

Abstract
Objective:
To characterize 2 novel TRPV4 mutations in 2 unrelated families exhibiting the Charcot-Marie-Tooth disease type 2C (CMT2C) phenotype.
Methods:
Direct CMT gene testing was performed on 2 unrelated families with CMT2C. A 4-fold symmetric tetramer model of human TRPV4 was generated to map the locations of novel TRPV4 mutations in these families relative to previously identified disease-causing mutations (neuropathy, skeletal dysplasia, and osteoarthropathy). Effects of the mutations on TRPV4 expression, localization, and channel activity were determined by immunocytochemical, immunoblotting, Ca2+ imaging, and cytotoxicity assays.
Results:
Previous studies suggest that neuropathy-causing mutations occur primarily at arginine residues on the convex face of the TRPV4 ankyrin repeat domain (ARD). Further highlighting the key role of this domain in TRPV4-mediated hereditary neuropathy, we report 2 novel heterozygous missense mutations in the TRPV4-ARD convex face (p.Arg237Gly and p.Arg237Leu). Generation of a model of the TRPV4 homotetramer revealed that while ARD residues mutated in neuropathy (including Arg237) are likely accessible for intermolecular interactions, skeletal dysplasia–causing TRPV4 mutations occur at sites suggesting disruption of intramolecular and/or intersubunit interactions. Like previously described neuropathy-causing mutations, the p.Arg237Gly and p.Arg237Leu substitutions do not alter TRPV4 subcellular localization in transfected cells but cause elevations of cytosolic Ca2+ levels and marked cytotoxicity.
Conclusions:
These findings expand the number of ARD residues mutated in TRPV4-mediated neuropathy, providing further evidence of the central importance of this domain to TRPV4 function in peripheral nerve.
Mutations in the transient receptor potential vanilloid 4 gene (TRPV4), encoding a Ca2+-permeable cation channel, result in 3 autosomal dominant diseases of peripheral nerve: Charcot-Marie-Tooth disease type 2C (CMT2C), congenital distal spinal muscular atrophy, and scapuloperoneal spinal muscular atrophy.1–9 While these diseases are clinically heterogeneous, a majority of patients have weakness of limb and vocal fold muscles.10,11 In addition to hereditary neuropathy, dominant mutations in TRPV4 are associated with forms of skeletal dysplasia and osteoarthropathy.12 TRPV4 mutations have also been described in individuals manifesting both skeletal dysplasia and either peripheral neuropathy or fetal akinesia.13,14 Our understanding of how TRPV4 mutations result in such diverse disease phenotypes is currently limited, although several in vitro studies suggest that neuropathy- and skeletal dysplasia–causing mutants exhibit normal expression levels and localization but increased channel activity.12
TRPV4 functions primarily as a homotetrameric channel expressed at the plasma membrane.15 The cytoplasmic N-terminus of each protomer (figure 1A) contains a prominent ankyrin repeat domain (ARD) comprising 6 ankyrin repeats, a motif mediating protein–protein/protein–ligand interactions.16 Structural analyses indicate that neuropathy-causing TRPV4 mutations occur primarily at arginine residues clustered on the ARD convex face (figure 1A).4,5,17 In contrast, mutations associated with skeletal dysplasia occur throughout the protein, with the exception of the ARD convex face.12 Osteoarthropathy-causing mutations reside within the third finger loop of the ARD.18
Figure 1. Two novel CMT2C-causing mutations identified at a highly conserved arginine residue in the TRPV4-ARD.

(A) Schematic illustrating the principal TRPV4 protein domains and the locations of neuropathy-causing mutations. *Novel mutations identified in the present study in 2 families with the Charcot-Marie-Tooth disease type 2C (CMT2C) phenotype. †A mutation found in a family with CMT2C and pronounced short stature.6 ‡A mutation described in a single patient with CMT2 and skeletal dysplasia,3 as well as in a single patient with axonal neuropathy and both the p.Val620Ile mutation and a second p.Arg151Trp TRPV4 variant.37 (B) Pedigrees of families 1 and 2 demonstrating affected individuals (white = unaffected, black = affected, gray = unclear disease status, *DNA collected). (C) The Arg237 residue of TRPV4 has been highly conserved through vertebrate evolution. ARD = ankyrin repeat domain.
In this article, we report 2 novel TRPV4 mutations in 2 families exhibiting the CMT2C phenotype. Both mutations occur at an arginine residue in the ARD (Arg237) not previously linked to peripheral neuropathy.
METHODS
Participants and molecular genetic analyses.
