

Abstract
Objective:
To identify the genetic cause in a large family with febrile seizures (FS) and temporal lobe epilepsy (TLE) and subsequently search for additional mutations in a cohort of 107 families with FS, with or without epilepsy.
Methods:
The cohort consisted of 1 large family with FS and TLE, 64 smaller French families recruited through a national French campaign, and 43 Italian families. Molecular analyses consisted of whole-exome sequencing and mutational screening.
Results:
Exome sequencing revealed a p.Glu402fs*3 mutation in the γ2 subunit of the GABAA receptor gene (GABRG2) in the large family with FS and TLE. Three additional nonsense and frameshift GABRG2 mutations (p.Arg136*, p.Val462fs*33, and p.Pro59fs*12), 1 missense mutation (p.Met199Val), and 1 exonic deletion were subsequently identified in 5 families of the follow-up cohort.
Conclusions:
We report GABRG2 mutations in 5.6% (6/108) of families with FS, with or without associated epilepsy. This study provides evidence that GABRG2 mutations are linked to the FS phenotype, rather than epilepsy, and that loss-of-function of GABAA receptor γ2 subunit is the probable underlying pathogenic mechanism.
Febrile seizures (FS) are the most frequent convulsive event in early infancy, affecting an estimated 2%–5% of children.1 They usually occur between 6 months and 5 years of age in the presence of fever. The prognosis is usually good, although children who have experienced FS have a higher risk of developing epilepsy later in life. Despite a clear genetic component, the genetics of FS is still poorly understood.2,3 Sodium channel and GABAA receptor gene mutations have been reported in familial syndromes including FS, such as genetic epilepsy with febrile seizures plus (GEFS+).4 Mutations in 3 ion channel genes collectively account for ∼15% of GEFS+ families: SCN1B encoding the voltage-gated sodium channel β1 subunit,5 SCN1A encoding the α1 subunit of the voltage-gated type I sodium channel,6 and GABRG2 encoding the γ2 subunit of the GABAA receptor gene.7 Mutations in the SCN1A gene are the most frequent cause of GEFS+ to date.8–10 Recently, mutations in STX1B, encoding the presynaptic protein syntaxin-1B, were found in 6 families with FS and epilepsy.11 We performed whole-exome sequencing in a large French family with FS and temporal lobe epilepsy (TLE) and identified a pathogenic frameshift mutation in the GABRG2 gene. We subsequently identified GABRG2 mutations in 5 of 107 families with FS.
METHODS
Patients.
The French cohort consisted of 64 families with FS with or without epilepsy. All families were recruited through a national French campaign on genetic studies of familial FS conducted by Généthon in 1998–2000 (J.F. Prud'homme, Evry, France). Inclusion criterion for this cohort was the presence of at least 3 family members with FS (with or without associated epilepsy) with a transmission compatible with an autosomal dominant trait. Family members underwent clinical assessment using a detailed questionnaire. Information was also obtained retrospectively from medical records. Probands of all families were negative for SCN1A and SCN1B point mutations.
The Italian cohort consisted of 43 families with at least 2 first-degree relatives with FS, with or without epilepsy. Patient evaluation included details on clinical features of seizures (semiology, precipitating factors, duration), family and personal history of seizures/epilepsy, and developmental milestones. All the probands were negative for SCN1A mutations.
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained from all participants (or the parents of minors). The study was performed with the approval of the Cochin-Port-Royal Hospital Ethics Committee and the G. Gaslini Institute (No. 003771-35).
Whole-exome sequencing.
Exome sequencing was performed on genomic DNA from peripheral blood of 3 affected members (A/II-1, A/II-4, and A/II-9) of family A. Targeted exome sequencing, library preparation, capture (Human All exon kit V4+UTRs, 70 Mb, Agilent, Interactive Biosoftware, Rouen, France), sequencing, and variant detection and annotation were performed by IntegraGen (Evry, France) using a previously described procedure.12 Variants with low quality (<20) and low VQSLOD (logarithm of odds ratio that a variant is real vs not under the trained gaussian mixture model) (<0) were filtered out. Variants with a read depth >30 and allele read frequency >0.30 were filtered. High-quality variants were further filtered for those predicted to affect the protein-coding sequence (nonsense, missense, frameshift, or essential splice-site) and with a minor allele frequency (<1%) in the exome variant server (http://evs.gs.washington.edu/EVS/).
Mutational screening of GABRG2.
