

Abstract
BACKGROUND. Recently, 1-octanol has been shown to have efficacy in treating patients with essential tremor (ET). The primary metabolite of 1-octanol is octanoic acid (OA), which is now thought to be the active substance that mediates tremor suppression. Our aim was to describe the maximum tolerated dose (MTD) of oral OA in patients with ET and assess the pharmacokinetics (PK) and pharmacodynamics (PD) profile of OA.
METHODS. The MTD was studied using an open-label, single-ascending 3 + 3 dose–escalation design. Predefined single doses ranged from 8 to 128 mg/kg, with grade 2 adverse events (AEs) defined as dose-limiting toxicity. Tremor was assessed using accelerometry, digital spiral analysis, and a standard clinical rating scale at baseline and up to 600 minutes after intake. Safety assessments and PK sampling were also performed.
RESULTS. Dose-limiting toxicity was not reached. The most frequent AE was mild abdominal discomfort. Exposure (AUC) increased linearly with the dose. Secondary efficacy measures suggested a dose-dependent reduction of tremor. Accordingly, a single unified PK/PD model with an effect compartment and sigmoid maximum effect (Emax) response could be built that accounted well for the time profiles of plasma concentrations as well as effects on tremor severity across the 5 dose levels.
CONCLUSION. Although our trial did not reach an MTD, a dose-dependent effect was demonstrated in the PK/PD model as well as in secondary efficacy outcomes. Future studies are needed to explore the safety in higher dose ranges and to confirm dose-dependent efficacy in a placebo-controlled design.
TRIAL REGISTRATION. Clinicaltrials.gov NCT01468948
FUNDING. NINDS Intramural Research Program; TG Therapeutics Inc.
Introduction
Essential tremor (ET) is a common movement disorder, with 3 of 4 patients reporting impaired activities of daily living (1). Less than 50% note tremor improvement with current pharmacotherapy, and there is a high rate of side effects, resulting in a need for novel treatment strategies (2). Many patients report significant reduction in tremor after administration of ethanol (3–5). Routine use of ethanol for tremor control is prohibitive due to the risks associated with intoxication and the potential medical, social, and legal consequences of chronic alcohol use. The long chain alcohol 1-octanol has been explored for potentially achieving effects comparable to those of ethanol without the risk of intoxication. Octanoic acid (OA), a medium chain fatty acid, is the primary metabolite of 1-octanol. As a relationship between OA pharmacokinetics (PK) and the timing of clinical improvement has been observed, OA itself was proposed to be the active substance mediating tremor suppression after 1-octanol intake (6). The presumed mechanism of action of OA acts via mechanisms similar to those of ethanol in reducing tremor; by including objectively proven ethanol responders in a screening visit in studies of ET, a potential bias of results related to safety and efficacy can be avoided. The efficacy and safety of a single oral OA dose was initially studied in a double-blind, placebo-controlled study, where 4 mg/kg of oral OA was demonstrated to be safe and potentially effective over placebo at time points of 150 minutes or more after administration (7). The aim of this phase 1/2 study was to determine the maximum tolerated dose (MTD) of a single-ascending oral dose of OA in patients with ET. High-dose OA (up to 710 mg/kg) was previously used in children with intractable epilepsy as part of a ketogenic diet treatment, with chronic administration for up to 2 years (8–10). Reported side effects were mild diarrhea and abdominal discomfort. As a secondary outcome in this study, a pharmacodynamic (PD) dose-response relationship of safety, PK, and efficacy was investigated.
Results
Safety.