Participants were evaluated at Stanford University Medical Center and the University of Washington Medical School. Weakness was graded as mild if the Medical Research Council scale score was ≥4/5, moderate if ≥3 and <4, and severe if ≤2. Sensory loss was determined to be moderate or mild by the examiner based on vibration testing. Genomic DNA was isolated from blood leukocytes using standard extraction protocols and examined by direct CMT gene testing.
Homology model generation.
The ARD is currently the only TRPV4 domain for which a high-resolution structure has been determined.5,17 In this study, SWISS-MODEL19 was used to generate a 4-fold symmetric tetramer model of human TRPV4 (residues 148–755 of 871) using the apo rat TRPV1 electron cryomicroscopy structure as a template (PDBID 3J5P20). Rat TRPV1-ARD and human TRPV4-ARD have 56% sequence identity, and their core Cα atoms have a root mean square deviation of 1.6 Å, allowing us to place the experimentally determined TRPV4-ARD crystal structure with high confidence within our homology model. Therefore, after generation of a full model, the SWISS-MODEL–generated ARD was removed and replaced with the experimentally determined x-ray crystal structure of the human TRPV4-ARD (PDBID 4DX2, chain B, residues 148–38917) after alignment to residues 350–389 (ankyrin repeat 6) in Coot.21 To relieve any resulting clashes, 10 rounds of geometry minimization of residues 390–470 and 635–666 were performed in phenix.refine22 with the 4-fold symmetry restrained. The C-terminal β-strand (residues 752–762 in the 3J5P TRPV1 structure) was then added to the homology model to illustrate the position of this structural feature. PyMOL was used for model analysis and image generation.
Immunocytochemistry.
HEK293T cells were cultured in Dulbecco's Modified Eagle's Medium supplemented with 10% (vol/vol) fetal calf serum at 37°C with 6% CO2. Cells were transfected with pcDNA3.1-TRPV4-FLAG (wild-type [WT] or mutant) constructs using Lipofectamine LTX (Life Technologies, Grand Island, NY) and then replated onto poly-l-ornithine–coated coverslips 4.5 hours after transfection in the absence or presence of the TRPV4-specific antagonist HC-067047 (Sigma-Aldrich, St. Louis, MO; 5 μM). At 24 hours after transfection, cells were fixed in 4% paraformaldehyde in 0.1 M Tris-buffered saline for 15 minutes at room temperature and then processed as described previously.5
Immunoblot analyses.
HEK293T cells were lysed 24 hours after transfection in radioimmunoprecipitation assay buffer (Sigma-Aldrich) supplemented with protease inhibitors (Sigma-Aldrich) and sonicated. Protein lysates were resolved on 4%–15% TGX gels (Bio-Rad Laboratories, Inc, Hercules, CA) and transferred to polyvinylidene fluoride membranes. Primary antibodies used were rabbit anti-FLAG (1:1,000; Cell Signaling Technology, Danvers, MA; #2368) and rabbit anti-β-actin (1:1,000; Cell Signaling Technology; #4970), followed by a donkey anti-rabbit-HRP–conjugated secondary antibody (1:200,000; GE Healthcare Life Sciences, Pittsburgh, PA). Membranes were developed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Waltham, MA).
Calcium imaging and cytotoxicity assays.
HEK293T and MN-1 (mouse motor neuron–neuroblastoma fusion) cells were cotransfected with pcDNA3-mCherry and pcDNA3.1-TRPV4-FLAG (WT or mutant) constructs using Lipofectamine LTX. HC-067047 (5 μM) was added 4.5 hours after transfection. Calcium imaging was performed on a Zeiss Axio Observer.Z1 inverted microscope equipped with a Lambda DG-4 (Sutter Instrument Company, Novato, CA) wavelength switcher. Cells were bath-loaded with Fura-2 AM (8 μM; Life Technologies) for 30 minutes at 37°C in calcium-imaging buffer (150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, 10 mM HEPES, pH 7.4) containing 0.04% Pluronic F-127. Transfected cells were identified for analysis by mCherry expression. Cell death assays were performed 24 hours after transfection using the Cytotoxicity Detection KitPLUS (LDH) kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions. Cytotoxicity in this assay is calculated by subtracting the absorbance of a background control (normal medium) from the absorbance of the experimental samples (supernatant from transfected cells). Results are expressed as a percentage relative to the absorbance of a high control (supernatant from cells treated with lysis buffer). Statistical significance was determined using the Mann-Whitney U test (calcium imaging) or unpaired Student t test (cytotoxicity assays).
RESULTS
Clinical findings.
