

Abstract
Objective:
To determine the genetic variants in patients with dystroglycanopathy (DGP) and assess the pathogenicity of these variants.
Methods:
A total of 20 patients with DGP were identified by immunohistochemistry or Western blot analysis. Whole-exome sequencing (WES) was performed using patient samples. The pathogenicity of the variants identified was evaluated on the basis of the phenotypic recovery in a knockout (KO) haploid human cell line by transfection with mutated POMGNT2 cDNA and on the basis of the in vitro enzymatic activity of mutated proteins.
Results:
WES identified homozygous and compound heterozygous missense variants in POMGNT2 in 3 patients with the milder limb-girdle muscular dystrophy (LGMD) and intellectual disability without brain malformation. The 2 identified variants were located in the putative glycosyltransferase domain of POMGNT2, which affected its enzymatic activity. Mutated POMGNT2 cDNAs failed to rescue the phenotype of POMGNT2-KO cells.
Conclusions:
Novel variants in POMGNT2 are associated with milder forms of LGMD. The findings of this study expand the clinical and pathologic spectrum of DGP associated with POMGNT2 variants from the severest Walker-Warburg syndrome to the mildest LGMD phenotypes. The simple method to verify pathogenesis of variants may allow researchers to evaluate any variants present in all of the known causative genes and the variants in novel candidate genes to detect DGPs, particularly without using patients' specimens.
Defects in the glycosylation of α-dystroglycan lead to a subgroup of muscular dystrophies and brain and eye malformations known as dystroglycanopathies (DGPs).1,2 These diseases exhibit a broad spectrum of severity, ranging from Walker-Warburg syndrome (WWS), muscle-eye-brain disease, and Fukuyama congenital muscular dystrophy to the milder forms of limb-girdle muscular dystrophy (LGMD) and asymptomatic hyperCKemia.3,4 To date, more than 17 causative genes have been identified,5–11 including GTDC2 identified as a cause of WWS,7 which was renamed as POMGNT2 after the elucidation of its enzymatic properties.12
POMGNT2 is an endoplasmic reticulum (ER)–resident protein that catalyzes the second step of the O-mannosyl glycosylation in the mucin-like domain of α-dystroglycan to produce functional laminin-binding glycans.12,13 The high expression levels of human POMGNT2 in brain, muscle, heart, and kidney in fetal as well as adult tissues suggest the importance of this gene during development.7 Three variants (p.Arg158His, p.Trp197*, and p.Arg445*) have been reported in patients with severe WWS.7,14 The mildest forms of muscular dystrophies have been reported in primary DGPs, which involves the mutated DAG1, and in secondary DGPs by mutations in FKRP and FKTN.15–19 In this study, we report 3 patients with milder types of LGMD with or without intellectual disability. We identified novel homozygous or compound heterozygous missense variants in POMGNT2 and demonstrated the pathogenicity of these variants using cell-rescue experiments and in vitro POMGNT assays.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by the ethics committee of the National Center of Neurology and Psychiatry, Japan. All of the participants were enrolled after obtaining their informed consent.
Patient selection.
We selected a cohort of 20 unrelated individuals who had been diagnosed with DGP based on their decreased immunoreactivity to an antibody against the glycoepitope and laminin binding according to Western blotting.20 We confirmed that all 20 patients had no 3-kb retrotransposal insertion in FKTN.21
Whole-exome sequencing.
WES and mapping and alignment of the data to the human genome chromosomal sequence were performed as reported previously.22 The data were filtered according to the following conditions: (1) mutation effect, i.e., splicing, start lost, exon deletion, frame shift, stop gained or lost, nonsynonymous codon change, codon insertion, or deletion; (2) variation frequency <0.01 in any of HapMap, ESP6500, 1000 Genomes Project, and Human Genetic Variation Database (HGVD); and (3) inheritance mode, i.e., homozygous variations, X-linked hemizygous variation, or more than 2 variations in the same gene. Variants were confirmed by Sanger sequencing. The compound heterozygosity of variants in P1 was confirmed by cloning and sequencing the PCR product, which spanned c.268–1138 in the genomic DNA.
Pathogenicity of the variants identified.