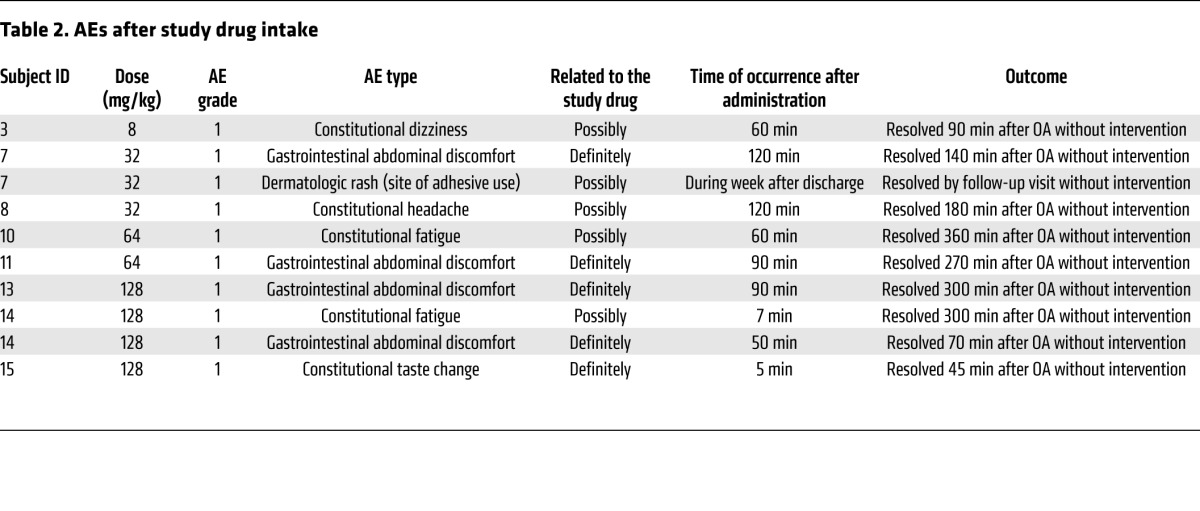
Fifteen adult ET patients (8 female, mean age 68.7 ± 9.8 yr) were enrolled for OA intake in 5 predefined dose groups (8, 16, 32, 64, and 128 mg/kg), (Figure 1). The baseline characteristics were balanced across treatment cohorts (Table 1). No dose-limiting toxicity (DLT) events or serious adverse events (AEs) were observed in any of the cohorts. The tolerability across all dose levels was good. In total, 10 nonserious AEs were documented. These nonserious AEs were mild grade (Common Terminology Criteria for Adverse Events v4.0 [CTCAE]; grade 1; ref. 11) and self limiting. The most frequent AE was mild abdominal discomfort (n = 4), which was deemed definitely related to the study substance by the investigators. All other AEs (dizziness, headache, taste change, rash under dressing of peripherally inserted central catheter [PICC] line insertion site, fatigue) were also of mild grade, and each occurred twice or less. Only taste change was determined as definitely related. All other AEs were determined as possibly related. All patients completed the study procedures per protocol. None of the nonserious AEs (Table 2) led to withdrawal from the study. No drug-related abnormalities were seen in the other safety measures.
Figure 1. Study flow chart.

Table 1. Baseline characteristics and PK parameters of ET patients.
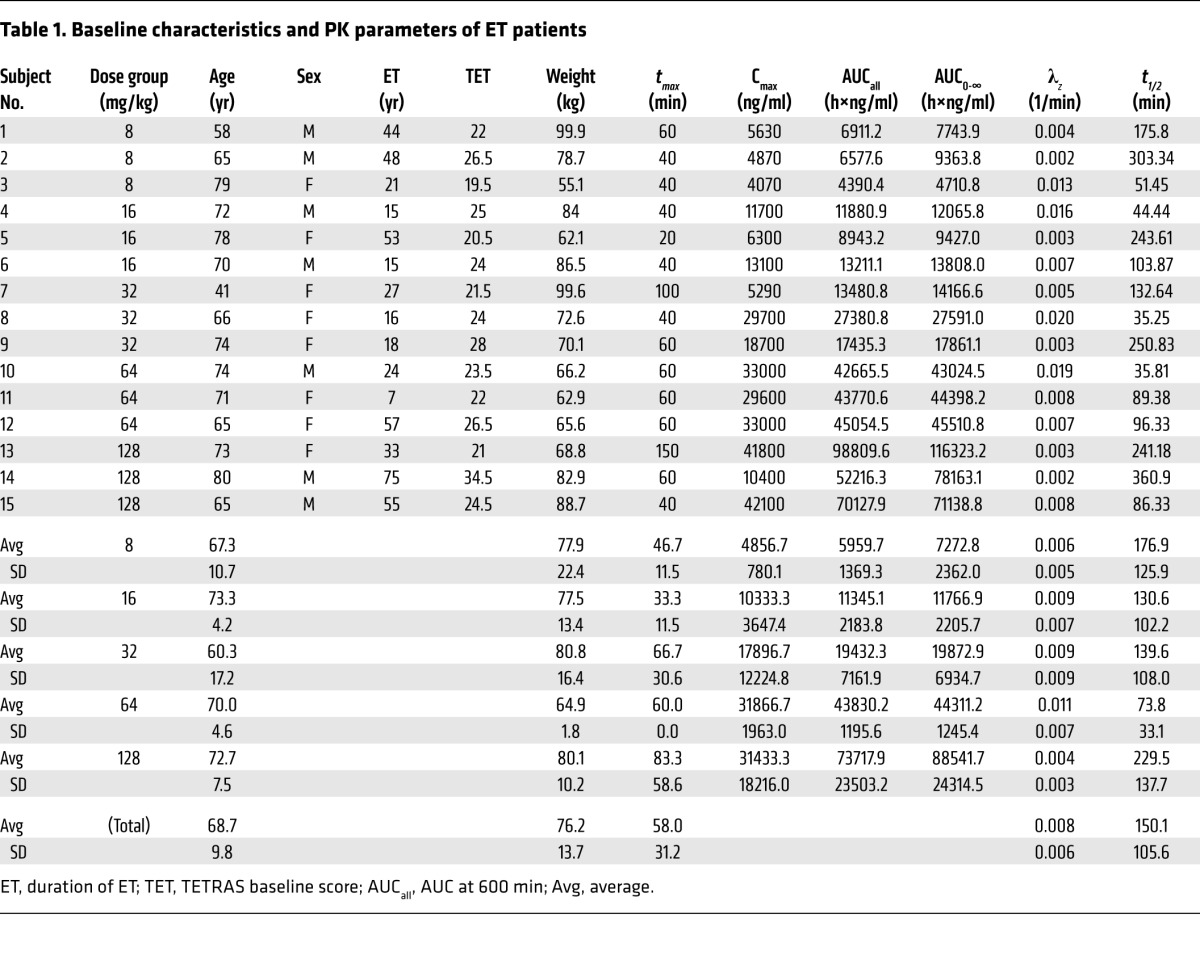
Table 2. AEs after study drug intake.
PK.
All measured OA values were above the lowest limit of quantitation. Average concentration-time profiles obtained for each dose level are summarized in Figure 2. OA exposure increased linearly with dose, especially for the AUC data (r2 = 0.991) despite the relatively low number of subjects per dose groups. For maximum plasma concentration (Cmax), the correlation was lower (r2 = 0.747), most likely due to possible absorption delays or problems at the highest dose