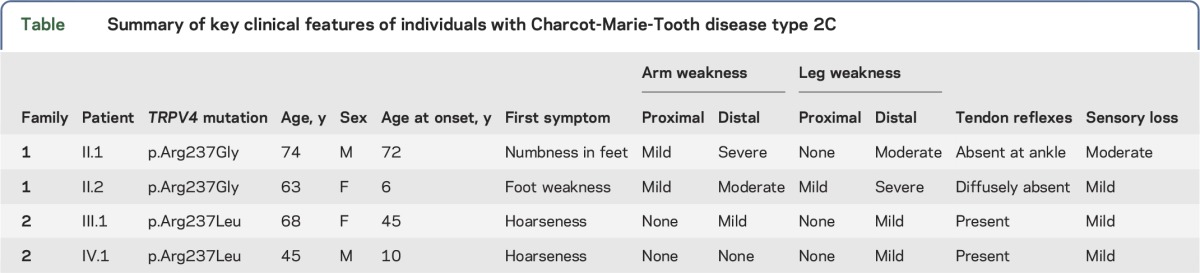
We examined 4 individuals from 2 families (figure 1B) exhibiting features consistent with the CMT2C phenotype,10,23 including distal limb weakness, hoarseness of voice, and vocal fold paresis (table). Three of the 4 individuals underwent nerve conduction studies, which revealed axonal neuropathy. Laryngoscopy in 2 patients confirmed vocal focal paresis. In individual IV.1 of family 2, laryngoscopy revealed bilateral vocal fold paresis with vocal fold movement at 15%–20% of normal. Vocal fold EMG in this individual confirmed a reduction in motor unit number, consistent with chronic denervation. None of the participants exhibited evidence of skeletal abnormalities. Family 2 has a 3-generation history of limb weakness and hoarseness on the maternal side (figure 1B).
Table.
Summary of key clinical features of individuals with Charcot-Marie-Tooth disease type 2C
Detection of novel mutations in TRPV4.
Genetic testing of family 1 revealed a heterozygous missense mutation (c.709C>G) in exon 4 of TRPV4 (RefSeq accession number NM_021625.4) in both examined individuals. This nucleotide change results in an arginine to glycine substitution at amino acid 237 (p.Arg237Gly) of TRPV4. A heterozygous sequence change (c.710G>T) affecting amino acid 237 of TRPV4 was also detected in the 2 examined individuals in family 2, resulting in an arginine to leucine substitution (p.Arg237Leu) at this residue. Neither nucleotide substitution is present in the NHLBI Exome Sequencing Project Exome Variant Server, the 1000 Genomes Project, Exome Aggregation Consortium, or dbSNP. Like most known neuropathy-causing TRPV4 mutations, Arg237 is located in the TRPV4-ARD (third ankyrin repeat; figure 1A) and is highly conserved across vertebrate species (figure 1C). In silico analyses predict both mutations to be deleterious (PolyPhen-2 HumVar scores: p.Arg237Gly, 0.785; p.Arg237Leu, 0.713) and disease-causing (MutationTaster scores: p.Arg237Gly, 1; p.ArgR237Leu, >0.999).
Structural analyses localize Arg237 to the convex face of the ARD.
Ankyrin repeats comprise inner and outer helices followed by a connecting finger loop, and the 6 repeats of the TRPV4-ARD together form a hand-shaped domain with a concave palm surface and a convex surface analogous to the back of the hand.16,17 Analysis of the high-resolution crystal structure of the human TRPV4-ARD17 reveals that Arg237 is situated within the second finger loop, near 2 other arginine residues mutated in CMT2C (Arg232 and Arg269; figure 2A). Like other neuropathy-causing mutations in the TRPV4-ARD, Arg237 is situated on the convex face of the domain.
Figure 2. Arg237 forms part of a neuropathy-causing mutation cluster, distinct from clusters of skeletal dysplasia–causing mutations.