Follow-up screening of GABRG2 was done by Sanger sequencing in the 43 probands of the Italian cohort as previously described.13 Screening in the cohort of 64 probands from French families was performed using a targeted epilepsy gene panel (SeqCapEZ custom design, Roche NimbleGen) including GABRG2, GABRA1, STX1B, PRRT2, CHRNA4, CHRNA2, CHRNB2, LGI1, DEPDC5, KCNT1, and EFHC1. Quality of raw sequencing data was assessed with the FASTQC tool. Reads were trimmed with Trimmomatic. BWA was used to align reads on human genome (hg19). Finally, GATK best practices were applied to call variants. Variants with low quality (<20) were excluded. Variants with a read depth >30 and allele read frequency >0.30 were filtered. We selected heterozygous nonsynonymous variants with frequency <1% in the exome variant server, affecting the protein-coding sequence (nonsense, missense, frameshift, or essential splice-site) and predicted to be damaging by at least 1 of 2 in silico prediction tools (Sift, http://sift.jcvi.org/; PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/). We followed the guidelines for assigning causality of sequence variants in human disease as outlined.14 Coverage of GABRG2 was satisfactory (>250×) except for exon 6, which was subsequently analyzed by traditional Sanger sequencing. Filtered variants were validated by Sanger sequencing, and segregation was analyzed in all available family members. Impact of mutations was assessed with Alamut version 2.4.3 (Interactive Biosoftware). GABRG2 mutations were mapped on the immature peptide, according to the current convention.15,16 We compared the frequency of rare nonsynonymous GABRG2 variants (including missense, nonsense, essential splice-site, in-frame Indels) identified in patients with epilepsy and those present in 33,370 European cases from the publicly available Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org) using Fisher exact tests (accessed July 2015).
Linkage analysis.
Pairwise LOD scores were calculated using the MLINK program of the FASTLINK package version 4.1 assuming an autosomal dominant trait for FS with disease allele frequency of 0.01 and incomplete penetrance of 85%. Because the frequency of FS in the general population is approximately 2%–5%, we used a phenocopy rate of 3% as previously reported.17 We considered as affected all individuals with FS.
CGH array.
CGH array was performed in the 43 probands of the Italian cohort using a custom 105K Agilent CGH array containing 44K backbone genome-wide probes and approximately 60K high-density probes spanning 440 genes encoding for ion channels (list available on request) according to previously described protocols and bioinformatics pipelines.18
RESULTS
Exome sequencing.
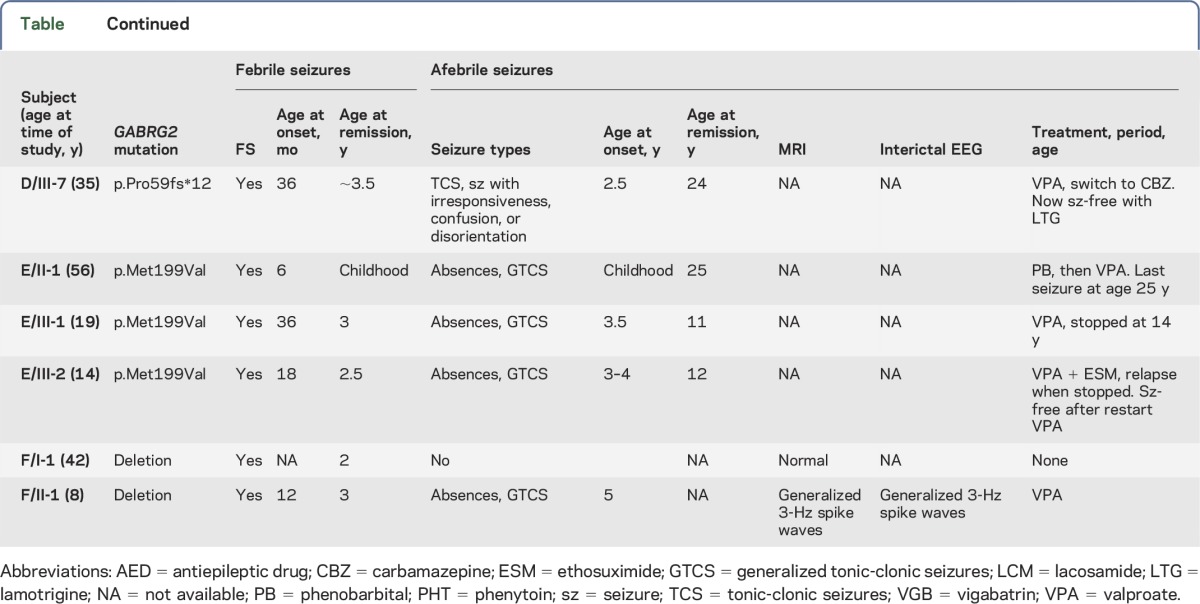
Exome sequencing revealed a pathogenic mutation in GABRG2. Clinical features of family A were described previously.19 This large 5-generation French family included 9 individuals with seizures (table). All 9 patients presented simple FS in early childhood (age at onset range: 12 months to 3 years), 7 of 9 individuals subsequently developed focal epilepsy with EEG and clinical features strongly suggestive of TLE, and 1 patient had only generalized tonic-clonic seizures (GTCS). Brain MRI showed normal hippocampal architecture and volume on both sides in 5 patients. Interictal EEGs showed epileptiform abnormalities localized in the temporal lobe in 6 patients, and was normal in 1 patient (A/II-6) with FS only.