that gave the largest deviation from linearity (Figure 3). Standard noncompartmental PK analysis of data from all subjects at all dose levels (n = 5 × 3) gave an average apparent volume of distribution of Vd/F = 398 ± 311 l, a time to maximum concentration of around 1 hour (tmax = 58.0 ± 31.2 min), and an elimination rate constant of λz = 0.0080 ± 0.0061 min–1, corresponding to an elimination half-life of t1/2 = 150.1 ± 105.6 min (for details see Table 1).
Figure 2. Concentration -time profiles and dose exposure relationships for OA as obtained in the present study.

Concentration-time profile for OA following 5 different oral doses (top). Corresponding dose-exposure relationships showing AUC and Cmax as a function of the administered dose (bottom). Data are shown as mean ± SEM for 3 subjects at each of the 5 dose levels.
Figure 3. Time profile of tremor rated by the TETRAS performance score following OA administration at 5 different oral doses.

Data are mean ± SEM for all subjects per each dose level as indicated and are shown in the left panel with absolute values and in the right panel normalized to baseline. Data are shown as mean ± SEM for 3 subjects at each of the 5 dose levels.
PD, PK/PD analysis.
Three different efficacy (PD) parameters were used to assess the effect on tremor severity: accelerometry, spirography, and The Essential Tremor Rating Assessment Scale (TETRAS) performance score (12, 13). Corresponding time profiles for the average values per dose levels (normalized to baseline) showed some dose response in all 3 secondary outcome parameters. Because of the nature of the data and the low number of subjects per group, there was considerable scatter. The TETRAS performance score data showed the least scatter as well as the most clear dose response (Figure 3). Therefore, for the present exploratory PK/PD analysis, the TETRAS data were used as the PD component (effect).