In all panels, Arg237 is represented in magenta; residues mutated in neuropathy, skeletal dysplasia, and osteoarthropathy are shown in blue (R232, R269, R315, R316), yellow (E183, K197, L199, Q239, E278, T295, I331, D333, V342, K407, F471, L523, Y591, F592, R594, L596, G600, Y602, I604, R616, F617, L618, M625, L709, A716), and orange (G270, R271, F273), respectively. Mutated residues reported as causing mixed nerve/bone disease are shown in green (A217, K276, E278, S542, V620, T740). (A) Ribbon diagram of the TRPV4-ARD (residues 148–392) with disease-causing mutation positions marked with spheres. Arg237 is located on the convex face of the ankyrin repeat domain (ARD) within the previously identified cluster of arginine residues (shown as sticks) mutated in hereditary neuropathy. (B) Homology model of human TRPV4 based on the TRPV1 structure.20 Known disease-causing mutation positions marked with spheres. One subunit of the TRPV4 tetramer is shown in cyan with the remainder shown in gray. The plasma membrane is denoted with black lines, and the arrow points to a cluster of skeletal dysplasia–causing mutations near the lower channel gate. As in figure 1, †indicates a mutation found in a family with Charcot-Marie-Tooth disease type 2C and pronounced short stature6 and ‡indicates a mutation described in a single patient with CMT2 and skeletal dysplasia,3 as well as in a single patient with axonal neuropathy and both the p.Val620Ile mutation and a second p.Arg151Trp TRPV4 variant.37 (C) Top-down view of the ion channel (as seen from the extracellular side) in surface representation. The transmembrane domain is colored cyan and the cytosolic regions gray. The cluster of neuropathy-causing mutations (indicated in 1 subunit by the arrow) is oriented toward the intracellular leaflet of the plasma membrane. (D) Expanded view of the region indicated in panel B showing 1 subunit in cyan ribbon representation and the neighboring subunit in surface representation. Several skeletal dysplasia–causing mutations occur at this intersubunit interface.
The positioning of the ARD relative to the rest of the assembled channel and the plasma membrane has not been previously investigated. Like other TRPV channels, functional TRPV4 channels are principally homotetramers.24 We used the recently described electron cryomicroscopy structure of the TRPV1 ion channel20 to generate a model of the human TRPV4 homotetramer (figure 2B) that incorporates the experimentally determined TRPV4-ARD. An overall high sequence identity between TRPV1 and TRPV4 (∼45%) allows us to map the positions of most disease-causing TRPV4 mutations in other regions onto this model with high confidence.25 Several interesting observations arise from this mapping. First, the convex face of the ARD, on which neuropathy-causing mutations cluster, is on the outer surface of the TRPV4 tetramer, facing toward the cytosol at an angle of ∼45° relative to the plasma membrane (figure 2, B and C). This orientation suggests that the ARD convex face would likely be readily accessible for protein–protein/protein–ligand interactions. Second, skeletal dysplasia–causing mutations form 2 main clusters within the channel (figure 2, B and D). The larger of these clusters is situated within the bottom half of the transmembrane region near the lower channel gate, consistent with the finding that many of these mutations increase constitutive channel activity.26 The second cluster occurs on the concave surface of the ARD. In the tetrameric channel, this surface participates in intersubunit interactions connecting the ARD of one protomer to a small β-sheet structure arising from the ARD-to-transmembrane domain linker and the C-terminus of the adjacent protomer (figure 2D). The location of this second cluster suggests that disruption of this intersubunit interface may also increase constitutive channel activity. Third, osteoarthropathy-causing mutants are clustered in the third finger loop of the ARD,18 which interacts with the β-sheet structure on one side but is exposed to the solvent on the other. Although no clear conclusion can be drawn from our model, this loop can be flexible,17 and it is plausible that the mutations affect interactions with a regulator or trafficking chaperone that results in reduced cell surface localization, as previously observed.18 No clear localization patterns were evident for mutations thought to cause both nerve and bone disease (green residues, figure 213,14).
p.Arg237Gly and p.Arg237Leu mutations alter TRPV4 function.
To gain insights into the functional consequences of the p.Arg237Gly and p.Arg237Leu amino acid changes, we performed a series of in vitro studies examining the effects of heterologous expression of these mutant channels. To assess the magnitude of any observed effects, experiments were performed in parallel with known disease-causing TRPV4 mutants (neuropathy, p.Arg269Cys; skeletal dysplasia, p.Ile331Phe; osteoarthropathy, p.Arg271Pro; the locations of these residues are shown in figure 2A). Immunocytochemical analyses revealed that each of the neuropathy-causing mutants examined exhibited a subcellular distribution comparable to that of the WT channel, localizing to both the cytoplasm and the plasma membrane (figure 3A). In contrast, the skeletal dysplasia– and osteoarthropathy-causing mutants localized primarily to the cytoplasm (figure 3Avi). Incubation of transfected cells in a TRPV4 channel antagonist (HC-067047) increased plasma membrane expression of both TRPV4WT, as described previously,27 and the mutant channels, including p.Ile331Phe and p.Arg271Pro (figure 3B). To examine effects on channel function, we performed ratiometric calcium imaging to measure cytosolic calcium levels ([Ca2+]i) of transiently transfected MN-1 (a motor neuron–like cell line) and HEK293T cells 24 hours after transfection. Expression of both p.Arg237Gly and p.Arg237Leu resulted in significant elevations of basal [Ca2+]i levels relative to the WT channel in both cell types, of comparable magnitude to known disease-causing TRPV4 mutants (figure 3, C and D). HEK293T cells expressing p.Arg237Gly and p.Arg237Leu also exhibited significantly increased levels of cytotoxicity relative to TRPV4WT-transfected cells (figure 3E). Both the effects on [Ca2+]i and cell viability were abrogated by application of HC-067047 (figure 3, C–E), consistent with a mutation-dependent increase in channel activity.4,5,7,28 It is interesting that despite causing significant elevations of basal [Ca2+]i, expression of the osteoarthropathy-causing mutant p.Arg271Pro was not associated with cytotoxicity (figure 3E). To confirm that the elevations in [Ca2+]i and cytotoxicity caused by the neuropathy- and skeletal dysplasia–causing mutants did not result from increased expression levels, we performed immunoblot analyses of whole-cell lysates 24 hours after transfection. These analyses revealed reduced expression of these mutants relative to TRPV4WT (figure 3F), likely reflecting their increased cytotoxicity. Immunoblots performed on cells incubated in HC-067047 to abrogate toxicity, however, demonstrated equivalent protein expression levels across the WT and mutant channels (figure 3G). Taken together, the above results suggest that the p.Arg237Gly and p.Arg237Leu mutants traffic normally to the plasma membrane but exhibit a gain of function in channel activity.