Table.
Clinical features of patients

We performed whole-exome sequencing on 3 affected family members (A/II-1, A/II-4, and A/II-9). Originally, a 20 cM spaced genome-wide microsatellite analysis was suggestive of a locus on chromosome 18qter and a probable second locus on chromosome 1q25-q31 with maximum pairwise LOD scores of 3.04 and 2.33, respectively.19 We first focused the analysis on these 2 regions, then on the rest of the exome dataset. According to our filtering criteria, only 7 variants were common to all 3 siblings, but none of the corresponding genes (ABL2, BOD1L1, B3GALT4, EXO1, HERC4, OR5K1, or SSH2) were functionally candidate for epilepsy or expressed in the brain. We therefore subsequently restricted the analysis to the exome datasets of individuals A/II-1 and A/II-4, who belong to the same branch of the family. We identified a heterozygous deletion/insertion mutation (c.1206delinsTTCAT) in exon 9 of the γ2 subunit of the GABAA receptor gene GABRG2 gene (located on chromosome 5) in both A/II-1 and A/II-4. Sanger sequencing indicated that the p.Glu402fs*3 variant was segregated within all affected family members of the first branch, but was not present in individual A/II-9 (figure 1). The variant created a frameshift at codon Glu402 leading to a new reading frame that ended in a stop codon 2 positions downstream (p.Glu402fs*3). Several arguments pointed toward the pathogenicity of the GABRG2 variant in the family: (1) the GABRG2 gene is known to be implicated in familial epilepsies; (2) this variant disrupts the reading frame leading to a loss-of-function mechanism, as previously reported for other mutations16; and (3) this truncating variant segregated among affected family members (apart from A/II-9). We therefore considered individual A/II-9 as a phenocopy. We reviewed the history of seizures of individual A/II-9, which consisted of few episodes of FS between ages 1 and 4 years. During adolescence, she presented a unique probable focal seizure with loss of consciousness and gestural automatisms and 2 GTCS (1 nocturnal and 1 diurnal), over a period of 6 months (period of emotional stress). We also noted that the mother of individual A/II-9 was asymptomatic, as well as her sister and her children, which confirms that this branch of the family was unlikely to bear the same genetic defect.
Figure 1. Pedigrees of the families with GABRG2 mutation segregation.

Pedigrees of family A (p.Glu402fs*3), family B (p.Arg136*), family C (p.Val462fs*33), family D (p.Pro59fs*12), family E (p.Met199Val), and family F (GABRG2 deletion) are represented. The respective GABRG2 mutations (NM_000816.3) are indicated. Individuals who carry a heterozygous GABRG2 mutation are denoted by m/+, and those negative for mutations are denoted by +/+. Arrows indicate index cases.
Mutational screening of GABRG2 in the follow-up cohort.
Sequencing of GABRG2 was performed in a follow-up cohort of 64 probands from French families and 43 from Italian families with FS with or without epilepsy (table and figure 1). CGH array was performed in the 43 probands of the Italian families.
Family B was a large multigenerational family, comprising 10 affected individuals, among which 7 patients had simple FS occurring between age 13 and 24 months. All patients had normal development. Two half brothers (B/IV-7 and B/IV-9) presented focal seizures in childhood with no history of FS. EEG and clinical features suggested benign rolandic epilepsy. Patient B/IV-5 presented at age 6 years a single brief afebrile GTCS 2 days after a fall while cycling with loss of consciousness of 10–15 minutes, suggesting a possible posttraumatic event. After the fall, her brain CT scan was normal and EEG 1 month after the accident was normal; she received no treatment. Gene panel sequencing identified a heterozygous nonsense mutation (c.406C>T, p.Arg136*, exon 4) in GABRG2 in the index case (B/III-10). Subsequent Sanger sequencing revealed that the p.Arg136* mutation segregated with the FS component and was absent in the 2 individuals with focal seizures without FS (B/IV-7 and B/IV-9) and in individual B/IV-5 with a single GTCS and no FS.