For PK/PD modeling, first, the PK data were fitted with a standard 1-compartment model (Vd) with first-order absorption (kabs, indicating absorption rate constant) and first-order elimination (kel, indicating elimination rate constant), as commonly done for orally administered drugs (Figure 4). For exploratory purposes, fitting of each dose group separately was also performed, but for the final model, fitting was done with a unified model, i.e., a single set of parameters for Vd, kabs, and kel with only the dose being varied among the different groups. As Figure 5 illustrates, the unified PK model gave acceptable fit of the data (overall r2 = 0.498) in agreement with the fact that there is good linearity in AUC, but somewhat less for Cmax.
Figure 4. Summary of the PK/PD model used.

C, concentration; D, dose; E, effect; EC50,concentration at which half-maximal response is achieved.
Figure 5. Fit of the PK and PD data with the present unified PK/PD model.

Measured and model-predicted PK (concentration) and PD (effect, TETRAS score, 25-fold rescaled) data are shown at left and right, respectively.
Corresponding parameters were Vd/F = 4.141 l/kg, kabs = 0.0288 min–1, and kel = 0.0072 min–1. These are in good agreement with the parameters of the noncompartmental analysis both for Vd/F, 4.141 vs. 390/76.2 = 5.116 l/kg (corrected with the average weight of 76.2 kg to a per kg basis), and for kel, 0.0072 vs. 0.0080 min–1. This provided a reassurance on the consistency of the PK approach.
Comparison of the PK (concentration) and PD (effect) time profiles revealed a clear delay in the onset of the effect compared with plasma concentration. For example, while the average tmax for concentration was around 1 hour (58 min), that for TETRAS was around 2 hours (128 min), and that for digital spiral analysis around 3 hours (209 min). Therefore, for the PK/PD modeling, an effect compartment model with a sigmoid (Hill-type) maximum effect (Emax) response function was selected (14, 15). A single unified PK/PD model was used for data from all dose levels, i.e., a single set of unified parameters (Vd, kabs, and kel for PK plus Emax, effect compartment rate constants [in][keff_in], and effect compartment rate constants [out] [keff_out] for PD) was used in the equations together with the 5 different doses. First, the PK component was fitted for all doses, and then this was coupled to an (indirect link) effect compartment model to fit the PD (effect) data of TETRAS. The best fit obtained with the simplest model (assuming a classic response function, i.e., Hill slope of unity n = 1) was already adequate, as it accounted for about 50% of the variability in all PK data (r2 = 0.498) and 80% of the variability in the PD data (r2 = 0.806). This fit was obtained with the following set of parameters: Vd/F = 4.141 l/kg, kabs = 0.0288 min–1, kel = 0.0072 min–1, Emax = 4.378, keff_in = 0.0135 min–1, keff_out = 0.0088 min–1. The fit of the PD data was further improved with a Hill slope that was different from unity, best fit obtained with n = 0.297 (r2 = 0.876; Figure 5).
Discussion
We demonstrated that OA was safe and well tolerated by ET patients up to a dose of 128 mg/kg. Therefore, the primary goal of the study to determine an MTD was not met. Gastrointestinal nonserious AEs were rated as definitely related to OA intake. In a previous study on OA given at 4 mg/kg to 18 ET patients in a placebo-controlled manner, taste change was the only gastrointestinal nonserious AE reported in 1 patient of each group, placebo and OA (7). Abdominal discomfort was not reported. OA in our study was administered orally in gel capsules under fasting conditions. The facts that patients had to take up to 26 capsules within a minute and that the study drug contained other compounds are alternative explanations for the self-limited abdominal discomfort. For future studies with higher doses, it will be necessary to improve the formulation. Further, gastrointestinal nonserious AEs are in line with reports of OA given in doses comparable to those given with a ketogenic diet (8–10). In studies on children using much higher doses than used in this study, an OA-containing diet was administered on a daily basis as an emulsion with the goal to supply 60% of the daily energy requirement using this diet. Diarrhea and abdominal pain were alleviated by temporary dose reduction or intake together with food. Of the 50 children, 6 were unable to tolerate the diet because of those side effects (9). Depending on the effect of OA on ET, those side effects will be relevant for the calculation of benefit/risk ratios in future studies in adult ET patients. It is not known whether there is a different metabolism for OA in children. This would also be an interesting question to answer in further studies using OA. All other nongastrointestinal, nonserious AEs in our study were not rated as definitely related to OA intake. Dizziness, headache, and fatigue may be rather nonspecific and also occurred in the placebo group as well as in the OA group in the previous low-dose OA study (7). Future high-dose studies conducted in a placebo-controlled manner could identify relatedness of these side effects to OA.