To examine the pathogenicity of the variants identified, we analyzed the functional recovery of dystroglycan in a POMGNT2-knockout (KO) haploid human cell line (HAP1) by transfection with lentiviral vectors, pLVSIN-IRES-ZsGreen (Clontech, Mountain View, CA), which harbored the wild-type or mutated human myc-tagged POMGNT2 cDNA as reported previously.15 The POMGNT2-KO HAP1 cells were provided by Thijn R. Brummelkamp, PhD, The Netherlands Cancer Institute, and cultured as reported previously.23 For analyzing the recovery of glycosylation in α-dystroglycan, the POMGNT2-KO HAP1 cell lines were transfected with myc-tagged wild-type or mutated POMGNT2 constructs and pUCV-BSD, and the transfected cells were then selected with blasticidin S. The glycosylation in α-dystroglycan was analyzed by Western blotting and laminin overlay after immunoprecipitation of the dystroglycan complex.20 The immunoprecipitation of the dystroglycan complex from HAP1 cells was performed according to previous reports with slight modifications in which 1% triton buffer and polyclonal antibody against β-dystroglycan were used for protein extraction and precipitation, respectively.24 The antibodies used in this study are listed in table e-1 at Neurology.org/ng. The GT20ADG antibody was provided by Kevin P. Campbell, PhD, University of Iowa Carver College of Medicine.
POMGNT2 assay.
To analyze the localization of POMGNT2, myc-tagged wild-type or mutated human POMGNT2 cDNA was transfected into HeLa cells. To determine the POMNT2 activity, the lysates were prepared from the membrane fractions of HEK293 cells and used on the POMGNT2 activity measurement based on the catalysis of GlcNAc transfer to a synthetic substrate, 4-methylumbelliferyl-α-D-mannose, as described previously.12 Statistical tests were performed using GraphPad Prism (GraphPad Software, La Jolla, CA). Paired data were analyzed using the Student t test. Graphs were plotted showing the mean ± SD.
RESULTS
Identification of POMGNT2 variants by WES.
After WES analysis in a cohort of 20 unrelated patients with DGP, we identified 3 male patients who harbored possible variants in POMGNT2. We hypothesized that recessive mutations were present in these patients and identified the following: 20, 32, and 9 genes with homozygous variations; 14, 15, and 21 genes with compound heterozygous variations; and 8, 6, and 4 genes with X-linked hemizygous variations, in P1, P2, and P3, respectively. In the known causative gene of DGP, we identified 2 heterozygous variants, c.494T>C and c.785C>T in POMGNT2 in P1, and homozygous variants of c.785C>T in P2 and P3. In addition, we identified variations in genes involved in glycosylation and metabolism pathways, i.e., compound heterozygous variations in MAN2B and B3GNTL1 in P2 (data not shown). Variations in muscle disease-causative genes were also identified, i.e., compound heterozygous variations in TTN in P3 (data not shown). However, we did not analyze the pathogenicity of these variations.
All of the POMGNT2 variants identified in the patients were confirmed by Sanger sequencing (figure 1A). After analyzing 24 clones of Escherichia coli based on PCR fragments spanning c.268–1138 in P1, we found that 12 clones possessed only the c.494T>C variant and the other 12 had the c.785C>T variant indicating compound heterozygosity. The variants c.494T>C and c.785C>T are predicted to lead missense substitutions, i.e., p.Met165Thr and p.Pro253Leu, respectively. In silico functional predictions showed that both substitutions would be deleterious in Polyphen2 and SIFT. Both residues are located in the putative glycosyltransferase domain, and they have been highly conserved during evolution (figure 1, B and C).
Figure 1. Genetic variations in POMGNT2.

(A) Electropherograms around the mutation sites in POMGNT2 based on Sanger sequencing. (B) Localizations of the identified mutations (in red) and known mutations (in black) in the domain structures of the POMGNT2 protein. This schema was modified from Ref. 14. (C) Amino acid conservation in the mutation sites among species.
Clinical features.
The clinical information is summarized in table 1.
Table 1.