Figure 3. p.Arg237Gly and p.Arg237Leu mutations do not alter the subcellular localization of TRPV4 but cause elevations of basal [Ca2+]i and significant cytotoxicity.

(A, B) Representative images of HEK293T cells transfected with wild-type (WT) or mutant TRPV4-FLAG and labeled immunocytochemically for FLAG (red) to reveal the subcellular distribution of TRPV4. Cells were costained for the transferrin receptor (green) and with DAPI (blue) to delineate individual cells and nuclei, respectively. These studies included examination of known neuropathy–causing (p.Arg269Cys), skeletal dysplasia–causing (p.Ile331Phe), and osteoarthropathy-causing (p.Arg271Pro) TRPV4 mutations. Transfected cells were cultured in the absence (A) or presence (B) of the TRPV4 antagonist HC-067047 (5 μM). All images are shown at the same magnification, and the scale bar represents 20 μm. (C, D) Quantification of baseline Fura-2 ratios in transiently transfected MN-1 (C) and HEK293T (D) cells reveals significant increases in basal [Ca2+]i with expression of each mutant channel that are blocked by the TRPV4 antagonist HC-067047 (5 μM). (E) Quantification of cell death in HEK293T cells 24 hours after transfection using an LDH cytotoxicity assay. Cells expressing TRPV4 mutants, with the exception of p.Arg271Pro, show significantly increased levels of cytotoxicity relative to TRPV4WT-expressing cells; this increase is abrogated by HC-067047 (5 μM). **p < 0.001, ***p < 0.0001. Data in C and D are averaged from >100 transfected cells, while data in E are averaged from 3 independent transfections. Error bars, SEM. (F, G) Immunoblotting of whole lysates from HEK293T cells 24 hours after transfection incubated in the absence (F) or presence (G) of HC-067047 (5 μM).
DISCUSSION
The structure of ARDs with a conserved hydrophobic core and variable exposed residues enables these domains to interact with binding partners (inter- and intramolecular) with high specificity and affinity.16 While these properties enable ARDs to perform diverse cellular functions, mutations in the ARDs of a number of proteins are associated with disease, including autism spectrum disorder (SHANK3), melanoma (p16), keratoderma (KANK2), and bare lymphocyte syndrome (RFXANK).29–32 To date, 8 of 12 neuropathy-causing mutations in TRPV4 occur within the TRPV4-ARD (figure 1A). The identification in the present study of 2 additional TRPV4-ARD mutations (p.Arg237Gly and p.Arg237Leu) in 2 families with the CMT2C phenotype further underscores the importance of this domain in TRPV4-mediated neuropathy.