Investigations of efficacy were mainly exploratory due to small cohort size and the open design. However, a tendency toward a dose-dependent reduction of tremor was seen in all efficacy outcome parameters. Overall, PK values in our study across the dose range investigated (8 to 128 mg/kg) were in good agreement with those obtained earlier in a single low-dose (4 mg/kg) trial previously published (7). Because of the expected longer elimination, we added another time point at 600 minutes after study drug intake. The estimated average elimination half-life in this study was longer than in the previous one (150 min vs. 83.5 min), but at least to some extent, this is due to the longer follow-up time. Most of the OA was eliminated after 600 minutes. If OA were to be given at higher doses, one should not only consider using a different formulation, but also using more frequent administration (2 or 3 times a day). Especially with the latter regimen, potential accumulation of the drug should also be considered.
The developed PK/PD model was able to account for the temporal dissociation between the time courses of concentration and effect, demonstrating a clear dose-effect relationship, with a delay in the onset of the effect compared with the PK time profile. A likely physiological explanation is that effects are likely mediated through critical OA levels in the CNS, which can be considered a separate compartment from plasma, requiring a cross-compartment distribution through the blood-brain barrier. Onset of the effect might therefore occur with a certain time delay compared with plasma concentrations. The main limitation of our study is that the efficacy was obtained in an uncontrolled, nonblinded manner. The benefit of adding a placebo arm to obtain more reliable safety and efficacy data was considered not to outweigh the risk of exposing additional subjects to the protocol or extending the inpatient stay for a crossover design (including placement of a PICC line), mainly due to the secondary nature of the efficacy outcomes and the small sample size.
In conclusion, our results are promising for the development of OA as a treatment of ET. Additional studies are needed to explore the safety of OA in ET patients at higher doses as well as to assess the dose dependence of the efficacy and the PK profile in a placebo-controlled, multiple-dose administration study. Nevertheless, a unified PK/PD model was developed that could provide an acceptable description of both the PK (concentration) and PD (effect) data across all doses, and this model should help guide the design of future studies.
Methods
Patients.
Patients with ET according to Movement Disorder Society consensus criteria were included (16). Tremor was the only allowed abnormality on neurological examination. Patients under pharmacotherapy for tremor were off antitremor medication for at least 4 plasma half-lives and were instructed to abstain from ethanol and/or caffeine for 48 hours prior to the experiment. Patients on primidone were excluded because of the long half-life of its metabolites. Other exclusion criteria were as follows: patients with active or past alcohol abuse; women who were pregnant or lactating; patients of age less than 21; and patients of East Asian or Native American descent, who may possess variant alleles influencing alcohol metabolism, resulting in higher sensitivity to toxic effects of alcohol. As part of the screening visit, an oral ethanol challenge was performed. Baseline tremor severity was measured using accelerometry and a clinical score, TETRAS (12, 13). The TETRAS performance score has been shown to be a valid scale with excellent inter- and intrarater reliabilities and sensitivity to change (17, 18). Patients then received a single oral serving of ethanol at a total dose of 0.8 g/l of total body water, which was administered together with a sugarless, uncaffeinated drink (19). Tremor response to ethanol was documented based on changes in spectral tremor power over time using accelerometry, which was performed in 20-minute intervals after administration for 2 hours. Cut-off for being considered as an ethanol responder was set at a minimum improvement of 35% at any time point within 2 hours after ethanol administration as compared with baseline measurements. ET can vary throughout the day in untreated subjects, with a diurnal variability up to 30% of a given mean (20). The criterion for response was therefore operationally defined in a dichotomized fashion as reduction of tremor intensities larger than the known diurnal variation of ET. Therefore, a patient was considered a responder if tremor decreased by 35% or more measured by accelerometry at the time point 60 minutes after an oral ethanol administration, as compared with baseline.
Study design.