Family C consisted of 5 affected individuals over 4 generations. Four patients (C/II-1, C/III-1, C/IV-1, and C/IV-2) had simple FS with an age at onset between 10 and 14 months. No information was available on the epilepsy type of the deceased patient (C/I-1) of the first generation. FS spontaneously remitted before age 3 years, except in individual C/IV-1, whose FS lasted up to age 8 years, therefore designated as FS+. None of the patients had subsequent epilepsy. Sequencing of the index case (C/III-1) revealed a heterozygous single base deletion (c.1382delG) in exon 9 of GABRG2, also present in his affected sons, but not in his father (C/II-1), which is another example of phenocopy. DNA of his mother was not available for genetic studies to investigate the mode of transmission of the mutation. This variant creates a frameshift, resulting in loss of the last 6 C-terminal amino acids and incorporation of 32 extra amino acids with a final predicted peptide of 493 amino acids instead of 467 (p.Val462fs*33).
Family D comprised 5 patients with FS or epilepsy. Medical reports were not available for review, but telephonic interviews with family members revealed that 3 of them (D/III-1, D/III-5, and D/III-7) had a history of FS, 2 of whom developed subsequent tonic-clonic seizures (TCS) and episodes of loss of consciousness (D/III-1 and D/III-7); 2 other family members had TCS (D/II-2) or TCS and episodes of loss of consciousness, both without a known history of FS (D/II-5). Insufficient information was available to classify the epilepsy in the family as generalized or focal. A c.174_175delinsA (p.Pro59fs*12) mutation in exon 2 of GABRG2 leading to a frameshift starting at codon Pro59 followed by a premature stop codon after 12 amino acids segregated in the 3 patients whose DNA was available.
Family E comprised a father and 2 daughters with FS, followed by early-onset absence seizures and GTCS. Development was normal in all 3 family members. Despite a high seizure frequency, especially in the father, all became seizure-free with valproate. A missense variant, c.595A>G/p.Met199Val in exon 5 of GABRG2, was found in the 3 patients. The variant was predicted deleterious by Sift, disease-causing by MutationTaster, and probably damaging by PolyPhen-2. The p.Met199Val mutation is located in the N-terminal extracellular loop of the protein (figure 2).
Figure 2. Schematic representation of the γ2 subunit of the GABAA receptor with the position of all reported point mutations.
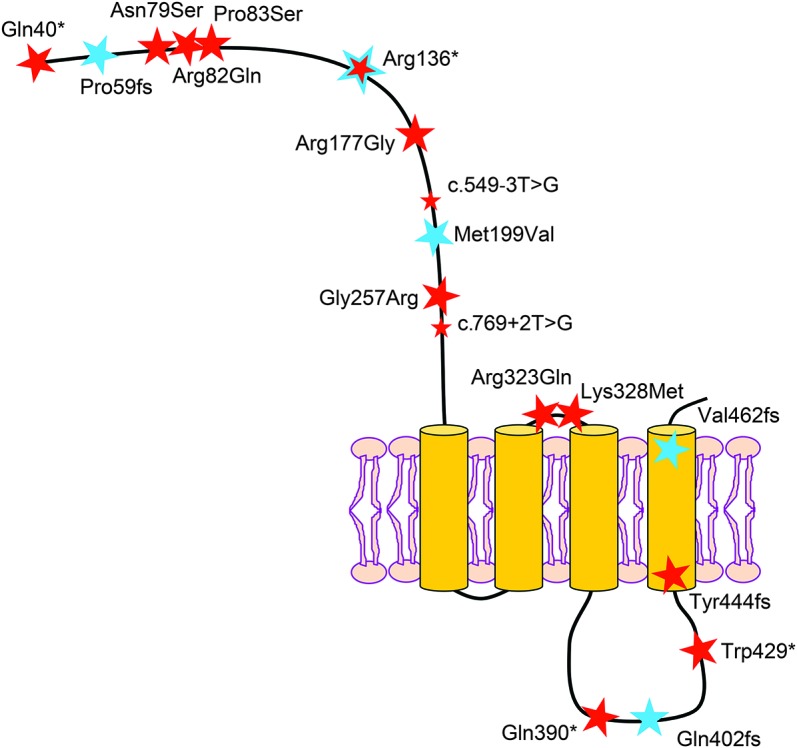
Disease-causing point mutations in GABRG2 include 8 missense mutations,7,22,24,26,27,30,31 4 nonsense mutations,20,23,29,41 4 frameshifts,25 and 2 splice-sites.28,31 Mutations indicated by a blue star were identified in this study; those indicated by a red star were previously reported. All mutations are reported on the immature peptide. Protein structure was designed according to UniProt database information (NP_000807.2).