Like previously described neuropathy-causing mutations in the TRPV4-ARD, p.Arg237Gly and p.Arg237Leu occur at an exposed arginine residue on the convex face of the ARD. Six of the 10 exposed arginine residues present on the convex face of TRPV4-ARD have now been associated with neuropathy. This cluster of arginine residues is highly conserved across vertebrate species but is not characteristic of the ARDs of other TRPV channel family members.1,3–5,8 We previously determined experimentally that the neuropathy-causing mutations R232C, R269C, R269H, R315W, and R316C (all surface-exposed arginine mutations) do not substantially alter the fold or stability of the human TRPV4-ARD, which suggests that the functional consequence of these mutations may instead lie in interactions with binding partners.17 While the concave surface of the TRPV4-ARD has been shown to bind ATP, calmodulin,5,17,33 and PI(4,5)P2,34 no interactors of the ARD convex face have yet been identified. By generating a model of the TRPV4 homotetramer, we can better understand the functional consequences of these mutations by understanding them in the context of a structural model of the full channel. In this study, we showed that the ARD convex face is directed toward the cytosol and would thus likely be readily accessible for protein–protein/protein–ligand interactions. Furthermore, the orientation of this surface toward the plasma membrane suggests that the ARD convex surface may interact principally with membrane-associated factors. As the TRPV1 electron cryomicroscopy structure20 on which our model is based did not include regions N-terminal to the ARD, it remains unclear how neuropathy-causing mutations in N-terminal regions of TRPV4 (figure 1A) localize relative to the ARD convex face. It is possible that the N-terminal region of TRPV4 influences access to binding partners of the ARD, as demonstrated for autism-associated mutations in the postsynaptic scaffolding protein SHANK3.35 It is also likely that the channel adopts other conformations not yet seen experimentally, but the relative orientation of the ARD in relation to the membrane is restrained by extensive interactions with other regions of the channel, suggesting that it is unlikely to be altered dramatically.
Generation of a model of the TRPV4 homotetramer also enabled the mapping of TRPV4 mutations associated with skeletal dysplasia and osteoarthropathy. Skeletal dysplasia–causing mutations were found to occur primarily within 2 clusters: a large cluster situated within the bottom half of the transmembrane region near the lower channel gate and a second cluster on the concave surface of the ARD that appears to participate in intersubunit interactions. Together, these findings suggest that skeletal dysplasia–causing mutations may influence TRPV4 channel activity by altering intramolecular interactions within the assembled channel. As described previously,18 osteoarthropathy-causing mutations localize to the tip of the ARD third finger loop. In the tetrameric channel model, the corresponding residues are part of a potentially flexible loop and could be accessible to regulatory partners. In addition, a lack of any clear localization patterns for mutations thought to cause both nerve and bone disease (green residues, figure 2) indicates that factors not accounted for by this model play an important role in TRPV4 protein function.
Heterologous expression studies indicate that p.Arg237Gly and p.Arg237Leu exhibit a subcellular distribution equivalent to TRPV4WT, suggesting that these mutations do not influence channel assembly or trafficking. Expression of these mutants, however, results in elevations of [Ca2+]i and significant cytotoxicity, both of which are abrogated by application of a TRPV4 antagonist, consistent with increased channel activity.4,5,7,8,28 The magnitude of these effects is comparable to that of known disease-causing TRPV4 mutants. Together, these results suggest that both the p.Arg237Gly and p.Arg237Leu mutations confer a gain of function in channel activity. While heterologous expression of both skeletal dysplasia– and osteoarthropathy-causing mutants (p.Ile331Phe and p.Arg271Pro, respectively) also resulted in significant increases in basal [Ca2+]i, this was not associated with significant cytotoxicity in p.Arg271Pro-expressing cells. Similarly, expression of the osteoarthropathy mutant p.Phe273Leu in porcine chondrocytes results in elevations of [Ca2+]i but does not cause the channel activity–dependent increases in gene expression characteristic of skeletal dysplasia–causing mutants.36 Together, these results suggest that factors in addition to, or independent of, increased Ca2+ influx may contribute to disease pathogenesis in some forms of TRPV4 channelopathy.
A clear understanding of how different mutations in TRPV4 engender the diverse disease phenotypes characteristic of patients is only slowly emerging.10,12 Elucidation of the etiologies underlying the different forms of TRPV4 channelopathy is important because TRPV4 is expressed at the plasma membrane and channel antagonism could therefore represent a therapeutic strategy, where appropriate. The identification of 2 novel mutations in the TRPV4-ARD highlights the key role of this domain in TRPV4-mediated neuropathy. Our findings suggest that molecular pathogenesis in these neuropathies may be driven by alterations in interactions of the ARD convex face with as-yet-unidentified binding partners. Identification of these interactors and characterization of their effects on channel function will be central to determining how mutations in TRPV4 precipitate peripheral axonal degeneration.