Clinical, laboratory, and genetic findings
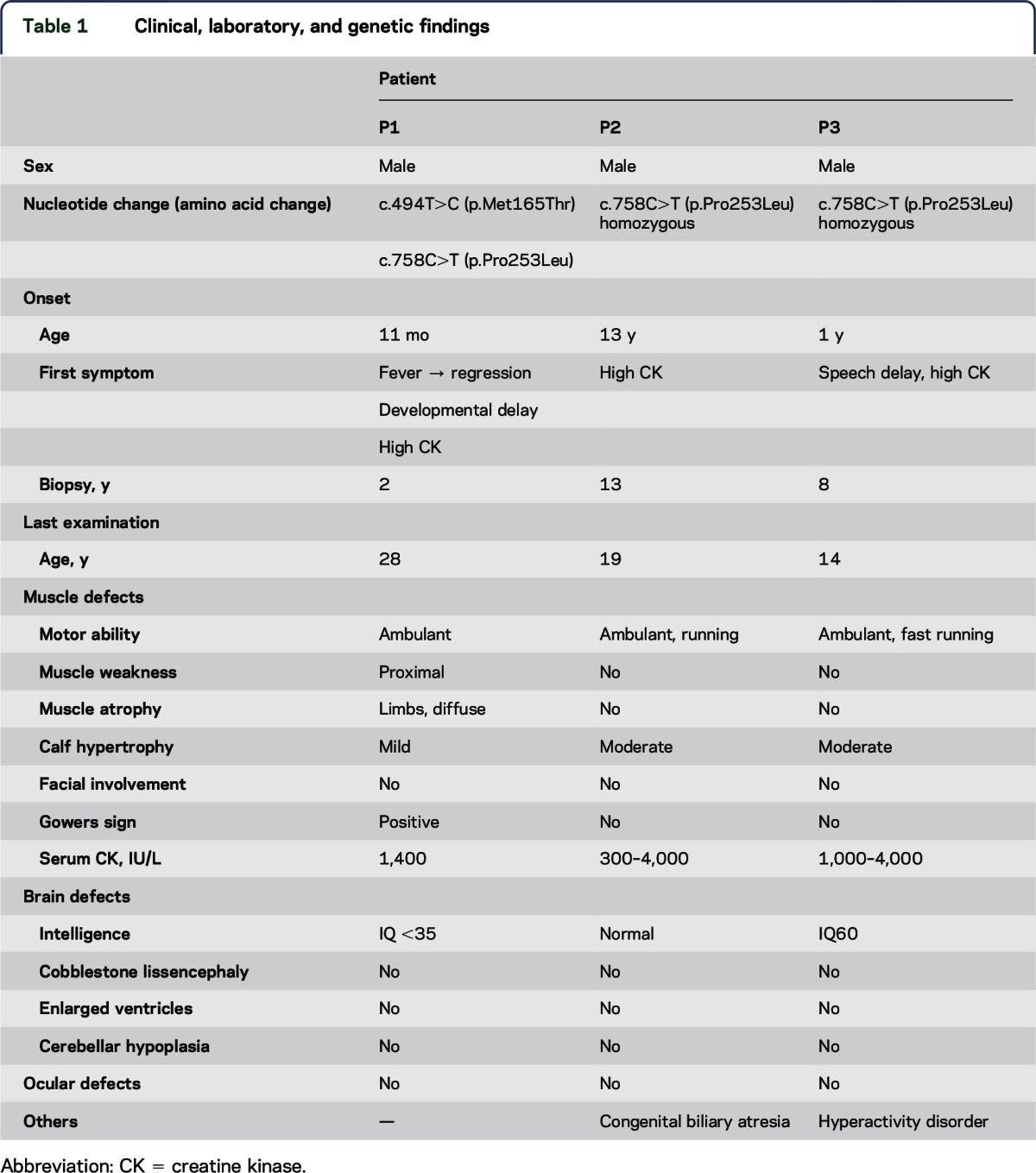
Family history.
All 3 patients were from a nonconsanguineous marriage and had no family history of neuromuscular diseases or intellectual disabilities.
Muscle defects.