An intragenic deletion in the proband of Italian family F and his affected mother was identified at 5q34 spanning 145K at chr5, 160.631.665–161.067.144, and affecting coding exons 1–4 of GABRG2. The proband (F/II-1) was an 8-year-old boy who had recurrent episodes of simple FS between ages 12 and 36 months, which remitted spontaneously. From age 5 years, he developed absence seizures and 2 GTCS, successfully treated with valproate. His mother (F/I-1) had 4 episodes of simple FS during her first 2 years of life. Both individuals had normal cognition. No additional affected relatives were reported.
Gene panel sequencing performed in the probands of family B, C, D, and E did not identify other rare variants in known epilepsy genes targeted in our panel. None of the GABRG2 mutations of this study were reported in the Exome Aggregation Consortium (ExAC) database, which includes the 1000 Genomes project database and the 6,503 exomes listed in the exome variant server. Although we acknowledge that the p value might be overestimated because of a lower coverage of exons 3 and 6 in ExAC control individuals, we observed a significant enrichment of rare nonsynonymous GABRG2 variants in our cohort of patients (6/108, 5.6%) compared with individuals of European ancestry from the ExAC browser (73/33,370 (0.22%), p < 2.2 × 10−7). To further demonstrate the causality of the GABRG2 variants in the FS phenotype by linkage analysis, we calculated 2-point LOD scores in the 6 families. The sum of the maximal pairwise LOD scores was 3.02, indicating significant likelihood that the GABRG2 variants are causal.
Four variants (p.Glu402fs*3, p.Arg136*, p.Val462fs*33, and p.Pro59fs*12) were securely considered pathogenic because they introduced an erroneous stop codon in the reading frame. While p.Arg136* and p.Pro59fs*12 were predicted to be degraded by the nonsense-mediated-decay system (NMD), p.Glu402fs*3 and p.Val462fs*33, located in the last exon of GABRG2, may escape the NMD and lead to a truncated or a longer γ2 subunit, respectively. The p.Met199Val missense variant was likely to be pathogenic according to 3 in silico prediction tools. All mutations were novel except p.Arg136*, which was previously reported in a family with GEFS+, learning difficulties, and autism spectrum disorder, although it was segregated mostly with the FS component.20
DISCUSSION
GABAA receptors mediate fast inhibitory synaptic transmission in the CNS. GABAA receptors are heteropentameric ligand-gated chloride channels combining 5 of 19 different subunits. The most common configuration in mammals requires 1 γ2 subunit in association with 2 α1-3 and 2 β2/3 subunits.21
Up to now, mutations in the GABRG2 gene, encoding the GABAA receptor γ2 subunit, have been reported in a subset of families, but their prevalence remained unknown. In the literature, individuals with GABRG2 mutations presented a variety of phenotypes ranging from pure FS,22 FS+ or FS and generalized epilepsy (absence seizures or GTCS),7,20,23–29 GTCS without FS,30 FS and rolandic epilepsy,31 to possibly Dravet syndrome (a nonsense mutation was reported in dizygotic twins with Dravet syndrome and their healthy father).32
In this study, we assessed the frequency of GABRG2 mutations in a large cohort of families with FS and reported pathogenic mutations in 6/108 (5.6%) families. First, we identified a GABRG2 disease-causing mutation (p.Glu402fs*3) in a multiplex family with FS and TLE, in which linkage analysis previously suggested a possible digenic inheritance on 1q25-q31 and 18qter.19 However, with the finding of a clear pathogenic mutation in GABRG2, leading to the introduction of a premature stop codon (p.Glu402fs*3), both loci were unlikely to cause the phenotype. This study provides a clear example of the limitation of linkage analysis and how phenocopies can distort linkage results (1 affected member of the large family did not carry the GABRG2 mutation). Unbiased next-generation sequencing approaches will undoubtedly unveil additional erroneous genetic claims in the literature. In the follow-up