MTD was studied using a 3 + 3 single-ascending dose–escalation design. A single, open-label dose of oral OA was administered during the second day of a 3-day inpatient stay. It was given to separate cohorts of 3 patients each at the levels of 8, 16, 32, 64, and 128 mg/kg. The dose of 4 mg/kg was demonstrated as being safe (7). Therefore, for the starting dose, 8 mg/kg was selected. Doses were rounded to the nearest available 50 mg increment. The plasma level of OA described as optimal for treatment in OA as ketogenic diet was 90 μg/ml Cmax, with actual measured Cmax values ranging from 98.2 to 258.9 μg/ml (8, 9). After 128 mg/kg of 1-octanol, the Cmax was 25 μg/ml (6). The dose versus plasma-concentration relation was suggested as following a linear relationship (8). Assuming a linear relation, a Cmax of roughly 40 μg/ml would be expected after the administration of 128 mg/kg OA in our study. In addition, regarding safety and feasibility, the highest predefined dose for our study, 128 mg/kg, was still well below the highest dose level that has been defined as safe in dietary studies in children, which was 710 mg/kg (10). The primary outcome measure was defined the rate of DLT events per dose level, determining MTD.
Toxicity was monitored and documented using CTCAE. As ET is a non–life threatening condition and OA would be a symptomatic and not curative treatment, the threshold for the definition of DLT was set particularly low (grade 2 level AEs). The protocol defined its escalation and stopping rules as follows: 1 grade 2 AE would prompt the addition of another cohort of 3 patients, and the presence of 2 or more grade 2 AEs at 1 particular dose level would stop the study. Besides the CTCAE rating, the investigators graded the AEs according to the relatedness to the study drug as not related, possibly related, or definitely related. Vital signs including blood pressure, pulse, and respiratory rate were monitored. Follow-up labs included complete blood count and serum chemistry, including electrolytes, glucose level, lipid levels, renal function, liver function, and prothrombin time (PT)/international normalized ratio (INR)/partial thromboplastin time (PTT). For any signs of intoxication, a questionnaire was given to the patients at the same intervals as the AE questionnaire (0, 60, 120, 180, 300, and 600 minutes). Patients returned for a follow-up visit for the same safety assessments 1 week later. After each cohort of 3 patients completed the study, an independent external data safety monitoring board (DSMB) reviewed any recorded AEs and decided whether the study could proceed per escalation design. In addition, site monitoring was performed by an external contract research organization (KAI Research Inc.). After each cohort, the IRB was informed and had to give approval for continuation of the study.
PK.
Plasma samples were drawn from a PICC at predefined time intervals (5, 20, 40, 60, 100, 150, 210, 300, and 600 minutes after administration) and analyzed for OA content using high-performance liquid chromatography (HPLC)/mass spectrometry (MS)/MS, which was methodologically similar to that used our previous studies (6, 7). The lowest limit of quantitation was 20 ng/ml.
PD.
Efficacy was assessed by quantifying tremor power by accelerometry, tremor scores derived from digital spiral analysis, and clinical severity using the TETRAS performance score, all at baseline (–30, –15, and 0), 20, 40, 60, 100, 150, 210, 300, and 600 minutes. Accelerometry was performed as previously reported (7). Efficacy data for PD modeling were normalized to baseline, with baseline defined as the mean of multiple separate measurements at –30, –15, and 0 min, to account for natural variation of tremor.
Statistics.
Analysis of PK data was performed using standard noncompartmental analyses with Phoenix WinNonlin 6.0 (Pharsight Corp.). Nominal times were used in all calculations. Values for plasma Cmax and tmax were obtained directly from the concentration-time data. The λz measurements were obtained using log-linear regression on the terminal phase, as selected by the default WinNonlin procedure that uses the largest number of last points with nonzero concentrations that maximize the adjusted r2 of the corresponding regression (14). For some subjects, especially in the lower dose groups, the last concentration values (at 600 min) were higher than the previous ones (300 min); these values were not used in the rate constant calculations. Elimination half-lives were then calculated using the standard t1/2 = ln2/λz relationship. AUCs were calculated using the linear trapezoidal method. The AUC to last concentration (AUC0-t) was calculated from time 0 to time of the last measurable concentration (Ct). The AUC to time infinity (AUC0-∞) was calculated as AUC0-∞ = AUC0-t + Ct/λz. The apparent Vd/F based on the terminal phase was calculated as Vd/F = dose/(λz × AUC0-∞), and the apparent clearance CL/F was calculated as dose/AUC0-∞. Data of each individual subject were fitted separately, and averages ± SD were calculated for each group.