P1 was noted to have become delayed in his motor development immediately after recovering from a respiratory tract infection at 11 months. Myositis was diagnosed because of elevated creatine kinase (CK) levels of 3,000 IU/L (reference range: 20–145 IU/L) and biopsy findings as described below. It was not certain whether a screening of antibodies was performed. He was treated with steroids and his motor development improved gradually, although the high CKemia continued. He walked at 2 years, but did not run. When he was a junior high school student, he could walk upstairs and squat from the sitting position. When he was reexamined at age 28 years, he could not walk uphill or downstairs without handrails and had difficulty standing from a squat because of weakness in the proximal lower limbs. His calves were mildly enlarged, which had not been noted at onset. He could raise his arms but not a heavy load. There was no prominent facial weakness or scapular winging. P2 was first found to have a serum CK level of 13,000 IU/L at age 13 years on a follow-up examination concerning congenital biliary atresia. He underwent muscle biopsy, although he had no muscle symptoms, and was diagnosed with DGP. He showed no fatigue or weakness at 19 years, but calf hypertrophy was detected and high CKemia continued. P3 had normal motor milestones and was found by chance to have high CKemia (3,000–9,000 IU/L) at age 4 years. Muscle biopsy was performed at age 8 years and he was diagnosed with DGP. At age 14 years, his calves were moderately enlarged, but the muscle power in his limbs was very strong and his whole-body muscle volume was sufficient.
Intellectual disabilities.
Intellectual development in P1 and P3 was slow. P1 spoke his first few words at 18 months, and he began to communicate using simple words before going to primary school. He could not keep up with classwork, so he went to a special school to support his learning. His IQ was less than 35 at age 28 years. P3 spoke meaningful words with no sentence formation at age 6 years. He had hyperactivity disorder, and Wechsler Intelligence Scale for Children–III testing showed that his IQ was 60 at age 14 years.
Ocular defects and others.
None of the 3 patients had problems with his eyes, heart, or kidneys. P2 had congenital biliary atresia at birth and vitamin K deficiency, which resulted in intracranial hemorrhage. He received a living liver transplantation at age 17 years.
Muscle imaging.
MRI of P1 detected diffuse atrophy in the skeletal muscles, especially in thigh muscles. Muscle images of P2 and P3 detected no atrophy or fatty change in the skeletal muscles (figure 2A).
Figure 2. Muscle imaging and hypoglycosylation of α-dystroglycan.

(A) Images of the thigh and calf muscles of the patients. P1 (at 28 years) exhibited atrophy and mild fatty infiltration in thigh muscles, but the triceps surae muscles were rather normal. P2 (at 19 years) and P3 (at 14 years) had sufficient skeletal muscle volumes and no fatty change. P1 and P3: T1-weighted image; P2: CT image. (B) Histology and immunostaining of skeletal muscle from the patients. The muscle histology in P1 exhibited a muscular dystrophy–like appearance, including necrotic and regenerating fibers, fibers with internal nuclei, and mild endomysial fibrosis, whereas those of P3 were mild and of P2 were almost normal except for some fibers with internal nuclei according to hematoxylin and eosin staining (HE). All muscles were positive for antibodies to β-dystroglycan (β-DG), but negative for the glycoepitope antibody to α-dystroglycan (α-DG VIA4-1). (C) Western blotting with VIA4-1 antibody and the laminin overlay assay of muscle proteins detected 2 reactive bands of α-DG at 150 and 120 kDa, whereas the GT20ADG antibody against the core peptide detected an additional band at 100 kDa. β-DG staining was normal.
Brain imaging and EEG.
Brain MRI showed no abnormality in P1 or P3 (data not shown). CT scanning of P2 detected no anomalies except for aftereffects of intracranial hemorrhage at birth. EEG of P1 at 11 months revealed asymmetric activity in sleep stage, i.e., decreased spindles in the right occipital region. There were no follow-up examination data. P2 had not been examined and P3 had normal EEG results.
Histologic features based on muscle biopsy.
Biopsy of P1 at 11 months detected numerous necrotic fibers, some regenerating fibers, marked cellular infiltration in the endomysium, perimysium, and perivascular regions, and moderate endomysial fibrosis. The muscle histology in P2 at age 13 years was almost normal except for some fibers with internal nuclei. P3 at age 14 years exhibited a muscular dystrophy–like appearance, including necrotic and regenerating fibers, fibers with internal nuclei, and mild endomysial fibrosis. Immunohistochemistry was positive for β-dystroglycan, dystrophin, merosin, and sarcoglycans, but negative for the glycoepitope of α-dystroglycan (figure 2B) in all 3 patients. Immunostaining in P1 was performed at 23 years but not at onset. Western blotting with the glycoepitope antibody and the laminin overlay assay of muscle proteins detected specific patterns with 2-weak reactive bands at 150 and 120 kDa, whereas 3 bands at 150, 120, and 90 kDa were detected with the GT20ADG antibody, which recognizes the polypeptide of α-dystroglycan (figure 2C). β-Dystroglycan was normal.