cohort consisting of 107 families with FS (with or without subsequent epilepsy), we identified 2 other frameshift mutations (p.Val462fs*33 and p.Pro59fs*12), 1 nonsense mutation (p.Arg136*), 1 missense mutation (p.Met199Val), and 1 deletion (exons 1–4) in the GABRG2 gene. Most patients carrying a GABRG2 mutation had a reported history of FS (22/23), associated with TLE in 1 family (family A), generalized epilepsy with absences and GTCS in 2 families (families E and F), and undefined epilepsy with TCS and episodes of loss of consciousness in 1 family (family D). GABRG2 mutations segregated predominantly with FS, indicating that GABRG2 mutations are drivers of the FS phenotype, rather than epilepsy. As expected with common traits, several affected family members did not carry a GABRG2 mutation: 3 family members without a history of FS (B/IV-5, B/IV-7, and B/IV-9) and 2 phenocopies (individuals A/II-9 and C/II-1). Overall, the penetrance of GABRG2 mutations was high in our cohort, with only one asymptomatic carrier (individual A/I-2). It should be noted though that a history of FS may have been underreported in this generation. The relatively high frequency (5.6%) of GABRG2 mutations compared with previously published data24,28,30 is most probably linked to the fact that FS segregating as an autosomal dominant trait was a study inclusion criterion. Additional genetic or epigenetic modifiers probably determine the associated epilepsy component, which can result either in no subsequent epilepsy or in the development of both generalized and focal seizures. Consistent with this hypothesis, 2 Gabrg2 knock-in (p.Arg82Gln) mouse models established in 2 different mouse strains exhibited similar temperature-seizure susceptibilities, mimicking the FS phenotype, whereas the epilepsy phenotype was only present in 1 strain.33 This suggested that FS are less sensitive to genetic background than the associated epileptic phenotypes. An emerging question is how to explain the link between FS and GABAergic defects. Reports have shown that the GEFS+-causing mutation p.Lys328Met affects the membrane dynamics and postsynaptic aggregation of the GABAA mutant receptor in conditions of increased temperature, confirming the susceptibility to temperature.6,34,35
Currently, a total of 19 different GABRG2 disease-causing mutations have been reported (including this study), consisting of 8 missense, 4 nonsense, 4 frameshifts, 2 splice-sites, and 1 deletion (figure 2). The high rate of truncating mutations (11/19, 57%), strengthened by this study, strongly suggests that haploinsufficiency resulting in loss of function of GABAA receptor γ2 subunit underlies GABRG2-related seizure phenotypes. This is concordant with numerous in vitro functional assays showing that GABRG2 mutations resulted in mRNA degradation by NMD, the translation of proteins retained in the endoplasmic reticulum with impaired surface-expression, or dysfunctional channel properties of GABAA receptors20,25,31,32,36–38 (for review, see references 15 and 16). However, 2 recent in vivo studies using Gln390*-Gabrg2 knock-in mice showed that dominant-negative effects on wild-type γ2 and partner subunits also certainly participated to reduce GABA-evoked currents.33,39 A reduction in cortical synaptic GABAergic inhibition was confirmed to cause seizures in another p.Arg82Gln-Gabrg2 knock-in mouse model.40 Nevertheless, the link between dysfunctional GABAA receptors and seizures is still poorly understood.
Mutations in GABRG2 are more frequent than initially reported, and screening of the gene should be included when considering genetic testing in families with an overall mild phenotype consisting of FS with or without epilepsy.
ACKNOWLEDGMENT
The authors thank the patients and families for their participation in the study and the sequencing platform of ICM (Y. Marie), the bioinformatics/biostatistics core facility of ICM (J. Guegan), the DNA and cell bank of ICM (C. Dussert and S. Forlani) and Genethon (J. Debusne) for providing technical assistance, and J. Lemke for discussion.
