding by an H7N9 influenza virus from humans. Nature. 2013;499(7459):496–499. doi: 10.1038/nature12372. [DOI] [PubMed] [Google Scholar]
- 14.Krause JC, et al. A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of influenza H1N1 virus hemagglutinin. J Virol. 2011;85(20):10905–10908. doi: 10.1128/JVI.00700-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsibane T, et al. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. PLoS Pathog. 2012;8(12): doi: 10.1371/journal.ppat.1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krause JC, et al. Epitope-specific human influenza antibody repertoires diversify by B cell intraclonal sequence divergence and interclonal convergence. J Immunol. 2011;187(7):3704–3711. doi: 10.4049/jimmunol.1101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krause JC, et al. Naturally occurring human monoclonal antibodies neutralize both 1918 and 2009 pandemic influenza A (H1N1) viruses. J Virol. 2010;84(6):3127–3130. doi: 10.1128/JVI.02184-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thornburg NJ, et al. Human antibodies that neutralize respiratory droplet transmissible H5N1 influenza viruses. J Clin Invest. 2013;123(10):4405–4409. doi: 10.1172/JCI69377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henry Dunand CJ, et al. Preexisting human antibodies neutralize recently emerged H7N9 influenza strains. J Clin Invest. 2015;125(3):1255–1268. doi: 10.1172/JCI74374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmeisser F, et al. Antibodies to antigenic site A of influenza H7 hemagglutinin provide protection against H7N9 challenge. PLoS One. 2015;10(1): doi: 10.1371/journal.pone.0117108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z, et al. Human monoclonal antibodies targeting the haemagglutinin glycoprotein can neutralize H7N9 influenza virus. Nat Commun. 2015;6: doi: 10.1038/ncomms7714. [DOI] [PubMed] [Google Scholar]
- 22.Bentebibel SE, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. 2013;5(176): doi: 10.1126/scitranslmed.3005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Groot AS, et al. Low immunogenicity predicted for emerging avian-origin H7N9: implication for influenza vaccine design. Hum Vaccin Immunother. 2013;9(5):950–956. doi: 10.4161/hv.24939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards KA, et al. Seasonal influenza can poise hosts for CD4 T-cell immunity to H7N9 avian influenza. J Infect Dis. 2015;212(1):86–94. doi: 10.1093/infdis/jiu662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith SA, et al. Persistence of circulating memory B cell clones with potential for dengue virus disease enhancement for decades following infection. J Virol. 2012;86(5):2665–2675. doi: 10.1128/JVI.06335-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30(2):E9–E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brochet X, Lefranc MP, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008;36(Web Server issue):W503–W508. doi: 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S, et al. Hemagglutinin (HA) proteins from H1 and H3 serotypes of influenza A viruses require different antigen designs for the induction of optimal protective antibody responses as studied by codon-optimized HA DNA vaccines. J Virol. 2006;80(23):11628–11637. doi: 10.1128/JVI.01065-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steel J, et al. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J Virol. 2009;83(4):1742–1753. doi: 10.1128/JVI.01920-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, et al. Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS) J Biol Chem. 2011;286(20):17889–17897. doi: 10.1074/jbc.M111.224535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu S, et al. Structural insights into glucan phosphatase dynamics using amide hydrogen-deuterium exchange mass spectrometry. Biochemistry. 2009;48(41):9891–9902. doi: 10.1021/bi9008853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamuro Y, et al. Mapping intersubunit interactions of the regulatory subunit (RIα) in the type I holoenzyme of protein kinase A by amide hydrogen/deuterium exchange mass spectrometry (DXMS) J Mol Biol. 2004;340(5):1185–1196. doi: 10.1016/j.jmb.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 34.Lu WD, Liu TT, Li SS, Woods VL, Jr, Hook VV. The prohormone proenkephalin possesses differential conformational features of subdomains revealed by rapid H-D exchange mass spectrometry. Protein Sci. 2012;21(2):178–187. doi: 10.1002/pro.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2008;2(4):522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu RR, et al. A recurring motif for antibody recognition of the receptor-binding site of influenza hemagglutinin. Nat Struct Mol Biol. 2013;20(3):363–370. doi: 10.1038/nsmb.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40(pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324(5924):246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16(3):265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333(6044):850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238(5):777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 44.Sheriff S, Hendrickson WA, Smith JL. Structure of myohemerythrin in the azidomet state at 1.7/1.3 A resolution. J Mol Biol. 1987;197(2):273–296. doi: 10.1016/0022-2836(87)90124-0. [DOI] [PubMed] [Google Scholar]
- 45.Abhinandan KR, Martin AC. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol Immunol. 2008;45(14):3832–3839. doi: 10.1016/j.molimm.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 46.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao Y, et al. The influenza virus resource at the National Center for Biotechnology Information. J Virol. 2008;82(2):596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1179. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.