Subcellular localization of mutated POMGNT2.
The transfected myc-tagged wild-type POMGNT2 protein colocalized with ER proteins (figure 3A). Similarly, the myc-tagged p.Met165Thr- and p.Pro253Leu-substituted POMGNT2 proteins were also localized in the ER.
Figure 3. Subcellular localization and enzymatic activities of mutated POMGNT2.

(A) Myc-tagged wild-type, p.Met165Thr-mutated, and p.Pro253Leu-mutated POMGNT2 cDNAs were transfected into HeLa cells. Both the myc-tagged wild-type and missense mutants of POMGNT2 (c-myc, red) were costained with endoplasmic reticulum (ER) protein, protein disulfide isomerase (PDI, green). Blue indicates the transfected cells with the marker protein, ZsGreen. The bar represents 20 μm. (B) Expression levels of myc-tagged wild-type (wild), Met165Thr-mutated (M165T), and Pro253Leu-mutated (P253L) POMGNT2 in HEK293 cells (arrow). (C) Specific POMGNT activity of wild-type and mutated POMGNT2 expressed in HEK293 cells. The relative POMGNT activity was calculated against that of wild-type POMGNT2. Data obtained from 5 individual measurements are shown as the mean, where the error bars indicate the SD. **p < 0.01 vs wild type according to the Mann-Whitney U test.
POMGNT activities of missense-substituted proteins.
The wild-type and missense-substituted POMGNT2 proteins had similar expression levels in human embryonic kidney cells, thereby indicating that these variations do not affect protein folding and stability (figure 3B). The specific POMGNT activities of both the missense-substituted proteins (p.Met165Thr and p.Pro253Leu) were greatly reduced to less than 10% of that of the wild-type POMGNT2 (figure 3C).
Rescue of POMGNT2-KO HAP1 cells by wild-type and missense POMGNT2 cDNA.
To determine the pathogenetic effects of the 2 missense variants identified in the patients, we transfected lentiviral vectors with wild-type or substituted POMGNT2 cDNAs (p.Met165Thr and p.Pro253Leu) into POMGNT2-KO HAP1 cells, which exhibited defects in their reactivity to the anti-α-dystroglycan antibody, IIH6. The POMGNT2-KO HAP1 cells were rescued by introducing the wild-type cDNA, thereby demonstrating the recovery of the strong immunoreactivity to IIH6, which was similar to that in the wild-type HAP1 cells. By contrast, the introduction of cDNAs with p.Met165Thr and p.Pro253Leu failed to rescue POMGNT2-KO HAP1 cells, which did not stain with the IIH6 antibody (figure 4A).
Figure 4. Mutant POMGNT2 proteins do not rescue hypoglycosylation of α-dystroglycan in POMGNT2-knockout (KO) cells.

(A) IIH6-4C2 staining of wild-type haploid human cell line (HAP1) cells, POMGNT2-KO cells (HAP1-POMGNT2-KO), and POMGNT2-KO cells transfected with wild-type (+WTPOMGNT2), Met165Thr-mutated (+M165T POMGNT2), and Pro253Leu-mutated (+P253L POMGNT2) POMGNT2 cDNA (in red). The transfected cells were positive for ZsGreen expression (in green). HAP1, wild-type haploid cells; HAP1-POMGNT2-KO, POMGNT2-KO HAP1 cells. The scale bar represents 20 μm. (B) Recovery of the glycosylation of α-dystroglycan in POMGNT2-KO HAP1 cells. α-Dystroglycan in POMGNT2-KO HAP1 cells was positive for IIH6 antibody- and laminin-binding after transfection with wild-type (wild) POMGNT2 cDNA, but not after transfection with M165T- and P253L-mutated POMGNT2 cDNA. The 43-kDa αDG band and its degraded products are shown by the arrowhead and asterisk (*), respectively. The immunoglobulin light chain band is indicated by the double asterisk (**).
The wild-type HAP1 cells produced 110-kDa glycosylated α-dystroglycan, which reacts with laminin. By contrast, the POMGNT2-KO HAP1 cells expressed the 90-kDa α-dystroglycan, which is recognized by the peptide-core antibody, GT20DAG, but it reacts negatively with IIH6 and laminin. The introduction of wild-type POMGNT2 cDNAs into POMGNT2-KO HAP1 cells recovered the production of the 110-kDa glycosylated α-dystroglycan, which was able to bind to laminin, whereas the introduction of the p.Met165Thr- or p.Pro253Leu-substituted POMGNT2 cDNAs failed to recover the glycosylation and laminin binding of α-dystroglycan (figure 4B).