GLOSSARY
- FS
febrile seizures
- GEFS+
genetic epilepsy with febrile seizures plus
- GTCS
generalized tonic-clonic seizures
- NMD
nonsense-mediated-decay system
- TCS
tonic-clonic seizures
- TLE
temporal lobe epilepsy
AUTHOR CONTRIBUTIONS
M. Boillot: drafting the manuscript, analysis and interpretation of genetic data. M. Morin-Brureau: acquisition, analysis, and interpretation of exome data. F. Picard: acquisition and interpretation of clinical data. S. Weckhuysen: interpretation of clinical and genetic data, drafting the manuscript. V. Lambrecq: acquisition and interpretation of clinical data. C. Minetti: acquisition of clinical data. P. Striano: acquisition and interpretation of clinical and genetic data. F. Zara: acquisition and interpretation of clinical and genetic data. M. Iacomino: acquisition and interpretation of genetic data. S. Ishida: acquisition of genetic data. I. An-Gourfinkel: acquisition and interpretation of clinical data. M. Daniau: acquisition of genetic data. K. Hardies: LOD score calculation, M. Baulac: supervising the acquisition and interpretation of clinical data. O. Dulac: supervising the acquisition and interpretation of clinical data. E. Leguern: study design. R. Nabbout: acquisition and interpretation of clinical data and study design. S. Baulac: analysis and interpretation of genetic data, study supervision, drafting the manuscript and obtaining funding. All authors critically reviewed and approved the final manuscript.
STUDY FUNDING
Supported by the French program “Investissements d'avenir” (ANR-10-IAIHU-06), the Fondation Française pour la Recherche sur l'Epilepsie (FFRE), and the Association de Recherche sur la Génétique des Epilepsies (ARGE).
DISCLOSURE
M. Boillot has received research support from the Ligue Française contre l'Epilepsie (LFCE). M. Morin-Brureau reports no disclosures. F. Picard has received a grant from the Swiss National Foundation (No. 320030-127608). S. Weckhuysen has received research support from the French program “Investissements d'avenir” (ANR-10-IAIHU-06). V. Lambrecq has received research support from the French program “Investissements d'avenir” (ANR-10-IAIHU-06). C. Minetti reports no disclosures. P. Striano has received research support from the Italian ministry of health (project GR-2009–1473821). F. Zara and M. Iacomino report no disclosures. S. Ishida has received research support from the Japan Society for the Promotion of Science (JSPS). I. An-Gourfinkel and M. Daniau report no disclosures. K. Hardies has been a PhD fellow of the Institute for Science and Technology (IWT)-Flanders. M. Baulac has served on the scientific advisory boards of UCB, Eisai, GSK, SAGE, GW Pharmaceuticals; has received travel support and/or honoraria from UCB and Eisai GSK; and has been a consultant for UCB, Eisai, and GSK. O. Dulac reports no disclosures. E. Leguern has received research support from Program “Investissements d'avenir” ANR-10-IAIHU-0, INSERM, and Assistance Publique des Hôpitaux de Paris. R. Nabbout has served on the scientific advisory board for Novartis; has received travel support from Shire and speaker honoraria from Novartis, Cyberonics, and Eisai; and has served on the editorial board for Epilepsia. S. Baulac has received research support from Program “Investissements d'avenir” ANR-10-IAIHU-06 and Carnot Institute (No. CT010). Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976;295:1029–1033. [DOI] [PubMed] [Google Scholar]
- 2.Johnson WG, Kugler SL, Stenroos ES, et al. Pedigree analysis in families with febrile seizures. Am J Med Genet 1996;61:345–352. [DOI] [PubMed] [Google Scholar]
- 3.Saghazadeh A, Mastrangelo M, Rezaei N. Genetic background of febrile seizures. Rev Neurosci 2014;25:129–161. [DOI] [PubMed] [Google Scholar]
- 4.Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev 2009;31:394–400. [DOI] [PubMed] [Google Scholar]
- 5.Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet 1998;19:366–370. [DOI] [PubMed] [Google Scholar]
- 6.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000;24:343–345. [DOI] [PubMed] [Google Scholar]
- 7.Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 2001;28:46–48. [DOI] [PubMed] [Google Scholar]
- 8.Baulac S, Gourfinkel-An I, Nabbout R, Huberfeld G, Serratosa J, Leguern E. Fever, genes, and epilepsy. Lancet Neurol 2004;3:421–430. [DOI] [PubMed] [Google Scholar]
- 9.Audenaert D, Van Broeckhoven C, De Jonghe P. Genes and loci involved in febrile seizures and related epilepsy syndromes. Hum Mutat 2006;27:391–401. [DOI] [PubMed] [Google Scholar]
- 10.Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 2010;51:1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet 2014;46:1327–1332. [DOI] [PubMed] [Google Scholar]
- 12.Ishida S, Picard F, Rudolf G, Noé E, Achaz G, Thomas P. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45:552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madia F, Gennaro E, Cecconi M, Buti D, Capovilla G, Dalla Bernardina B. No evidence of GABRG2 mutations in severe myoclonic epilepsy of infancy. Epilepsy Res 2003;53:196–200. [DOI] [PubMed] [Google Scholar]