For the PK/PD modeling, which was performed as an exploratory outcome measure, a first-order absorption, first-order elimination 1-compartmental PK model linked to an effect compartment model and a sigmoid (Hill-type) Emax concentration response curve was chosen (Figure 4 and refs. 14, 15). First, concentration (PK) data from all doses were fitted with a single unified equation describing a 1-compartment model (Vd) with kabs and kel. Then an indirect link effect compartment model was used to model the effect (PD) data. This was needed to account for the temporal dissociation between the time courses of concentration and effect, as there was a clear delay in the onset of the effect compared with the time profile of the measured plasma concentration. The corresponding effect compartment concentration (Ceff) was described by a previously derived equation (21) and linked to the effect with a classical Hill-type sigmoid Emax equation (Figure 4). Because of the low number of subject per dose levels, average values per dose levels were used for all fittings — this also allowed better visual presentation.
Study approval.
The study was approved by NINDS Combined Neurosciences IRB and conducted under an FDA investigational new drug (IND) application (IND 103,671). Patients gave written informed consent prior to inclusion in the study.
Author contributions
BV was responsible for conception, organization, execution of the research project, and writing of the first draft of the manuscript. EL was responsible for organization, execution of the research project, and writing of the first draft of the manuscript. GM was responsible for organization and execution of the research project. ST and CL were responsible for execution of the research project, review, and critique of the manuscript. GG, JS, and GP were responsible for organization and execution of the research project, review and critique of the manuscript. PB was responsible for execution, review, and critique of PK/PD modeling and statistical analysis and of the manuscript. DH and MH were responsible for conception, organization of the research project and design, review and critique of the statistical analysis and of the manuscript.
Supplementary Material
Acknowledgments
The study was supported by the NINDS Intramural Program with supplemental funding from TG Therapeutics Inc. B. Voller and E. Lines were both sponsored by TG Therapeutics Inc. B. Voller worked as a contractor and as a special volunteer at the NIH/NINDS in accordance with the Cooperative Research and Development Agreement (CRADA 02036). E. Lines worked through the Postbaccalaureate Intramural Research Training Award Program at NIH/NINDS.
Footnotes
Role of funding source: The sponsor TG Therapeutics Inc. (formerly Manhattan Pharmaceuticals Inc.) only provided financial support. They had no role in designing the study; in collection, analysis, or interpretation of data; in writing the manuscript; or in the decision to submit the paper for publication. The sponsor had the right to give permission before publication, but there was no change in the manuscript after review by the sponsor.
Conflict of interest: M. Hallett may accrue revenue on US Patent no. 6,780,413 B2 and US Patent no. 7,407,478, and he has received licensing fee payments from the NIH for the latter patent. Additionally, he has received research funds from BCN Peptides SA, Medtronic, the Parkinson Alliance, UniQure, Merz, and Allergan.
Reference information:J Clin Invest. 2016;126(4):1451–1457. doi:10.1172/JCI83621.
Contributor Information
Bernhard Voller, Email: bernhard.voller@meduniwien.ac.at.
Emily Lines, Email: elines@stanford.edu.
Gayle McCrossin, Email: mccrossing@ninds.nih.gov.
Sule Tinaz, Email: aysesule.tinaz@nih.gov.
Codrin Lungu, Email: codrin.lungu@nih.gov.
George Grimes, Email: george.grimes@nih.gov.
Judith Starling, Email: JStarling@cc.nih.gov.
Gopal Potti, Email: gopal.potti@nih.gov.
Peter Buchwald, Email: pbuchwald@med.miami.edu.
Dietrich Haubenberger, Email: dietrich.haubenberger@nih.gov.
Mark Hallett, Email: hallettm@ninds.nih.gov.