DISCUSSION
In this study, we identified 3 patients with DGP, 1 who had compound heterozygous and 2 others who had homozygous variants in POMGNT2. According to the DGP classification,25 P1 had LGMD with mental retardation, whereas P2 and P3 had high CKemia without muscle weakness with or without intellectual disability. The clinical phenotypes of our patients were much milder compared with those of previously reported patients with WWS.7 The pathologic findings were in proportion to the clinical severity. Our findings expand the clinical and pathologic spectrum of DGP associated with POMGNT2 variants from the severest WWS to the mildest LGMD phenotypes.
P1 was believed to have myositis for over 20 years because immunohistochemical analysis was not performed during the first pathologic analysis. P2 was diagnosed as normal based only on a set of routine examinations without immunohistochemistry. Thus, some patients with DGP associated with POMGNT2 mutations with milder phenotypes may have escaped diagnosis, or they could have been diagnosed with myositis or asymptomatic high CK. Our patients experienced no disturbance to their cardiac, respiratory, or renal functions, whereas cardiac involvement has been reported in ambulant patients with DGP.
One of the identified variants, c.785C>T, was found in the ESP6500 database with an allele frequency of 0.000077, but we could not find this variant in HGVD, the genetic variation database for the Japanese population. If we assume that the frequency rate is the same in the Japanese population as ESP6500, then the prevalence of homozygous patients is expected to be 1 in 168 million individuals. However, we found 2 homozygous patients with this variant, although the overall Japanese population is 120 million, so the allele frequency of this variant in the Japanese population may be higher than that in western countries.
As reported previously, the hypoglycosylation levels of α-dystroglycan are not correlated consistently with the clinical severity.26 Nevertheless, our patients exhibited a specific pattern of α-dystroglycan, with 3 bands of different sizes based on Western blotting with a peptide-core recognition antibody. Two larger isoforms also reacted with the glycoepitope antibody and laminin, whereas the smaller one did not, thereby suggesting that there were at least 3 glycoforms of α-dystroglycan in these patients. The remaining laminin-binding abilities of α-dystroglycan may be related to the milder symptoms of the patients, as suggested previously by a DGP model in mice, where smaller amounts of the functional α-dystroglycan were sufficient for maintaining muscle function. Moreover, this specific pattern with 3 bands of α-dystroglycan differs from those in milder forms of DGP with FKTN or DAG1 mutations, which we have reported previously.15,16 Thus, we hypothesize that a reduction, rather than the complete loss, of the enzymatic activities due to missense mutations in FKTN and POMGNT2 may generate intermediate glycoforms of α-dystroglycan. However, it is important that the pattern of α-dystroglycan glycosylation in patients will be determined by the residual function of the affected enzyme. POMGNT2 functions in the ER, where it transfers the second GlcNAc residue during the synthesis of the core M3 chain (GalNAc-β3-GlcNAc-β4-Man-αSer/Thr), which is believed to be a platform structure that is essential for the initiation of heteropolymer “LARGE glycan” synthesis,12,27 whereas FKTN functions in LARGE glycan extension in the Golgi.28,29 Further analyses should clarify the relationship between genetic variants and the glycosylation pattern of residual α-dystroglycan.
Both of the variants that we identified (p.Met165Thr and p.Pro253Leu) were located in the putative glycosyltransferase domain and they were predicted to be deleterious by in silico. Our results showed that these missense variants did not affect the subcellular localization of protein products in the ER,14 and that the PONGNT2 enzymatic activities were greatly reduced, but not lost. These results also suggest that these missense variants may be the cause of milder symptoms in the patients because of the reduced activity of POMGNT2. According to a previous report and the present study, all of the identified missense variants are located in the putative glycosyltransferase domain. Based on comparisons of the patients' symptoms, we suggest that the c.758C>T (p.Pro253Leu) mutation is associated with milder phenotypes in both the muscles and brain, compared with c.494T>C (p.Met165Thr) and other reported mutations, although we found no differences in the in vitro enzymatic activities and glycosylation recovery in the complementation tests between 2 mutated POMGNT2 proteins. Furthermore, P2 and P3 had the same variants and exhibited similar phenotypes in their skeletal muscles, but there were slight differences in their clinical phenotypes in other organs because P2 had congenital biliary atresia but had normal intelligence, whereas P3 exhibited intellectual disability and hyperactivity. However, the congenital biliary atresia in P2 and hyperactivity disorder in P3 may not have been due to the variation in POMGNT2.