GLOSSARY
- ARD
ankyrin repeat domain
- CMT2C
Charcot-Marie-Tooth disease type 2C
- WT
wild-type
AUTHOR CONTRIBUTIONS
J.M. Sullivan: study conceptualization and design, acquisition of data, analysis and interpretation, and drafting and revision of manuscript. C.M. Zimanyi: acquisition of data, analysis and interpretation, and revision of manuscript. W. Aisenberg and B. Bears: acquisition of data and revision of manuscript. D.-H. Chen and J.W. Day: acquisition of clinical data and revision of manuscript. T.D. Bird: study conceptualization and revision of manuscript. C.E. Siskind: study conceptualization and revision of manuscript. R. Gaudet: study conceptualization and design, analysis and interpretation, and revision of manuscript. C.J. Sumner: primary investigator of the study, study conceptualization and design, analysis and interpretation, and drafting and revision of manuscript.
STUDY FUNDING
Supported by NIH grant R21NS08757902 to C.J.S., MDA grant 277551 to C.J.S., NIH grant R01GM081340 to R.G., and a Postdoctoral Fellowship from the Jane Coffin Childs Memorial Fund for Medical Research to C.M.Z.
DISCLOSURE
J.M. Sullivan reports no disclosures. C.M. Zimanyi has received research support from Jane Coffin Childs Fund. W. Aisenberg and B. Bears report no disclosures. D.-H. Chen holds a patent for identifying genetic mutations in subjects susceptible to ataxia neurologic diseases; has received research support from NIH (R01 NS069719-01) and National Ataxia Foundation (FA100588); and has received license fee payments from Athena Diagnostics, Inc. J.W. Day has received travel funding from Cytokinetics, Biogen, Roche Inc, Isis Pharmaceuticals, the Spinal Muscular Atrophy Foundation, Parent's Project Muscular Dystrophy, the Myotonic Dystrophy Foundation, and the American Association of Pediatrics; holds patents for Myotonic Dystrophy type 2 genetic testing and Spinocerebellar Ataxia type 5 genetic testing; has been an employee of Stanford University; has been a consultant for Isis Pharmaceuticals, Biogen, Cytokinetics Inc, and Sarepta Therapeutics; has received research support from Genzyme Corporation, Isis Pharmaceuticals, Sarepta Pharmaceuticals, NIH/NINDS (NS058901), the Muscular Dystrophy Association, the Myotonic Dystrophy Foundation, and the Spinal Muscular Atrophy Foundation; and has received royalty payments for DM2 genetic testing and SCA5 genetic testing from Athena Diagnostics. T.D. Bird holds a patent for Genetic testing technology for CMT1C and SCA14 and has received research support from the Department of Veterans' Affairs. C.E. Siskind has received speaker honoraria from the National Society of Genetic Counselors and Invitae and has received research support from the Muscular Dystrophy Association. R. Gaudet has served on the editorial boards of Pflügers Archiv—European Journal of Physiology and the Journal of Molecular Biology and has received research support from NIH grant 1R01GM081340 and NIH grant 2R01DC002281. C.J. Sumner has served on the scientific advisory boards of Families of Spinal Muscular Atrophy and Avexis; has received travel funding/speaker honoraria from the Massachusetts General Hospital, the European NeuroMuscular Centre, Thomas Jefferson University, NYU School of Medicine, Carver College of Medicine, and the University of California Los Angeles School of Medicine; has served on the editorial boards of Experimental Neurology and the Annals of Clinical and Translational Neurology; coholds a patent for Modulation of SMN expression; has been a consultant for Avexis, Pfizer, Isis Pharmaceuticals, Roche, PTC Pharmaceuticals, Biogen Idec, and the SMA Foundation; and has received research support from NIH/NINDS R211NS087579, Muscular Dystrophy Association 2777551, Fight SMA Foundation and Gwendolyn Strong Foundation Emerging Investigator Award, Spinal Muscular Atrophy Research Team (SMART), Families of SMA Research Award, and the Spinal Muscular Atrophy Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Auer-Grumbach M, Olschewski A, Papić L, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet 2010;42:160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiorillo C, Moro F, Brisca G, et al. TRPV4 mutations in children with congenital distal spinal muscular atrophy. Neurogenetics 2012;13:195–203. [DOI] [PubMed] [Google Scholar]
- 3.Zimoń M, Baets J, Auer-Grumbach M, et al. Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies. Brain 2010;133:1798–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landouré G, Sullivan JM, Johnson JO, et al. Exome sequencing identifies a novel TRPV4 mutation in a CMT2C family. Neurology 2012;79:192–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landouré G, Zdebik AA, Martinez TL, et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet 2010;42:170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen DH, Sul Y, Weiss M, et al. CMT2C with vocal cord paresis associated with short stature and mutations in the TRPV4 gene. Neurology 2010;75:1968–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein CJ, Shi Y, Fecto F, et al. TRPV4 mutations and cytotoxic hypercalcemia in axonal Charcot-Marie-Tooth neuropathies. Neurology 2011;76:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng HX, Klein CJ, Yan J, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet 2010;42:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Echaniz-Laguna A, Dubourg O, Carlier P, et al. Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy. Neurology 2014;82:1919–1926. [DOI] [PubMed] [Google Scholar]