References
- 1.Louis ED, Ottman R, Hauser WA. How common is the most common adult movement disorder? Estimates of the prevalence of essential tremor throughout the world. Mov Disord. 1998;13(1):5–10. doi: 10.1002/mds.870130105. [DOI] [PubMed] [Google Scholar]
- 2.Louis ED, et al. Correlates of functional disability in essential tremor. Mov Disord. 2011;16(5):914–920. doi: 10.1002/mds.1184. [DOI] [PubMed] [Google Scholar]
- 3.Lou JS, Jankovic J. Essential tremor: clinical correlates in 350 patients. Neurology. 1991;41(2 pt 1):234–238. doi: 10.1212/wnl.41.2_part_1.234. [DOI] [PubMed] [Google Scholar]
- 4.Koller WC, Busenbark K, Miner K. The relationship of essential tremor to other movement disorders: report on 678 patients. Essential Tremor Study Group. Ann Neurol. 1994;35(6):717–723. doi: 10.1002/ana.410350613. [DOI] [PubMed] [Google Scholar]
- 5.Deuschl G, Wenzelburger R, Löffler K, Raethjen J, Stolze H. Essential tremor and cerebellar dysfunction clinical and kinematic analysis of intention tremor. Brain. 2000;123(pt 8):1568–1580. doi: 10.1093/brain/123.8.1568. [DOI] [PubMed] [Google Scholar]
- 6.Nahab FB, et al. An open-label, single-dose, crossover study of the pharmacokinetics and metabolism of two oral formulations of 1-octanol in patients with essential tremor. Neurotherapeutics. 2011;8(4):753–762. doi: 10.1007/s13311-011-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haubenberger D, et al. Octanoic acid in alcohol-responsive essential tremor: A randomized controlled study. Neurology. 2013;80(10):933–940. doi: 10.1212/WNL.0b013e3182840c4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sills MA, Forsythe WI, Haidukewych D. Role of octanoic and decanoic acids in the control of seizures. Arch Dis Child. 1986;61(12):1173–1177. doi: 10.1136/adc.61.12.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sills MA, Forsythe WI, Haidukewych D, MacDonald A, Robinson M. The medium chain triglyceride diet and intractable epilepsy. Arch Dis Child. 1986;61(12):1168–1172. doi: 10.1136/adc.61.12.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Traul KA, Driedger A, Ingle DL, Nakhasi D. Review of the toxicologic properties of medium-chain triglycerides. Food Chem Toxicol. 2000;38(1):79–98. doi: 10.1016/S0278-6915(99)00106-4. [DOI] [PubMed] [Google Scholar]
- 11. [May 27, 2010]; [February 18, 2016];CTCAE Files. http://evs.nci.nih.gov/ftp1/CTCAE/About.html
- 12.Tintner R, Tremor Research Group The Tremor R¡ating Scale (TRS) Mov Disord. 2004;19(9):1131–1132. [Google Scholar]
- 13.Elble R, et al. The essential tremor rating assessment scale (TETRAS) Mov Disord. 2008;23(suppl 1): doi: 10.1002/mdc3.12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gabrielsson J, Weiner D, eds. Pharmacokinetic and Pharmacodynamic Analysis: Concepts and Applications. 3rd ed. Boca Raton, Florida, USA: CRC Press; 2001. [Google Scholar]
- 15.Derendorf H, Meibohm B. Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives. Pharm Res. 1999;16(2):176–185. doi: 10.1023/A:1011907920641. [DOI] [PubMed] [Google Scholar]
- 16.Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord. 1998;13(suppl 3):2–23. doi: 10.1002/mds.870131303. [DOI] [PubMed] [Google Scholar]
- 17.Elble R, et al. Reliability of a new scale for essential tremor. Mov Disord. 2012;27(12):1567–1569. doi: 10.1002/mds.25162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voller B, et al. Alcohol challenge and sensitivity to change of the Essential Tremor Rating Assessment Scale. Mov Disord. 2014;29(4):555–558. doi: 10.1002/mds.25667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watson PE. Total body water and blood alcohol levels: updating the fundamentals. In: Crow K, Batt R, eds. Human Metabolism of Alcohol. Vol. 1. Oxon, United Kingdom: Taylor & Francis; 1989:53–54. [Google Scholar]
- 20.Cleeves L, Findley LJ. Variability in amplitude of untreated essential tremor. J Neurol Neurosurg Psychiatry. 1987;50(6):704–708. doi: 10.1136/jnnp.50.6.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colburn WA. Simultaneous pharmacokinetic and pharmacodynamic modeling. J Pharmacokinet Biopharm. 1981;9(3):367–388. doi: 10.1007/BF01059272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.