In this study, we also performed phenotypic rescue experiments using POMGNT2-KO HAP1 cells based on lentivirus-mediated expression of mutated cDNA in the same manner as DAG1 variants. This method was sensitive in evaluating the pathogenic effects of the identified mutations rapidly and easily, even when the mutations were milder. We extended this mutation-based assay to Western blotting and laminin-overlay analyses to obtain objective and semiquantitative results. This method is technically as simple as immunostaining or Western blotting using muscle samples from patients; therefore, it may allow researchers to evaluate any variants present in all of the known causative genes and the variants in novel candidate genes to detect DGPs, particularly without using patients' specimens.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Nozomi Matsuyama and Kanako Goto for technical support, Thijn R. Brummelkamp for supplying HAP1 cells, and Kevin P. Campbell for supplying the GT20ADG antibody.
GLOSSARY
- CK
creatine kinase
- DGP
dystroglycanopathy
- ER
endoplasmic reticulum
- HAP1
haploid human cell line
- HGVD
Human Genetic Variation Database
- KO
knockout
- LGMD
limb-girdle muscular dystrophy
- WES
whole-exome sequencing
- WWS
Walker-Warburg syndrome
Footnotes
AUTHOR CONTRIBUTIONS
Y.E. made WES pipeline; analyzed and interpreted the data; drafted and edited the manuscript; and performed statistical analysis. M.D. conducted acquisition, analysis, and interpretation of data; drafted and edited the manuscript. S. Noguchi supervised all aspects of this study including study design and data interpretation; drafted and edited the manuscript; and performed statistical analysis. M.O. contributed on technical aspects. Y.K.H. selected patients and performed WES. S.K., K.S., S. Nagai, and S.O. collected clinical information of the patient. I. Nonaka and I. Nishino supervised manuscript preparation and edited the manuscript.
STUDY FUNDING
This study supported by Intramural Research Grant (23-5, 25-5 and 26-8) for Neurological and Psychiatric Disorders of NCNP (to S. Noguchi, Y.K.H., and I. Nishino); grants from Research on Applying Health Technology from the Ministry of Health, Labor, and Welfare (H23-Jitsuyouka (Nanbyou)-Ippan-008) (to Y.K.H. and I. Nishino); grants from Research on rare and intractable diseases from the Ministry of Health, Labour, and Welfare (H26-Itaku (Nan)-Ippan-081) (to S. Noguchi and I. Nishino); and grants from MEXT-Supported Program (S141101) for the Strategic Research Foundation at Private Universities from The Ministry of Education, Culture, Sports, Science and Technology (MEXT) (to Y.K.H.).