- 10.McEntagart M. TRPV4 axonal neuropathy spectrum disorder. J Clin Neurosci 2012;19:927–933. [DOI] [PubMed] [Google Scholar]
- 11.Sullivan JM, Lloyd TE, Sumner CJ. Hereditary channelopathies caused by TRPV4 mutations. In: Weiss N, Koschak A, eds. Pathologies of Calcium Channels. Berlin, Heidelberg: Springer Verlag; 2014:413–440. [Google Scholar]
- 12.Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep 2013;14:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho TJ, Matsumoto K, Fano V, et al. TRPV4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genet A 2012;158A:795–802. [DOI] [PubMed] [Google Scholar]
- 14.Unger S, Lausch E, Stanzial F, et al. Fetal akinesia in metatropic dysplasia: the combined phenotype of chondrodysplasia and neuropathy? Am J Med Genet A 2011;155A:2860–2864. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Elias A, Mrkonjic S, Jung C, Pardo-Pastor C, Vicente R, Valverde MA. The TRPV4 channel. Handb Exp Pharmacol 2014;222:293–319. [DOI] [PubMed] [Google Scholar]
- 16.Gaudet R. A primer on ankyrin repeat function in TRP channels and beyond. Mol Biosyst 2008;4:372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inada H, Procko E, Sotomayor M, Gaudet R. Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry 2012;51:6195–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamandé SR, Yuan Y, Gresshoff IL, et al. Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat Genet 2011;43:1142–1146. [DOI] [PubMed] [Google Scholar]
- 19.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006;22:195–201. [DOI] [PubMed] [Google Scholar]
- 20.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013;504:107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams P, Afonine P, Bunkóczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindler A, Sumner C, Hoover-Fong JE. TRPV4-associated disorders. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. Seattle, WA: University of Washington; 2014. [Google Scholar]
- 24.Hellwig N, Albrecht N, Harteneck C, Schultz G, Schaefer M. Homo- and heteromeric assembly of TRPV channel subunits. J Cell Sci 2005;118:917–928. [DOI] [PubMed] [Google Scholar]
- 25.Baker D, Sali A. Protein structure prediction and structural genomics. Science 2001;294:93–96. [DOI] [PubMed] [Google Scholar]
- 26.Loukin S, Su Z, Kung C. Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by TRPV4 mutations. PLoS One 2011;6:e19533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mrkonjić S, Garcia-Elias A, Pardo-Pastor C, et al. TRPV4 participates in the establishment of trailing adhesions and directional persistence of migrating cells. Pflugers Arch 2015;467:2107–2196. [DOI] [PubMed] [Google Scholar]
- 28.Fecto F, Shi Y, Huda R, Martina M, Siddique T, Deng HX. Mutant TRPV4-mediated toxicity is linked to increased constitutive function in axonal neuropathies. J Biol Chem 2011;286:17281–17291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenkins NC, Jung J, Liu TT, Wilde M, Holmen SL, Grossman D. Familial melanoma-associated mutations in p16 uncouple its tumor-suppressor functions. J Invest Dermatol 2012;133:1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nekrep N, Geyer M, Jabrane-Ferrat N, Peterlin BM. Analysis of ankyrin repeats reveals how a single point mutation in RFXANK results in bare lymphocyte syndrome. Mol Cell Biol 2001;21:5566–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007;39:25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramot Y, Molho-Pessach V, Meir T, et al. Mutation in KANK2, encoding a sequestering protein for steroid receptor coactivators, causes keratoderma and woolly hair. J Med Genet 2014;51:388–394. [DOI] [PubMed] [Google Scholar]
- 33.Phelps CB, Wang RR, Choo SS, Gaudet R. Differential regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. J Biol Chem 2010;285:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi N, Hamada-Nakahara S, Itoh Y, et al. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P2. Nat Commun 2014;5:4994. [DOI] [PubMed] [Google Scholar]
- 35.Mameza MG, Dvoretskova E, Bamann M, et al. SHANK3 gene mutations associated with autism facilitate ligand binding to the Shank3 ankyrin repeat region. J Biol Chem 2013;288:26697–26708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leddy HA, McNulty AL, Lee SH, et al. Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations. FASEB J 2014;28:2525–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medne L, Bönnemann C, Scherer S, et al. Expanding phenotype of TRPV4 related neuropathies with notable intrafamilial variability. Neuromuscul Disord 2014;24:888–889. [Google Scholar]