- 14.MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR. Guidelines for investigating causality of sequence variants in human disease. Nature 2014;508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macdonald RL, Kang JQ, Gallagher MJ. GABAA receptor subunit mutations and genetic epilepsies. In: Noebels JL, Avoli M, Rogawski MA, et al., eds. Jasper's Basic Mechanisms of the Epilepsies, 4th ed Bethesda: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- 16.Hirose S. Mutant GABA(A) receptor subunits in genetic (idiopathic) epilepsy. Prog Brain Res 2014;213:55–85. [DOI] [PubMed] [Google Scholar]
- 17.Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M, Prud'homme JF, Baulac M. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet 1999;65:1078–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Striano P, Coppola A, Paravidino R, Malacarne M, Gimelli S, Robbiano A. Clinical significance of rare copy number variations in epilepsy: a case-control survey using microarray-based comparative genomic hybridization. Arch Neurol 2012;69:322–330. [DOI] [PubMed] [Google Scholar]
- 19.Baulac S, Picard F, Herman A, Feingold J, Genin E, Hirsch E. Evidence for digenic inheritance in a family with both febrile convulsions and temporal lobe epilepsy implicating chromosomes 18qter and 1q25-q31. Ann Neurol 2001;49:786–792. [DOI] [PubMed] [Google Scholar]
- 20.Johnston AJ, Kang JQ, Shen W, Pickrell WO, Cushion TD, Davies JS. A novel GABRG2 mutation, p.R136*, in a family with GEFS+ and extended phenotypes. Neurobiol Dis 2014;64:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 2005;6:215–229. [DOI] [PubMed] [Google Scholar]
- 22.Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006;67:687–690. [DOI] [PubMed] [Google Scholar]
- 23.Sun H, Zhang Y, Liang J, Liu X, Ma X, Wu H. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J Hum Genet 2008;53:769–774. [DOI] [PubMed] [Google Scholar]
- 24.Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H. Novel alpha1 and gamma2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci 2011;34:237–249. [DOI] [PubMed] [Google Scholar]
- 25.Tian M, Mei D, Freri E, Hernandez CC, Granata T, Shen W. Impaired surface alphabetagamma GABA(A) receptor expression in familial epilepsy due to a GABRG2 frameshift mutation. Neurobiol Dis 2013;50:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 2001;28:49–52. [DOI] [PubMed] [Google Scholar]
- 28.Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol 2002;59:1137–1141. [DOI] [PubMed] [Google Scholar]
- 29.Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet 2002;70:530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi X, Huang MC, Ishii A, Yoshida S, Okada M, Morita K. Mutational analysis of GABRG2 in a Japanese cohort with childhood epilepsies. J Hum Genet 2010;55:375–378. [DOI] [PubMed] [Google Scholar]
- 31.Reinthaler EM, Dejanovic B, Lal D, Semtner M, Merkler Y, Reinhold A. Rare variants in gamma-aminobutyric acid type A receptor genes in rolandic epilepsy and related syndromes. Ann Neurol 2015;77:972–986. [DOI] [PubMed] [Google Scholar]
- 32.Ishii A, Kanaumi T, Sohda M, Misumi Y, Zhang B, Kakinuma N. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res 2014;108:420–432. [DOI] [PubMed] [Google Scholar]
- 33.Reid CA, Kim T, Phillips AM, Low J, Berkovic SF, Luscher B. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology 2013;80:1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eugene E, Depienne C, Baulac S, Baulac M, Fritschy JM, Le Guern E. GABA(A) receptor gamma 2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J Neurosci 2007;27:14108–14116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouthour W, Leroy F, Emmanuelli C, Carnaud M, Dahan M, Poncer JC. A human mutation in Gabrg2 associated with generalized epilepsy alters the membrane dynamics of GABAA receptors. Cereb Cortex 2012;22:1542–1553. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Tian M, Hernandez CC. The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol Dis 2012;48:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang JQ, Shen W, Macdonald RL. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J Neurosci 2009;29:2845–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang JQ, Shen W, Macdonald RL. Trafficking-deficient mutant GABRG2 subunit amount may modify epilepsy phenotype. Ann Neurol 2013;74:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang JQ, Shen W, Zhou C, Xu D, Macdonald RL. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat Neuroscience 2015;18:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A 2007;104:17536–17541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res 2006;70(suppl 1):S206–S217. [DOI] [PubMed] [Google Scholar]