DISCLOSURE
Dr. Endo and Dr. Dong report no disclosures. Dr. Noguchi holds patent WO 2010/131712A1 and has received research support from Novartis Pharmaceuticals Japan, the Ministry of Health, Labour, and Welfare. Ms. Ogawa reports no disclosures. Dr. Hayashi has served on the editorial board of Neuromuscular Disorders and has received grants from The Ministry of Education, Culture, Sports, Science and Technology (MEXT), the Ministry of Health, Labor, and Welfare, Japan Society for the Promotion of Science (JSPS), and The Promotion and Mutual Aid Corporation for Private Schools of Japan. Dr. Kuru, Dr. Sugiyama, Dr. Nagai, Dr. Ozasa, and Dr. Nonaka report no disclosures. Dr. Nishino has served on scientific advisory board(s) for Genzyme; has served on the editorial board of Neuromuscular Disorders, Neurology and Clinical Neuroscience, Therapeutic Advances in Neurological Disorders, the Journal of the Neurological Sciences, Skeletal Muscle, and the Journal of Neuromuscular Diseases; holds a patent for Efficacious agents to pathologic condition due to GNE protein dysfunction; and has received research support from Astellas Pharma Inc., Genzyme Japan, Japan Society for the Promotion of Science (JSPS), and the Ministry of Health, Labor, and Welfare. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Hayashi YK, Ogawa M, Tagawa K, et al. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001;57:115–121. [DOI] [PubMed] [Google Scholar]
- 2.Muntoni F, Brockington M, Brown SC. Glycosylation eases muscular dystrophy. Nat Med 2004;10:676–677. [DOI] [PubMed] [Google Scholar]
- 3.Brown SC, Torelli S, Brockington M, et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol 2004;164:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006;9:427–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willer T, Lee H, Lommel M, et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 2012;44:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roscioli T, Kamsteeg EJ, Buysse K, et al. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat Genet 2012;44:581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manzini MC, Tambunan DE, Hill RS, et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet 2012;91:541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carss KJ, Stevens E, Foley AR, et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan. Am J Hum Genet 2013;93:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buysse K, Riemersma M, Powell G, et al. Missense mutations in β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum Mol Genet 2013;22:1746–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaheen R, Faqeih E, Ansari S, Alkuraya FS. A truncating mutation in B3GNT1 causes severe Walker-Warburg syndrome. Neurogenetics 2013;14:243–245. [DOI] [PubMed] [Google Scholar]
- 11.Di Costanzo S, Balasubramanian A, Pond HL, et al. POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum Mol Genet 2014;23:5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida-Moriguchi T, Willer T, Anderson ME, et al. SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 2013;341:896–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida-Moriguchi T, Yu L, Stalnaker SH, et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science 2010;327:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogawa M, Nakamura N, Nakayama Y, et al. GTDC2 modifies O-mannosylated α-dystroglycan in the endoplasmic reticulum to generate N-acetyl glucosamine epitopes reactive with CTD110.6 antibody. Biochem Biophys Res Commun 2013;440:88–93. [DOI] [PubMed] [Google Scholar]
- 15.Dong M, Noguchi S, Endo Y, et al. DAG1 mutations associated with asymptomatic hyperCKemia and hypoglycosylation of α-dystroglycan. Neurology 2015;84:273–279. [DOI] [PubMed] [Google Scholar]
- 16.Murakami T, Hayashi YK, Noguchi S, et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol 2006;60:597–602. [DOI] [PubMed] [Google Scholar]
- 17.de Paula F, Vieira N, Starling A, et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: the other end of the spectrum. Eur J Hum Genet 2003;11:923–930. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez C, de Paula AM, Figarella-Branger D, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006;66:1585–1587. [DOI] [PubMed] [Google Scholar]
- 19.Fiorillo C, Moro F, Astrea G, et al. Novel mutations in the fukutin gene in a boy with asymptomatic hyperCKemia. Neuromuscul Disord 2013;23:1010–1015. [DOI] [PubMed] [Google Scholar]
- 20.Michele DE, Barresi R, Kanagawa M, et al. Posttranslational disruption of dystroglycan–ligand interactions in congenital muscular dystrophies. Nature 2002;418:417–422. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998;394:388–392. [DOI] [PubMed] [Google Scholar]
- 22.Endo Y, Noguchi S, Hara Y, et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2+ channels. Hum Mol Genet 2015;24:637–648. [DOI] [PubMed] [Google Scholar]
- 23.Jae LT, Raaben M, Riemersma M, et al. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 2013;340:479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noguchi S, Wakabayashi E, Imamura M, Yoshida M, Ozawa E. Formation of sarcoglycan complex with differentiation in cultured myocyte. Eur J Biochem 2000;267:640–648. [DOI] [PubMed] [Google Scholar]
- 25.Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130:2725–2735. [DOI] [PubMed] [Google Scholar]
- 26.Jimenez-Mallebrera C, Torelli S, Feng L, et al. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol 2009;19:596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hara Y, Kanagawa M, Kunz S, et al. Like-acetylglucosaminyltransferase (LARGE)-dependent modification of dystroglycan at Thr-317/319 is required for laminin binding and arenavirus infection. Proc Natl Acad Sci U S A 2011;108:17426–17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willer T, Inamori K, Venzke D, et al. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated α-dystroglycan functional glycosylation. Elife 2014;3. 10.7554/eLife.03941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Praissman JL, Live DH, Wang S, et al. B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of α-dystroglycan. Elife 2014;3. 10.7554/eLife.03943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.